

Abstract
Recently, automated robotic systems have become very efficient, thanks to improved coupling between sensor systems and algorithms, of which the latter have been gaining significance thanks to the increase in computing power over the past few decades. However, intelligent automated chemistry platforms for discovery orientated tasks need to be able to cope with the unknown, which is a profoundly hard problem. In this Outlook, we describe how recent advances in the design and application of algorithms, coupled with the increased amount of chemical data available, and automation and control systems may allow more productive chemical research and the development of chemical robots able to target discovery. This is shown through examples of workflow and data processing with automation and control, and through the use of both well-used and cutting-edge algorithms illustrated using recent studies in chemistry. Finally, several algorithms are presented in relation to chemical robots and chemical intelligence for knowledge discovery.
Short abstract
Advances in algorithms and automatic data acquisition systems promise to help chemists improve productivity and to construct systems to work autonomously leading to new discoveries in chemical space.
An algorithm is a set of rules that determines the execution of a sequence of operations. As they are fundamental theoretical constructs they are of great use, and the earliest recorded algorithms detailing procedures to solve mathematical problems date back almost 4000 years.1 In the field of chemistry, the desire for repeatability, control, and correlation of sensor outputs with inputs exemplifies the need for well-defined control and decision-making systems. Algorithms in chemistry are often implemented in real-world chemical systems, and so their development is affected by hardware, physical and computational resources, as well as chemical handling constraints. This leads to new technologies being quickly utilized for chemical purposes. An early case is the use of punch cards at the advent of digital computing for analysis of mass spectra.2 With the increase of computing power at an ever-diminishing cost, chemistry has gained much from new instrumentation, data collection and analysis, better scientific communication, and many other avenues of improvement. In recent years there have been breakthroughs in the ability of computers to complete tasks that once seemed the exclusive purview of humans, such as image processing3 and playing games.4 In this Outlook we describe, through real-case examples, how algorithms could assist in current chemical research through increased productivity and also how the proper use of algorithms coupled with integrated platforms can expand the ability to search for new chemical knowledge.
Current Uses of Algorithms in Chemistry
Algorithms for use in chemistry can be separated into three classes: menial, assistive, and enabling. The menial are mainly low-level algorithms such as those controlling syringe pumps for liquid handling, whose primary purpose is to replace manual technical work. Other algorithms that belong to this group are higher-level algorithms for monitoring and control. The assistive class primarily improves the intellectual productivity of the human chemist; fundamentally, these algorithms reduce the cognitive load on the user. A common usage case is in the evaluation and processing of analytical measurements,5 for example, using wavelet transforms to treat and extract data from spectra.6 In this case, an algorithm interprets the data and assigns peaks based on the available database. Other algorithms help to visualize, manipulate, and extract chemical information from representations of molecules.7,8 The integration of these algorithms allows for sophisticated platforms to be built which perform chemistry without human intervention;9−11 a plot of a simulated optimization sequence undergone in such a system is shown in Figure 1. The optimization algorithm used is called the Stable Noisy Optimization by Branch and Fit (SNOBFIT).11 It combines both local and global searching to find the maximal value in the available search space in the most efficient manner. In this example the maximal value sought was the highest yield, the search space defined over ranges of concentration, pH, and temperature, and efficiency in this case is conducting the least amount of experiments. The enabling algorithms are the most powerful as they accomplish tasks that humans are incapable of. This is often due to the amount of chemical data available reaching levels beyond the ability of any human to process (e.g., chemical databases such as Reaxys). Therefore, many algorithms are being designed or co-opted to deal with such a large wealth of information and data.
Figure 1.
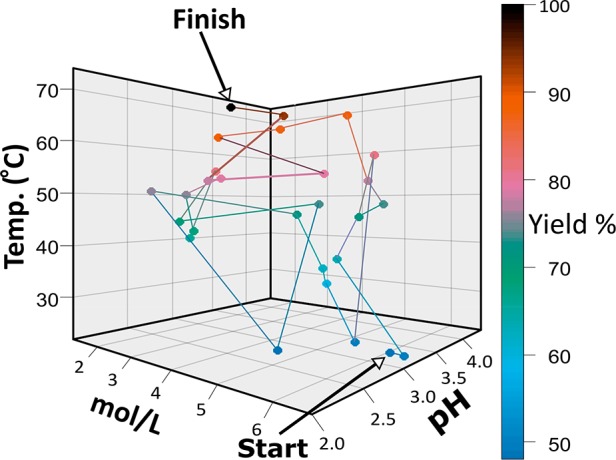
Simulation of a SNOBFIT optimization algorithm displayed for three parameters: pH, mol/L, and temperature. The system is optimized for the highest product yield. The optimization initiates from a random starting point (yield ∼50%) until the final point (yield ∼95%). The line shows the steps taken between points during the optimization.
Big Data and Automatic Data Analysis Including Feature Extraction
“Big data” is a growing area of science with great significance in the field of chemistry (i.e., drug discovery).12−14 Big data means not only a large amount of data but also usually more varied data. The Web provides access to a rich selection of diverse chemical data sources (some of the most common can be found in Table 1 or in the literature15). A crucial factor is the availability of representations of chemical data, predominantly molecular structure; notably simplified molecular input line entry specification (SMILES),8 a line notation for molecules; Mol,16 property information about atoms, bonds, and connectivity of molecules; structure data format (SDF), text format representing multiple chemical structures; and many more as described in the literature.17,18 The fundamental benefits of using such databases are the huge number of samples presented in a consistent manner and scalable with clear barriers to access, if any. An important caveat is that the quality of the data can vary greatly as most of the data is a collection of reported results, most of which are not independently verified. Because of the large amounts of available data, scientists must identify which data to mine and how to preprocess it for their research purposes. In addition to existing stored data, the combination of experimental chemical platforms with digitization produces large amounts of new data with the potential to promote cooperation with business and academia on the characterization and interpretation of the data.19 The tasks have growing significance for computational and statistical analysis arising from the size, complexity, and heterogeneity of available data sets, and could be aided using adequate algorithms.20 One such common task in databases is knowledge discovery which can refer to the use of methodologies from virtual screening, machine learning, statistics, and pattern recognition. For example, the retrosynthetic software Chematica uses chemical reaction information to search for new synthesis reaction routes.19,21 A different approach for the same task has recently also shown inference of chemical reactivity from knowledge graphs.22
Table 1. Examples of Some Available Databases and Software Packages Used in Chemistry.
