

Abstract
The serine/threonine phosphatase calcineurin (CaN) is a unique but confounding calcium/calmodulin-mediated enzyme. CaN has shown to play essential roles from regulating calcium homeostasis to being an intricate part of learning and memory formation. Neurological disorders, despite differing in their etiology, share similar pathological outcomes, such as mitochondrial dysfunction and apoptotic signaling brought about by excitotoxic elements. CaN, being deeply integrated in vital neuronal functions, may be implicated in various neurological disorders. Understanding the enzyme and its physiological niche in the nervous system is vital in uncovering its roles in the spectrum of brain disorders. By reviewing the crosstalk in different neurological pathologies, a possible grasp of CaN’s complex signaling may lead to forming better neurotherapy. This Outlook attempts to explore the various neuronal functions of CaN and investigate its pervasive role through the gamut of neurological disorders.
Short abstract
Calcineurin has vital biological functions with significant roles across different neuropathies. Herein, we present its involvement under varying neuropathological conditions.
1. Calcineurin: A Prologue
Even 40 years after its initial discovery by Wang and Desai as a column fraction, calcineurin (CaN) continues to intrigue the scientific community with its vast biological role.1 In its initial findings, CaN was often referred to as an “affinity-purified phosphodiesterase”.2 Klee and Krinks were the first to purify CaN and presumed it to be a regulatory subunit of a phosphodiesterase based on its ability to inhibit the same.3 Wallace and Lynch further purified CaN from bovine brain extracts using a two-column approach and found its activity to hinder the adenylate cyclase and nucleotide phosphodiesterases.4,5 Due to similarity in biological activity traced by various research groups, it was therefore speculated that CaN may possibly be a regulatory enzyme. Its abundant allocation in the nervous system prompted Klee et al. to coin the term “calcineurin”.6 Klee’s version of CaN was corroborated by the work of Philip Cohen in which he found a fraction in cellular extracts that dephosphorylated the α-/β-subunits of phosphorylase kinase; the fraction revealed to be nearly identical to the CaN discovered by Klee.7,8
As per consensus, CaN is deemed as a calcium/calmodulin-dependent (Ca2+/CaM) serine-threonine phosphatase, also known as protein phosphatase 2B (PP2B), ubiquitous in most mammalian tissues but predominating in the brain.9 It is the only reported phosphatase thus far known to be completely dependent on CaM. From the initial presumption as being a calmodulin-dependent cyclic nucleotide phosphodiesterase inhibitor to the radical revelation of being the target of immunosuppressant drugs such as cyclosporin A in organ transplants, CaN seems to manifest itself in key biological pathways throughout various tissues in the body and necessitates a deeper investigation in its intricate physiological function.10 Its prevalence in the nervous tissue has recently made it a target of investigation for many neurological disorders, particularly those constituted by calcium excitotoxicity and neurotransmitter imbalance.11,12 It is believed that CaN plays a major role in bridging Ca2+ homeostasis to brain function; studies even point to CaN being the link between aging and brain dysregulation.13
Though discussing the vast role of CaN in all the tissues is far beyond the realm of this Outlook, an attempt shall be made to delve into the CaN’s role in the brain and across the spectrum of neurological disorders such as stroke, Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative disorders involving CaN. This crosstalk may clarify the extent of CaN’s involvement in these perplexing disorders.
2. Calcineurin: Classification and Structure
The classification of serine/threonine phosphatases was established by Cohen in which he proposed two categories: type 1 and type 2 protein phosphatases (PPs).14 Type 1 PPs dephosphorylate the β-subunit of phosphorylase kinase while type 2 act on the α-subunit. The two PPs can be distinguished by their inhibition study in which phosphopeptide inhibitors act on type 1 but are unable to inhibit type 2 PPs. Cohen also later subclassified type 2 PPs according to the dependence on divalent ions for activity.15 He designated PP2A, PP2B, and PP2C as having no requirement for divalent ions, requiring Ca2+ (CaN), and requiring Mg2+ for activity, respectively. Evolutionarily speaking, PP1, PP2A, and PP2B (CaN) are some of the most highly conserved PP families throughout nature.16
Human CaN is a heterodimeric serine/threonine phosphatase, comprising a unique structure composed of two subunits connected jointly: a 61 kDa catalytic A subunit (though variations between species ranges from 58 to 64 kDa) and a 19 kDa regulatory B subunit.17 The B subunit, although smaller in size, consists of a helical B-binding segment, a calmodulin-binding (CaM-binding) segment, and an autoinhibitory domain.18 The B-binding segment tightly adheres to the A subunit. Half of CaN appears to be cytosolic while the amino terminal glycine of the B subunit is believed to be myristoylated for greater association with the plasma membrane.19 Furthermore, the B segment consists of four Ca2+-binding sites (EF-motifs).20,21 Two Ca2+ sites reside on the N-terminal and have lower affinity (dissociation constant in the micromolar range). These seemingly serve as sensors for the fluctuations in intracellular Ca2+ levels and thereby serve a regulatory role, especially for the binding of CaM to the CaM-binding segment of subunit B. Another two Ca2+ sites reside on the C-terminal of the B subunit. These have much higher affinity (have dissociation constants in the nanomolar range) and act as stabilizers to keep both subunits intact and confer structural stability to the entirety of CaN.22 The CaM-binding segment relies on Ca2+ to help associate with subunit A, the catalytic site. This structure therefore stands to reason how CaN is able to couple Ca2+ signals to cellular responses. However, the enzyme undergoes complicated conformations to reach the active state. In previous studies, the conformational shift from inactive to active enzymatic states raised several questions. After all, the autoinhibitory site of subunit B (residues 467–486) lies at a relative distance from the B-binding site (341–372) and the CaM-binding segment (391–414). These questions were answered through studies done by Shen et al. in which the group discovered there are a separate sequence of conformational shifts that eventually lead to the fully activated enzyme.23 They found that, during elevated levels, Ca2+ initially binds to the lower-affinity Ca2+ sites, thereby causing a conformation which exposes the CaM-binding site. This fulfills subunit B’s main role as a regulator. When CaM binds to its respective site, another conformational change stabilizes subunit B by releasing the autoinhibitory peptide and renders the enzyme fully active. In summary, when the enzyme is inactive (resting Ca2+ levels), influx (micromolar) of Ca2+ binding to subunit B causes a conformational change to reveal the CaM-binding site on subunit A (partial activation), which upon binding to CaM displaces the autoinhibitory peptide and stabilizes and fully activates the enzyme (see Figure 1).
Figure 1.

Activation of calcineurin (CaN). The phosphatase is activated through the sequence of two subsequent conformations. Initially, the influx of calcium (Ca2+) raises the intracellular calcium load. At the nanomolar range, Ca2+ first binds to the two higher-affinity EF motifs (Hx) of subunit B, tightly adhering subunit B to subunit A and stabilizing the enzyme. As Ca2+ levels rise to the micromolecular threshold, it binds to the lower-affinity EF motifs (Lx), spurring a conformational change that unveils the calmodulin (CaM) site to which CaM binds and initiates a second conformational change responsible for displacing the autoinhibitory peptide. The final conformation stabilizes and fully renders CaN active.
3. Calcineurin: Vital Homeostatic Mechanisms
3.1. Ca2+ Homeostasis
As CaN is ubiquitous throughout the body (several isoforms exist among different tissues), its roles are accordingly highly diversified. However, this Outlook will focus on CaN’s roles solely in the brain, where the highest levels can be found.24 CaN is known to constitute slightly more than 1% of total protein in the brain,25 and it is most densely populated in the caudate putamen and hippocampus but distributed throughout other regions of the brain.26 CaN is also found at the level of dendrites, axons, neuronal spines, presynaptic terminals, and postsynaptic densities. Magnifying the grandeur of CaN’s role in neuronal homeostasis is its very existence as the sole phosphatase heavily dependent on Ca2+/CaM signaling. It is well-established that calcium and its various calcium channels play a major and intricate role in neuronal function and activity. Calcium is arguably one of the most important secondary messengers capable of initiating pivotal cellular events.27 Calcium plays an essential role in many biological pathways that may be operating simultaneously within the neuron; drastic changes in its levels are associated with numerous neurological disorders.28,29 As Ca2+ relays synaptic signals to the nucleus then to synapse again, it is immensely responsible for turning on and off the various genetic expressions necessary for normal neuronal activity.30 Therefore, dysregulation (excess and deficient Ca2+ stimulation) in the Ca2+ synapse-to-nucleus signaling can cause breakdown in essential gene expressions that underlie most neurological pathologies, both degenerative and psychiatric in nature.
CaN is an integral member of the Ca2+ signaling pathway on both sides of the synapse.31 It behaves as a sensor and is vigilant of the intracellular levels of Ca2+ and maintains the proper equilibria necessary for cell maintenance and operation. There are several mechanisms CaN deploys to regulate intracellular Ca2+ levels and the downstream activities that maintain an optimum neuronal microenvironment.
3.2. Synaptic Exocytosis
Presynaptically, CaN and cyclin-dependent kinase 5 (CDK5) oversee vesicular release of neurotransmitters and prevent excessive exocytosis to maintain an adequate amount of neurotransmitters reaching the postsynaptic sites.32 These exocytotic processes require an ensemble of proteins that coordinate action potential and Ca2+ signaling to neurotransmitter release. CDK5 is able to phosphorylate proteins, like synapsin 1 which attaches to synaptic vesicles and sequesters them to the release-reluctant resting pool.33 Studies have shown that CDK5 knockout accelerates synaptic cycling and exocytotic kinetics; similarly, CaN knockout has shown to slow down exocytosis rates.34 CaN counteracts this effect, dephosphorylates synapsin 1, and is able to reverse the suppression of exocytosis and synaptic cycling induced by CDK5.35 Therefore, during greater Ca2+ influx, CaN is able to increase the amount of neurotransmitters released into the synaptic cleft36 (see Figure 2a).
Figure 2.

