

Abstract
Background
Galloway–Mowat syndrome (GAMOS) is a rare hereditary renal–neurological disease characterized by early-onset steroid-resistant nephrotic syndrome in combination with microcephaly and brain anomalies. Recently, novel causative mutations for this disease have been identified in the genes encoding the four KEOPS subunits: OSGEP, TP53RK, TPRKB, and LAGE3.
Case presentation
We detected a novel homozygous TP53RK mutation (NM_033550, c.194A > T, p.Lys65Met) using whole exome sequencing in a familial case of GAMOS with three affected siblings. All three patients manifested similar phenotypes, including very early-onset nephrotic syndrome (8 days, 1 day, and 1 day after birth, respectively), microcephaly, dysmorphic faces, and early fatality (10 months, 21 days, and 25 days of age, respectively). One patient also showed hiatal hernia with gastric volvulus. Renal biopsy performed on one patient revealed focal segmental glomerulosclerosis with severe tubulo-interstitial changes.
Conclusion
We report on a familial case of GAMOS with three affected siblings carrying a novel homozygous TP53RK mutation. To our knowledge, this is only the second report on GAMOS in association with a TP53RK mutation.
Keywords: Galloway–Mowat syndrome, KEOPS complex, TP53RK mutation
Background
Galloway-Mowat syndrome (GAMOS) is a rare hereditary renal–neurological disease characterized by early-onset steroid-resistant nephrotic syndrome combined with microcephaly and brain anomalies [1, 2]. There have been several reports of candidate genes for this disease, namely WDR73 [3–8], NUP107 [9], and WHAMM [10]. Recently, Hildebrandt and colleagues [11] identified novel causative mutations in the genes encoding the four KEOPS (Kinase, Endopeptidase and Other Proteins of small Size) subunits, OSGEP, TP53RK, TPRKB, and LAGE3, in 37 individuals from 32 families with GAMOS. They also revealed that knocking down genes encoding KEOPS subunits in human podocytes results in impaired cell proliferation, translational attenuation, endoplasmic reticulum (ER) stress, activation of DNA damage response (DDR) signaling, increased apoptosis, and defects in actin regulation, which are possible pathogenic features of GAMOS [11]. Another study independently reported a familial case of GAMOS with a homozygous OSGEP mutation [12].
Here, we report the identification of a homozygous TP53RK mutation in a familial case of GAMOS with three affected siblings.
Case presentations
The parents of the patients were nonconsanguineous and had four offspring (II-1–II-4), three of which exhibited very similar phenotypes (Table 1). This study was approved by the Seoul National University Hospital’s Institutional Review Board (IRB No. 0812–002-264). Informed consent was obtained from the parents.
Table 1.
Clinical features of affected individuals
| Case | II-1 | II-2 | II-4 |
|---|---|---|---|
| Gender | Female | Male | Male |
| Age of death | 10 months | 21 days | 25 days |
| Ethnicity | Korean | Korean | Korean |
| Neonatal Profile | |||
| Gestational period (weeks) | 39+ 3 | 36+ 6 | 36+ 4 |
| Apgar score (5 min) | 7 | 7 | 7 |
| Pregnancy course | fetal distress | fetal distress, C/S | fetal distress, C/S |
| Birth height (cm) | 46 (5–10th percentile) | 47.5 (10–25th percentile) | 45 (3rd–5th percentile) |
| Birth weight (g) | 2250 (< 3rd percentile) | 1960 (< 3rd percentile) | 1780 (< 3rd percentile) |
| Head circumference (cm) | 29 (< 3rd percentile) | 28 (< 3rd percentile) | 27 (< 3rd percentile) |
| Renal Phenotypes | |||