| name | properties | URL |
|---|---|---|
| Databases | ||
| PubChem23 | substance, compound, and BioAssay | http://pubchem.ncbi.nlm.nih.gov/ |
| ChEMBL17 | bioactivity data from the medicinal chemistry literature | https://www.ebi.ac.uk/chembldb/ |
| DrugBank18 | chemical, pharmacological, and pharmaceutical data | http://www.drugbank.ca/ |
| SDBS | integrated spectral database system for organic compounds | http://sdbs.db.aist.go.jp/ |
| ChemSpider24 | chemical structure database | http://www.chemspider.com/ |
| Beilstein25 | organic-chemistry-related reactions and substances | http://www.reaxys.com/ |
| Software | ||
| Vls3d26 | ligand-based, structure-based, screening utilities, binding pockets | http://www.vls3d.com/ |
| PyRx27 | virtual screening for computational drug discovery that can be used to screen libraries of compounds against potential drug targets | http://pyrx.sourceforge.net/ |
| DOCK Blaster28 | service for structure-based ligand discovery | http://blaster.docking.org/ |
| Open Babel8 | chemical toolbox | http://openbabel.org/wiki/Main_Page |
| ChemDraw29 | molecule editor | https://www.cambridgesoft.com/ |
One class of algorithms that can process so-called “big data” are neural nets. A neural net is made of highly connected nonlinear logical units where each connection has parameter that is adjusted as part of the training phase. The number of connections and therefore also parameters can reach into the thousands. Following a period of training where the network is taught a known relation between inputs and outputs it can be used to make prediction on new inputs. This approach allows the algorithm to implement mathematical operations such as classification of chemicals based on their chemical structure/behavior; modeling of relationships between different structures; and storage and retrieval of given information. Indeed, chemists have been working with neural nets for decades,30 and with the recent resurgence of neural nets in deep learning,31 new prospects and applications are again gaining traction.32,33 Large amounts of data improve the ability of neural nets, and so the growing amount of available chemical information allows researchers to construct new ways of performing and analyzing chemistry.34,35 Some algorithms even build up their own information about the space of chemistry from first-principles with little guidance from established chemical knowledge.15
One of the major uses of big data-driven chemistry is virtual screening (VS), which describes the usage of computational algorithms and models for identification of bioactive molecules. Generally, compounds with common physicochemical properties are combined into assembled libraries/databases. This allows for classification of big data sets of chemical compounds according to their probability to match a criterion, for example, bioactivity where top performing compounds are tested in bioassays.36 Most VS approaches depend on the application of descriptors of molecular structure and properties. The accumulated knowledge from VS techniques can be used to propose many possible molecules according to chosen criteria. VS has been successfully applied together with high-throughput screening (HTS). HTS allows for more cost-effective research and development in chemical laboratories by running a large number of experiments.37,38
The combination of chemical experiments alongside with virtual screening allows for a more targeted and efficient use of the large number of experiments that can be conducted by HTS. Nevertheless, as vast areas of chemical compound space, which is the relevant search space, do not contain useful molecules, it is vital to filter chemical space in order to identify the molecules with a high likelihood of selectivity. Filtering out molecules that are not likely to be of use can be achieved by a similarity search. In this process, defined search criteria allow for the identification of compounds that are similar in their required properties to those stored in a database. Other methods that could expedite and increase the efficiency and accuracy of screening include the following: privileged structures,39 fingerprints,40,41 single similarity measure,42 pharmacophore-based methods (centered on geometric and topological constraints),43 quantitative structure–activity relationship (QSAR),44 “forward” and “backward” filtering as described by Klebe,45 and many more as described in refs (46−48).
One of the objectives of chemical research is to produce reliable data to enable knowledge discovery. The main challenges to achieving this goal are validating the data and giving a statistically significant interpretation. For the former using data of bad quality will at best yield nothing and at worst produce an erroneous result. The latter is important since in chemistry the analysis of data is in service of increased understanding which must rely on statistically significant results. Substantial work on these issues is being done in the field of chemometrics.49,50 This discipline utilizes statistical approaches to demonstrate, interpret, and rationalize the results of measurements of chemical data.51 Various multivariate data analysis (MVA) or pattern recognition52 algorithms are covered by chemometrics, which can be divided into two groups: unsupervised, which allows searching for hidden structures from unlabeled data, and supervised, which mainly focuses on classification or prediction of new samples based on categorized samples. These algorithms can assist in interpreting the outputs at various stages of processing pipelines (Figure 2) thereby making it easier for the user to focus on a higher level of abstraction.6,53
Figure 2.
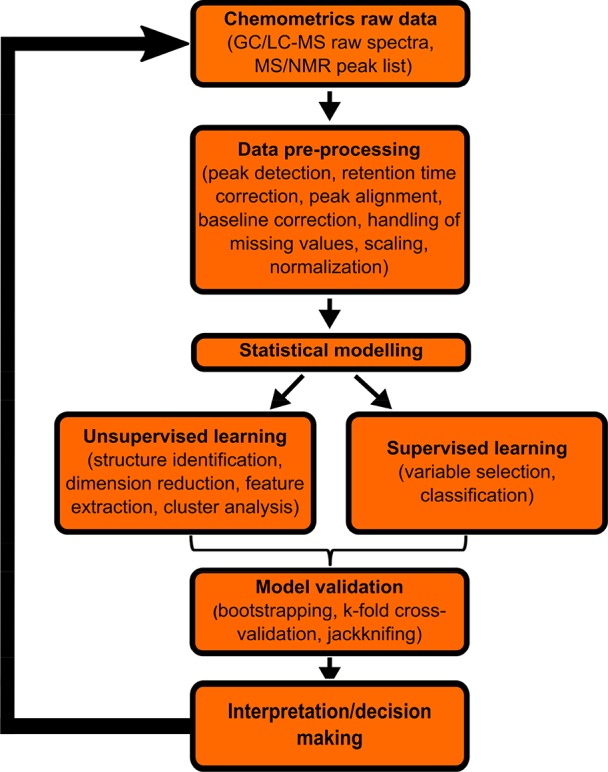
Diagram demonstrating a standard chemometrics workflow including data processing. Different data inputs are first preprocessed into compatible data matrices, followed by specific problem-related algorithms that are applied for data modeling and validation. At the end of a given analysis, the results go through interpretation followed by decision making.
Chemometrics approaches such as principal component analysis (PCA), cluster analysis,54 multidimensional scaling (MDS), and partial least-squares (PLS)5,55 allow chemists to recognize potential outliers and specify whether there are any patterns or trends in the data. All these methods reframe the space representation of the data according to criteria which are different for each method. PCA attempts to relate the variance in the data; MDS rearranges the data by similarity, and PLS finds a linear relation between the input and output variables. Furthermore, methods like PCA and MDS can be used for feature selection and dimensionality reduction of large and complex data sets. Alternatively, regression algorithms such as principal component regression, ridge regression, stepwise regression, robust regression, and partial least-squares regression,56 which deal with outputs that are continuous, could be helpful in decision making involving online monitoring or in process control of a given metric.5 A major focus in this area is on feature extraction. Feature extraction is a critical step in knowledge. For this process, a variety of algorithms are used to transform a large data set into reduced features called “latent variables”. A selection of latent variables is expected to cover essential information derived from the original data, so that the chosen goal can be achieved by using the reduced representation of the original data set (Figure 3). In other words, the process reduces the influence of certain parameters/variables and focuses on those that provide most of the information captured by the first several latent variables. The automatic or manual mining of features can represent the conclusion of the research question or a processing step in understanding the observed chemical system.57 When the data is labeled, the chemical classification problem can be solved by application of supervised methods which cover traditional discriminatory algorithms [linear discriminant analysis (LDA), partial least-squares-discriminant analysis (PLS-DA)] and various machine learning methods (e.g., support vector machines, random forests).58−61 Other knowledge discovery algorithms successfully applied in chemistry include k-nearest neighbors, neural networks,62 genetic algorithms,63,64 Gaussian mixture models, and many more as described in refs (65−67). Additionally, the subject has been repeatedly reported in the literature.68,69
Figure 3.

Feature extraction scheme. The original data with two classes (red squares vs blue circles) and multiple variables [left-hand side; as an example, two variables (V2 and V5) are plotted against each other resulting in no separation/pattern, and in fact no other selection would show a clear separation] are transformed using an unsupervised method (here PCA) to a new feature space (right-hand side; Scores plot) such that the first two latent variables encompass most relevant information (variance) that clearly separates two groups in a far reduced number of variables (indicating separation/pattern). In this example plotting of original data and finding which variables plotted against each other provides any pattern would be very difficult whereas with PCA requires only first two variables to answer the question if there is any pattern/separation.
Automation and Control
The advantages of automating chemical processes are numerous. They include a substantial increase in scale, improved precision, a reduction in the amount and effect of uncontrollable variables, better reproducibility, and continuous feedback. The desirability of these traits has brought investment from large pharmaceutical companies to build highly automated systems.70,71 Automating chemical processes is also prominent in chemical research, enabling faster and more precise scientific inquiries.72,73 The abilities gained by automation lend themselves to be combined with statistical methods for optimization of chosen chemical parameters in chemical space.