(a) Presynaptic homeostatic functions of calcineurin (CaN). (A) CaN plays an integral role in vesicle exocytosis by regulating the release in coordination with neurotransmission signaling. The influx of calcium (Ca2+) activates CaN and causes it to dephosphorylate synapsin 1, removing its suppression of vesicles and allowing them to move from the release-reluctant resting pool toward the axonal terminal for release. The process is reversed by the phosphorylation by cyclin-dependent kinase 5 (CDK5). (B) CaN also plays a role in endocytosis of vesicles after activation by Ca2+ and then dephosphorylation of dynamin 1. CaN and dynamin 1 form a complex and move toward the endocytotic packaging assembly where involved proteins allow for the packaging of the vesicle at the axonal terminal, its uptake, and delivery of the vesicle toward the vesicle pool for recycling. This series of synaptic cycling and recycling by CaN is tightly coordinated and coupled to the Ca2+-dependent signaling along with the crosstalk of involved proteins. Interruptions in the processes can disturb the equilibria of neurotransmission and often lead to neuronal impair. (b) Postsynaptic homeostatic functions of calcineurin (CaN). Postsynaptically, CaN tightly regulates several functions. It controls intracellular Ca2+ levels through modulation of voltage-gated calcium channels (VGCCs), NMDA (N-methly-d-aspartate), and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors. Influx of Ca2+ through VGCCs tends to trigger the release of more Ca2+ from the endoplasmic reticulum (calcium-induced calcium release) via ryanodine and inositol triphosphate receptors (RyRs and IP3Rs). The increased Ca2+ load activates CaN, and through a negative feedback control, it dephosphorylates VGCCs, RyRs, and IP3Rs, decreasing the duration of opening and frequency as well as weakening the incoming cationic currents. Ca2+ influx via NMDA receptors (NMDARs) and the Ca2+-permeable AMPA receptors (AMPARs) follows a similar pathway of Ca2+ regulation and decreases long-term potentiation (LTP) through internalization and reduced NMDAR and AMPAR expression and increasing long-term depotentiation (LTD). This Ca2+-dependent inactivation (CDI) reinitiates the opening of these channels by triggering kinases, like protein kinase A (PKA) which complexes with A-kinase anchoring protein 79/150 (AKAP79/150) after guided by microtubule-associated protein 2B (MAP2B), that cause rephosphorylation. In the case of voltage-gated A-type potassium 4.2 (Kv4.2) channels, however, phosphorylation of the channels results in internalization and increased long-term potentiation (LTP) while the process is reversed after dephosphorylation by CaN.
3.3. Synaptic Endocytosis
CaN also participates in endocytosis as it facilitates clathrin-dependent recycling by targeting different dephosphins, endocytic proteins, one in particular called dynamin 1.37 The interaction with dynamin 1 is based on CaN’s sensitivity toward Ca2+ and spurs the depolarization-induced vesicular recycling; disturbance to such an interaction blocks endocytosis.38 However, such a role has been under controversy as CaN has shown to be implicated at different endocytic sites catalyzing different and opposing events.39 Regardless, studies continue to confirm that CaN is a key role player in endocytotic processes at both nerve terminals as well as in non-neuronal cells40 (see Figure 2a).
3.4. Ca2+ Influx and Regulation
Postsynaptically, CaN manages the Ca2+ homeostasis in the neuron by modulating its influx rates. The entry of Ca2+ activates the cell and triggers a cascade of a wide variety of biomolecular events. This intracytoplasmic Ca2+ influx is either via voltage-gated calcium channels (VGCCs) or ligand-gated channels such as the N-methyl-d-aspartate receptors (NMDARs).41,42 For NMDARs, CaN is located in the postsynaptic densities, near the cytoplasm and colocalized with NMDA receptors.43 The high affinity of CaN for the incoming Ca2+ activates it to dephosphorylate the C-terminal of the NR2A subunits of NMDARs, leading to glycine-independent desensitization and weakening of Ca2+ currents into the cell by decreasing the average duration of opened Ca2+ channels as well as their frequency.44
It has also been documented that VGCCs are directly involved in synaptic plasticity and dendritic excitability.45,46 VGCCs, specifically the L-type CCs and to some extent the N-type CCs, are phosphorylated.47 CaN is targeted toward these channels through A-kinase anchoring protein 79/150 (AKAP) when calcium-dependent inactivation (CDI) is stimulated by the Ca2+ influx. CDI then further triggers microtubule-associated protein 2B (MAP2B) to guide protein kinase A (PKA) toward the VGCCs for priming and reinitiating Ca2+ current.48,49 CaN is able to reduce the Ca2+ influx through this negative feedback effect and utilizes it to keep a check on the amount of Ca2+ entering the cell.
CaN also regulates the Ca2+-induced Ca2+ release (CICR), a universal mechanism by which cells are able to couple the inositol triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) to Ca2+ channels to trigger the release of Ca2+ from intracellular stores.50,51 Intracellular stores of Ca2+ reside in the sarcoplasmic reticulum and are mediated by IP3Rs and RyRs.30 It is believed this is another way CDI can be initiated alternatively to VGCC dephosphorylation due to the intimate association of VGCCs and the CCs opened through RyRs located on the sarcoplasmic reticulum (SR).52 CaN is able to dephosphorylate a phosphokinase C (PKC) phosphosite on both IP3Rs and RyRs and reduce their sensitivity toward alterations in intracellular Ca2+ concentrations and the subsequent affinity for their ligands (IP3 and Ry, respectively), thereby preventing the mechanism of CICR53(see Figure 2b).
4. Synaptic Plasticity
In conjunction to maintaining homeostatic Ca2+ levels throughout the neuronal network, CaN also facilitates homeostasis to synaptic plasticity, a term that denotes the flexibility and versatility of the neuronal network to change the synaptic strength in a direction that opposes the elevation or reduction in activity.54 This synaptic scaling often tends to be a negative feedback process and responds to altered synaptic strength to stabilize the neuronal network.55,56 CaN has been shown to be an important mediator of synaptic plasticity as its activation or inhibition can block or initiate synaptic scaling which further tends to enhance synaptic strength. CaN is able to regulate this through Ca2+ signaling, but at times even Ca2+ signaling can be disrupted and in turn can disrupt CaN activity. For the neuronal network to function properly synaptic plasticity must be sustained, and in such cases restoring Ca2+ signals becomes imperative. Studies suggest that chronic Ca2+-dependent inhibition (CDI) can lead to an attenuation of neuronal firing and decrease neuronal activity. As Ca2+ influx decreases, CaN activity also decreases and sets off a compensatory mechanism involving the phosphorylation of serine 845 of GluA1 by PKA bound to AKAP79/150 and the elevated expression of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) which are more sensitive and permeable to Ca2+, thereby restoring Ca2+ signaling57 (see Figure 2b). Upon restoration of Ca2+ signals, CaN is reactivated and reverts to facilitating proper neuronal signaling.
CaN has proven to modulate synaptic activity through Ca2+ influx through VGCCs and NMDARs.58 Furthermore, CaN has proven to confer bidirectional plasticity toward memory and learning while its deficiency has shown to impair working memory.59 This faculty of CaN to control bidirectional synaptic plasticity has great implications in memory and learning.60 Both phosphorylation and dephosphorylation play vital roles in memory formation and, respectively, trigger long-term potentiation (LTP) and long-term depression (LTD).61 Phosphokinases such as CaMKII, PKA, PKC, and MAPK have been involved in producing LTP, a phenomenon marked by both presynaptic and postsynaptic stimulations occurring at higher than normal frequencies or basically an enhanced synaptic transmission (the presynaptic stimulation often precedes the postsynaptic by varying milliseconds).62 Phosphatases, such as CaN, tend to depress this high-frequency stimulation and work in an opposing manner. Overexpression of CaN has shown to impair LTP while its inhibition can disrupt depotentiation.63 Whereas LTP will produce greater NMDARs/AMPARs expression, LTD will tend to decrease the same.64 CaN can indirectly also enhance LTP by reducing the LTD through its extensive manipulation of inhibitory synapses. Its affinity for GABA-A receptors and their subsequent dephosphorylation renders them for disinhibition as less GABA firing leads to less inhibition of the excitatory neurotransmission, thereby raising the LTP and overall synaptic efficacy.65−67 This is exemplary of the sophistication of CaN and its unique command over its own actions and the ability to adjust them as per subcellular requirements via downstream dephosphorylation. However, even though LTP may offer a larger collection of studies supporting its role in memory formation (particularly in the CA1 region of the hippocampus), LTD may be just as integral.68,69 Therefore, regardless of which direction the debate on LTP and LTD swings, CaN has forged its own niche in the continuous quest to elucidate memory formation and learning.70
Although mainly associated with LTD, CaN has shown its ability to control neuronal excitability via regulation of voltage-gated A-type potassium (K+) Kv4.2 channels.71,72 These channels are highly populated in postsynaptic terminals as well as dendrites.73,74 Their role is repolarization after the passage of action potentials and preventing their reverse propagation into dendritic regions.75,76 Overexpressed Kv4.2 channels have shown to shorten action potentials and mitigate LTP; this negative regulation of LTP resonates with that of CaN’s. Increase in NR2B/NR2A subunit ratio has been shown to facilitate synaptic plasticity, and LTP ensues. Overexpressed Kv4.2 channels reduce this ratio and trigger LTD.77 It has been shown that CaN modulates the activity of these channels in arms with the PKA/AKAP79/150 complex. The dephosphorylation by CaN stabilizes Kv4.2 channels at the postsynaptic terminal, dampening of neurotransmission (LTD). PKA counters this activity through phosphorylation of the channels by binding to AKAP79/150 to induce their internalization into the cytoplasm.78 This presents fewer Kv4.2 channels at the postsynaptic terminal and leads to LTP. Deficits in Kv4.2 channels have shown to produce spatial learning deficits and memory impairment79 (see Figure 2b).
5. Receptor Signaling
AMPARs are the most abundant excitatory receptors throughout the nervous system when it comes to fast synaptic transmission.80 Their expansive distribution and association with postsynaptic protein complexes grant them a high-level priority in regards to synaptic transmission and the electrophysiology of neurons.81 Their dynamic nature is conducive for changes in synaptic plasticity and therefore generates acute responsiveness within cells toward extracellular signals. Similar to metabotropic glutamate receptors and NMDARs, these receptors are also governed by phosphorylation (by PKA bound to AKAP79/150) and dephosphorylation (CaN), both targeting the highly sensitive s845 residue of the receptor.82 Phosphorylation increases greater expressions of the GluA1 on the postsynaptic membrane and the amount of current flowing through the AMPARs, thereby strengthening synaptic transmission.83,84 Dephosphorylation of s845 sequesters the receptor and induces LTD.85 PKA and CaN regulate synaptic plasticity at AMPARs and serve a vital role in maintaining the optimum signaling throughout the neural circuitry.
