

Abstract
Background
Loxoscelism is a severe human envenomation caused by Loxosceles spider venom. To the best of our knowledge, no study has evaluated the presence of antibodies against Loxosceles venom in loxoscelism patients without treatment with antivenom immunotherapy. We perform a comparative analysis for the presence of antibodies capable of recognizing Loxosceles venom in a group of patients diagnosed with loxoscelism and in a group of people without loxoscelism.
Methods
The detection of L. laeta venom, Sicarius venom and recombinant phospholipases D from Loxosceles (PLDs) in sera from people with loxoscelism (Group 1) and from healthy people with no history of loxoscelism (Group 2) was evaluated using immuno-dot blot, indirect ELISA, and Western blot.
Results
We found naturally heterophilic antibodies (IgG-type) in people without contact with Loxosceles spiders or any clinical history of loxoscelism. Either serum pools or single sera from Group 1 and Group 2 analyzed by dot blot tested positive for L. laeta venom. Indirect ELISA for venom recognition showed titles of 1:320 for Group 1 sera and 1:160 for Group 2 sera. Total IgG quantification showed no difference in sera from both groups. Pooled sera and purified IgG from sera of both groups revealed venom proteins between 25 and 32 kDa and the recombinant phospholipase D isoform 1 (rLlPLD1), specifically. Moreover, heterophile antibodies cross-react with PLDs from other Loxosceles species and the venom of Sicarius spider.
Conclusions
People without contact with the spider venom produced heterophilic antibodies capable of generating a cross-reaction against the venom of L. laeta and Sicarius spiders. Their presence and possible interference should be considered in the development of immunoassays for Loxosceles venom detection.
Electronic supplementary material
The online version of this article (10.1186/s40409-018-0155-x) contains supplementary material, which is available to authorized users.
Keywords: Loxosceles laeta, Heterophilic antibodies, Natural antibodies, Sicarius
Background
Loxoscelism is a clinical picture of poisoning produced by Loxosceles spiders venom that has a considerable impact on the population living in the Americas, from the United States to Chile [1, 2]. In countries such as Brazil, it is a serious public health problem, with a high number of cases reported annually, with some of them corresponding to fatal cases [3–5]. It is also considered a public health problem in Chile, where the most recent data from the Center for Toxicological Information at the Pontificia Universidad Católica de Chile (CITUC), showed that of 2831 possible cases in a year roughly 10% were confirmed as loxoscelism [6]. Loxoscelism is provoked by bites of spiders from the genus Loxosceles and its clinical presentation can progress from the local and most frequently dermal necrosis lesion, called cutaneous loxoscelism (CL), to a systemic condition less frequent, but more severe, called systemic or viscero-cutaneous loxoscelism (VCL). The clinical manifestations of loxoscelism (CL or VCL) depend on different factors, such as the amount and concentration of inoculated venom, the anatomical location of the bite, the susceptibility of the host, and the species and gender of the spider [7–9].
The diagnosis of loxoscelism is usually clinical and presumptive. This often depends on capture of the arachnid by the patient for later taxonomic identification in the laboratory, which helps guide an effective diagnosis [10]. However, this is rare, since patients do not bring the arachnid with them, making diagnoses dependent upon histological findings, epidemiology, signs and symptoms [11, 12]. In addition, there are no commercial diagnostic tests available, so the diagnosis is made according to the evolution of clinical symptoms [11, 13]. In this regard, the designing of these diagnostic assays may involve the careful attention in specificity, sensitivity, dynamic range, reproducibility and accuracy, but also require the identification of factors that may interfere with the assay leading to erroneous results [14].
Few efforts have been made in the development of a specific detection method for Loxosceles venom, which include a sandwich ELISA test for detection of L. intermedia venom on mice inoculated with L. intermedia venom and distinguish them from those inoculated with venom from L. gaucho, L. laeta, P. nigreventer, scorpions, and snakes. The test was able to detect 0.8 ng of venom per assay and could detect L. intermedia antigens in clinical serum samples from loxoscelism patients [15]. A second sandwich ELISA was developed for detecting L. reclusa venom using polyclonal rabbit antibodies, and could detect 0.1 ng of Loxosceles venom. However, cross-reactivity was detected with venom from other arachnid species not related to Loxosceles genus [16]. Despite this, its clinical efficacy was documented using a noninvasive sample from the lesions of loxoscelism patients [17, 18]. More recently, immunoconjugates of LimAb7 monoclonal antibody – specific against toxic components of L. intermedia venom – were evaluated for developing a competitive ELISA and sandwich ELISA for detecting L. intermedia venom. This assay had a detection limit of 39 ng/mL, however, was not able to detect venom from other species of Loxosceles as L. laeta or L. gaucho venom [19].
Although the specificity and sensitivity of immunoassays are important aspects to be considered and overcome in order to implement a diagnostic test for loxoscelism, other aspects such as the type of sample to be used and the presence of interfering factors should also be considered. Therefore, endogenous agents present in serum samples may interfere and cause false positive or false negative results. Important interfering agents in immunoassays are the endogenous antibodies, including: heterophilic antibodies (HA), human anti-animal antibodies (HAAA), and autoantibodies (AA). Heterophilic antibodies are naturally occurring antibodies present in individuals with no known exposure to specific antigens [20, 21]. These antibodies have low affinity and broad specificity, and their immunoassay interference mechanism occurs generally by cross-linking with captured antibodies or detection antibodies [22].
Up to this moment, only one study evaluated the presence of antibodies capable of recognizing Loxosceles venom present in loxoscelism patients who received antivenom therapy [23]. However, a detailed analysis of the presence of antibodies anti-Loxosceles venom has not been performed in patients diagnosed with loxoscelism and without antivenom therapy in order to rule out the participation in the venom detection of antibodies introduced in the antivenom therapy. Also, there has not been performed an evaluation of the possible presence of antibodies with cross-reaction potential for Loxosceles venom, or the presence of antibodies capable of interfering in specific immunoassays in people without loxoscelism.
In this study we performed a comparative analysis of the presence of antibodies capable of recognizing Loxosceles venom in a group of patients diagnosed with loxoscelism and in a group of people without contact with the spider or clinical history of loxoscelism to determine the relevance of using serum samples in the development of a rapid inmunotest for detection of Loxosceles venom. We found the presence of heterophilic antibodies capable of detecting the venom of Loxosceles and Sicarius spiders in both groups and discussed its origins and importance as a potential interference in diagnostic immunoassays for loxoscelism.
Methods
Serum sample collection from people with or without a clinical history of loxoscelism
Ten serum samples from people with a clinical history of loxoscelism and 30 serum samples from people without clinical history of loxoscelism or other arachnid-bite symptoms were collected during the period from March to December 2012, in the city of Antofagasta, Chile. Ten milliliters of peripheral blood were collected by venipuncture (informed consent was obtained from donors). Serum was collected through centrifugation at 2000×g for 10 min at 4 °C, and stored in aliquots of 500 μL at − 80 °C until use. Protocols for sample collection and informed consent were approved by the Ethics Committee in Scientific Research of the University of Antofagasta (CEIC-UA).
For the purpose of the study, samples from people with a clinical history of loxoscelism were part of the loxoscelism study group (Group 1), and samples from people without clinical history of loxoscelism were part of the control group (Group 2).
Group 1 was defined based on:
loxoscelism diagnosis according to a clinical history;
a patient’s own assertion of having been bitten by some arachnid (association with biting by L. laeta) with and without dermonecrotic or visceral loxoscelism;
visual assessment of current or previous dermonecrotic lesions. People who reported being bitten by an arachnid were identified and confirmed for loxoscelism dermonecrotic lesions at the Molecular Parasitology Laboratory of the Faculty of Health Sciences of the Universidad de Antofagasta, according to the clinical guide for handling the bites of the corner spider from the Chilean Ministry of Health [24].
People in Group 2 were defined based on:
no verifiable clinical history of loxoscelism or having previously suffered a bite from L. laeta or other type of arachnid;
no presentation of chronic diseases or allergies;
no evidence of autoimmune diseases, rheumatoid arthritis, or any known physical illness affecting their immunological status;
no acute infectious processes at the time of sample extraction; and
no presence of skin lesions attributable to infectious bacterial processes.
Among the samples in Group 1, 60% were from patients with cutaneous loxoscelism and 40% were from patients with viscero-cutaneous loxoscelism. The age range of people of groups 1 and 2 was 18 to 60 years old, and both groups had equal proportions of men and women (Table 1).
Table 1.