The complex tasks of identifying significant parameters for optimizing outcomes and exploring regions of interest in chemical space are required for effective experiments and knowledge discovery. In essence a given chemical space is being searched either to find an optimal point or to gather more information about the areas of interest in the space. A tool for that task is design of experiments (DoE), which helps in recognizing the most relevant parameters. The numerous statistical methods in use today for DoE are linked to the work of R. A. Fisher starting from 1935. Fisher demonstrated the importance of effective randomization, repetition, blocking, orthogonality, and factorial experiments in order to increase the sensitivity of designed experiments. Fisher indicated that the key factor in DoE is to apply valid and efficient experiments that will produce quantitative results to support decision making.74 One of the biggest advantages of DoE is that it allows researchers to decide which reactions and conditions to focus on. This can be achieved through the generation of a mathematical model/design space which exposes a relationship between factors affecting a process and the output of that process. In other words, DoE (Figure 4) could reveal which factors impact the outcome and determine optimal parameters (time, temp, quantity, pH, etc.).69
Figure 4.

Schematic example of an experimental design in which the samples are selected according to a full factorial design. Hypotheses are based on limited information (red dots) which is then used by a model to predict the response (blue space) for a combination of the different factors (arrows). Subsequent samples (red dots) are chosen according to a scheme where all factors of interest are varied simultaneously. First the maximum and minimum values for examined factors are selected including center point for both (left-hand side plot), followed by estimation of the center points for selected factors (middle figure). This allows for estimating factor directions (right-hand side figure), which facilitate the use and interpretation of multivariate statistical models. The important impacts from single factors and relations between factors can subsequently be estimated. As more data is collected the model becomes more precise.
However, one also needs to take into account that, in DoE, no one method offers a complete solution, and significant work is needed to find the many factors required for discovery. Hence, the algorithms used for searching the space may be simple (e.g., screening design of experiment such as a fractional factorial design) or verbose (e.g., full factorial design).75 A good DoE will allow for the robust comparison of experimental outputs and provide good sample size requirements. Various DoE algorithms have been applied in chemistry such as 2-level factorial, Plackett-Burman, full factorial, Box–Behnken, Doehlert, Mixture, and many more.74 A selection of other search algorithms such as simplex, multidirectional search, parallel simplex search, and more are described in a report by Dixon and Lindsey.76 The report also shows that such approaches have been used effectively in chemistry-related studies to maximize the output of information with a minimal amount of computing power and experimental resources. Performing experiments in a given chemical space and validating the results can benefit greatly from using DoE techniques.
Chemical Robots
Recent advances in the design and application of algorithms, big data, and automation and control systems may allow the development of intelligent chemical robots that can target discovery. A “chemical robot” can be defined as any controllable agent capable of performing chemistry. Under this definition, there are several different types of systems that fall into this category. This would include simple systems that are static, yet offer the capability of using their inherent properties to modify the chemical system by performing the experiments in designed 3D printed devices.77,78 More complex systems use integration of analytical instruments into the experimental platform at the cost of requiring bespoke fabrication and construction.79 At the other end of the spectrum, there are many different commercial systems available today80,81 which offer modularity, reliability and ease of use, at the cost of high expense and lack of integrability. However, most systems in use in research are built in-house to avoid these shortcomings. They offer flexibility and a focus on making a robot that is as close as possible to the right tool for the job; there are no superfluous abilities or complexity, as that would waste resources. An example model of such a system for flow chemistry can be seen in Figure 5.
Figure 5.
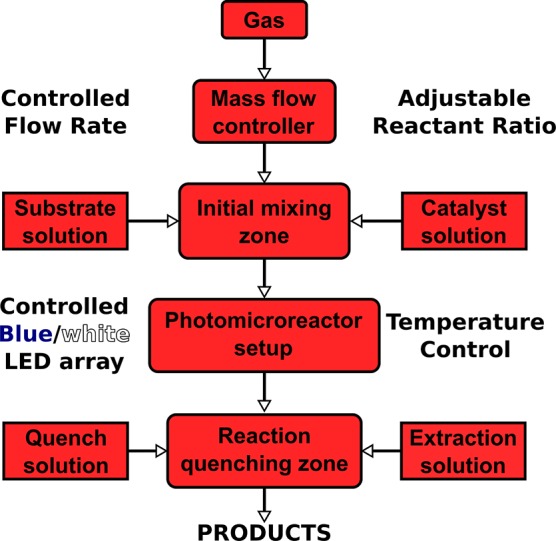
Representation of a typical gas–liquid photoredox continuous flow system for gas–liquid photocatalytic transformations. The system starts from the top with a reactant gas with a mass flow controller. The gas enters a mixing zone before entering a photo-microreactor, often assembled from a coiled PFA capillary with an LED array as the light source. After the reaction quenching zone the product solution can be obtained.
Figure 6 shows a scheme for an automated system for the exploration of an inorganic polyoxometalate chemical space involving many possible input materials.81 The computer controls the pumps dispensing the starting materials and so can perform an array of reactions with different starting material ratios which resulted in the discovery of several new inorganic compounds. The drawbacks of the systems81−84 include technical expense and numerous engineering challenges. Beyond solving the specific problems required by the various chemical operations, a major hurdle is the difficulty in integration of the various kinds of subsystems. Many subsystems, such as analytical tools and material handling, do not offer an industry-wide standard for control or even a physical interface. Thus, much work is required to integrate these devices into a larger system, especially across different vendors. It is hoped that, with time, the demand for simpler subsystems with the ability to easily integrate between vendors, as well as different kinds of modules, will result in more integration-focused products with cross-industry standardization.
Figure 6.

Illustration of an automated system for the exploration of an inorganic polyoxometalate chemical space with a high number of possible input materials.
A lot of work is being done to develop robots with ever-increasing complexity and ability. Recently systems with differing modules of chemical operations have been created38,85,86 that enable several automated chemical reactions, including workup of products. Some robots are even able to conduct end-to-end pharmaceutical processes, including purification and formulation,87−89 as depicted in Figure 7. Despite the high level of engineering in these systems and their expense, they have a lot of potential if they could be generalized. However, looking past the improvements in engineering, many systems are not reaching the fullest potential of chemical robots. Robots should not be merely a combination of modules that perform chemistry. They can be enablers of improved chemistry, which in turn can enable better chemical robots.90 If fit-for-purpose chemistry can be coupled to enhanced capabilities of the robotic system, then the capabilities of chemical robots can be advanced. Instead of performing the same chemistry but only in an automated manner the chemistry can be adapted to the abilities of the chemical robots and thereby acting as a multiplier for its effectiveness.
Figure 7.

Flowchart of a flexible modular chemical synthesis system (left diagram). There are seven different modules used in four different synthesis procedures. The modularity enables the use of only a subset of the modules as needed to perform the synthesis. Examples of chemicals made by such a system are diphenhydramine hydrochloride (A) and diazepam (B).
Chemical Intelligence
There is an ongoing drive toward improved automation. On one front, systems are becoming cheaper, more common, and easier to use. On a different front, researchers are working to extend the capabilities of such automated systems.91 Beyond the engineering effort going into this field there is a more profound enhancement that automated systems require: autonomy. In addition to the layers of systems, components, and algorithms capable of automatic operation there is scope to add another layer of algorithms that will give the overall system the ability to decide on its own which experiments to execute once it is set in motion.