CaN also serves another regulatory role by dephosphorylating the metabotropic glutamate receptor 5 (mGluR5) which is responsible for augmenting the responsiveness of neurons to different signals from their microenvironment.86 In other words, mGluR5 elevates LTP. Belonging to group I of mGluRs and located postsynaptically, mGluR5 works via the Gq pathway: it stimulates phospholipase C to hydrolyze phosphatidylinositol biphosphate (PIP2) into IP3 and diacylglycerol (DAG) and further leads to the opening of Ca2+ stores via IP3Rs and RyRs and alteration in activities of various voltage-gated channels.87 In the process, it triggers a Ca2+ influx through NMDARs and becomes desensitized. CaN, after being activated by the NMDAR-mediated influx of Ca2+, resensitizes mGluR5 by dephosphorylation of its C-terminus and prolongs its activity; this is an important aspect of synaptic plasticity.88
6. Transcription Regulation
One of the major associations of CaN involves the nuclear factor for activated T-cell (NFAT) transcription factor. This family consists of five NFAT downstream targets residing in the cytoplasm in a hyperphosphorylated idle state (NFATc1-4, NFAT5/TonEBP).89,90 The CaN-NFAT signaling contributes to a wide array of biological developments such as cardiac and lung morphogenesis, differentiation and the growth of skeletal muscle, and immune responses like T-cell activation.91 As CaN is enriched throughout the brain regions, the tightly regulated signaling makes NFAT as proportionally and deeply integrated in many neuronal responses and activities such as axonal growth, memory formation, and neuronal apoptosis.92 Furthermore, the pathway is heavily involved in the development of the nervous system (synaptogenesis, corticogenesis), maintenance (myelination, neuronal survival), and nervous system operation (synaptic connectivity, plasticity, and neurotransmission).93 CaN-NFAT pathway begins when Ca2+ influx activates CaN to dephosphorylate the CaN-phosphosites from the N-terminal of NFATc subunits, exposing their nuclear sites.94 Once within the nucleus, these NFATc subunits partner with their NFATn counterparts on their specific response elements and promote various transcription sequences necessary for the homeostatic functioning of the neuron. However, the Ca2+ that activates CaN may not be sufficient to trigger this pathway. Rather, a greater influx of Ca2+ is provided by unique Ca2+ release activated Ca2+ (CRAC) channels.95 Some studies also suggest that the NFAT complexes induce a positive feedback loop to increase the intracellular Ca2+ levels via IP3R-RyRs Ca2+ release.96 Their job is to sustain the Ca2+ threshold necessary to maintain the NFATc–NFATn complexes within the nucleus for extended transcription to fulfill cellular needs at specified time points. A drop-off in intracellular Ca2+ levels signals the rephosphorylation of the NFAT complexes by initial priming by dual-specificity tyrosine-phosphorylation regulated kinases (DYRKs) particularly DYRK1A and DYRK2, recently discovered regulators of NFAT.97 Once primed, they are further subjected to rephosphorylation for export to the cytoplasm by glycogen synthase kinase 3 β (GSK-3), which acts to fine-tune the NFAT signaling98 (see Figure 3).
Figure 3.
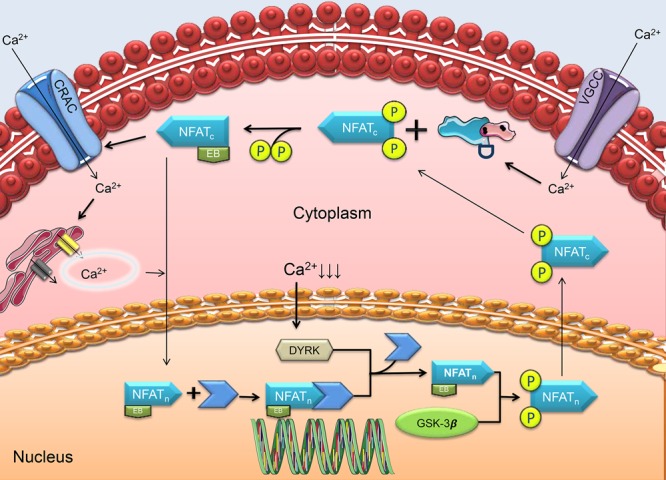
Regulation of NFAT by CaN. CaN drives the transcriptional role of the nuclear factor of activated T-cells (NFAT) following activation by the calcium (Ca2+) influx via voltage-gated calcium channels (VGCCs). NFAT is hyperphosphorylated and therefore idle in the cytoplasm (hence, NFATc). CaN dephosphorylates its N-terminal phosphosites and reveals its element binding domain (EB). The Ca2+ influx triggers further release of Ca2+ from the endoplasmic reticulum, driving the translocation of NFATc into the nucleus to partner with its nuclear counterpart, NFATn. The complex binds to its DNA site and transcribes neuronal requisites essential for growth, maintenance, and survival. The transcription remains as long as intracellular Ca2+ levels remain high (further maintained by calcium release activated Ca2+ channels or CRAC). The fall of Ca2+ levels activates dual-specificity tyrosine-phosphorylation regulated kinases (DYRKs) and triggers the cessation of transcription by the NFATc–NFATn complex. DYRKs dissociate the complex and prime NFATc for rephosphorylation by glycogen synthase kinase 3 β (GSK-3β) and relocation to the cytoplasm, where the next cycle begins contingent to the next calcium drive.
7. CaN–Mitochondria Connection
The intrinsic and paramount role of mitochondria in orchestrating cell survival and apoptosis has been well-documented.99−101 Studies over time have delineated several mechanistic pathways of mitochondria and their tightly regulated control over cell viability.102 Members of the Bcl-2 family, BAX and BAD, remain in an inactive state after phosphorylation by phosphokinases such as PKA, Akt, and Raf-1.103−105 Therefore, phosphorylation promotes cell survival and is employed by various growth factors. CaN has been postulated to be a key element in several of these pathways, particularly those favoring apoptosis.106 Its ability to dephosphorylate BAD activates the pro-apoptotic factor to bind and complex with the mitochondrial membrane-bound anti-apoptotic factors, Bcl-2 and Bcl-xL.107 The dephosphorylation occurs at primarily two serine residues of human BAD, s75 and s99.108 This dephosphorylation causes BAD to dissociate from the protein 14-3-3 and relocate to the outer mitochondrial membrane (OMM). The dimerization of BAD with Bcl-2 and Bcl-xL (suppression of pro-survival signals) prompt the translocation of BAX to the OMM to initiate the formation of mitochondrial permeability transition pores (mPTP) which exacerbate the apoptotic cascade through the release of cytochrome c from the mitochondria and result in execution of cell death by caspases 3, 6, and 7.109
As it has been elucidated that CaN plays a major role in programmed cell death, would the changes in CaN expression affect this role? Such a query arises from the relatively higher concentrations of the PP2B distributed in the brain. What role would a denser population of constitutive CaN play in influencing neuronal survival? Asai et al. went on to address this in a study where they investigated how deeply integrated CaN was in the intrinsic cell death pathway.110 What the group discovered was that CaN predisposes neurons to the cyt c-caspase 3 dependent pathway more frequently in comparison to other cells in the body. The elevated CaN levels (due to Ca2+ overload) render neurons to undergo apoptosis under even normal cell conditions. Thus, Ca2+-induced excitotoxicity through NMDARs increased the probability of inducing cell death due to the greater expression and sensitivity of CaN in neurons.
Despite establishing CaN’s role in apoptosis, its inhibition alone is not enough to prevent the same, and therefore other mechanisms independent of CaN’s dephosphorylation of BAD must exist.107 This curiosity fueled the search for alternative CaN-linked pathways related to apoptosis. Falling in line with CaN’s role in the intrinsic pathway of apoptosis is another recently discovered component of mitochondrial dysfunction also facilitated by CaN during cellular duress: dynamin-related peptide 1 (DRP1). A cytosolic dynamin GTPase, DRP1 was one of the initial fission proteins to be discovered, but an understanding of its function still remains unclear.111 Mitochondrial fission is often necessary during biogenesis of new mitochondria, but it may also indicate dysfunction in the organelle and trigger apoptosis.112 DRP1 is proposed to trigger mitochondrial fission by transferring to the OMM surface and forming puncta of DRP1 to form an oligomeric chain that constricts mitochondria to facilitate fission.113 This event resembles to the way dynamin cleaves endocytic vessels.114 Being a GTPase in nature, DRP1 uses GTP-hydrolysis to cause scission of mitochondria through the twisting movement of the chain.115 CaN mediates the dephosphorylation at s637 required to activate DRP1 and translocate it to the OMM to initiate fission.116 However, controversy surrounds whether it is BAD-Bcl-xL dimerization or mitochondrial fission that transpires prior to eliciting cell death.117 Some studies suggest that BAX activation and cytochrome c release follow fission in mammalian cells.118 Other studies point to the overexpression of Bik, triggering CICR that fission occurs following Ca2+.119 The lack of evidence of elongated mitochondrial void of cytochrome c resulting from mitochondrial fusion suggests that fission, not fusion, is more involved in apoptosis.120 This makes sense as healthy cells require the fusion process to create a dynamic interconnected network and therefore take on an elongated morphology in contrast to the spherical shapes encountered in mitochondrial fission121 (see Figure 4).
Figure 4.

Calcineurin–mitochondrial (CaN–mitochondrial) connection. (A) At abnormally high calcium (Ca2+) influx rates, such as during elevated glutamate signaling, calcineurin (CaN) becomes highly active and triggers the pathway of apoptosis by dephosphorylating the pro-apoptotic factor Bcl-2-associated death promoter (BAD) which translocates to the outer mitochondrial membrane (OMM) and dimerizes with the anti-apoptotic factors B-cell lymphphoma-2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL). The event spurs another pro-apoptotic factor, Bcl-2-associated-X (BAX), to transition to the OMM and catalyze the formation of the mitochondrial permeability transition pore (mPTP). The sustained high Ca2+ load continues to maintain the opening of the mPTP, leading to the release of cytochrome c and formation of the apoptosome, culminating in the caspase cascade leading to apoptosis. (B) CaN has also shown to be involved in triggering mitochondrial fission by dephosphorylating the dynamin-related protein 1 (DRP1). DRP1 translocates to the OMM, forms puncta, and constricts the mitochondria with an oligomeric chain and splices the organelle similar to vesicular cleavage by dynamin. The fission of mitochondria is thought to come before the release of cytochrome c, leading to dysfunction and triggering apoptosis or necrosis based on the rate and severity of the event. Evidence has shown that mitochondrial fission is indicative of mitochondrial dysfunction, and the spherical mitochondrial debris void of cytochrome c implicates the occurrence of cell death pathways.
This pivotal role of CaN in the intrinsic apoptotic signaling pathway further emphasizes the importance of the balance between phosphorylating and dephosphorylating events that dictate the fate of a cell.
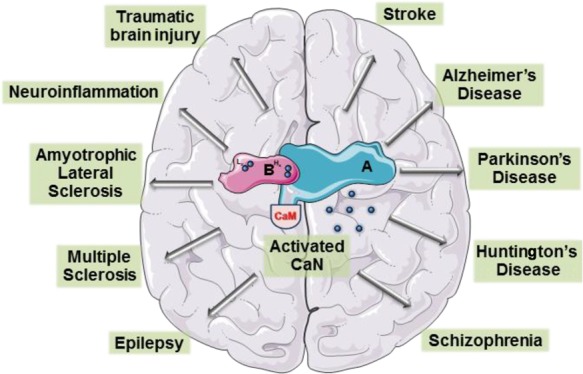
8. Calcineurin: Role in Neurological Disorders
The complexity of neurological disorders makes it difficult to dissect and diagnose the underlying etiology. However, certain etiologies ranging from dysfunctional VGCCs to disturbed mitochondrial dynamics have been common hallmarks of several neurological disorders.122,123 Keeping in perspective that Ca2+ is the engine the drives nearly all neuronal processes it is therefore worth investigating mediators that are intimately and heavily dependent on Ca2+ signaling, such as CaN.41 CaN, being an integral sensor and regulator of Ca2+ flux, may be deeply involved in many neurodegenerative disorders. The following sections explore the involvement of CaN through the spectrum of neurological disorders.