Distribution of serum samples from patients groups with and without loxoscelism
| Age group | Without loxoscelisma (n = 30) | With loxoscelisma | ||||
|---|---|---|---|---|---|---|
| Cutaneousb (n = 6) | Systemicb (n = 4) | |||||
| Male | Female | Male | Female | Male | Female | |
| 18–29 | 11/36.7% | 16/53.3% | 1/16.7% | 2/33.3% | 2/50% | 1/25% |
| 30–60 | 3/10% | – | 2/33.3% | 1/16.7% | – | 1/25% |
aSamples of serum obtained under informed consent (procedure approved by the Research Ethics Committee of the Universidad de Antofagasta – CEIC-UA)
bAccording to clinical history and/or direct observation of dermonecrotic lesion. Samples are represented as number and percentage (n/%)
Spider venom, recombinant phospholipase D expression and purification
The recombinant protein rLlPLD1 was expressed and purified as previously described [25]. In addition, the nucleotide sequences for different phospholipase D isoforms of the four representative species of Loxosceles (L. laeta, L. intermedia, L. reclusa and L. gaucho) available in GeneBank (NCBI) were used for expression of their ORFs in E. coli BL21 DE3 and purified as fusion proteins with a 6His tag at the N-terminus (LrSMD1 and LgDerProt1) or at the C-terminus (rLlPLD2 and LiDerTox1) by GenScript (GenScript Inc., USA). GenBank accession numbers for nucleotide sequences used and the molecular masses of the respective recombinant proteins were: L. laeta PLD isoform 2 (LlPLD2), access n° GU121906 [25], 32,055 Da with C-His tag; L. reclusa sphingomyelinase D isoform 1 (LrSMD1), access n° AY559846.1 [26], 31,219 Da with N-His tag; L. intermedia sphingomyelinase P1 (LiSMD P1), access n° AY304471.2 [27], 34,982 Da and C-His tag; and L. gaucho dermonecrotic protein 1 (LgDerProt1), access n° AY974250.1, 31,172 Da with N-His tag. Moreover, venom from 20 female L. laeta and 20 Sicarius adults was extracted by electrostimulation and collected as previously reported [28]. Polyclonal mouse anti-L. laeta venom antibodies were prepared as previously documented [25].
Dot blot to determine L. laeta antivenom antibodies
Dot blot for L. laeta venom antibody detection was assembled in our laboratory, with 1 μg of L. laeta venom being adsorbed onto a nitrocellulose membrane using a 96-well Dot-Blot Filtration Manifold System (Gibco BRL). The presence of adsorbed proteins on the membrane was evaluated by staining with Ponceau red. The membrane was then blocked for 1 h at 22–25 °C with 5% non-fat milk in PBS/0.1% Tween20 (PBS-T). Each dot was then incubated with a pool of Group 1 or Group 2 sera (1:1000 dilution), and alternatively with single serums of Group 1 or Group 2 at a 1:10 dilution. The membranes were washed three times with PBS-T and incubated for 1 h at 22–25 °C with the anti-human IgG-HRP secondary antibody in 1:50,000 dilution, then washed again three times with PBS-T and developed by ECL.
PBS or pre-immune mouse serum (1:1000 dilution) was used as the negative control. As positive control, mouse anti-L. laeta venom serum (1:10,000 dilution) and monoclonal antibody 7E4-D2 anti-rL1PLD1 (1:50,000 dilution) were used [25]. BSA was used as unrelated antigen to evaluate specificity of reaction. Images were captured on a ChemiBis 2.0 DNR photo-documenter (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel). Intensity of dots was evaluated by densitometry and expressed as relative density percentage.
Indirect ELISA for detection of L. laeta venom antibodies
The titration of different sera was carried out using an indirect ELISA for detecting specific circulating antibodies against L. laeta venom, based on previously published protocols [29], and mounted in our laboratory. On 96-well ELISA microtiter plates (Nunc MaxiSorp™, Thermo Fisher Scientific), 1 μg of L. laeta venom in 0.02 M sodium carbonate/bicarbonate buffer (pH 9.6) per well was adsorbed, incubated at 37 °C for 1 h, and then at 4 °C for the overnight. Wells with L. laeta venom were blocked with 5% non-fat milk in PBS/0.05% Tween20 (PBS-T) for 1 h. The titer of sera from Group 1 and Group 2 was determined by using a two-fold serial dilution of serum samples from 1:10 to 1:5120 and incubated for 1 h at 37 °C.
Column 11 of the microplate was incubated with only PBS-T and used as a blank, while column 12 was used as specificity control with BSA adsorbed to the well. Subsequently, each well was washed four times with PBS-T and incubated for 1 h at 37 °C with anti-human IgG bound to peroxidase at 1:50,000 dilution (Sigma Aldrich Co, USA). After four washes with PBS-T and two washes with only PBS, 100 μL of the tetramethylbenzidine (TMB) substrate were added and incubated for 30 min at room temperature in the dark. The reaction was stopped by the addition of 100 μL of 3 N sulfuric acid (stop solution) and the absorbance of each well was measured at 450 nm in a BioRad model 550 microplate reader (BioRad, Hercules, USA). The background cut-off point was determined by the mean value of the blank absorbance (PBS-T) for at least 30 negative control replicates, plus three standard deviations (0.055 + 0.0088 = 0.0814). Reaction titers were determined as the inverse of the last positive reaction at the cut off value for each serum dilution. Samples from both groups showed normal distributions, according to the D’Agostino & Pearson omnibus normality test.
For IgG avidity ELISA, 100 μL of pooled serum of group 1 or 2 diluted 1:100 in buffer was added to each well coated with L. laeta venom in triplicate. After incubation for 1 h at 37 °C, wells were incubated with 6 M urea solution or PBS for 10 min. After four washes, wells were incubated with peroxidase labeled anti-human IgG for 1 h at 37 °C, then substrate was added, and the reactions were stopped after 30 min by addition of 100 μL of stop solution per well. Reactions were read at 450 nm, and avidity index was calculated by dividing absorbance of the wells treated with urea by that of untreated wells.
IgG quantification in serum samples
Serum sample IgG antibody quantification was performed using radial immunodiffusion (RID) quantification, using the Diffu-Plate® kit for total IgG (Biocientifica S.A, Buenos Aires, Argentina), following the manufacturer’s instructions. For this, 5 μL of serum from each individual was seeded into each well of the immunodiffusion plate and incubated at room temperature (22–25 °C) for 48 h. The measurement of each immunoprecipitation halo was performed using a ruler with accuracy of 0.01 mm, and the total IgG concentration was determined by comparison against data provided by the manufacturer (batch 1157, plate range: 201.8–3645.7 mg/dL; adult reference value: 710–1520 mg/dL).
Purification of IgGs and immunoadsorption of antibodies against L. laeta venom
Purification of IgG antibodies from sera was performed using the Pierce™ Protein G Agarose kit (Thermo Fisher Scientific, Inc. Waltham, MA, USA), following manufacturer’s instructions. Protein G Agarose resin in a 3:1 ratio with binding buffer (0.1 M sodium acetate, pH 5.0) was incubated with serum pools from groups 1 and 2, both previously diluted 1:1 in binding buffer and subsequently incubated in an orbital shaker at room temperature for 1 h and centrifuged at 500×g for 1 min. Then, each purification was washed twice with two volumes of PBS and again centrifuged at 500×g for 1 min, and the supernatant was discarded.
Finally, 1 mL of the elution buffer (0.1 M glycine-HCl buffer, pH 2.8) was added twice and incubated for 10 min at room temperature, with gentle mixing. The purification was neutralized with 50 μL of 1 M Tris, pH 8.0, per mL of obtained eluate, then concentrated in a Microcon® centrifugal filter (Merck-Millipore, Burlington, MA, USA) with a cut off of 10 kDa, and the elution buffer was exchanged for PBS (pH 7.3). The purified IgG antibodies were stored at − 20 °C. The purified IgG antibodies were evaluated by SDS-PAGE in gel at 10% and measuring absorbance at 280 nm in a TECAN® Infinite M200® PRO spectrofluorometer (Tecan Group Ltd., Männedorf, Switzerland). Subsequently, purified IgG antibodies were immunoselected using 2 μg of L. laeta venom adsorbed onto a nitrocellulose membrane in a 96-well Dot Blot Filtration Manifold system (Gibco BRL).
The presence of the adsorbed proteins to the membrane was evaluated by staining with Ponceau red. The membrane was then blocked with 5% non-fat milk in PBS-T for 1 h at 22–25 °C. Subsequently the membrane was washed with PBS-T and incubated with 10 mL (1:10 diluted) of purified IgG from groups 1 or 2 in PBS for 2 h at 4 °C. Next, IgGs that did not recognize L. laeta venom were washed three times in Borate-Saline wash buffer (0.1 M boric acid, 0.25 mM sodium tetraborate, 0.5 M NaCl, 0.05% Tween-20, pH 8.0) for 10 min with gentle mixing. IgG antibodies immunoselected against L. laeta venom were eluted during incubation with elution buffer (0.1 M glycine, 0.15 M NaCl, pH 2.6) for 5 min. Immediately, the IgG antibodies elution was neutralized with 50 μL of 1 M Tris-HCl, pH 8.0. Antibodies were concentrated using an Amicon® Ultra-15 centrifugal filter (Merck-Millipore, Burlington, MA, USA) with a cut-off of 10 kDa and the elution buffer was exchanged for PBS (pH 7.3). The L. laeta venom immunoselected IgG antibodies were stored at − 20 °C.