An obvious approach to introducing autonomy is by giving the system some level of chemical understanding. To do so, first the standard chemical representation of information needs to be digitized. Efforts to standardize this fundamental requirement have produced several previously mentioned widely used representations such as SMILES,8 InChI,92,93 and Mol.16 Once this information is digitized, it becomes possible to use supervised learning for prediction. In chemistry, many types of systems, also called expert systems, use accumulated knowledge to evaluate the likely outcomes of human or computer generated hypotheses. A recent example of this approach is to use a large database of experimental results along with digital features of the chemicals involved to predict possible reactions;94 this work is also noteworthy for using data about negative results as well as positive results. Another clear yet difficult usage of these techniques is in retrosynthesis, finding synthesis routes that match given criteria. These efforts began many decades ago95 and are still ongoing.96,97 The operations performed by these systems are computationally intensive and therefore are often considered independently of a running experimental system.19 We, however, envision the use of these systems in close coupling with a running system in real-time so that the theoretical predictions are used to direct experiments, and the feedback from real-world data can be used to give fine-grained information for the expert system to improve its output.
However, not every chemical system is reliable. This is particularly true for scientific research as there cannot be experimental information for chemicals, reactions, and methods created as part of the research. In fact, the push to expand scientific understanding demands that we investigate systems with partial or no information. In that case, understanding of the chemical system is comparable to conducting a search within the accessible chemical space with no prior knowledge. We can define the parameters of a chemical system a set of input parameters and associate their relation to the resulting state of the chemical system. This allows us to map any set of input parameters of a given size. Different sets of input variables can have the same output, yet the reverse is not allowed; there cannot be more than one output from the same set of input values. All the states and the definition of their inputs comprise a space which can be viewed as a surface (see left plot in Figure 8), for which each point has an outcome associated with it, which is the chemical and physical state. The point on the surface is the chemical space resulting from performing an experiment with specific input parameters.
Figure 8.

Surface of a model function with two continuous input parameters. The left shows the real space where the outcome is the chemical system, and on the right are three different plots originating from approximating the real system with different utility functions M1–M3, where M1 is the difference between peaks in the mass spectrum, M2 is the amplitude of the UV/vis spectrum at a given wavelength, and M3 is a combination of the former two.
The outcome of any experiment in a chemical system is the physical and chemical state of the all constituent parts of the system from the lowest level of molecules up to clusters, micelles, and any other compound structure. The full richness and information about these systems often cannot be evaluated exactly. First, there is a matter of output variability, as even conducting a repetition of an experiment with the same input parameter values will likely yield an outcome that is within a distribution of outcomes. Second, the chemical and physical state of a system is difficult to know exactly down to the individual molecule level, thus introducing experimental uncertainty. Although the entire complete chemical state of a system is likely hard to measure, there is a practical level of knowledge that can be reached. For a desired level of knowledge about the chemistry, there is undoubtedly a set of measurements that contain the relevant information about the state. The measurements represent the real outcome by a mapping function. This mapping function relates the results of the measurements to the desired information about the outcome. A schematic example of the results from different utility functions can be seen in Figure 8. When the input parameters are designed or otherwise known, understanding the chemical system is the same as learning these two functions: the space function which would give the results of measurements for any given input point, and the mapping function which ties the measurements to the representative chemical outcome. Presenting the experimental chemical system in this way is a prerequisite for an autonomous system to be able to conduct experiments that improve the chemical understanding of the system especially when aiming at discovery.
Algorithm Design for Chemical Discovery
The choice of algorithms to use for discovery in chemical systems can lead in many different directions with many forks in the road. By understanding the characteristics of the different chemical spaces and algorithms we can make the selection easier. Given the vast size of organic chemical space (mw up to 500), it is estimated that more than 1060 molecules98 might be stable, with a limited range of conditions for reactions between these molecules; the space is in essence extremely sparse. This stems from a basic truth that most molecules, under most conditions, do not react with most other molecules. This leaves many possible combinations of reaction conditions and starting materials empty. The main problem with sparsity is that it becomes difficult to get statistically significant understanding about the space with which to make better decisions. An additional problem in this type of space is that for chemical systems we have additional constraints such as time, expense, and availability. Given that the clear majority of chemical experiments are destructive to the starting materials, this forms a hard limit on the total number of experiments that can be done. If possible, the design of the chemistry to use in a system should use heuristics to focus on the options that reduce sparsity. In fact, in most chemical systems this is an intuitive method. A chemist uses their knowledge of chemical reactivity to choose a set of chemicals and conditions that constitute a portion of the chemical space that is dense. In experimental terms, that means that a significant portion of the possible reactions produce a measurable result. Some spaces, however, either cannot be designed or cannot be guaranteed to be dense. Fortunately, there are also some systems that are not only dense, but also convex, or in other words, the space function has a single global maximum. A common case would be the yield of a reaction as a function of temperature; from the peak of yield at a certain temperature the yield will decrease continuously in both directions. These types of systems lend themselves easily to optimization, and it is common in chemistry to solve these problems with various DoE algorithms, as discussed earlier. However, most interesting scientific problems stand to be more complex than that. For instance if there are a number of combinations of variables that lead to high yields then it is not trivial to find which of these regions is the best without measuring the entire space.
A simple way to tackle the search problem is the application of random experiments in order to explore the space. This has proven to be useful in combination with clever heuristics to improve search efficiency.99,100 However, the process is not robust, and it is hard to statistically validate the outcome since it would require many repetitions of different sets of random experiments over the same space. On the other hand, brute-force algorithms cover the entire possible space.101 This allows one to reduce the odds of missing interesting outcomes, but which would be impractical for many systems given resource constraints. A comparison between random and brute-force algorithms can be seen in Figure 9A,B. Many optimization algorithms for solving complicated systems are instead stochastic. These are divided into two classes of algorithm: instance-based, and model-based. Both classes of algorithm choose the next experiment based on the previously performed experiments. This means they use closed-loop feedback to iterate over performing experiments to gather more information which is used to choose the next experiments and so on. For instance-based algorithms such as simulated annealing,102,103 particle swarm optimization,98 and genetic algorithms,63,99 the sequence of chosen experiments aims to follow a general direction of improvement of the outcomes, yet there is no model being constructed or updated. On the other hand, a model-based algorithm builds and updates the model that it was trained on. The model can be seen as an approximation of the space function. This function can be constructed using an additional algorithm such as support vector machines,101,102 self-organizing maps,104 and kriging.105 As the models built during the search are closely related to the surface function, they are more useful in terms of discovery.
Figure 9.
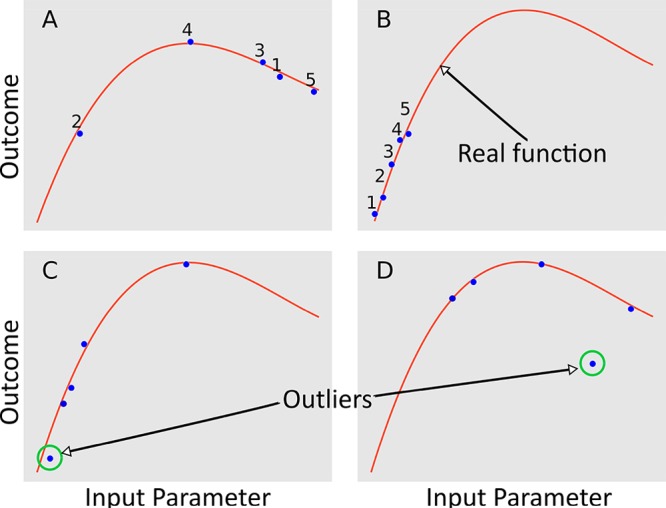
Model of the five first experiments conducted in a 1D system, whose surface is the red line, randomly (A) and with a brute-force approach (B). (C, D) Examples of outliers where the former has an outcome that is a statistical outlier from the three experiments, and the latter is an outlier due to a deviation from the real outcome surface.