8.1. Ischemic Stroke
The global burden of stroke is a major medical concern that continues to rise in the oncoming years.124 It currently ranks as the second highest cause of mortality worldwide for the elderly population aged 60 years or older.125 Ischemic stroke accounts for nearly 85% of strokes (the rest being hemorrhagic and transient in nature).126 Needless to say, more efficient therapy is in dire need to treat patients facing a narrow therapeutic window. A glance at the etiology shows that stroke, particularly ischemic stroke, is encompassed by excitotoxicity, oxidative and nitrosative stress, inflammation, and edema.127−129 Cerebral ischemia expresses a cumbersome pathology that threatens neuronal viability not only during ischemia but after reperfusion as well; the underlying mechanisms have been well-delineated over the years.127,130−133 The ischemic phase sets the stage for cell damage as acidic conditions trigger various membrane pumps to dysfunction and thereby elicit heightened depolarization (Na+–H+ exchanger pumps more Na+, the Na+/K+ ATPase slows down, while Na+/Ca2+ exchanger reverses and raises intracellular Ca2+ levels). Due to the elevated intracellular Ca2+ load, CaN has been reported to be elevated during cerebral ischemia.110 Reperfusion also entails Ca2+ overload (by reduction of extracellular H+ levels and therefore accelerating Na+/H+ exchange and in turn more Ca2+ loading via Na+/Ca2+ exchange) and consequently further elevates CaN activation. There is also greater Ca2+ uptake by the mitochondria beyond the capacity via the mitochondrial Ca2+ uniporter (MCU) due to restoration of the electrochemical potential.133 Often referred to as the “oxygen paradox”, this oxidative burst has been believed to be the culprit behind mPTP formation, and reperfusion often sends the cell mainly through necrotic/necroptotic signaling along with apoptotic signaling contingent on the extent of Ca2+ loading along with duration and severity of the ischemic milieu.134
During the reperfusion phase, studies have discerned CaN’s ability to dephosphorylate BAD and induce apoptosis.107 Furthermore, CaN also dephosphorylates DRP-1 to induce mitochondrial fission and trigger apoptosis (see Table 1).116 Many of the mechanistic pathways still remain cryptic, and an understanding of CaN’s behavior throughout not only cerebral ischemia but also other neurodegenerative pathologies is difficult to pinpoint. However, a crosstalk of CaN with other biological molecules during cerebral ischemia may be proposed. In summary, CaN seems to play an integral role in ischemic stroke through the elevated Ca2+ influx, by virtue of activating several markers involved in mitochondrial and subsequent apoptotic signaling.
Table 1. Summary of Calcineurin’S Role in Neurodegenerative Disorders.
| neurodegenerative disorder | pathology involved | potential role of calcineurin (CaN) |
|---|---|---|
| stroke | ischemia/reperfusion injury, excitotoxicity, oxidative stress, inflammation, mitochondrial dysfunction | calcium-driven BAD and DRP1 dephosphorylation leading to mitochondrial dysfunction and cell death pathways; elevated neurotransmitter exocytotic kinetics107,116 |
| Alzheimer’s disease | proteinopathy: neurofibrillary tangles, β-amyloid (Aβ) plaques, neurotransmitter imbalance, inflammation | Aβ lesions trigger greater glutamate signaling and calcium excitotoxicity and drives CaN toward mitochondrial dysfunction and neuronal death; CaN accelerates dendritic spine loss and neuritic dystrophy; CaN increases CaMKII dephosphorylation, decreasing AMPAR-mediated LTP139,141 |
| Parkinson’s disease | proteinopathy: α-synuclein, neurotransmitter imbalance | elevated nuclear translocation of NFAT by CaN and increased inflammatory cascade98,145 |
| Huntington’s disease | CAG expansion repeats leading to mutant huntingtin accumulation | mitochondrial dysfunction due to excessive NMDAR activation by huntingtin; greater dephosphorylation at s421 of huntingtin shows disrupted axonal disruption and BDNF transport148,150 |
| amyotrophic lateral sclerosis | proteinopathy: TDP-43, C9orf72, and mutant superoxide dismutase (SOD 1G93A), excitotoxicity | mitochondrial dysfunction driven by CaN via calcium overload; CaN fails to dephosphorylate TDP-43 after poorly binding to mutant SOD 1155−157 |
| multiple sclerosis | autoimmune inflammatory demyelination, | CaN is involved in a the cycle of autoimmune T-cell-induced glutamate excitotoxicity that exacerbates neuroinflammation through CaN-activated NFAT transcription of cytokines; mitochondrial dysfunction and apoptosis; CaN may also drive exocytosis of glutamate at axonal terminals36,107,159 |
| schizophrenia | neurotransmitter imbalance (dopamine, glutamate, and GABA), mitochondrial dysfunction | altered synaptic cycling and exocytotic kinetics of neurotransmitters like dopamine, glutamate, and GABA by CaNγ subunit absence37 |
| epilepsy | neurotransmitter imbalance, cerebrovascular injury and impairment | CaN may sequester GABAA receptors and increase neuronal excitability; mitochondrial dysfunction through greater glutamate signaling and calcium excitotoxicity160,162 |
| traumatic brain injury | focal/global physical injury, axonal injury, cerebrovascular injury and impairment, consequential neurotransmitter imbalance | CaN initiates astrocyte activation by NFAT transcription and worsens the inflammation; it may also be involved in the subsequent excessive neurotransmitter due to contributions to exo-/endocytosis processes161 |
| neuroinflammation | astrocytic and microglial activation, cytokine release and T-cell activation, oxidative and nitrative stress | elevated NFAT exacerbates the inflammatory milieu by signaling for cytokine release and astrocyte and microglial activation9 |
8.2. Alzheimer’s Disease (AD)
The global burden of Alzheimer’s disease (AD) is a major concern as 47 million AD patients have been diagnosed worldwide.135 The most prevalent neurodegenerative disease (and predominant cause of dementia) is projected to grow to 76 million cases by 2030. Often reported to impair brain histology, cognitive functions, and behavior as well as deeply impacting the physical, emotional, and financial lifestyle of patients and their close ones, AD is the most lethal and complicated cause of dementia.136 The ever-controversial pathology of AD has been hallmarked by accumulative lesions of two degenerate proteins, amyloid-β (Aβ) and τ, and have been extensively studied.137,138 As AD is a disease where memories become lost, and CaN is a crucial protein deeply associated with learning and memory, this link therefore warrants a deeper investigation. Several studies honing on amyloidosis have detected abnormally high levels of CaN localized around the Aβ lesions and furthermore have detected the same in astrocytic processes enveloping amyloid deposits.139 Certain studies report CaN to become aberrant due to the calcium overload triggered by Aβ oligomer manipulation of glutamate-dependent cascades, though the mechanisms remain unclear.140 CaN seems to become dysregulated, upregulating the NFAT translocation and transcription, causing dendritic spine loss and neuritic dystrophy, changes that lie in complete contrast to homeostatic CaN-dependent NFAT that underlies axonal growth, elevated dendritic branching, and overall neuronal survival. Deregulated CaN also increases CaMKII dephosphorylation, which downregulates AMPAR-dependent LTP. This imbalance between the two enzymes results in greater LTD, synapse weakening, and memory loss (see Table 1).141 Thus, deregulated CaN may spur dendritic spine loss, dysregulate AMPAR-mediated LTP, and trigger apoptosis through mitochondrial-mediated pathways.
8.3. Parkinson’s Disease (PD)
PD follows AD as the second most burdensome neurodegenerative disease worldwide affecting a wide range of age groups: from 41 per 100 000 in the fourth decade to over 1900 per 100 000 for elderly above 80 years of age.142
PD is the most notorious movement disorder plagued by inclusions of aberrant α-synuclein in both the perikaryon and the processes of dopaminergic distributed throughout the substantia nigra pars compacta region.143 Studies have reported altered and abnormal Ca2+ signaling induced by the synucleinopathy en route to weakening of LTP and memory impairment.144 CaN, deeply involved with LTP and memory formation, compellingly points to a link in PD. A study elicited CaN’s confounding switch between protective Ca2+ regulation to a cascade of downstream sequences that exacerbate the toxicity perpetuated by α-synuclein.145 The study also witnessed enhanced nuclear localization of NFATc4 in the brains of PD patients. This association of NFAT to PD further bolsters elevated cytosolic Ca2+ load and the failure of CaN. This may be due to several mechanisms unexplored at the time. However, it may be proposed that elevated Ca2+ due to α-synuclein may be responsible for overactivating CaN which drives it toward LTP weakening.44 Meanwhile, sustained Ca2+ load prevents the rephosphorylation of NFAT and may explain its retention in the nucleus.98 The consequent inflammatory signaling interferes with synaptic connectivity and neurotransmission, exacerbating neuronal survival (see Table 1). Thus, CaN seems to entail LTP weakening, prolonged NFAT-mediated inflammation, and the consequential breakdown of neurotransmission.
8.4. Huntington’s Disease (HD)
Hallmarked by the death of medium spiny neurons of the striatum and the consequential aggressive progression of motor, cognitive, and behavioral decline, HD is now the most common monogenic brain disorder in developed nations (Europe and North America) with a prevalence of nearly 10 times more than that of Asian nations.146 The CAG expansion (beyond 35 repeats) in the gene responsible for the production of huntingtin (HTT) protein has been discovered in nearly 90% of HD patients. Pathology manifests through the aberrant splicing pattern of HTT fragments, particularly those accumulating in HTT exon 1 protein.147 However, the pathology comprises proteins involved in a vast array of neuronal functions, from regulating the cell cycle to cell signaling, emphasizing the complexity of HD and the quest for more promising therapeutic targets. A link between CaN and HD, associating elevated levels of the phosphatase in the disease after NMDAR activation triggering the intrinsic apoptotic pathway, has been reported.148 Studies have pointed to the phosphorylation of s421 evincing an inverse relationship with disease susceptibility.149 Greater dephosphorylation of s421 seems to disrupt the axonal transport and diminished BDNF transport, promoting neurodegeneration; the inhibition of CaN increased phosphorylation of s421 thereby restoring antero- and retrograde axonal transport in mutant HTT.150 Mutant HTT has also shown to downregulate CaN and trigger hyperphosphorylation of τ protein in another study without any noticeable changes in τ kinases (see Table 1).151 It can therefore be proposed that elevated CaN, due to hyperactivation of NMDARs (induced by mutant HTT), seems to disrupt axonal and BDNF transport and triggers mitochondrial-mediated apoptotic pathways.