Two-dimensional electrophoresis
Two-dimensional (2D) electrophoresis was performed using 100 μg of electrostimulated venom from L. laeta, which was first precipitated and resuspended in C1 buffer (8 M Urea, 1 M Thiourea, 4% CHAPS, 66 mM DTT, 0.5% ampholytes, pH range 3–10 NL). IPG strips (7 cm, pH 3–10 NL, Bio-Rad, Hercules, CA, USA) were rehydrated with samples in C1 buffer for 12 h at 20 °C. Isoelectric focusing was performed in the PROTEAN IEF Cell (Bio-Rad, Hercules, CA, USA) system until a total of 11,000 Vh− 1 was reached. After the first dimension was run, the strips were stored at − 80 °C until used. For the second dimension, the IPG strips were thawed at room temperature, then the proteins were subjected to a reduction treatment by incubating for 15 min in equilibrium buffer (50 mM Tris-HCl, pH 8.8, 6 M urea, 2% SDS, and 30% glycerol) containing DTT and then alkylated by incubating for 15 min in equilibrium buffer with iodoacetamide.
Finally, IPG strips were placed in 12% SDS-PAGE gels. Gels were stained with Coomassie Brilliant Blue G-250. To perform Western blot on the 2D electrophoresis-separated venom, 20 μg of venom was used and detected using 1 μg/mL of IgG antibodies purified from sera of Groups 1 and 2, washed, and then incubated with goat anti-human HRP-IgG antibody (1:50,000 dilution) in TBS-T for 1 h at room temperature, and the membranes were developed using the ECL™ Western blotting detection reagent kit (GE Healthcare, Chicago, IL, USA).
Immunoblotting
Immunoblotting was performed by separating 5 μg of the different recombinant proteins (rLlPLD1 and rLlPLD2 from L. laeta, LiSMDP1 from L. intermedia, LrSMD1 from L. reclusa, and LgDerProt1 from L. gaucho), or 5 μg of L. laeta and Sicarius venom, using a 12% SDS-PAGE gels under non-reducing conditions. Additionally, 5 μg of phospholipase A2 (PLA2) from bee venom (Apis mellifera) (Sigma-Aldrich Co, St Louis, MO, USA) and phospholipase C (PLC) from Bacillus cereus (Sigma-Aldrich, USA) were tested. Gels were stained with Coomassie Brilliant Blue or transferred to a nitrocellulose membrane. After transfer, the membranes were blocked for 2 h with 5% non-fat milk in TBS/0.1% Tween20 (TBS-T) and incubated for 1 h at room temperature with pooled sera from Groups 1 and 2 (1:1000 dilution) or with purified and immunoselected IgGs from both groups at 1 μg/mL. Membranes were washed six times for 10 min each with TBS-T and incubated with goat anti-human HRP-IgG antibody (1:50,000 dilution) in TBS-T for 1 h at room temperature. After another six washes with TBS-T, the membranes were developed with the ECL™ Western blotting detection reagent kit (GE Healthcare, Chicago, IL, USA).
Immunoprecipitation
Immunoprecipitation of L. laeta venom was done using Pierce™ Protein G Agarose (Thermo Fisher Scientific, Inc. Waltham, MA, USA), according the manufacturer’s instructions. For this, 100 μg of pure L. laeta venom was incubated with 5 μL (1:20 dilution) of pooled serum from Group 1 or Group 2 in 100 μL of IP buffer (25 mM Tris, 150 mM NaCl, pH 7.2) overnight at 4 °C. Another 100 μg of venom was incubated with 5 μL of mouse anti-L. laeta venom immune serum, 5 μL of pre-immune mouse serum, or 5 μL of unrelated antibody anti-BSA and used as IP control. Subsequently, 100 μL of protein G agarose slurry was added to the venom-antibody complex and incubated for 2 h at room temperature with gentle mixing. Then, 0.5 mL of IP buffer was added and centrifuged at 2500×g for 3 min, and the supernatant was discarded. The immune complex was neutralized with 50 μL of neutralization buffer (1 M Tris, pH 8.0), centrifuged again at 2500×g for 3 min, and 50 μL of 2X SDS-PAGE loading buffer was added for evaluation by SDS-PAGE electrophoresis in a 12% gel. The presence of the immunoprecipitated venom was determined by immunoblot using rabbit polyclonal anti-L. laeta venom serum (1:10,000 dilution) or anti-rLlPLD1 monoclonal antibody 7E4-D2 (1:50,000 dilution), then developed by incubating with goat anti-mouse IgG antibodies labeled with HRP (1:50,000) or goat anti-rabbit IgG antibody labeled with HRP (1:50,000) and by ECL.
Hemolytic neutralization assay
The human erythrocyte hemolysis assay was performed as previously described [25]. Human erythrocytes were washed three times with veronal buffered saline (VBS2+ − pH 7.4; 10 mM sodium barbitone, 0.15 mM CaCl2, 0.5 mM MgCl2, and 145 mM NaCl) and resuspended at 2% in VBS2+. The cells were sensitized for 30 min at 37 °C with 25 μg/mL venom of L. laeta in 100 μL of VBS2+ in presence or absence of pooled serum from Group 1 and pooled serum from Group 2 at (1:1, 1:10 and 1:100 dilutions). Negative controls were incubated only with VBS2+. After incubation, the sensitized erythrocytes were washed three times with VBS2+ and were analyzed in a complement dependent hemolytic assay. Then, 100 μL of sensitized erythrocytes were mixed with 100 μL of normal human serum (NHS; 1:2 in VBS2+). The negative control was evaluated by incubating the erythrocytes with VBS2+ (without complement control) and total hemolysis control was incubated with H2O. After incubation for 1 h at 37 °C, the non-lysed cells were centrifuged at 440×g for 5 min, the supernatant was collected and measured at 414 nm. Results were expressed as a percentage of hemolysis. The assays were made in duplicate for a total of two independent experiments. The erythrocytes and normal serum were obtained from the same donor.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 5.00 for Mac OS X (GraphPad Software Inc., La Jolla, CA, USA). Student t-test and One-Way ANOVA with Bonferroni Multiple Comparison post-hoc test was used to determine the statistical significance of differences among mean values. A statistical significance criterion significance level of p < 0.05 was used.
Results
Sera of people with no clinical history of loxoscelism recognize L. laeta venom
In order to get a general view of immunoreactivity, the serum samples from individuals with loxoscelism (Group 1) and without loxoscelism (Group 2) were evaluated. Initially, the serum pools from Group 1 and Group 2 were used, and the detection of L. laeta venom by both groups was evaluated using dot blot. The pool of sera from the Group 1 was able to detect L. laeta venom. Meanwhile, the pool of sera from Group 2 was also able to recognize the venom (Fig. 1a). Incubation with PBS and pre-immune mouse serum did not show reactivity, whereas mouse anti-L. laeta venom serum showed a marked reaction.
Fig. 1.

Detection of Loxosceles spider venom by sera from Group 1 and Group 2 by dot blot and ELISA. a Dot blot for detection of L. laeta venom incubated with serum pools from Group 1 and Group 2 (1:1000 dilution). b Representative dot blot of L. laeta venom incubated with individual serum from Group 1 (blue line, dots A1-B2) and Group 2 (black line, dots C2-H5); controls (red line): monoclonal antibody 7E4-D2 anti-rLlPLD1 (1:50,000 dilution) (dot A6), polyclonal mouse anti- L. laeta venom serum (1:10,000 dilution) (dot B6), pre-immune mouse serum (1:1000 dilution) (dot C6). c Indirect ELISA for the titration of Group 1 sera that recognize L. laeta venom. d Indirect ELISA for titration of sera from Group 2 that recognize L. laeta venom
In order to determine whether venom detection by the Group 2 pool of samples was due to the presence of individual serum that could present specific antibodies against L. laeta venom, we evaluated the detection of each individual’s serum using dot blot. All ten samples from patients with loxoscelism could detect L. laeta venom (Fig. 1b). In comparison, among the 30 samples from the group without loxoscelism, 18 of them showed detection levels similar to the sera from the loxoscelism group. In addition, five samples (dots C2, D2, B3, C3, C4, and C5) had higher detection levels than those observed in the loxoscelism group. In contrast, dots F3, A4, G4, H4, and H5 showed lower levels of detection (Fig. 1b; Additional file 1A). Strong detection was observed with mouse anti-L. laeta venom immune serum (dot A6) and monoclonal anti-rLlPLD1 (dot B6). Detection was not observed with pre-immune mouse serum (dot C6).