Discovery does not mean that the chemical system is described in its entirety by a model. Rather, it is the new information gained from a new experiment. In other words, a discovery occurs when the model needs to be updated by a substantial amount to better match the real space function. Finding new results that differ from previous data in a statistically significant way is called outlier or anomaly detection. It is an area of significant research106,107 as it is in many settings important to know when new data is different enough to merit special attention. Figure 9C and 9D shows examples of outliers. Outliers indicate a statistical difference from expectation and as such can indicate either a positive discovery or a worsening of the outcome. It is the mapping function that must be able to distinguish between these possibilities. Such a mapping function should give an outlier for a real discovery receiving a high value, whereas an outlier with a negative outcome should receive a low value. Both positive and negative values should represent a significant deviation from expectation which means that they both add substantially more information about the chemical space.
Performing experiments to completely understand the function describing a chemical system is in many cases impossible. Even if it is possible, it may be impractical, and even if practical, it is likely to be inefficient. The shape of the model that any algorithm would be able to produce depends on the mapping function. Even for the same surface function, if the desired outcome from the mapping function is changed, so would the shape of the surface as depicted in Figure 8. Therefore, even if the space is fully explored, the shape of the resulting function may not match the real system, as the mapping function must always be an approximation. Furthermore, using a static mapping function will block an avenue of discovery and limit the possible discoveries to the shape of the surface exclusively. It can therefore be useful for discovery that the algorithm to understand the space function and the algorithm to define the mapping function are connected and coevolving. As the exploration of the system progresses, the mapping function needs to be updated as well, so both move together to gain a better understanding of the system and the outliers that should be of most interest.
Conclusions
While algorithms are very widely used in the chemical sciences, the potential to expand the use beyond data processing to decision making and active searching of chemical space is possible.108 By exploring the types of algorithms that are needed to accomplish different goals, it is possible to build on those used in standard chemical work as well as classes for extending the possibilities of research that could otherwise not be accessible. The key excitement should be focusing on the potential of developments for chemical discovery. By explaining the inherent problems of conducting research in the scope of chemical space, we have shown that such scientific problems can be related to optimization and searching methods.109 We have shown the importance of the definition of the space function and the utility function. Finally, we have explained how the coupled exploration of space and utility function might assist in real discovery, and this might also be applicable to more complex chemical systems.110 As such we feel there are two directions of development for the use of algorithms in chemistry. The first is employing algorithms into standard chemical science. An increasing selection of algorithms is finding a use in chemical research over different levels of operation. However, these algorithms need to have suitable frameworks and software foundations for integration in chemical systems. Thus, they can be implemented as a tool by nonexperts. The second direction is improving the algorithms used for development of systems capable of new discoveries. Here, new algorithms are being implemented along with existing algorithms being modified to suit the chemical world. Many of these algorithms will be used for discovery and the expansion of chemical space to search new undiscovered possibilities. Finally, the use of algorithms helps scientists to set up entirely new models of interactions, behaviors, and expectations of discovery. Consequently, this allows to define a new area of chemistry, that of “meta-chemistry”. This might be compared to “meta-physics”, whereby radical new models of reality emerge from making logical arguments with existing data.
Acknowledgments
We would like to thank Dr. Jonathan Grizou for useful discussions and Naomi A. B. Johnson for her artistic depiction of the robot for the Table of Contents graphic. This work was supported by the University of Glasgow, the EPSRC Grants (No. EP/J015156/1; EP/L023652/1; EP/I033459/1; EP/K023004/1). L.C. thanks the Royal Society/Wolfson Foundation for a Merit Award and the ERC for an Advanced Grant (ERC-ADG, 670467 SMART-POM).
The authors declare no competing financial interest.
References
- Knuth D. E. Ancient Babylonian Algorithms. Commun. ACM 1972, 15 (7), 671–677. 10.1145/361454.361514. [DOI] [Google Scholar]
- Chen W. L. Chemoinformatics: past, present, and future. J. Chem. Inf. Model. 2006, 46 (6), 2230–2255. 10.1021/ci060016u. [DOI] [PubMed] [Google Scholar]
- Krizhevsky A.; Sutskever I.; Hinton E. H.. ImageNet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. Conference, 2012; Vol. 25, pp 1–9.
- Silver D.; Huang A.; Maddison C. J.; Guez A.; Sifre L.; van den Driessche G.; Schrittwieser J.; Antonoglou I.; Panneershelvam V.; Lanctot M.; Dieleman S.; Grewe D.; Nham J.; Kalchbrenner N.; Sutskever I.; Lillicrap T.; Leach M.; Kavukcuoglu K.; Graepel T.; Hassabis D. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529 (7587), 484–489. 10.1038/nature16961. [DOI] [PubMed] [Google Scholar]
- Escandar G. M.; Faber N. M.; Goicoechea H. C.; Munoz de la Pena A.; Olivieri A. C.; Poppi R. J. Second- and third-order multivariate calibration: data, algorithms and applications. TrAC, Trends Anal. Chem. 2007, 26 (7), 752–765. 10.1016/j.trac.2007.04.006. [DOI] [Google Scholar]
- Barclay V. J.; Bonner R. F.; Hamilton I. P. Application of wavelet transforms to experimental spectra: smoothing, denoising, and data set compression. Anal. Chem. 1997, 69 (1), 78–90. 10.1021/ac960638m. [DOI] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeerschd T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4 (8), 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3 (10), 1–33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott A. J.; Bourne R. A.; Akien G. R.; Irvine D. J.; Poliakoff M. Self-Optimizing continuous reactions in supercritical carbon dioxide. Angew. Chem., Int. Ed. 2011, 50 (16), 3788–3792. 10.1002/anie.201100412. [DOI] [PubMed] [Google Scholar]
- Krishnadasan S.; Brown R. J. C.; DeMello A. J.; DeMello J. C. Intelligent routes to the controlled synthesis of nanoparticles. Lab Chip 2007, 7 (11), 1434–1441. 10.1039/b711412e. [DOI] [PubMed] [Google Scholar]