8.5. Amyotrophic Lateral Sclerosis (ALS)
ALS is an age-dependent insidious and fatal motor neuron (MN) disease of the cortex, brain stem, and spinal cord, encompassing damage to both upper and lower motoneurons. It has a current global prevalence of 1.9 per 100 000 that threatens to amass by 69% by 2040.152 Along with the recent breakthrough discovery of a genetic culprit of ALS (an intronic hexanucleotide expansion in the C9orf72 gene), the predominance of pathological studies revolve around proteinopathies and Ca2+ overload excitotoxicity brought about hyperactivated glutamatergic signaling, mainly a ramification of the lack of GluR2 subunits and heightened permeability in AMPARs.153,154 The altered intracellular Ca2+ milieu sets off a Ca2+-dependent cascade, still not clearly understood, which culminates in cell death. The affected neurons eventually lose their Ca2+ buffering capacities, and therefore, mitochondrial dysfunction and apoptosis via the intrinsic pathway have been reported.155 CaN has shown to be highly involved in triggering this pathway during Ca2+ overload. Plaques of trans-activating response region DNA binding protein (TDP-43) have been depicted in nearly 90% of ALS cases, and Liachko et al. have found that dephosphorylation of TDP-43 at sites s409/410 by CaN helps to limit toxic protein accumulation.156 Kim et al. shed further light on the interaction between TDP-43 and CaN when the group observed weakened dephosphorylation of TDP-43 by CaN after its poor binding with mutated superoxide dismutase 1 (SOD 1G93A).157 Furthermore, TDP-43 pathology is now believed to drive ALS progression along with being a factor in other proteinopathies like AD and PD (see Table 1). CaN seems to therefore exacerbate neuronal damage through its role in mitochondrial dysfunction as well through its failure of limiting the intracellular toxic TDP-43 levels due to interfering binding by SOD 1G93A
8.6. Multiple Sclerosis (MS)
MS is a chronic, autoimmune inflammatory neurodegenerative disease that plagues about 2.5 million people worldwide.158 Glutamate-induced excitotoxicity has been implicated to be a driving force in neuronal death. Studies report that inflammatory cells in foci ensuing autoimmune inflammatory insult are a major source of extracellular glutamate, releasing it via cysteine/glutamate transporters Xc–.159 The surplus glutamate not only further activates more T-cells via their upregulated expression of NMDARs, but it also triggers the rapid influx of Ca2+ in neuronal cells that accelerates pro-apoptotic signaling. CaN is speculated to play a role in MS by virtue of the large intracellular Ca2+ load driving it to facilitate the intrinsic pathway and mitochondrial dysfunction.107 It also can be postulated that excitotoxicity drives the exocytotic kinetics through the activated CaN and synapsin 1 complex to release greater glutamate, which fuels the vicious T-cell-mediated autoimmunity and exacerbates excitotoxic milieu in neighboring neurons (see Table 1).36,159 CaN, by virtue of Ca2+ overload, seems to accelerate pro-apoptotic signaling as well drive the excess exocytosis of glutamate into the synapse, exacerbating inflammation-induced neurodegeneration. Such a cascade may deserve deeper investigation in future studies concerning MS and the role of CaN.
9. Other Neurodegenerative Disorders
CaN is involved in other intricate neurological pathologies (Table 1). Knockdown of the CaNγ subunit has shown to alter synaptic vesicle cycling and has shown to cause psychiatric disorders like schizophrenia.37 In an epileptic study, CaN has shown to augment neuronal excitability by greater endocytosis and internalization of GABAA receptors, disturbing the excitatory/inhibitory equilibrium to prevent inhibitory actions of GABA that mitigate the synchronous neuronal discharge responsible for seizure activity.160 The same study also showed the prevention of mitochondrial dysfunction by inhibition of CaN with cyclosporin A (CsA), a cyclophilin D binding drug that complexes with CaN to render it inactive and thereby preventing the opening of mPTP. A study based on traumatic brain injury (TBI) showed how the CaN/NFAT pathway following TBI induced astrocyte activation that further compounded the inflammatory environment and escalated neuronal damage.161 Neuroinflammation is a major component in CNS disorders and acute and chronic brain injury. Inhibition of CaN mitigated the detrimental activation of NFAT that transcribes inflammatory markers and recruits astrocytes and glial cells to the site of injury. In neuroinflammation studies, CaN plays both anti- and pro-inflammatory roles based on its interactions with transcription factors and displays a complex but unique dichotomy in mediating inflammatory milieu. For instance, it can associate with NFAT and activator protein 1 (AP1) to activate T-cells while inducing their anergy and tolerance by associating NFAT with forkhead box P3 (FOXP3).9 In conjunction, activation of Nuclear Factor κB (NFκB) and NFAT triggers the transcription of cytokines, activating astrocytes and microglia while promoting T-cell expansion in the CNS tissue, ultimately leading to inflammation-mediated neuronal damage and cell death.162
10. Immunosuppression of CaN and Its Effects on the Brain
CaN inhibitors (CNIs), namely, cyclosporine A and tacrolimus, have been immensely utilized in the field of organ transplantation, becoming the cornerstone therapy for preventing immune rejections.163 However, their effects on the brain remain a concern. CNI therapy used in kidney transplant patients has elicited neurological symptoms such as recurrent headaches, tremors, seizures, extrapyramidal syndrome, encephalopathy, and posterior encephalopathy syndrome.164 Other clinical studies have shown neurotoxicity with the initiation of CNI therapy.165,166 A recent study focusing on the neural effects of CNIs in orthotopic liver transplant patients by Pflugrad et al. showcased cognitive decline and altered brain structure.165 The neurological adverse reactions observed in CNI therapy further emphasize the complex role of CaN in the brain. Inhibiting CaN with CNIs alone to treat neurological disorders is still up for debate and requires extensive investigation to overcome the associated neurotoxicity. Furthermore, safer drugs void of neurotoxic side effects are desperately required in organ transplantation.
There are certain risk factors that can be assessed prior to organ transplantation that may help avoid possible neurologic complications. Encephalopathy, although often indicated in solid organ transplant patients, can be assessed by varying characteristic abnormalities observed in magnetic resonance imaging (MRI). For instance, altered signal changes can be observed in the insular and cingulated cortices with relatively lesser changes in occipital and perirolandic cortex may indicate possible neurotoxicity.166 Other diagnostic suggestions involve the requirement for more sensitive Doppler devices to monitor intracranial pressure noninvasively, monitoring of EEG levels post-transplant, and impaired verbal and motor responsiveness without electrolyte imbalance, tinnitus, or hearing loss (improved after discontinuation of tacrolimus).167Alternative therapies to CNI involve monoclonal antibodies (belatacept),168 combinations of mycophenolate mofetil and sirolimus,169 along with newer CNI-free therapies in the pipeline.170 CNI-free immunosuppressant therapy may prove to be beneficial in organ transplant patients in the future.171
11. Conclusion
Neurodegenerative disorders present a major conundrum in medical science. The complex neuropathologies, despite sharing common pathological manifestations, arise from dysregulated crosstalk and genetic expressions that are difficult to trace. Some biological molecules involved in regulating much of the neuronal functions may be honed in on as potential therapeutic targets. CaN is one such target that is inveterate to running essential neuronal components such as regulating Ca2+ homeostasis and Ca2+-dependent downstream processes, synaptic plasticity, neurotransmission release, and synaptic cycling. Furthermore, it is involved in regulating transcription processes and the overall neuronal survival, operation, and maintenance. Due to such a diversified and crucial biological role, CaN may lie at the crux of many neurodegenerative disorders. From stroke to proteinopathies to inflammatory pathologies, CaN has been reported in underlying mechanistic pathways. However, it is rather perplexing to pinpoint whether CaN serves to protect and salvage neurons during neurological disorders or whether it perpetuates neuronal damage. The convoluted pathways of CaN exhibit its rescue of one aspect of the cell while deteriorating another during neuropathic conditions. Furthermore, some pathways are not truly delineated due to their erratic and volatile dispositions. Retentive in mind should also be that simple inhibition of an endogenous molecule as vital as CaN is to neuronal function alone may bring more harm than good in neurotherapy. Although many of the signaling cascades remain to be elucidated, and manipulating CaN may seem daunting, CaN warrants a deeper investigation. By bringing a greater focus to understanding its roles, it may be possible to strategize methods to modulate CaN as a potential therapeutic target to alleviate neurological disorders.
Acknowledgments
The authors acknowledge Department of Science and Technology (DST), Government of India, for their financial support through Grant (SB/YS/LS-196/2014), International Society for Neurochemistry (ISN) Return Home Grant, Department of Pharmaceuticals, Ministry of Chemical and Fertilizers, Government of India, and National Institute of Pharmaceutical Education and Research (NIPER) Ahmedabad, Gandhinagar, India. The authors also want to express their thanks to Prof. Larry Benowitz, Boston Children’s Hospital, Harvard Medical School, Boston, MA, United States, and the Director, NIPER Ahmedabad, for providing necessary support.
Author Contributions
J.S., P.B., K.R.D., and D.R.Y. conceived and designed the study. J.S., D.S., H.K., K.R.D., and P.B. outlined the performed rigorous literature search. J.S., D.S., H.K., K.K., and P.B. conceived and designed the figures and images. J.S., P.B., K.R.D., K.K., and D.R.Y. wrote the manuscript.
The authors declare no competing financial interest.