Additionally, titers of serums from both groups were evaluated by indirect ELISA. Briefly, each serum was diluted in the range of 1:10 to 1:5120. Absorbance values under the cut-off point (0.081) were considered to be non-specific or negative reactions. The detection of L. laeta venom for Group 1 serum sample titration media was 160 (n = 10) (Fig. 1c). Similarly, the media of titer for Group 2 sera was 80 (n = 30). Group 1 sera had absorbance values over the cut-off point for L. laeta venom detection at low dilutions (Fig. 1c). However, at 1:160 dilution, samples 1 (dot A1), 2 (dot A2), 8 (dot H1), and 10 (dot B2) presented lower values of the cut-off point. The majority of Group 2 sera had absorbance values over the cut-off (Fig. 1d). Samples 11 (dot G4), 14 (dot F3), 18 (dot A4), 19 (dot H5), and 26 (dot H4) had lower values of the cut-off point at 1:40 dilution. This is consistent with the results of the dot blot. However, the trend of the samples from the non-loxoscelism group (Group 2) remained above the cut-off point up to the titer 80. Additionally, avidity index for both pooled serums showed a high affinity with antibodies present in sera of groups 1 and 2 for detection of L. laeta venom (Additional file 2A).
In contrast, the total IgG concentration (mg/dL) of Group 1 and Group 2 sera was similar and within the reference range of the assay (Table 2). Total serum IgG levels of Group 1 samples ranged from 825 to 1622.5 mg/dL, and the total serum IgG levels of Group 2 samples ranged from 993.6 to 1902.9 mg/dL. The sample with lowest level of total IgG of group 1 sera was the sample 8 (dot H1), with a concentration of 825 mg/dL, while the sample with lowest level of total IgG from group 2 was the sample 14 (dot F3), with a concentration of 993.6 mg/dL. The latter correlates with data showed by dot blot and indirect ELISA, in which both samples had the lowest detection levels of L. laeta venom observed for both groups. Moreover, no differences in total IgG level were observed related to gender among samples.
Table 2.
Human IgG levels in serum samples from patients with and without loxoscelism
| Serum samples | IgG a (mg/dL) | Reference range (mg/dL) | p-value# |
|---|---|---|---|
| Loxoscelism group (n = 10) | 1355 ± 117.1 | 710–1520 | 0.7445 |
| Without loxoscelism group (n = 30) | 1385 ± 36.14 | 710–1520 | 0.7445 |
aValues of media ± SEM. Human IgG concentration in serum were determined by radial immunodiffusion (RID). # t-test; α = 0.05
Sera from individuals with no history of loxoscelism recognize phospholipase D family proteins from L. laeta
In order to confirm the above results, the L. laeta venom component that was specifically recognized by sera from groups 1 and 2 was evaluated. Each serum was assessed by immunoblot of L. laeta venom separated by electrophoresis, showing that all sera from Group 1 and Group 2 recognized a protein component between 25 and 35 kDa (Additional file 3). Sera of Group 2 that could recognize different bands of Loxosceles venom, compared to those in the range of 25–35 kDa proteins, were excluded on suspicion of previous contact with the Loxosceles venom.
Additionally, considering that pooled sera from both groups recognized BSA in dot blot, and to discard reactions from antibodies other than anti-L. laeta venom in sera, we proceeded to purify IgG antibodies from the Group 1 and Group 2 serum pools and immunoselected against L. laeta venom. They were later evaluated by immunoblot for recognizing L. laeta venom separated by 1D and 2D electrophoresis (Fig. 2). As a detection control, mouse anti-L. laeta venom serum was used, noting that it recognizes a protein band pattern of L. laeta venom in the range of 25–35 kDa, while pre-immune mouse serum does not detect L. laeta venom (Fig. 2a), which was corroborated by densitometry analysis of bands (Additional file 1B).
Fig. 2.

Immunoblot detection of L. laeta venom using pooled sera of Group 1 and Group 2. a Immunoblot detection of L. laeta venom with mouse anti-L. laeta venom immune serum. Lane 1: 12% SDS-PAGE of L. laeta venom stained with Coomassie brilliant blue. Lane 2: L. laeta venom immunoblot incubated with pre-immune mouse serum (1:1000 dilution). Lane 3: L. laeta venom immunoblot incubated with mouse L. laeta antivenom immune serum (1:10,000 dilution). b L. laeta venom immunoblot detected by pooled serum and purified IgGs of Group 1 or Group 2. Lanes 1 and 2: Serum pools for Group 1 and Group 2, respectively. Lanes 3 and 4: purified IgG antibodies (1 μg/mL) of Group 1 and Group 2 sera, respectively. Lane 5: pre-immune mouse serum. c Immunoblot of L. laeta venom separated by 2D electrophoresis
Similar detection patterns of L. laeta venom were observed in Group 1 and Group 2 serum pools, as well as with purified IgG antibodies for both groups (Fig. 2b). By means of 2D venom electrophoresis, it was possible to see that the IgG antibodies of both study groups recognize a similar pattern of spots of L. laeta venom proteins, within the range of 25 and 35 kDa (Fig. 2c).
The protein components of Loxosceles venom between 25 and 35 kDa have been considered members of the phospholipase D family and are present in different Loxosceles species [30]. Therefore, we evaluated whether purified IgG antibodies from both groups could recognize the L. laeta phospholipase D1 protein (rLlPLD1), showing detection of the recombinant PLD with purified IgGs from both groups (Fig. 3a). In addition, L. laeta venom immunoprecipitation with pooled sera from groups 1 and 2 and subsequent immunoblot with a rabbit polyclonal anti-L. laeta venom serum (Fig. 3b, upper panel) or with monoclonal antibody anti-LlPLD1 (Fig. 3b, bottom panel) showed that PLD was the major protein immunoprecipitated from the venom. Moreover, since pooled sera from groups 1 and 2 could detect BSA in dot blot (Fig. 1a), we also carried out immunoprecipitation of L. laeta venom with an anti-BSA antibody as an unrelated antibody, which was not detected, corroborating the hypothesis that detection of L. laeta venom was a consequence of the presence of anti-PLDs antibodies in the serum samples of both groups. Additionally, mouse pre-immune serum was not able to immunoprecipitate L. laeta venom, and mouse polyclonal anti-L. laeta venom serum has only a low immunoprecipitation capacity of PLD of L. laeta venom. Based on these data, we can assume that the antibodies present in individuals with no clinical history of loxoscelism could correspond to heterophilic IgG-type antibodies, since there is no known previous exposure to the Loxosceles venom antigens in this group.
Fig. 3.
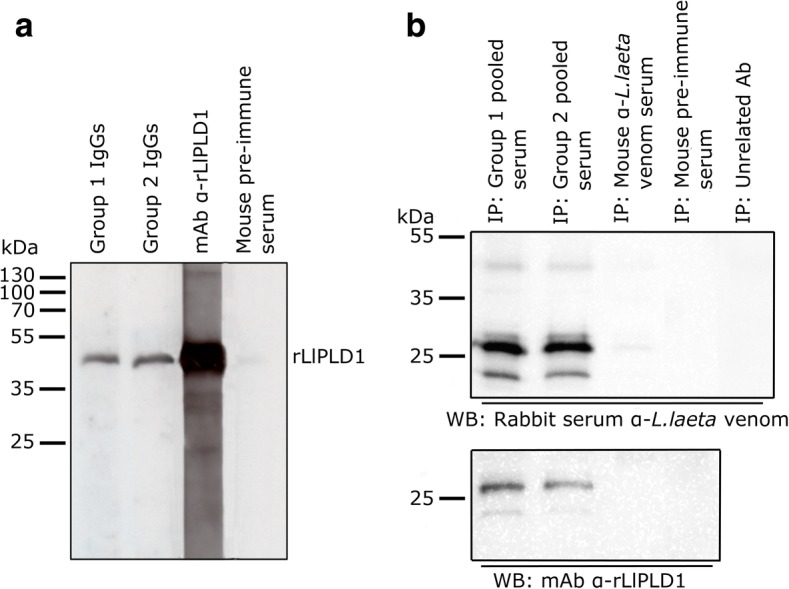
Immunoblot detection of recombinant LlPLD1 from L. laeta venom by heterophilic antibodies of groups 1 and 2. a Immunoblot detection of recombinant protein rLlPLD1 (5 μg) with purified IgGs (1 μg/mL) of sera from groups 1 and 2. Positive control comprised mAb anti-rLlPLD1-7E4-D2 (1:50,000 dilution) and negative control was pre-immune mouse serum (1:1000 dilution). b Immunoprecipitation (IP) of L. laeta venom with antibodies from groups 1 and 2 sera, and detection by (upper panel) immunoblot with rabbit polyclonal L. laeta antivenom serum (1:10,000 dilution) or (below panel) anti-rLlPLD1 monoclonal antibody 7E4-D2 (1:50,000 dilution). Mouse L. laeta antivenom serum, pre-immune mouse serum, and unrelated antibody anti-BSA were used as IP control
Multispecificity of IgGs anti-PLD antibodies present in serum from individuals without history of loxoscelism
A second characteristic of heterophilic antibodies is it multispecificity. Consequently, we evaluated the multispecificity of these possibly heterophilic antibodies on the recognition of other phospholipase D isoforms from L. intermedia, L. reclusa, and L. gaucho through immunoblot. Thus, the purified and immunoselected IgG antibodies from both groups were able to detect different PLDs from other Loxosceles species, with a strongest detection against L. intermedia and L. gaucho PLDs (Fig. 4). This indicates a strong cross-immunoreaction of serum IgG antibodies from individuals with loxoscelism (Group 1) (Fig. 4a) and without loxoscelism (Group 2) (Fig. 4b) with the PLDs of these species. In addition, IgGs from both groups were able to strongly recognize phospholipase A2 (PLA2) from Apis mellifera venom, and weakly recognize phospholipase C (PLC) from Bacillus cereus (Fig. 4).