| Onset of nephrotic syndrome | 8 days | 1 day | 1 day |
| Renal biopsy (age) | FSGS (2 weeks) | NP | NP |
| Urinary tract abnormalities | (−) | (−) | (−) |
| Neurological Features | |||
| Brain MRI | microcephaly simplified gyral pattern progressive brain atrophy |
microcephaly simplified gyral pattern cerebellar hypoplasia pontine hypoplasia |
microcephaly simplified gyral pattern |
| Cerebellum atrophy | (−) | (−) | (−) |
| Others | facial dysmorphism hiatal hernia gastric volvulus |
facial dysmorphism skeletal deformities |
facial dysmorphism |
C/S Caesarean section, MRI magnetic resonance imaging, NP not performed
Case II-1
This female baby was born after a gestational period of 39+ 3 weeks. The birth weight was 2250 g (< 3rd percentile), height was 46 cm (5–10th percentile), head circumference was 29 cm (< 3rd percentile), and the Apgar scores at 1 and 5 min were 5 and 7, respectively. A diaphragmatic hernia was noted in the delivery room. The initial serum albumin level at day one was 3.3 g/dL and the initial serum creatinine level, which reflects the mother’s renal function, was 0.58 mg/dL. Imaging revealed a hiatal hernia with gastric volvulus (Fig. 1). She also had facial dysmorphism including ocular hypertelorism and low-set ears, and a brain magnetic resonance image (MRI) revealed microcephaly with a simplified gyral pattern (Fig. 2a). Surgical repair of the hiatal hernia was performed 6 days after birth without any serious events. The baby started to feed on a mix of breast milk and formula. Two days after surgery, generalized edema developed with a decrease in urine volume. Serum albumin levels decreased to 2.0 g/dL, serum creatinine levels increased to 1.29 mg/dL, and 24-h urinary protein excretion was 2871 mg/day. Renal ultrasonography revealed increased echogenicity of both kidneys with poor differentiation between the peripheral cortex and central echogenic complex. At the age of 2 weeks, an open kidney biopsy was performed (Fig. 3). Thirty-eight (44%) of 87 glomeruli exhibited segmental lobular collapse and sclerosis, and some of the nonsclerotic glomeruli showed features of immature fetal glomeruli. Tubules displayed severe focal atrophy and loss. Infiltration of mononuclear cells and fibrosis were observed in the interstitium. A follow-up brain MRI at 4 months of age showed the progression of diffuse brain atrophy with subarachnoid space widening (Fig. 2b). Massive proteinuria persisted and serum creatinine levels began to rise rapidly at the age of 9 months. However, the baby received conservative treatment only, including intermittent albumin replacement, because the parents did not want immunosuppressive treatment or any other aggressive renal replacement therapy. The baby died at the age of 10 months.
Fig. 1.

Upper gastrointestinal examination of Case II-1. Anteroposterior (a) and lateral (b) projections revealed an upward dislocation of the stomach into the mediastinum, which is compatible with a hiatal hernia
Fig. 2.

Patients’ brain magnetic resonance images (MRIs). Axial T2-weighted MRI (a) of Case II-1 taken at 2 weeks of age shows a simplified gyral pattern with too few and shallow sulci and normal cortical thickness. Follow-up axial T2-weighted MR image (b) acquired at 4 months of age shows a progression of diffuse brain atrophy with subarachnoid space widening. Axial T2-weighted MRI (c) of Case II-2 obtained at 3 weeks of age shows a similar pattern of simplified sulcation as the sibling. Note extensive fluid collection in the scalp (arrows)
Fig. 3.