References
- Wang J. H.; Desai R. A brain protein and its effect on the Ca2+-and protein modulator-activated cyclic nucleotide phosphodiesterase. Biochem. Biophys. Res. Commun. 1976, 72 (3), 926. 10.1016/S0006-291X(76)80220-3. [DOI] [PubMed] [Google Scholar]
- Watterson D. M.; Vanaman T. C. Affinity chromatography purification of a cyclic nucleotide phosphodiesterase using immobilized modulator protein, a troponin C-like protein from brain. Biochem. Biophys. Res. Commun. 1976, 73 (1), 40. 10.1016/0006-291X(76)90494-0. [DOI] [PubMed] [Google Scholar]
- Klee C.; Krinks M. Purification of cyclic 3′, 5′-nucleotide phosphodiesterase inhibitory protein by affinity chromatography on activator protein coupled to Sepharose. Biochemistry 1978, 17 (1), 120. 10.1021/bi00594a017. [DOI] [PubMed] [Google Scholar]
- Wallace R. W.; Lynch T. J.; Tallant E. A.; Cheung W. Y. An endogenous inhibitor protein of brain adenylate cyclase and cyclic nucleotide phosphodiesterase. Arch. Biochem. Biophys. 1978, 187 (2), 328. 10.1016/0003-9861(78)90042-5. [DOI] [PubMed] [Google Scholar]
- Wallace R. W.; Lynch T. J.; Tallant E. A.; Cheung W. Y. Purification and characterization of an inhibitor protein of brain adenylate cyclase and cyclic nucleotide phosphodiesterase. J. Biol. Chem. 1979, 254 (2), 377. [PubMed] [Google Scholar]
- Klee C.; Crouch T.; Krinks M. Calcineurin: a calcium-and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. U. S. A. 1979, 76 (12), 6270. 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A. A.; Ingebritsen T. S.; Manalan A.; Klee C. B.; Cohen P. Discovery of A Ca2+-and calmodulin-dependent protein phosphatase. FEBS Lett. 1982, 137 (1), 80. 10.1016/0014-5793(82)80319-0. [DOI] [PubMed] [Google Scholar]
- INGEBRITSEN T. S.; COHEN P. The protein phosphatases involved in cellular regulation. Eur. J. Biochem. 1983, 132 (2), 255. 10.1111/j.1432-1033.1983.tb07357.x. [DOI] [PubMed] [Google Scholar]
- Furman J. L.; Norris C. M. Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 2014, 11 (1), 158. 10.1186/s12974-014-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Farmer J. D. Jr; Lane W. S.; Friedman J.; Weissman I.; Schreiber S. L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66 (4), 807. 10.1016/0092-8674(91)90124-H. [DOI] [PubMed] [Google Scholar]
- Carriedo S. G.; Yin H. Z.; Sensi S. L.; Weiss J. H. Rapid Ca2+ entry through Ca2+-permeable AMPA/Kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J. Neurosci. 1998, 18 (19), 7727. 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.; Fass D. M.; Reynolds I. J. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca2+] i increases and NMDA receptor mRNA levels. Brain Res. 1999, 849 (1–2), 97. 10.1016/S0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Foster T. C.; Sharrow K. M.; Masse J. R.; Norris C. M.; Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 2001, 21 (11), 4066. 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingebritsen T. S.; Cohen P. Protein phosphatases: properties and role in cellular regulation. Science 1983, 221 (4608), 331. 10.1126/science.6306765. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58 (1), 453. 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Cohen P. Protein phosphatases come of age. J. Biol. Chem. 1989, 264 (36), 21435. [PubMed] [Google Scholar]
- Klee C.; Draetta G.; Hubbard M. Calcineurin. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 61, 149. 10.1002/9780470123072.ch4. [DOI] [PubMed] [Google Scholar]
- Griffith J. P.; Kim J. L.; Kim E. E.; Sintchak M. D.; Thomson J. A.; Fitzgibbon M. J.; Fleming M. A.; Caron P. R.; Hsiao K.; Navia M. A. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995, 82 (3), 507. 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- Coghlan V. M.; Perrino B. A.; Howard M.; Langeberg L. K.; Hicks J. B.; Gallatin W. M.; Scott J. D. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 1995, 267 (5194), 108. 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- Kissinger C. R.; Parge H. E.; Knighton D. R.; Lewis C. T.; Pelletier L. A.; Tempczyk A.; Kalish V. J.; Tucker K. D.; Showalter R. E.; Moomaw E. W. Crystal structures of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature 1995, 378 (6557), 641. 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- Feng B.; Stemmer P. M. Interactions of calcineurin A, calcineurin B, and Ca2+. Biochemistry 1999, 38 (38), 12481. 10.1021/bi990492w. [DOI] [PubMed] [Google Scholar]
- Gallagher S. C.; Gao Z.-H.; Li S.; Dyer R. B.; Trewhella J.; Klee C. B. There is communication between all four Ca2+-bindings sites of calcineurin B. Biochemistry 2001, 40 (40), 12094. 10.1021/bi0025060. [DOI] [PubMed] [Google Scholar]
- Shen X.; Li H.; Ou Y.; Tao W.; Dong A.; Kong J.; Ji C.; Yu S. The secondary structure of calcineurin regulatory region and conformational change induced by calcium/calmodulin binding. J. Biol. Chem. 2008, 283 (17), 11407. 10.1074/jbc.M708513200. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Xiong F.; Kong S.; Ogawa T.; Kobayashi M.; Liu J. O. Distinct tissue and cellular distribution of two major isoforms of calcineurin. Mol. Immunol. 1997, 34 (8–9), 663. 10.1016/S0161-5890(97)00054-0. [DOI] [PubMed] [Google Scholar]
- Solà C.; Tusell J. M.; Serratosa J. Comparative study of the distribution of calmodulin kinase II and calcineurin in the mouse brain. J. Neurosci. Res. 1999, 57 (5), 651.. [DOI] [PubMed] [Google Scholar]
- Yakel J. L. Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol. Sci. 1997, 18 (4), 124. 10.1016/S0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- Miller R. J. Multiple calcium channels and neuronal function. Science 1987, 235 (4784), 46. 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- Kennedy M. B. Regulation of neuronal function by calcium. Trends Neurosci. 1989, 12 (11), 417. 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- Berridge M. J.; Lipp P.; Bootman M. D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1 (1), 11. 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bading H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14 (9), 593. 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Mansuy I. M. Calcineurin in memory and bidirectional plasticity. Biochem. Biophys. Res. Commun. 2003, 311 (4), 1195. 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Verstegen A. M.; Tagliatti E.; Lignani G.; Marte A.; Stolero T.; Atias M.; Corradi A.; Valtorta F.; Gitler D.; Onofri F. Phosphorylation of synapsin I by cyclin-dependent kinase-5 sets the ratio between the resting and recycling pools of synaptic vesicles at hippocampal synapses. J. Neurosci. 2014, 34 (21), 7266. 10.1523/JNEUROSCI.3973-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabi A. A.; Tsien R. W. Synaptic vesicle pools and dynamics. Cold Spring Harbor Perspect. Biol. 2012, 4 (8), a013680. 10.1101/cshperspect.a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell J. R.; Levenson J. M.; Kim S. H.; Gibson H. E.; Richardson K. A.; Sivula M.; Li B.; Ashford C. J.; Heindl K. A.; Babcock R. J. Working memory impairment in calcineurin knock-out mice is associated with alterations in synaptic vesicle cycling and disruption of high-frequency synaptic and network activity in prefrontal cortex. J. Neurosci. 2013, 33 (27), 10938. 10.1523/JNEUROSCI.5362-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H.; Ryan T. A. CDK5 serves as a major control point in neurotransmitter release. Neuron 2010, 67 (5), 797. 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H.; Ryan T. A. Balance of calcineurin Aα and CDK5 activities sets release probability at nerve terminals. J. Neurosci. 2013, 33 (21), 8937. 10.1523/JNEUROSCI.4288-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell J. R.; Li B.; Kyung J. W.; Ashford C. J.; Mann J. J.; Horvath T. L.; Ryan T. A.; Kim S. H.; Gerber D. J. Calcineurin Aγ is a functional phosphatase that modulates synaptic vesicle endocytosis. J. Biol. Chem. 2016, 291 (4), 1948. 10.1074/jbc.M115.705319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S. M.; De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012, 13 (2), 75. 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton E. L.; Anggono V.; Smillie K. J.; Chau N.; Robinson P. J.; Cousin M. A. The phospho-dependent dynamin–syndapin interaction triggers activity-dependent bulk endocytosis of synaptic vesicles. J. Neurosci. 2009, 29 (24), 7706. 10.1523/JNEUROSCI.1976-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.-S.; Zhang Z.; Zhao W.-D.; Wang D.; Luo F.; Wu L.-G. Calcineurin is universally involved in vesicle endocytosis at neuronal and nonneuronal secretory cells. Cell Rep. 2014, 7 (4), 982. 10.1016/j.celrep.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. J. Neuronal calcium signaling. Neuron 1998, 21 (1), 13. 10.1016/S0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Ross W. N. Understanding calcium waves and sparks in central neurons. Nat. Rev. Neurosci. 2012, 13 (3), 157. 10.1038/nrn3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-Y.; Orser B. A.; Brautigan D. L.; MacDonald J. F. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature 1994, 369 (6477), 230. 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- Krupp J. J.; Vissel B.; Thomas C. G.; Heinemann S. F.; Westbrook G. L. Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology 2002, 42 (5), 593. 10.1016/S0028-3908(02)00031-X. [DOI] [PubMed] [Google Scholar]
- Fino E.; Paille V.; Cui Y.; Morera-Herreras T.; Deniau J. M.; Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J. Physiol. 2010, 588 (16), 3045. 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin J. L.; Day M.; Surmeier D. J. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat. Neurosci. 2011, 14 (7), 881. 10.1038/nn.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria S. F.; Dell’Acqua M. L.; Sather W. A. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 2007, 55 (2), 261. 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T.; Meuth S.; Pape H.-C. Calcium-dependent inactivation of neuronal calcium channels. Nat. Rev. Neurosci. 2002, 3 (11), 873. 10.1038/nrn959. [DOI] [PubMed] [Google Scholar]
- Dittmer P. J.; Dell’Acqua M. L.; Sather W. A. Ca2+/calcineurin-dependent inactivation of neuronal L-type Ca2+ channels requires priming by AKAP-anchored protein kinase A. Cell Rep. 2014, 7 (5), 1410. 10.1016/j.celrep.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron A. M.; Steiner J. P.; Roskams A. J.; Ali S. M.; Ronnettt G. V.; Snyder S. H. Calcineurin associated with the inositol 1, 4, 5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell 1995, 83 (3), 463. 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Sun H.; Leblanc N.; Nattel S. Mechanisms of inactivation of L-type calcium channels in human atrial myocytes. American Journal of Physiology-Heart and Circulatory Physiology 1997, 272 (4), H1625. 10.1152/ajpheart.1997.272.4.H1625. [DOI] [PubMed] [Google Scholar]
- Sham J.; Cleemann L.; Morad M. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (1), 121. 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. American Journal of Physiology-Cell Physiology 1983, 245 (1), C1. 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Arendt K. L.; Zhang Z.; Ganesan S.; Hintze M.; Shin M. M.; Tang Y.; Cho A.; Graef I. A.; Chen L. Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (42), E5744. 10.1073/pnas.1510239112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor Perspect. Biol. 2012, 4 (1), a005736. 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D.; Malenka R. C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440 (7087), 1054. 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Kim S.; Ziff E. B. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 2014, 12 (7), e1001900. 10.1371/journal.pbio.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef I. A.; Mermelstein P. G.; Stankunas K.; Neilson J. R.; Deisseroth K.; Tsien R. W.; Crabtree G. R. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 1999, 401 (6754), 703. 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Chattarji S.; Barbarosie M.; Rondi-Reig L.; Philpot B. D.; Miyakawa T.; Bear M. F.; Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 2001, 107 (5), 617. 10.1016/S0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Malleret G.; Haditsch U.; Genoux D.; Jones M. W.; Bliss T. V.; Vanhoose A. M.; Weitlauf C.; Kandel E. R.; Winder D. G.; Mansuy I. M. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 2001, 104 (5), 675. 10.1016/S0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Giese K. P.; Mizuno K. The roles of protein kinases in learning and memory. Learn. Mem. 2013, 20 (10), 540. 10.1101/lm.028449.112. [DOI] [PubMed] [Google Scholar]