Fig. 4.

Heterophilic antibodies can detect PLD from other Loxosceles species and the venom from Sicarius spiders. A quantity of 5 μg of rLlPLD1, rLlPLD2, L. intermedia PLD (LiPLD), L. reclusa PLD (LrPLD), and L. gaucho PLD (LgPLD), together with 5 μg PLA2 of Apis mellifera venom (AmPLA2), and PLC of Bacillus cereus (BcPLC) were separated by SDS-PAGE in 12% gel and transferred to a nitrocellulose membrane. Then, each protein was detected by incubation with immunoselected IgGs from both groups at a concentration of 1 μg/mL, followed by incubated with goat anti-human HRP-IgG antibody (1:50,000 dilution), and developed with ECL. a IgG purified from Group 1 sera. b IgG purified from Group 2 sera. c Immunoblot from the venom of Sicarius, with serum pools and purified IgG antibodies of groups 1 and 2. (Left) SDS-PAGE in 12% gel of L. laeta venom and Sicarius venom stained with Coomassie blue. (Right) Immunoblot of Sicarius venom detected using serum pools from Group 1 and Group 2, and IgG antibodies purified from Group 1 and Group 2 sera
Then, we evaluated whether these IgG-type heterophilic antibodies could recognize venom of Sicarius spiders, a genus closely related to Loxosceles, which has paralogue PLD enzymes to those present in Loxosceles venom [31, 32]. The L. laeta venom and the Sicarius venom presented similar protein patterns (Fig. 4c), and both serum pools from groups 1 and 2, as well as the purified and immunoselected IgGs from both groups, were able to recognize the Sicarius venom (Fig. 4c).
Finally, we evaluated the neutralizing effect of these antibodies through a neutralization assay of L. laeta venom hemolytic activity, in which the non-neutralizing effect was observed for both pooled sera at different dilutions (Additional file 2B).
Discussion
In the present study, we evaluated the presence of antibodies capable of detecting the venom of Loxosceles in serum samples from patients with (Group 1) and without (Group 2) loxoscelism. Surprisingly, it was possible to detect the presence of heterophilic antibodies capable of recognizing L. laeta venom in a group of control sera (people with no history of loxoscelism).
The antibodies found in people with no clinical history of loxoscelism not only were able to detect L. laeta venom in dot blot and ELISA tests, but also presented similar titers to serums from patients diagnosed with loxoscelism. In this latter group, the differences in observed detection level for individual serum could be due to different factors that influence in the severity of the clinical symptoms, and the developed immune response, as factors related to the spider, such as inter- and intra-species variations, spider developmental status, gender, and quantity of venom inoculated [7, 8, 33, 34]. In addition, there are patient factors, such as venom inoculation site, age and nutritional status [35].
Animal models inoculated with recombinant phospholipase D (the main immunogenic component of the venom), showed a significant increase of antibodies against venom in sera [25]. This antibody production is considered the basis for the development of neutralizing sera used as specific treatment [36], therefore, an increasing in IgG levels in patients with loxoscelism can be expected. However, the total IgG content in different analyzed sera showed no significant variations between both groups, and any differences due to sex. Therefore, exposure to L. laeta venom does not generate a significant increase in total IgG production in patients with loxoscelism. The latter is consistent with studies indicating that there is no relationship between the clinical picture of loxoscelism and IgG antibody levels in patient sera [23], and points towards the presence of natural or endogenous antibodies against the Loxosceles venom. In addition, our data showed that antibodies found in serum samples of people from both study groups had no neutralizing capacity. Consequently, they do not seem to influence the severity of the clinical picture of loxoscelism at hemolytic way. However, such observation requires further studies to determine the role of antibodies in the severity of the clinical picture, as for example, dermonecrosis.
From the three possible endogenous antibodies – heterophilic antibodies (HA), human anti-animal antibodies (HAAA), and autoantibodies – we believe that the antibodies present in sera from persons without loxoscelism do not appear to be autoantibodies, since the selection of individuals in this group included a criterion that would exclude people with a history of autoimmune diseases, especially rheumatoid factor. Also, the detection of L. laeta venom, both by dot blot and indirect ELISA, led us to believe that these antibodies do not correspond to human anti-animal antibodies, since these antibodies are known to be produced against animal immunoglobulins in people with history of immunotherapy. In addition, the serum samples from loxoscelism patient group used in our study were taken from patients who received no antivenom therapy, since Chilean guidelines for loxoscelism treatment does not suggest the use of antivenom therapy [2]. Thus, the detection of Loxosceles venom observed in this group was as a consequence of the presence of anti L. laeta venom antibodies produced by themselves and not the presence of antibodies from antivenom treatment, which could lead to production of HAAAs.
Certain future considerations and cautions should be taken about Loxosceles antivenom immunotherapy, since it involves the use of an anti-arachnid serum produced in horses [36], which could lead to the production of human anti-animal antibodies. In this regard, it has been documented the presence of anti-horse IgG antibodies in healthy volunteers without treatment with a horse antivenom used for the treatment of snakebites [37]. The presence and specificity of IgG antibodies in patients with loxoscelism undergoing serotherapy has been previously studied [23], showing that only results from four patients out of twenty that underwent serotherapy were able to detect the L. gaucho venom protein component of ~ 35 kDa by immunoblot. The authors indicate that the low number of patients able to recognize the venom was due to an inhibitory effect that sequesters the circulating immunogenic material. The authors also evaluated the sera through ELISA, in which the highest recognition titer of the venom was 1:640 and the lowest was 1:80 [23]. In our study, the mean titer for loxoscelism sera was 1:320, while the sera of patients without loxoscelism was 1:160, which is double the lowest titer reported by Barbaro et al. [23] for L. gaucho venom detection among loxoscelism patients. This indicates that the antibodies present in this group were possibly produced against an antigen similar to one of the components of the venom of Loxosceles, which present cross-reactivity immunodetection.
The third class of endogenous antibodies are the heterophilic ones, which are produced without exposure to a specific immunogen, so they can be considered as naturally occurring [21]. These antibodies are characterized by their multispecificity, being multireactive against heterogeneous or poorly defined antigens, and generally are often presumed to be low affinity antibodies, but this rule have exceptions [22]. Thus, the avidity index of antibodies in sera of both groups showed high affinity and led us to believe that these antibodies were produced early by an antigen with similar epitopes as Loxosceles PLDs. Despite having a high affinity for L. laeta venom, we think that antibodies found in sera from people without loxoscelism are heterophilic IgG antibodies, due to the unknown exposure to the antigen, which is considered a major criterion to consider an antibody as heterophile [20]. Therefore, we evaluated which Loxosceles venom components were detected by these possible heterophilic antibodies.
In our study, pooled sera from Group 1 (loxoscelism) and Group 2 (without loxoscelism), as well as IgG antibodies purified from both groups and immunoselected against L. laeta venom, could detect L. laeta venom proteins between 25 and 35 kDa, both in 1D and 2D immunoblot. The latter technique detected different proteins in this range, presumable indicating the multispecificity of these antibodies. Among the different protein components of the Loxosceles venom, the family of phospholipase D proteins (PLD) are capable of producing dermonecrosis, neutrophil activation, complement dependent red blood cell hemolysis, platelet aggregation, blood vessel permeability changes, kidney cytotoxicity, and recently it was demonstrated its role in monocyte recruitment [38, 39]. PLD molecular mass varies between 30 kDa and 35 kDa, and different isoforms of Loxosceles PLD have been documented for the different Loxosceles species [30, 40]. Our data showed that these IgG class antibodies detected the L. laeta recombinant protein phospholipase D1 (rLlPLD1), and other PLD isoforms of L. intermedia, L. gaucho, and L. reclusa indicating an important cross-immunoreactivity of antibodies present in the sera of individuals without loxoscelism, as well as that the specificity of the reaction was due to the presence of IgG antibodies capable of recognizing Loxosceles PLDs.