- Skilton R. A.; Parrott A. J.; George M. W.; Poliakoff M.; Bourne R. A. Real-time feedback control using online attenuated total reflection Fourier transform infrared (ATR FT-IR) spectroscopy for continuous flow optimization and process knowledge. Appl. Spectrosc. 2013, 67 (10), 1127–1131. 10.1366/13-06999. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Lowe D. M.; Sayle R. A.; Tarselli M. A.; Landrum G. A. Big data from pharmaceutical patents: a computational analysis of medicinal chemists’ bread and butter. J. Med. Chem. 2016, 59 (9), 4385–4402. 10.1021/acs.jmedchem.6b00153. [DOI] [PubMed] [Google Scholar]
- Lynch C. Big data: How do your data grow?. Nature 2008, 455 (7209), 28–29. 10.1038/455028a. [DOI] [PubMed] [Google Scholar]
- Gibb B. C. Big (chemistry) data. Nat. Chem. 2013, 5 (4), 248–249. 10.1038/nchem.1604. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. L. The chemical space project. Acc. Chem. Res. 2015, 48 (3), 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Dalby A.; Nourse J. G.; Hounshell W. D.; Gushurst A. K. I.; Grier D. L.; Leland B. A.; Laufer J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Model. 1992, 32 (3), 244–255. 10.1021/ci00007a012. [DOI] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40 (D1), D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Shrivastava S.; Hassanali M.; Stothard P.; Chang Z.; Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh S.; Wei Y. H.; Kowalczyk B.; Gothard C. M.; Baytekin B.; Gothard N.; Grzybowski B. A. Estimating chemical reactivity and cross-influence from collective chemical knowledge. Chem. Sci. 2012, 3 (5), 1497–1502. 10.1039/c2sc00011c. [DOI] [Google Scholar]
- Gardiner E. J.; Gillet V. J. Perspectives on knowledge discovery algorithms recently introduced in chemoinformatics: rough set theory, association rule mining, emerging patterns, and formal concept analysis. J. Chem. Inf. Model. 2015, 55 (9), 1781–1803. 10.1021/acs.jcim.5b00198. [DOI] [PubMed] [Google Scholar]
- Fialkowski M.; Bishop K. J. M.; Chubukov V. A.; Campbell C. J.; Grzybowski B. A. Architecture and evolution of organic chemistry. Angew. Chem., Int. Ed. 2005, 44 (44), 7263–7269. 10.1002/anie.200502272. [DOI] [PubMed] [Google Scholar]
- Segler M. H. S.; Waller M. P. Modelling chemical reasoning to predict and invent reactions. Chem. Eur. J. 2017, 23, 6118–6128. 10.1002/chem.201604556. [DOI] [PubMed] [Google Scholar]
- Wang Y. L.; Xiao J. W.; Suzek T. O.; Zhang J.; Wang J. Y.; Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pence H. E.; Williams A. ChemSpider: an online chemical information resource. J. Chem. Educ. 2010, 87 (11), 1123–1124. 10.1021/ed100697w. [DOI] [Google Scholar]
- Fink T.; Reymond J. L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007, 47 (2), 342–353. 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- Villoutreix B. O.; Renault N.; Lagorce D.; Sperandio O.; Montes M.; Miteva M. A. Free resources to assist structure-based virtual ligand screening experiments. Curr. Protein Pept. Sci. 2007, 8 (4), 381–411. 10.2174/138920307781369391. [DOI] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K.; Mysinger M. M.; Huang N.; Colizzi F.; Wassam P.; Cao Y. Q. Automated docking screens: a feasibility study. J. Med. Chem. 2009, 52 (18), 5712–5720. 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills N. ChemDraw Ultra 10.0. J. Am. Chem. Soc. 2006, 128 (41), 13649–13650. 10.1021/ja0697875. [DOI] [Google Scholar]
- Gasteiger J.; Zupan J. Neural networks in chemistry. Angew. Chem. Int. Ed. 1993, 32 (4), 503–527. 10.1002/anie.199305031. [DOI] [Google Scholar]
- Goh G. B.; Hodas N. O.; Vishnu A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38 (16), 1291–1307. 10.1002/jcc.24764. [DOI] [PubMed] [Google Scholar]
- Maltarollo V. G.; Honorio K. M.; da Silva A. B. F. Applications of artificial neural networks in chemical problems. Artificial Neural Networks - Architecture and Applications 2013, 203–223. 10.5772/51275. [DOI] [Google Scholar]
- Gawehn E.; Hiss J. A.; Schneider G. Deep learning in drug discovery. Mol. Inf. 2016, 35 (1), 3–14. 10.1002/minf.201501008. [DOI] [PubMed] [Google Scholar]
- Bickerton G. R.; Paolini G. V.; Besnard J.; Muresan S.; Hopkins A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4 (2), 90–98. 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2012, 64, 4–17. 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Scior T.; Bender A.; Tresadern G.; Medina-Franco J. L.; Martinez-Mayorga K.; Langer T.; Cuanalo-Contreras K.; Agrafiotis D. K. Recognizing pitfalls in virtual screening: a critical review. J. Chem. Inf. Model. 2012, 52 (4), 867–881. 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- Collins K. D.; Gensch T.; Glorius F. Contemporary screening approaches to reaction discovery and development. Nat. Chem. 2014, 6 (10), 859–871. 10.1038/nchem.2062. [DOI] [PubMed] [Google Scholar]
- Santanilla A. B.; Regalado E. L.; Pereira T.; Shevlin M.; Bateman K.; Campeau L. C.; Schneeweis J.; Berritt S.; Shi Z. C.; Nantermet P.; Liu Y.; Helmy R.; Welch C. J.; Vachal P.; Davies I. W.; Cernak T.; Dreher S. D. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 2015, 347 (6217), 49–53. 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- Ortholand J.-Y.; Ganesan A. Natural products and combinatorial chemistry: back to the future. Curr. Opin. Chem. Biol. 2004, 8 (3), 271–280. 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Eckert H.; Bojorath J. Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches. Drug Discov. Today 2007, 12 (5–6), 225–233. 10.1016/j.drudis.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Hert J.; Willett P.; Wilton D. J.; Acklin P.; Azzaoui K.; Jacoby E.; Schuffenhauer A. Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org. Biomol. Chem. 2004, 2 (22), 3256–3266. 10.1039/b409865j. [DOI] [PubMed] [Google Scholar]
- Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11 (23–24), 1046–1053. 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Sun H. M. Pharmacophore-based virtual screening. Curr. Med. Chem. 2008, 15 (10), 1018–1024. 10.2174/092986708784049630. [DOI] [PubMed] [Google Scholar]
- Tropsha A. Best Practices for QSAR Model Development, Validation,and Exploitation. Mol. Inf. 2010, 29 (6–7), 476–488. 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today 2006, 11 (13–14), 580–594. 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11 (5), 494–502. 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Virtual screening of chemical libraries. Nature 2004, 432 (7019), 862–865. 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwoski G.; Kothiwale S.; Meiler J.; Lowe E. W. Jr Computational methods in drug discovery. Pharmacol. Rev. 2014, 66 (1), 334–395. 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brereton R. G. The evolution of chemometrics. Anal. Methods 2013, 5 (16), 3785–3789. 10.1039/c3ay90051g. [DOI] [Google Scholar]
- Hopke P. K. The evolution of chemometrics. Anal. Chim. Acta 2003, 500 (1–2), 365–377. 10.1016/S0003-2670(03)00944-9. [DOI] [Google Scholar]
- Bro R. Multivariate calibration. What is in chemometrics for the analytical chemist?. Anal. Chim. Acta 2003, 500 (1–2), 185–194. 10.1016/S0003-2670(03)00681-0. [DOI] [Google Scholar]