- Lisman J.; Yasuda R.; Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13 (3), 169. 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel K.; Mansuy I. M. Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 2012, 19 (9), 375. 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- Beattie E. C.; Carroll R. C.; Yu X.; Morishita W.; Yasuda H.; von Zastrow M.; Malenka R. C. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 2000, 3 (12), 1291. 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Jones M. V.; Westbrook G. L. Shaping of IPSCs by endogenous calcineurin activity. J. Neurosci. 1997, 17 (20), 7626. 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. M.; Mansuy I. M.; Kandel E. R.; Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 2000, 26 (1), 197. 10.1016/S0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu S.; Haditsch U.; Tu W.; Cochrane K.; Ahmadian G.; Tran L.; Paw J.; Wang Y.; Mansuy I. Interaction of calcineurin and type-A GABA receptor γ2 subunits produces long-term depression at CA1 inhibitory synapses. J. Neurosci. 2003, 23 (3), 826. 10.1523/JNEUROSCI.23-03-00826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder D. G.; Mansuy I. M.; Osman M.; Moallem T. M.; Kandel E. R. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell 1998, 92 (1), 25. 10.1016/S0092-8674(00)80896-X. [DOI] [PubMed] [Google Scholar]
- Lüscher C.; Malenka R. C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspect. Biol. 2012, 4 (6), a005710. 10.1101/cshperspect.a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R. C.; Bear M. F. LTP and LTD: an embarrassment of riches. Neuron 2004, 44 (1), 5. 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Lin L.; Sun W.; Kung F.; Dell’Acqua M. L.; Hoffman D. A. AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phospho-regulation of A-type K+ channel trafficking. J. Neurosci. 2011, 31 (4), 1323. 10.1523/JNEUROSCI.5383-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J.-j.; Zhao Q.-R.; Liu D.-D.; Chow C.-W.; Mei Y.-A. Neuritin Up-regulates Kv4. 2 α-Subunit of Potassium Channel Expression and Affects Neuronal Excitability by Regulating the Calcium-Calcineurin-NFATc4 Signaling Pathway. J. Biol. Chem. 2016, 291 (33), 17369. 10.1074/jbc.M115.708883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S.; Jeromin A.; Kosaka T. Postsynaptic and extrasynaptic localization of Kv4. 2 channels in the mouse hippocampal region, with special reference to targeted clustering at gabaergic synapses. Neuroscience 2005, 134 (2), 483. 10.1016/j.neuroscience.2005.04.065. [DOI] [PubMed] [Google Scholar]
- Alonso G.; Widmer H. Clustering of KV4. 2 potassium channels in postsynaptic membrane of rat supraoptic neurons: an ultrastructural study. Neuroscience 1997, 77 (3), 617. 10.1016/S0306-4522(96)00561-1. [DOI] [PubMed] [Google Scholar]
- Kim J.; Wei D. S.; Hoffman D. A. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J. Physiol. 2005, 569 (1), 41. 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman D. A.; Magee J. C.; Colbert C. M.; Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 1997, 387 (6636), 869. 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Yashiro K.; Philpot B. D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55 (7), 1081. 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader L. A.; Anderson A. E.; Mayne A.; Pfaffinger P. J.; Sweatt J. D. PKA modulation of Kv4. 2-encoded A-type potassium channels requires formation of a supramolecular complex. J. Neurosci. 2002, 22 (23), 10123. 10.1523/JNEUROSCI.22-23-10123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockridge A.; Yuan L. L. Spatial learning deficits in mice lacking A-type K+ channel subunits. Hippocampus 2011, 21 (11), 1152. 10.1002/hipo.20877. [DOI] [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010, 62 (3), 405. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd J. D.; Huganir R. L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613. 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Lee H.-K.; Takamiya K.; Han J.-S.; Man H.; Kim C.-H.; Rumbaugh G.; Yu S.; Ding L.; He C.; Petralia R. S. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 2003, 112 (5), 631. 10.1016/S0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Man H.-Y.; Sekine-Aizawa Y.; Huganir R. L. Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (9), 3579. 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche K. W.; O’Brien R. J.; Mammen A. L.; Bernhardt J.; Huganir R. L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 1996, 16 (6), 1179. 10.1016/S0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Lee H.-K.; Kameyama K.; Huganir R. L.; Bear M. F. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 1998, 21 (5), 1151. 10.1016/S0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Jong Y.-J. I.; Kumar V.; O’Malley K. L. Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J. Biol. Chem. 2009, 284 (51), 35827. 10.1074/jbc.M109.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagni L.; Chavis P.; Ango F.; Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000, 23 (2), 80. 10.1016/S0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S.; Saugstad J.; Warren L.; Mansuy I. M.; Gereau IV R. W.; Conn P. J. NMDA-induced potentiation of mGluR5 is mediated by activation of protein phosphatase 2B/calcineurin. Neuropharmacology 2005, 49, 135. 10.1016/j.neuropharm.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G. R.; Olson E. N. NFAT signaling: choreographing the social lives of cells. Cell 2002, 109 (2), S67. 10.1016/S0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5 (6), 472. 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Crabtree G. R.; Schreiber S. L. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell 2009, 138 (1), 210. 10.1016/j.cell.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.-Y.; Yang H. W.; Kim J.-R.; Do Heo W.; Cho K.-H. A hidden incoherent switch regulates RCAN1 in the calcineurin–NFAT signaling network. J. Cell Sci. 2011, 124 (1), 82. 10.1242/jcs.076034. [DOI] [PubMed] [Google Scholar]
- Kipanyula M. J.; Kimaro W. H.; Etet P. F. The emerging roles of the calcineurin-nuclear factor of activated T-lymphocytes pathway in nervous system functions and diseases. Journal of Aging Research 2016, 2016, 5081021. 10.1155/2016/5081021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.-Y.; Choo S.-M.; Kim D.; Baek S. J.; Wolkenhauer O.; Cho K.-H. Switching feedback mechanisms realize the dual role of MCIP in the regulation of calcineurin activity. FEBS Lett. 2006, 580 (25), 5965. 10.1016/j.febslet.2006.09.064. [DOI] [PubMed] [Google Scholar]
- Parekh A. B.; Putney J. W. Jr Store-operated calcium channels. Physiol. Rev. 2005, 85 (2), 757. 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Nguyen T.; Di Giovanni S. NFAT signaling in neural development and axon growth. Int. J. Dev. Neurosci. 2008, 26 (2), 141. 10.1016/j.ijdevneu.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y.; Sharma S.; Nardone J.; Tanasa B.; Iuga A.; Srikanth S.; Okamura H.; Bolton D.; Feske S.; Hogan P. G. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006, 441 (7093), 646. 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- Beals C. R.; Sheridan C. M.; Turck C. W.; Gardner P.; Crabtree G. R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997, 275 (5308), 1930. 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Green D. R.; Reed J. C. Mitochondria and apoptosis. Science 1998, 281 (5381), 1309–1312. 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Rossé T.; Olivier R.; Monney L.; Rager M.; Conus S.; Fellay I.; Jansen B.; Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature 1998, 391 (6666), 496. 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Nicholls D. G.; Budd S. L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80 (1), 315. 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Green D. R.; Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004, 305 (5684), 626. 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Guyton K. Z.; Liu Y.; Gorospe M.; Xu Q.; Holbrook N. J. Activation of mitogen-activated protein kinase by ho role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271 (8), 4138. 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Philpott K. L.; McCarthy M. J.; Klippel A.; Rubin L. L. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J. Cell Biol. 1997, 139 (3), 809. 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Fujii K.; Zhang L.; Roberts T.; Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK–ERK independent mechanism. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (14), 7783. 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. R.; Ranger A. M.; Lin M. Z.; Sturgill J. F.; Ma Y.-C.; Cowan C. W.; Dikkes P.; Korsmeyer S. J.; Greenberg M. E. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell 2002, 3 (5), 631. 10.1016/S1534-5807(02)00326-X. [DOI] [PubMed] [Google Scholar]
- Wang H.-G.; Pathan N.; Ethell I. M.; Krajewski S.; Yamaguchi Y.; Shibasaki F.; McKeon F.; Bobo T.; Franke T. F.; Reed J. C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284 (5412), 339. 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Niemi N. M.; MacKeigan J. P. Mitochondrial phosphorylation in apoptosis: flipping the death switch. Antioxid. Redox Signaling 2013, 19 (6), 572. 10.1089/ars.2012.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.; Thompson C. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000, 7 (12), 1182. 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- Asai A.; Qiu J.-h.; Narita Y.; Chi S.; Saito N.; Shinoura N.; Hamada H.; Kuchino Y.; Kirino T. High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 1999, 274 (48), 34450. 10.1074/jbc.274.48.34450. [DOI] [PubMed] [Google Scholar]
- Slupe A. M.; Merrill R. A.; Flippo K. H.; Lobas M. A.; Houtman J. C.; Strack S. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J. Biol. Chem. 2013, 288 (17), 12353. 10.1074/jbc.M113.459677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M.; Reddy P. H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21 (11), 2538. 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X.; Qvit N.; Su Y.-C.; Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126 (3), 789. 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faelber K.; Held M.; Gao S.; Posor Y.; Haucke V.; Noé F.; Daumke O. Structural insights into dynamin-mediated membrane fission. Structure 2012, 20 (10), 1621. 10.1016/j.str.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Fröhlich C.; Grabiger S.; Schwefel D.; Faelber K.; Rosenbaum E.; Mears J.; Rocks O.; Daumke O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32 (9), 1280. 10.1038/emboj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S. L. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369 (23), 2236. 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; Karbowski M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6 (8), 657. 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- Breckenridge D. G.; Stojanovic M.; Marcellus R. C.; Shore G. C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160 (7), 1115. 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M.; Mathai J. P.; McBride H. M.; Shore G. C. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005, 24 (8), 1546. 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou J.-C.; Youle R. Which came first, the cytochrome c release or the mitochondrial fission?. Cell Death Differ. 2006, 13 (13), 1291–1295. 10.1038/sj.cdd.4401985. [DOI] [PubMed] [Google Scholar]