Likewise, this multispecificity was corroborated by cross-detection with other types of phospholipases, such as Apis mellifera phospholipase A2 (PLA2) and Bacillus cereus phospholipase C (PLC), which could indicate a common pattern of antigenicity among different types of phospholipases. Together with this, the IgG heterophilic antibodies from Group 2 recognized the venom of Sicarius spiders, which presented a venom protein pattern similar to that of L. laeta. These spiders are a closely related genus to Loxosceles, both belonging to the Sicariidae family [31, 41]. Both spider genera share important characteristics, such as similar venom protein patterns in the size range corresponding to known sphingomyelinase D (SMase D or PLD) proteins (31–35 kDa) and presence of active [30, 32]. Also, Sicarius ornatus exhibits venom interspecies differences at the gender level and has been documented as having active PLDs with complement dependent hemolytic activity in human red blood cells and cytotoxic activity in keratinocytes [42], similar to those reported for Loxosceles spiders [8]. In addition to this, serum anti-PLD of L. intermedia recognizes the 33 kDa component of Sicarius venom, which is a molecular mass also associated with PLDs in Loxosceles venom [42].
A relevant characteristic to consider an antibody as heterophilic is its unknown exposure to a specific immunogen [21]. In this regard, the possible origin of these natural or heterophilic antibodies in individuals without contact with Loxosceles venom is still unclear. However, the interspecies similarity between amino acid sequences of Loxosceles PLDs and the similarity in the venom protein patterns between the genus Loxosceles and Sicarius suggest that contact with the venom proteins of spiders closely related with Loxosceles could produce these antibodies [31]. Similarly, it cannot be ruled out that other arachnid genus could present PLDs capable of stimulating the production of these heterophilic antibodies. In addition, the antibodies could be produced due to previous exposure to bacterial PLDs, since the PLDs of Loxosceles spiders and bacteria such as Corynebacterium pseudotuberculosis possess similar molecular mass (31–32 kDa), have roughly 30% of sequence identity and have phospholipase activity on lysophosphatidylcholine (LPC) [43, 44].
The presence of SMase D (PLDs) in different pathogenic organisms, such as arachnids (genera Acanthoscurria and Stegodyphus), acarus (genera Dermatophagoides, Varroa, Psoroptes, and Tetranychus), ticks (Ixodes scapularis), bacteria (genera Burkholderia, Streptomyces, and Austwickia), and fungi (genera Aspergillus, Fusarium, Coccidioides, and Trichophyton, among others) has been reported and they share the same tridimensional structure as Loxosceles PLDs [45]. This would indicate that such a broad diversity of organisms with similar PLDs would facilitate the contact and production of antibodies capable of reacting with Loxosceles venom. This hypothesis is strengthened by the results observed in the present study for the immunodetection of heterophilic IgGs to Bacillus cereus PLC. Additionally, the origin of these heterophilic antibodies may be the exposure to other phospholipases, such as PLA2 from bee venom (Apis mellifera), since it has been reported that IgG4 antibodies present cross-reactivity for secreted PLA2s from different species, such as Bos taurus (cattle), Apis mellifera (honey bee), Daboia russelii (Russell’s viper), and Naja mossambica (spitting cobra) in patients allergic to A. mellifera venom and in control subjects [46].
The results presented in this study corroborate the presence of heterophilic IgG-class antibodies in the sera of individuals without loxoscelism. Interference caused by endogenous antibodies in sandwich immunoassays can occur by binding to, bridging, or blocking binding sites in capture or detection antibodies [22]. However, in the particular case of serum samples with the presence of anti-PLD antibodies, these could interfere in venom detection in a different way by directly binding to PLDs from Loxosceles venom. This would limit the binding of these proteins to the capture antibodies of sandwich ELISA or competitive type ELISA assays, due to a potential sequestration effect of venom components, which may lead to false negatives and underestimation of the presence of Loxosceles venom in patients. For this reason, considerations must be taken when blood and serum samples are used. Indeed, differences in venom detection, based on sample origin, have been documented in rabbits, in which detection of Loxosceles venom in hair samples, aspiration, and skin biopsy was possible for up to 7 days post-inoculation, but was undetectable in serum [47]. Along with our data, this observation point towards the use of skin samples over serum as a sample source for immunodetection tests of Loxosceles venom.
Conclusions
In conclusion, in the present study we demonstrated the presence of IgG-class heterophilic antibodies directed against PLDs of Loxosceles and Sicarius spiders, present in people without contact with Loxosceles spider venom. The presence of these antibodies in serum samples should be considered as a possible interference in immunoassays for the specific detection of Loxosceles spider venom in humans.
Additional files
Densitometry analysis for dot blot and Western blot shown in Figs. 1b, 2a and b. Intensity of dots and bands were realized using ImageJ program, verifying for non-saturation and subtracting background. (A) Values from dots of Fig. 1b were expressed as relative density percentage calculated for each dot and normalized against control dot intensity with anti-L. laeta venom antibodies. Values are means ± S.E.M (n = 3). In addition, values of Western blot from Fig. 2a and Fig. 2b were expressed as relative density calculated from area mean density of each band and (B) normalized against the control band with mouse anti-L. laeta venom serum, (C) or normalized against control band with pool of serums of Group 1. Significance was evaluated with an ANOVA one-way with Bonferroni post-hoc test; (ns) indicates not statistically significant, and *** indicates significant differences between dots and control with p < 0.05. (TIF 27016 kb)
Avidity index of pooled serums from Group 1 and Group 2, and neutralizing capacity of serums against hemolytic activity of venom of L. laeta. (A) Comparison of avidity index of pooled serums of Group 1 and Group 2 (1:100 diluted), treated with 6 M urea by IgG avidity ELISA. (ns) indicates not statistically significant. (B) Human erythrocytes were sensitized for 1 h at 37 °C with 25 μg/mL of venom of L. laeta in the presence or absence of pooled sera of Group 1 or Group 2 at 1:1, 1:10 and 1:100 dilutions, and evaluated in a complement-dependent hemolysis assay. Negative control was incubated only with VBS and with not presence of complement serum (control without complement). Results were expressed as percentage of hemolysis. The assays were made in duplicate for a total of two independent experiments and results are expressed as mean ± SEM. Significance was evaluated with an ANOVA one-way with Bonferroni post-hoc test; (ns) indicates not statistically significant. (TIF 9838 kb)
Detection of L. laeta venom by immunoblot using single serums of Group 1 and Group 2. (A – Right) SDS-PAGE in 12% gel of L. laeta venom stained with Coomassie brilliant blue. (A – Left) Immunoblot detection of L. laeta venom incubated with mouse L. laeta antivenom immune serum (1:10,000 dilution) (CP). Immunoblot incubated with pre-immune mouse serum (1:1000 dilution) (CN). (B) L. laeta venom immunoblot detected by single serum from loxoscelism group (Group 1). (C) L. laeta venom immunoblot detected by individual serum from without loxoscelism group (Group 2). (TIF 2986 kb)
Acknowledgments
The authors would like to thank to the company BioPub for language corrections and translations.
Funding
This study was supported by National Fund for Scientific and Technological Development (FONDECYT) Initiation into Research (Project n°. 11130020), Fund for Promotion of Scientific and Technological Development (FONDEF) (Project n°. D04I1247) and partially by doctoral fellowship D-21151173, from the National Commission for Scientific and Technological Research (CONICYT), Chile. Also, grants were received from FIC-R “Ideas Innovativas”, BIP n° 30111017–0 and Research Fund for the Development of Undergraduate Thesis or Degree Activity n° ATI15–2-03 from the Vice-Rectorate of Research, Innovation and Postgraduate Studies of the University of Antofagasta (VRIIP-UA).
Availability of data and materials
The dataset used and/or analyzed during this study are included in the published article (and its Additional files).
Abbreviations
- 2D
two-dimensional
- AA
autoantibodies
- AmPLA2
PLA2 of Apis mellifera venom
- BcPLC
PLC of Bacillus cereus
- CL
cutaneous loxoscelism
- HA
heterophilic antibodies
- HAAA
human anti-animal antibodies
- IP
immunoprecipitation
- LgPLD
Loxosceles gaucho PLD
- LiPLD
Loxosceles intermedia PLD
- LPC
lysophosphatidylcholine
- LrPLD
Loxosceles reclusa PLD
- PLA2
phospholipase A2
- PLC
phospholipase C
- PLD
phospholipases D
- RID
radial immunodiffusion
- rLlPLD1
recombinant phospholipase D isoform 1
- TMB
tetramethylbenzidine
- VBS
veronal buffered saline
- VCL
viscero-cutaneous loxoscelism
Authors’ contributions
Authors TA-S and JR contributed equally to this work. AC, JG, JEA conceived and designed the experiments. JMR, TSA, MSM, VC, LO performed the experiments. TSA, JMR, LO, WC, and AC. analyzed the data. TSA, JMR, JEA, and AC wrote and corrected the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
All procedures involving collection of human serum samples and informed consent were approved by the Ethics Committee in Scientific Research of the University of Antofagasta (CEIC-UA), Chile.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Tomás Arán-Sekul and José M. Rojas contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s40409-018-0155-x) contains supplementary material, which is available to authorized users.