- Jain A. K.; Duin R. P. W.; Mao J. C. Statistical pattern recognition: A review. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22 (1), 4–37. 10.1109/34.824819. [DOI] [Google Scholar]
- Brereton R. G. Consequences of sample size, variable selection, and model validation and optimization, for predicting classification ability from analytical data. TrAC, Trends Anal. Chem. 2006, 25 (11), 1103–1111. 10.1016/j.trac.2006.10.005. [DOI] [Google Scholar]
- Jain A. K.; Murty M. N.; Flynn P. J. Data clustering: a review. ACM Comput. Surv. 1999, 31 (3), 264–323. 10.1145/331499.331504. [DOI] [Google Scholar]
- Brereton R. G.; Lloyd G. R. Partial least squares discriminant analysis: taking the magic away. J. Chemom. 2014, 28 (4), 213–225. 10.1002/cem.2609. [DOI] [Google Scholar]
- Wold S.; Sjostrom M.; Eriksson L. PLS-regression: a basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58 (2), 109–130. 10.1016/S0169-7439(01)00155-1. [DOI] [Google Scholar]
- Geladi P.; Sethson B.; Nystrom J.; Lillhonga T.; Lestander T.; Burger J. Chemometrics in spectroscopy - Part 2. Examples. Spectrochim. Acta, Part B 2004, 59 (9), 1347–1357. 10.1016/S0584-8547(04)00167-3. [DOI] [Google Scholar]
- Hengl T.; de Jesus J. M.; Heuvelink G. B. M.; Gonzalez M. R.; Kilibarda M.; Blagotic A.; Shangguan W.; Wright M. N.; Geng X.; Bauer-Marschallinger B.; Guevara M. A.; Vargas R.; MacMillan R. A.; Batjes N. H.; Leenaars J. G. B.; Ribeiro E.; Wheeler I.; Mantel S.; Kempen B. SoilGrids250m: Global gridded soil information based on machine learning. PLoS One 2017, 12 (2), e0169748. 10.1371/journal.pone.0169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutihac L.; Mutihac R. Mining in chemometrics. Anal. Chim. Acta 2008, 612 (1), 1–18. 10.1016/j.aca.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zomer S.; Brereton R. G. Support Vector Machines: A recent method for classification in chemometrics. Crit. Rev. Anal. Chem. 2006, 36 (3–4), 177–188. 10.1080/10408340600969486. [DOI] [Google Scholar]
- Zomer S.; Sanchez M. D. N.; Brereton R. G.; Pavon J. L. P. Active learning support vector machines for optimal sample selection in classification. J. Chemom. 2004, 18 (6), 294–305. 10.1002/cem.872. [DOI] [Google Scholar]
- Schuett K. T.; Arbabzadah F.; Chmiela S.; Mueller K. R.; Tkatchenko A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz J. E.; Shukhaev A.; Du W. B.; Druskin S.; Daugulis O.; Ismagilov R. F. Evolution of catalysts directed by genetic algorithms in a plug-based microfluidic device tested with oxidation of methane by oxygen. J. Am. Chem. Soc. 2010, 132 (9), 3128–3132. 10.1021/ja909853x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leardi R. Genetic algorithms in chemometrics and chemistry: a review. J. Chemom. 2001, 15 (7), 559–569. 10.1002/cem.651. [DOI] [Google Scholar]
- Markou M.; Singh S. Novelty detection: a review - part 1: statistical approaches. Signal Processing 2003, 83 (12), 2481–2497. 10.1016/j.sigpro.2003.07.018. [DOI] [Google Scholar]
- Markou M.; Singh S. Novelty detection: a review - part 2: neural network based approaches. Signal Processing 2003, 83 (12), 2499–2521. 10.1016/j.sigpro.2003.07.019. [DOI] [Google Scholar]
- Lang T.; Flachsenberg F.; von Luxburg U.; Rarey M. Feasibility of active machine learning for multiclass compound classification. J. Chem. Inf. Model. 2016, 56 (1), 12–20. 10.1021/acs.jcim.5b00332. [DOI] [PubMed] [Google Scholar]
- Gromski P. S.; Muhamadali H.; Ellis D. I.; Xu Y.; Correa E.; Turner M. L.; Goodacre R. A tutorial review: Metabolomics and partial least squares-discriminant analysis-a marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. 10.1016/j.aca.2015.02.012. [DOI] [PubMed] [Google Scholar]
- Leardi R. Experimental design in chemistry: A tutorial. Anal. Chim. Acta 2009, 652 (1–2), 161–172. 10.1016/j.aca.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Bernlind C.; Urbaniczky C. An efficient laboratory automation concept for process chemistry. Org. Process Res. Dev. 2009, 13 (6), 1059–1067. 10.1021/op900154c. [DOI] [Google Scholar]
- Weber A.; Von Roedern E.; Stilz H. U. SynCar: An approach to automated synthesis. J. Comb. Chem. 2005, 7 (2), 178–184. 10.1021/cc049838z. [DOI] [PubMed] [Google Scholar]
- Goodell J. R.; McMullen J. P.; Zaborenko N.; Maloney J. R.; Ho C.-X.; Jensen K. F.; Porco J. A.; Beeler A. B. Development of an automated microfluidic reaction platform for multidimensional screening: reaction discovery employing bicyclo[3.2.1]octanoid scaffolds. J. Org. Chem. 2009, 74 (16), 6169–6180. 10.1021/jo901073v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heublein N.; Moore J. S.; Smith C. D.; Jensen K. F. Investigation of Petasis and Ugi reactions in series in an automated microreactor system. RSC Adv. 2014, 4 (109), 63627–63631. 10.1039/C4RA13626H. [DOI] [Google Scholar]
- Hibbert D. B. Experimental design in chromatography: A tutorial review. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 910, 2–13. 10.1016/j.jchromb.2012.01.020. [DOI] [PubMed] [Google Scholar]
- Lundstedt T.; Seifert E.; Abramo L.; Thelin B.; Nystrom A.; Pettersen J.; Bergman R. Experimental design and optimization. Chemom. Intell. Lab. Syst. 1998, 42 (1–2), 3–40. 10.1016/S0169-7439(98)00065-3. [DOI] [Google Scholar]
- Dixon J. M.; Lindsey J. S. Performance of search algorithms in the examination of chemical reaction spaces with an automated chemistry workstation. JALA 2004, 9 (6), 364–374. 10.1016/j.jala.2004.08.004. [DOI] [Google Scholar]
- Symes M. D.; Kitson P. J.; Yan J.; Richmond C. J.; Cooper G. J. T.; Bowman R. W.; Vilbrandt T.; Cronin L. Integrated 3D-printed reactionware for chemical synthesis and analysis. Nat. Chem. 2012, 4 (5), 349–354. 10.1038/nchem.1313. [DOI] [PubMed] [Google Scholar]
- Kitson P. J.; Symes M. D.; Dragone V.; Cronin L. Combining 3D printing and liquid handling to produce user-friendly reactionware for chemical synthesis and purification. Chem. Sci. 2013, 4 (8), 3099–3103. 10.1039/C3SC51253C. [DOI] [Google Scholar]
- Mawatari K.; Kazoe Y.; Aota A.; Tsukahara T.; Sato K.; Kitamori T. Microflow systems for chemical synthesis and analysis: Approaches to full integration of chemical process. J. Flow Chem. 2012, 1 (1), 3–12. 10.1556/jfchem.2011.00003. [DOI] [Google Scholar]
- Jensen K. F.; Reizman B. J.; Newman S. G. Tools for chemical synthesis in microsystems. Lab Chip 2014, 14 (17), 3206–3212. 10.1039/C4LC00330F. [DOI] [PubMed] [Google Scholar]
- Richmond C. J.; Miras H. N.; de la Oliva A. R.; Zang H.; Sans V.; Paramonov L.; Makatsoris C.; Inglis R.; Brechin E. K.; Long D.; Cronin L. A flow-system array for the discovery and scale up of inorganic clusters. Nat. Chem. 2012, 4 (12), 1037–1043. 10.1038/nchem.1489. [DOI] [PubMed] [Google Scholar]
- Ley S. V.; Fitzpatrick D. E.; Myers R. M.; Battilocchio C.; Ingham R. J. Machine-assisted organic synthesis. Angew. Chem., Int. Ed. 2015, 54 (35), 10122–10136. 10.1002/anie.201501618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastre J. C.; Browne D. L.; Ley S. V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42 (23), 8849–8869. 10.1039/c3cs60246j. [DOI] [PubMed] [Google Scholar]