- Chen H.; Detmer S. A.; Ewald A. J.; Griffin E. E.; Fraser S. E.; Chan D. C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160 (2), 189. 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discovery 2016, 15 (1), 19. 10.1038/nrd.2015.5. [DOI] [PubMed] [Google Scholar]
- Burté F.; Carelli V.; Chinnery P. F.; Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11 (1), 11. 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Kaur H.; Sarmah D.; Saraf J.; Vats K.; Kalia K.; Borah A.; Yavagal D. R.; Dave K. R.; Ghosh Z.; Bhattacharya P. Noncoding RNAs in ischemic stroke: time to translate. Ann. N. Y. Acad. Sci. 2018, 1421 (1), 19–36. 10.1111/nyas.13612. [DOI] [PubMed] [Google Scholar]
- Feigin V. L.; Forouzanfar M. H.; Krishnamurthi R.; Mensah G. A.; Connor M.; Bennett D. A.; Moran A. E.; Sacco R. L.; Anderson L.; Truelsen T. Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet 2014, 383 (9913), 245. 10.1016/S0140-6736(13)61953-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmah D.; Agrawal V.; Rane P.; Bhute S.; Watanabe M.; Kalia K.; Ghosh Z.; Dave K. R.; Yavagal D. R.; Bhattacharya P. Mesenchymal Stem Cell therapy in Ischemic stroke: A meta-analysis of preclinical studies. Clin. Pharmacol. Ther. 2018, 103 (6), 990–998. 10.1002/cpt.927. [DOI] [PubMed] [Google Scholar]
- Chamorro Á.; Dirnagl U.; Urra X.; Planas A. M. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15 (8), 869. 10.1016/S1474-4422(16)00114-9. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P.; Pandey A. K.; Paul S.; Patnaik R. Melatonin renders neuroprotection by protein kinase C mediated aquaporin-4 inhibition in animal model of focal cerebral ischemia. Life Sci. 2014, 100 (2), 97. 10.1016/j.lfs.2014.01.085. [DOI] [PubMed] [Google Scholar]
- Sarmah D.; Kaur H.; Saraf J.; Pravalika K.; Goswami A.; Kalia K.; Borah A.; Wang X.; Dave K. R.; Yavagal D. R.. Getting Closer to an Effective Intervention of Ischemic Stroke: The Big Promise of Stem Cell. Transl. Stroke Res. 2017, 10.1007/s12975-017-0580-0. [DOI] [PubMed] [Google Scholar]
- Bhosale G.; Sharpe J. A.; Sundier S. Y.; Duchen M. R. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350 (1), 107. 10.1111/nyas.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanero S.; Santro T.; Arumugam T. V. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem. Int. 2013, 62 (5), 712. 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Niatsetskaya Z. V.; Sosunov S. A.; Matsiukevich D.; Utkina-Sosunova I. V.; Ratner V. I.; Starkov A. A.; Ten V. S. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia–ischemia in neonatal mice. J. Neurosci. 2012, 32 (9), 3235. 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J.; Tan Y.-W.; Hagenston A. M.; Martel M.-A.; Kneisel N.; Skehel P. A.; Wyllie D. J.; Bading H.; Hardingham G. E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeris T.; Baines C. P.; Krenz M.; Korthuis R. J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos Picanc L.; Ozela P.; Pinheiro A.; Padilha E.; Braga F.; Dos C. S.; Rosa J. Alzheimer’s disease: A review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2016, 23, 1. 10.2174/0929867323666161213101126. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association. Alzheimer's Dementia 2017, 13 ( (4), ), 325. 10.1016/j.jalz.2017.02.001. [DOI] [Google Scholar]
- Ittner L. M.; Götz J. Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12 (2), 67. 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Bloom G. S. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology 2014, 71 (4), 505. 10.1001/jamaneurol.2013.5847. [DOI] [PubMed] [Google Scholar]
- Abdul H. M.; Sama M. A.; Furman J. L.; Mathis D. M.; Beckett T. L.; Weidner A. M.; Patel E. S.; Baig I.; Murphy M. P.; LeVine H. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 2009, 29 (41), 12957. 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012, 15 (3), 349. 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Popugaeva E.; Pchitskaya E.; Bezprozvanny I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease–A therapeutic opportunity?. Biochem. Biophys. Res. Commun. 2017, 483 (4), 998. 10.1016/j.bbrc.2016.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacabelos R. Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18 (3), 551. 10.3390/ijms18030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. J.; Lee V. M.-Y.; Trojanowski J. Q. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14 (9), 626. 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Z. S.; Neugebauer V.; Dineley K. T.; Kayed R.; Zhang W.; Reese L. C.; Taglialatela G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J. Neurochem. 2012, 120 (3), 440. 10.1111/j.1471-4159.2011.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraveo G.; Auluck P. K.; Whitesell L.; Chung C. Y.; Baru V.; Mosharov E. V.; Yan X.; Ben-Johny M.; Soste M.; Picotti P. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (34), E3544. 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringsheim T.; Wiltshire K.; Day L.; Dykeman J.; Steeves T.; Jette N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27 (9), 1083. 10.1002/mds.25075. [DOI] [PubMed] [Google Scholar]
- Bates G. P.; Dorsey R.; Gusella J. F.; Hayden M. R.; Kay C.; Leavitt B. R.; Nance M.; Ross C. A.; Scahill R. I.; Wetzel R. Huntington disease. Nature reviews Disease primers 2015, 1, 15005. 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- Xifró X.; García-Martínez J. M.; Del Toro D.; Alberch J.; Pérez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J. Neurochem. 2008, 105 (5), 1596. 10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- Kratter I. H.; Zahed H.; Lau A.; Tsvetkov A. S.; Daub A. C.; Weiberth K. F.; Gu X.; Saudou F.; Humbert S.; Yang X. W. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J. Clin. Invest. 2016, 126 (9), 3585. 10.1172/JCI80339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda J. R.; Pardo R.; Zala D.; Yu H.; Humbert S.; Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington’s disease. Mol. Brain 2009, 2 (1), 33. 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M.; Noël A.; Julien C.; Cisbani G.; Milot-Rousseau P.; Morin F.; Dickler M.; Goupil C.; Bezeau F.; Poitras I. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum. Mol. Genet. 2015, 24 (1), 86. 10.1093/hmg/ddu456. [DOI] [PubMed] [Google Scholar]
- Arthur K. C.; Calvo A.; Price T. R.; Geiger J. T.; Chio A.; Traynor B. J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. 10.1038/ncomms12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E.; Majounie E.; Waite A.; Simón-Sánchez J.; Rollinson S.; Gibbs J. R.; Schymick J. C.; Laaksovirta H.; Van Swieten J. C.; Myllykangas L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72 (2), 257. 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W.; Philips T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14 (4), 248. 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Turner M. R.; Hardiman O.; Benatar M.; Brooks B. R.; Chio A.; De Carvalho M.; Ince P. G.; Lin C.; Miller R. G.; Mitsumoto H. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12 (3), 310. 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko N. F.; Saxton A. D.; McMillan P. J.; Strovas T. J.; Currey H. N.; Taylor L. M.; Wheeler J. M.; Oblak A. L.; Ghetti B.; Montine T. J. The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol. 2016, 132 (4), 545. 10.1007/s00401-016-1600-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. M.; Billington E.; Reyes A.; Notarianni T.; Sage J.; Agbas E.; Taylor M.; Monast I.; Stanford J. A.; Agbas A.. Impaired Cu–Zn Superoxide Dismutase (SOD1) and Calcineurin (Cn) Interaction in ALS: A Presumed Consequence for TDP-43 and Zinc Aggregation in Tg SOD1 G93A Rodent Spinal Cord Tissue. Neurochem. Res. 2018. 10.1007/s11064-017-2461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez R.; Stys P. K.; Vivien D.; Lipton S. A.; Docagne F. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15 (10), 1089. 10.1016/S1474-4422(16)30165-X. [DOI] [PubMed] [Google Scholar]
- Evonuk K. S.; Baker B. J.; Doyle R. E.; Moseley C. E.; Sestero C. M.; Johnston B. P.; De Sarno P.; Tang A.; Gembitsky I.; Hewett S. J. Inhibition of system xc– transporter attenuates autoimmune inflammatory demyelination. J. Immunol. 2015, 195 (2), 450. 10.4049/jimmunol.1401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S.; Yang H.; Kim B. S.; Chu K.; Lee S. K.; Jeon D. The immunosuppressant cyclosporin A inhibits recurrent seizures in an experimental model of temporal lobe epilepsy. Neurosci. Lett. 2012, 529 (2), 133. 10.1016/j.neulet.2012.08.087. [DOI] [PubMed] [Google Scholar]
- Furman J. L.; Sompol P.; Kraner S. D.; Pleiss M. M.; Putman E. J.; Dunkerson J.; Abdul H. M.; Roberts K. N.; Scheff S. W.; Norris C. M. Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci. 2016, 36 (5), 1502. 10.1523/JNEUROSCI.1930-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C.; Yao X.; Engel T.; Tiwari D.; Xing L.; Rowley S.; Danielson S. W.; Thomas K. T.; Jimenez-Mateos E. M.; Schroeder L. M. MicroRNA-mediated downregulation of the potassium channel Kv4. 2 contributes to seizure onset. Cell Rep. 2016, 17 (1), 37. 10.1016/j.celrep.2016.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi J. R.; Sayegh M. H.; Mallat S. G. Calcineurin inhibitors: 40 years later, can’t live without···. J. Immunol. 2013, 191 (12), 5785. 10.4049/jimmunol.1390055. [DOI] [PubMed] [Google Scholar]
- Ishikura K.; Ikeda M.; Hamasaki Y.; Hataya H.; Shishido S.; Asanuma H.; Nishimura G.; Hiramoto R.; Honda M. Posterior reversible encephalopathy syndrome in children: its high prevalence and more extensive imaging findings. Am. J. Kidney Dis. 2006, 48 (2), 231. 10.1053/j.ajkd.2006.04.076. [DOI] [PubMed] [Google Scholar]
- Pflugrad H.; Schrader A. K.; Tryc A. B.; Ding X.; Lanfermann H.; Jäckel E.; Schrem H.; Beneke J.; Barg-Hock H.; Klempnauer J. Longterm calcineurin inhibitor therapy and brain function in patients after liver transplantation. Liver Transplantation 2018, 24 (1), 56. 10.1002/lt.24984. [DOI] [PubMed] [Google Scholar]
- Bindu P. S.; Sinha S.; Taly A. B.; Christopher R.; Kovoor J. M. Cranial MRI in acute hyperammonemic encephalopathy. Pediatric neurology 2009, 41 (2), 139. 10.1016/j.pediatrneurol.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Pruitt A. A.; Graus F.; Rosenfeld M. R. Neurological complications of solid organ transplantation. Neurohospitalist 2013, 3 (3), 152. 10.1177/1941874412466090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; LeCorchick S.; Gupta G. Belatacept as an alternative to calcineurin inhibitors in patients with solid organ transplants. Front. Med. 2017, 4, 60. 10.3389/fmed.2017.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperman L.; Moonka D.; Sebastian A.; Sher L.; Marotta P.; Marsh C.; Koneru B.; Goss J.; Preston D.; Roberts J. P. Calcineurin Inhibitor–Free Mycophenolate Mofetil/Sirolimus Maintenance in Liver Transplantation: The Randomized Spare-the-Nephron Trial. Liver transplantation 2013, 19 (7), 675. 10.1002/lt.23658. [DOI] [PubMed] [Google Scholar]
- Webber A. B.; Vincenti F. An Update on Calcineurin Inhibitor–Free Regimens: The Need Persists, but the Landscape has Changed. Transplantation 2016, 100 (4), 836. 10.1097/TP.0000000000000872. [DOI] [PubMed] [Google Scholar]
- Goede L.; Pflugrad H.; Schmitz B.; Tryc A. B.; Barg-Hock H.; Klempnauer J.; Weissenborn K.; Lanfermann H.; Ding X.-Q. Neurotoxic Side Effects of Calcineurin Inhibitors in Patients After Liver Transplantation: Preliminary Results of a Quantitative MRI Study of the Brain. J. Clin. Exp. Hepatol. 2017, 7, S31. 10.1016/j.jceh.2017.01.042. [DOI] [Google Scholar]