Contributor Information
Tomás Arán-Sekul, Email: tomas.aran@uantof.cl.
José M. Rojas, Email: jose.rojas@uantof.cl
Mario Subiabre, Email: mesubiabre@uc.cl.
Victoria Cruz, Email: victoria.cruzb@gmail.com.
William Cortés, Email: william.cortes.araya@gmail.com.
Luis Osorio, Email: lraosorio@gmail.com.
Jorge González, Email: jorge.gonzalez@uantof.cl.
Jorge E. Araya, Email: jorge.araya@uantof.cl
Alejandro Catalán, Phone: +56 55 2637376, Email: alejandro.catalan@uantof.cl.
References
- 1.Swanson DL, Vetter RS. Bites of brown recluse spiders and suspected necrotic arachnidism. N Engl J Med. 2005;352(7):700–707. doi: 10.1056/NEJMra041184. [DOI] [PubMed] [Google Scholar]
- 2.MINSAL . Guía para el manejo de la mordedura de araña de los rincones (Departamento de Enfermedades transmisibles DdPyCdE, Ministerio de Salud ed) Santiago de Chile: Ministerio de Salud de Chile; 2016. [Google Scholar]
- 3.Chippaux JP. Epidemiology of envenomations by terrestrial venomous animals in Brazil based on case reporting: from obvious facts to contingencies. J Venom Anim Toxins Incl Trop Dis. 2015;21:13. doi: 10.1186/s40409-015-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brasil. Ministério da Saúde. Fundação Nacional de Saúde (FUNASA) Manual de diagnóstico e tratamento de acidentes por animais peçonhentos. 2. Brasília: Fundação Nacional de Saúde; 2001. [Google Scholar]
- 5.Lucas SM. The history of venomous spider identification, venom extraction methods and antivenom production: a long journey at the Butantan institute, Sao Paulo, Brazil. J Venom Anim Toxins Incl Trop Dis. 2015;21:21. doi: 10.1186/s40409-015-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ríos JC, Pérez M, Sánchez P, Bettini M, Mieres JJ, Paris E. Prevalence and epidemiology of Loxosceles laeta bite. Analysis of consultations to a poison control center. Rev Med Chil. 2007;135(9):1160–1165. doi: 10.4067/S0034-98872007000900010. [DOI] [PubMed] [Google Scholar]
- 7.de Oliveira KC, Goncalves de Andrade RM, Piazza RM, Ferreira JM, Jr, van den Berg CW, Tambourgi DV. Variations in Loxosceles spider venom composition and toxicity contribute to the severity of envenomation. Toxicon. 2005;45(4):421–429. doi: 10.1016/j.toxicon.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Cristina de Oliveira K, Goncalves de Andrade RM, Giusti AL, Dias da Silva W, Tambourgi DV. Sex-linked variation of Loxosceles intermedia spider venoms. Toxicon. 1999;37(1):217–221. doi: 10.1016/S0041-0101(98)00130-5. [DOI] [PubMed] [Google Scholar]
- 9.McGlasson DL, Harroff HH, Sutton J, Dick E, Elston DM. Cutaneous and systemic effects of varying doses of brown recluse spider venom in a rabbit model. Clin Lab Sci. 2007;20(2):99–105. [PubMed] [Google Scholar]
- 10.Schenone H. Toxic pictures produced spiders bites in Chile: latrodectism and loxoscelism. Rev Med Chil. 2003;131(4):437–444. doi: 10.4067/S0034-98872003000400013. [DOI] [PubMed] [Google Scholar]
- 11.Malaque CM, Castro-Valencia JE, Cardoso JL, Francca FO, Barbaro KC, Fan HW. Clinical and epidemiological features of definitive and presumed loxoscelism in Sao Paulo, Brazil. Rev Inst Med Trop Sao Paulo. 2002;44(3):139–143. doi: 10.1590/S0036-46652002000300005. [DOI] [PubMed] [Google Scholar]
- 12.Vetter RS, Bush SP. Reports of presumptive brown recluse spider bites reinforce improbable diagnosis in regions of North America where the spider is not endemic. Clin Infect Dis. 2002;35(4):442–445. doi: 10.1086/341244. [DOI] [PubMed] [Google Scholar]
- 13.Malaque CM, Santoro ML, Cardoso JL, Conde MR, Novaes CT, Risk JY, Franca FO, de Medeiros CR, Fan HW. Clinical picture and laboratorial evaluation in human loxoscelism. Toxicon. 2011;58(8):664–671. doi: 10.1016/j.toxicon.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Cox KL, Devanarayan V, Kriauciunas A, Manetta J, Montrose C, Sittampalam S: Immunoassay Methods. 2012 May 1 [Updated 2014 Dec 24]. In Assay Guidance Manual [Internet]. Edited by Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Chung TDY, et al. Bethesda (MD); Eli Lilly & Company and the National Center for Advancing Translational Sciences. 2004. [PubMed]
- 15.Chávez-Olórtegui C, Zanetti VC, Ferreira AP, Minozzo JC, Mangili OC, Gubert IC. ELISA for the detection of venom antigens in experimental and clinical envenoming by Loxosceles intermedia spiders. Toxicon. 1998;36(4):563–569. doi: 10.1016/S0041-0101(97)00159-1. [DOI] [PubMed] [Google Scholar]
- 16.Gomez HF, Krywko DM, Stoecker WV. A new assay for the detection of Loxosceles species (brown recluse) spider venom. Ann Emerg Med. 2002;39(5):469–474. doi: 10.1067/mem.2002.122914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoecker WV, Green JA, Gomez HF. Diagnosis of loxoscelism in a child confirmed with an enzyme-linked immunosorbent assay and noninvasive tissue sampling. J Am Acad Dermatol. 2006;55(5):888–890. doi: 10.1016/j.jaad.2006.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akdeniz S, Green JA, Stoecker WV, Gomez HF, Keklikci SU. Diagnosis of loxoscelism in two Turkish patients confirmed with an enzyme-linked immunosorbent assay (ELISA) and non-invasive tissue sampling. Dermatol Online J. 2007;13(2):11. [PubMed] [Google Scholar]
- 19.Jiacomini I, Silva SK, Aubrey N, Muzard J, Chavez-Olortegui C, De Moura J, Billiald P, Alvarenga LM. Immunodetection of the “brown” spider (Loxosceles intermedia) dermonecrotoxin with an scFv-alkaline phosphatase fusion protein. Immunol Lett. 2016;173:1–6. doi: 10.1016/j.imlet.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan IV, Levinson SS. When is a heterophile antibody not a heterophile antibody? When it is an antibody against a specific immunogen. Clin Chem. 1999;45(5):616–618. [PubMed] [Google Scholar]
- 21.Levinson SS, Miller JJ. Towards a better understanding of heterophile (and the like) antibody interference with modern immunoassays. Clin Chim Acta. 2002;325(1–2):1–15. doi: 10.1016/S0009-8981(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 22.Bolstad N, Warren DJ, Nustad K. Heterophilic antibody interference in immunometric assays. Best Pract Res Clin Endocrinol Metab. 2013;27(5):647–661. doi: 10.1016/j.beem.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Barbaro KC, Cardoso JL, Eickstedt VR, Mota I. IgG antibodies to Loxosceles sp. spider venom in human envenoming. Toxicon. 1992;30(9):1117–1121. doi: 10.1016/0041-0101(92)90057-C. [DOI] [PubMed] [Google Scholar]
- 24.Ríos JC, Paris E, Apt W, Ristori L, Ramírez G. Guía clínica para el manejo de la mordedura de araña de rincón. Loxosceles laeta. Chile: Ministerio de Salud; 2004. [Google Scholar]
- 25.Catalán A, Cortés W, Sagua H, González J, Araya JE. Two new phospholipase D isoforms of Loxosceles laeta: cloning, heterologous expression, functional characterization, and potential biotechnological application. J Biochem Mol Toxicol. 2011;25(6):393–403. doi: 10.1002/jbt.20399. [DOI] [PubMed] [Google Scholar]
- 26.Ramos-Cerrillo B, Olvera A, Odell GV, Zamudio F, Paniagua-Solis J, Alagon A, Stock RP. Genetic and enzymatic characterization of sphingomyelinase D isoforms from the north American fiddleback spiders Loxosceles boneti and Loxosceles reclusa. Toxicon. 2004;44(5):507–514. doi: 10.1016/j.toxicon.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Tambourgi DV, de F Fernandes Pedrosa M, van den Berg CW, Goncalves-de-Andrade RM, Ferracini M, Paixão-Cavalcante D, Morgan BP, Rushmere NK. Molecular cloning, expression, function and immunoreactivities of members of a gene family of sphingomyelinases from Loxosceles venom glands. Mol Immunol. 2004;41(8):831–840. doi: 10.1016/j.molimm.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Catalán A, Espoz MC, Cortés W, Sagua H, González J, Araya JE. Tetracycline and penicillin resistant Clostridium perfringens isolated from the fangs and venom glands of Loxosceles laeta: its implications in loxoscelism treatment. Toxicon. 2010;56(6):890–896. doi: 10.1016/j.toxicon.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Theakston RD, Lloyd-Jones MJ, Reid HA. Micro-ELISA for detecting and assaying snake venom and venom-antibody. Lancet. 1977;2(8039):639–641. doi: 10.1016/S0140-6736(77)92502-8. [DOI] [PubMed] [Google Scholar]