- Straathof N. J. W.; Su Y.; Hessel V.; Noël T. Accelerated gas-liquid visible light photoredox catalysis with continuous-flow photochemical microreactors. Nat. Protoc. 2015, 11 (1), 10–21. 10.1038/nprot.2015.113. [DOI] [PubMed] [Google Scholar]
- Ingham R. J.; Battilocchio C.; Fitzpatrick D. E.; Sliwinski E.; Hawkins J. M.; Ley S. V. A systems approach towards an intelligent and self-controlling platform for integrated continuous reaction sequences. Angew. Chem., Int. Ed. 2015, 54 (1), 144–148. 10.1002/anie.201409356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislieri D.; Gilmore K.; Seeberger P. H. Chemical assembly systems: layered control for divergent, continuous, multistep syntheses of active pharmaceutical ingredients. Angew. Chem., Int. Ed. 2014, 54 (2), 678–682. 10.1002/anie.201409765. [DOI] [PubMed] [Google Scholar]
- Adamo A.; Beingessner R. L.; Behnam M.; Chen J.; Jamison T. F.; Jensen K. F.; Monbaliu J.-C. M.; Myerson A. S.; Revalor E. M.; Snead D. R.; Stelzer T.; Weeranoppanant N.; Wong S. Y.; Zhang P. On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system. Science 2016, 352 (6281), 61–67. 10.1126/science.aaf1337. [DOI] [PubMed] [Google Scholar]
- Heider P. L.; Born S. C.; Basak S.; Benyahia B.; Lakerveld R.; Zhang H.; Hogan R.; Buchbinder L.; Wolfe A.; Mascia S.; Evans J. M. B.; Jamison T. F.; Jensen K. F. Development of a multi-step synthesis and workup sequence for an integrated, continuous manufacturing process of a pharmaceutical. Org. Process Res. Dev. 2014, 18 (3), 402–409. 10.1021/op400294z. [DOI] [Google Scholar]
- Mascia S.; Heider P. L.; Zhang H.; Lakerveld R.; Benyahia B.; Barton P. I.; Braatz R. D.; Cooney C. L.; Evans J. M. B.; Jamison T. F.; Jensen K. F.; Myerson A. S.; Trout B. L. End-to-end continuous manufacturing of pharmaceuticals: integrated synthesis, purification, and final dosage formation. Angew. Chem., Int. Ed. 2013, 52 (47), 12359–12363. 10.1002/anie.201305429. [DOI] [PubMed] [Google Scholar]
- Li J.; Ballmer S. G.; Gillis E. P.; Fujii S.; Schmidt M. J.; Palazzolo A. M. E.; Lehmann J. W.; Morehouse G. F.; Burke M. D. Synthesis of many different types of organic small molecules using one automated process. Science 2015, 347 (6227), 1221–1226. 10.1126/science.aaa5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner J.; Ceylan S.; Kirschning A. Flow Chemistry – A key enabling technology for (multistep) organic synthesis. Adv. Synth. Catal. 2012, 354 (1), 17–57. 10.1002/adsc.201100584. [DOI] [Google Scholar]
- Heller S.; McNaught A.; Stein S.; Tchekhovskoi D.; Pletnev I. InChI - the worldwide chemical structure identifier standard. J. Cheminf. 2013, 5 (7), 1–9. 10.1186/1758-2946-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller S. R.; McNaught A.; Pletnev I.; Stein S.; Tchekhovskoi D. InChI, the IUPAC International Chemical Identifier. J. Cheminformatics 2015, 7 (23), 1–34. 10.1186/s13321-015-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raccuglia P.; Elbert K. C.; Adler P. D. F.; Falk C.; Wenny M. B.; Mollo A.; Zeller M.; Friedler S. A.; Schrier J.; Norquist A. J. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533 (7601), 73–76. 10.1038/nature17439. [DOI] [PubMed] [Google Scholar]
- de Souza R. O. M. A.; Miranda L. S. M.; Bornscheuer U. T. A retrosynthesis approach for biocatalysis in organic synthesis. Chem. - Eur. J. 2017, 23 (50), 12040–12063. 10.1002/chem.201702235. [DOI] [PubMed] [Google Scholar]
- Szymkuć S.; Gajewska E. P.; Klucznik T.; Molga K.; Dittwald P.; Startek M.; Bajczyk M.; Grzybowski B. A. Computer-assisted synthetic planning: the end of the beginning. Angew. Chem., Int. Ed. 2016, 55 (20), 5904–5937. 10.1002/anie.201506101. [DOI] [PubMed] [Google Scholar]
- Bøgevig A.; Federsel H.-J.; Huerta F.; Hutchings M. G.; Kraut H.; Langer T.; Löw P.; Oppawsky C.; Rein T.; Saller H. Route Design in the 21st Century: The ICSYNTH software tool as an idea generator for synthesis prediction. Org. Process Res. Dev. 2015, 19 (2), 357–368. 10.1021/op500373e. [DOI] [Google Scholar]
- Chen X.; Du W.; Qi R.; Qian F.; Tianfield H. Hybrid gradient particle swarm optimization for dynamic optimization problems of chemical processes. Asia-Pac. J. Chem. Eng. 2013, 8 (5), 708–720. 10.1002/apj.1712. [DOI] [Google Scholar]
- Corma A.; Serra J.; Serna P.; Valero S.; Argente E.; Botti V. Optimisation of olefin epoxidation catalyst applying high-throughput and genetic algorithms assisted by artificial neural networks (soft computing techniques). J. Catal. 2005, 229 (2), 513–524. 10.1016/j.jcat.2004.11.024. [DOI] [Google Scholar]
- Li H. D.; Liang Y. Z.; Xu Q. S. Support vector machines and its applications in chemistry. Chemom. Intell. Lab. Syst. 2009, 95 (2), 188–198. 10.1016/j.chemolab.2008.10.007. [DOI] [Google Scholar]
- Baumes L. A.; Serra J. M.; Serna P.; Corma A. Support vector machines for predictive modeling in heterogeneous catalysis: A comprehensive introduction and overfitting investigation based on two real applications. J. Comb. Chem. 2006, 8 (4), 583–596. 10.1021/cc050093m. [DOI] [PubMed] [Google Scholar]
- Sutter J. M.; Dixon S. L.; Jurs P. C. Automated descriptor selection for quantitative structure-activity relationships using generalized simulated annealing. J. Chem. Inf. Model. 1995, 35 (1), 77–84. 10.1021/ci00023a011. [DOI] [Google Scholar]
- Suman B. Study of simulated annealing based algorithms for multiobjective optimization of a constrained problem. Comput. Chem. Eng. 2004, 28 (9), 1849–1871. 10.1016/j.compchemeng.2004.02.037. [DOI] [Google Scholar]
- Reker D.; Rodrigues T.; Schneider P.; Schneider G. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (11), 4067–4072. 10.1073/pnas.1320001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg S.; Stutz B.; Schmidt T.; Hamprecht F.; Maier W. F. A QCAR-approach to materials modeling. J. Mol. Model. 2006, 12 (5), 611–619. 10.1007/s00894-005-0068-9. [DOI] [PubMed] [Google Scholar]
- Kandhari R.; Chandola V.; Banerjee A.; Kumar V.; Kandhari R. Anomaly detection. ACM Comput. Surv. 2009, 41 (3), 1–6. 10.1145/1541880.1541882. [DOI] [Google Scholar]
- Goldstein M.; Uchida S. A Comparative evaluation of unsupervised anomaly detection algorithms for multivariate data. PLoS One 2016, 11 (4), e0152173. 10.1371/journal.pone.0152173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duros V.; Grizou J.; Xuan W.; Hosni Z.; Long D.-L.; Miras H. N.; Cronin L. Human versus robots in the discovery and crystallization of gigantic polyoxometalates. Angew. Chem., Int. Ed. 2017, 56, 10815–10820. 10.1002/anie.201705721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M.; Hinkley T.; Tsuda S.; Abul-Haija Y. M.; McBurney R. T.; Kulikov V.; Mathieson J. S.; Galinanes Reyes S.; Castro M. D.; Cronin L. Using evolutionary algorithms and machine learning to explore sequence space for the discovery of antimicrobial peptides. Chem. 2018, 4 (3), 533–543. 10.1016/j.chempr.2018.01.005. [DOI] [Google Scholar]
- Points L. J.; Taylor J. W.; Grizou J.; Donkers K.; Cronin L. Artificial intelligence exploration of unstable protocells leads to predictable properties and discovery of collective behavior. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (5), 885–890. 10.1073/pnas.1711089115. [DOI] [PMC free article] [PubMed] [Google Scholar]