- 30.Binford GJ, Wells MA. The phylogenetic distribution of sphingomyelinase D activity in venoms of Haplogyne spiders. Comp Biochem Physiol B Biochem Mol Biol. 2003;135(1):25–33. doi: 10.1016/S1096-4959(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 31.Binford GJ, Callahan MS, Bodner MR, Rynerson MR, Nunez PB, Ellison CE, Duncan RP. Phylogenetic relationships of Loxosceles and Sicarius spiders are consistent with western Gondwanan vicariance. Mol Phylogenet Evol. 2008;49(2):538–553. doi: 10.1016/j.ympev.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Binford GJ, Bodner MR, Cordes MH, Baldwin KL, Rynerson MR, Burns SN, Zobel-Thropp PA. Molecular evolution, functional variation, and proposed nomenclature of the gene family that includes sphingomyelinase D in sicariid spider venoms. Mol Biol Evol. 2009;26(3):547–566. doi: 10.1093/molbev/msn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Futrell JM. Loxoscelism. Am J Med Sci. 1992;304(4):261–267. doi: 10.1097/00000441-199210000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Goncalves de Andrade RM, De Oliveira KC, Giusti AL, Dias da Silva W, Tambourgi DV. Ontogenetic development of Loxosceles intermedia spider venom. Toxicon. 1999;37(4):627–632. doi: 10.1016/S0041-0101(98)00200-1. [DOI] [PubMed] [Google Scholar]
- 35.Tambourgi DV, Goncalves-de-Andrade RM, van den Berg CW. Loxoscelism: from basic research to the proposal of new therapies. Toxicon. 2010;56(7):1113–1119. doi: 10.1016/j.toxicon.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 36.Pauli I, Puka J, Gubert IC, Minozzo JC. The efficacy of antivenom in loxoscelism treatment. Toxicon. 2006;48(2):123–137. doi: 10.1016/j.toxicon.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Leon G, Segura A, Herrera M, Otero R, Franca FO, Barbaro KC, Cardoso JL, Wen FH, de Medeiros CR, Prado JC, et al. Human heterophilic antibodies against equine immunoglobulins: assessment of their role in the early adverse reactions to antivenom administration. Trans R Soc Trop Med Hyg. 2008;102(11):1115–1119. doi: 10.1016/j.trstmh.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 38.Rojas JM, Arán-Sekúl T, Cortés E, Jaldín R, Ordenes K, Orrego PR, González J, Araya JE, Catalán A. Phospholipase D from Loxosceles laeta spider venom induces IL-6, IL-8, CXCL1/GRO-alpha, and CCL2/MCP-1 production in human skin fibroblasts and stimulates monocytes migration. Toxins (Basel). 2017;9(4):e125. [DOI] [PMC free article] [PubMed]
- 39.Gremski LH, Trevisan-Silva D, Ferrer VP, Matsubara FH, Meissner GO, Wille AC, Vuitika L, Dias-Lopes C, Ullah A, de Moraes FR, et al. Recent advances in the understanding of brown spider venoms: from the biology of spiders to the molecular mechanisms of toxins. Toxicon. 2014;83:91–120. doi: 10.1016/j.toxicon.2014.02.023. [DOI] [PubMed] [Google Scholar]
- 40.Chaves-Moreira D, Senff-Ribeiro A, Wille AC, Gremski LH, Chaim OM, Veiga SS. Highlights in the knowledge of brown spider toxins. J Venom Anim Toxins Incl Trop Dis. 2017;23:6. doi: 10.1186/s40409-017-0097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platnick NI. Advances in spider taxonomy 1988-1991. New York: The New York Entomological Society; 1993. [Google Scholar]
- 42.Lopes PH, Bertani R, Goncalves-de-Andrade RM, Nagahama RH, van den Berg CW, Tambourgi DV. Venom of the Brazilian spider Sicarius ornatus (Araneae, Sicariidae) contains active sphingomyelinase D: potential for toxicity after envenomation. PLoS Negl Trop Dis. 2013;7(8):e2394. doi: 10.1371/journal.pntd.0002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Meeteren LA, Frederiks F, Giepmans BN, Pedrosa MF, Billington SJ, Jost BH, Tambourgi DV, Moolenaar WH. Spider and bacterial sphingomyelinases D target cellular lysophosphatidic acid receptors by hydrolyzing lysophosphatidylcholine. J Biol Chem. 2004;279(12):10833–10836. doi: 10.1074/jbc.C300563200. [DOI] [PubMed] [Google Scholar]
- 44.Bernheimer AW, Campbell BJ, Forrester LJ. Comparative toxinology of Loxosceles reclusa and Corynebacterium pseudotuberculosis. Science. 1985;228(4699):590–591. doi: 10.1126/science.3983643. [DOI] [PubMed] [Google Scholar]
- 45.Dias-Lopes C, Neshich IA, Neshich G, Ortega JM, Granier C, Chavez-Olortegui C, Molina F, Felicori L. Identification of new sphingomyelinases D in pathogenic fungi and other pathogenic organisms. PLoS One. 2013;8(11):e79240. doi: 10.1371/journal.pone.0079240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sin BA, Akdis M, Zumkehr J, Bezzine S, Bekpen C, Lambeau G, Akdis CA. T-cell and antibody responses to phospholipase A2 from different species show distinct cross-reactivity patterns. Allergy. 2011;66(12):1513–1521. doi: 10.1111/j.1398-9995.2011.02689.x. [DOI] [PubMed] [Google Scholar]
- 47.Krywko DM, Gomez HF. Detection of Loxosceles species venom in dermal lesions: a comparison of 4 venom recovery methods. Ann Emerg Med. 2002;39(5):475–480. doi: 10.1067/mem.2002.123551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Densitometry analysis for dot blot and Western blot shown in Figs. 1b, 2a and b. Intensity of dots and bands were realized using ImageJ program, verifying for non-saturation and subtracting background. (A) Values from dots of Fig. 1b were expressed as relative density percentage calculated for each dot and normalized against control dot intensity with anti-L. laeta venom antibodies. Values are means ± S.E.M (n = 3). In addition, values of Western blot from Fig. 2a and Fig. 2b were expressed as relative density calculated from area mean density of each band and (B) normalized against the control band with mouse anti-L. laeta venom serum, (C) or normalized against control band with pool of serums of Group 1. Significance was evaluated with an ANOVA one-way with Bonferroni post-hoc test; (ns) indicates not statistically significant, and *** indicates significant differences between dots and control with p < 0.05. (TIF 27016 kb)
Avidity index of pooled serums from Group 1 and Group 2, and neutralizing capacity of serums against hemolytic activity of venom of L. laeta. (A) Comparison of avidity index of pooled serums of Group 1 and Group 2 (1:100 diluted), treated with 6 M urea by IgG avidity ELISA. (ns) indicates not statistically significant. (B) Human erythrocytes were sensitized for 1 h at 37 °C with 25 μg/mL of venom of L. laeta in the presence or absence of pooled sera of Group 1 or Group 2 at 1:1, 1:10 and 1:100 dilutions, and evaluated in a complement-dependent hemolysis assay. Negative control was incubated only with VBS and with not presence of complement serum (control without complement). Results were expressed as percentage of hemolysis. The assays were made in duplicate for a total of two independent experiments and results are expressed as mean ± SEM. Significance was evaluated with an ANOVA one-way with Bonferroni post-hoc test; (ns) indicates not statistically significant. (TIF 9838 kb)
Detection of L. laeta venom by immunoblot using single serums of Group 1 and Group 2. (A – Right) SDS-PAGE in 12% gel of L. laeta venom stained with Coomassie brilliant blue. (A – Left) Immunoblot detection of L. laeta venom incubated with mouse L. laeta antivenom immune serum (1:10,000 dilution) (CP). Immunoblot incubated with pre-immune mouse serum (1:1000 dilution) (CN). (B) L. laeta venom immunoblot detected by single serum from loxoscelism group (Group 1). (C) L. laeta venom immunoblot detected by individual serum from without loxoscelism group (Group 2). (TIF 2986 kb)
Data Availability Statement
The dataset used and/or analyzed during this study are included in the published article (and its Additional files).