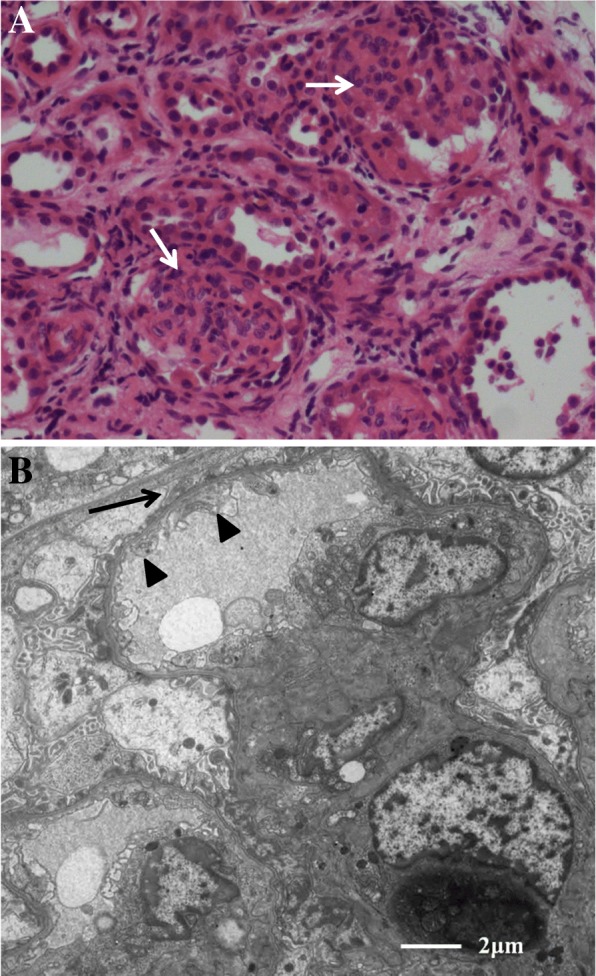
Renal pathological findings from Case II-1 at 2 weeks of age. a About half of the glomeruli showed segmental sclerosis (arrows). Hematoxylin and eosin staining, original magnification × 400. b Ultrastructurally, the glomerular basement membranes exhibit normal thickness, but show focal subendothelial widening (arrowheads). No electron-dense deposits were found. Epithelial cell foot processes show focal marked effacement (arrows). Original magnification × 5000
Case II-2
This male baby was born by cesarean section due to fetal distress at the gestational age of 36+ 6 weeks. Birth weight was 1960 g (< 3rd percentile), height was 47.5 cm (10–25th percentile), head circumference was 28 cm (< 3rd percentile), and the Apgar scores at 1 and 5 min were 6 and 7, respectively. The baby displayed various features of facial dysmorphism (microcephaly, almond-shaped eye, abnormally high but narrow forehead, ocular hypertelorism, depressed nasal bridge, large and low-set ears, micrognathia, and high-arched cleft palate), and skeletal deformities (arachnodactyly and clasped thumb). Massive proteinuria, hypoalbuminemia, and generalized edema were noted immediately after birth. Renal ultrasonography revealed bilaterally small echogenic kidneys with poor cortico-medullary differentiation. A brain MRI revealed microlissencephaly, small cerebellar hemisphere size, and decreased volume of the ventral pons (Fig. 2c). At the age of 15 days, serum albumin and creatinine levels were 2.6 g/dL and 2.88 mg/dL, respectively. The parents did not want any active treatment at this time, and the patient died at the age of 21 days due to rapidly decreasing renal function.
Case II-3
Cases II-3 and II-4 were dizygotic twin babies born after in vitro fertilization. They were born by cesarean section after a gestational period of 36+ 4 weeks. The birth weight of Case II-3 was 2830 g (10–25th percentile), height was 46 cm (5–10th percentile), and head circumference was 34 cm (25–50th percentile). Physical examination revealed no abnormalities, and there were no perinatal or neonatal problems. At the age of 6 weeks, serum albumin and creatinine levels were 4.1 g/dL and 0.22 mg/dL, respectively. Urinalysis revealed no proteinuria.
Case II-4
This male baby was the second of the dizygotic twins. Intrauterine growth retardation was detected by fetal ultrasonography. Birth weight was 1780 g (< 3rd percentile), height was 45 cm (3rd–5th percentile), head circumference was 27 cm (< 3rd percentile), and the Apgar scores at 1 and 5 min were 4 and 7, respectively. He exhibited ocular hypertelorism, micrognathia, and low-set ears. A brain MRI revealed lissencephaly. Generalized edema with massive proteinuria developed soon after birth. Serum creatinine levels at 3 days and 7 days after birth were 1.11 mg/dL and 1.27 mg/dL, respectively. Renal ultrasonography performed at 1 week after birth revealed small echogenic kidneys with poor differentiation between the peripheral cortex and the central echogenic complex. The patient was treated conservatively and died 25 days after birth.
Mutational analyses
Whole exome sequencing was performed for Case II-2 as previously described [13], which revealed a novel homozygous c.194A > T (p.Lys65Met) mutation in exon 1 of the TP53RK gene (NM_033550). This was later confirmed using traditional Sanger sequencing. A study of all family members using Sanger sequencing revealed that Case II-4 carried the same homozygous mutation, and that both parents and Case II-3 were heterozygous for the mutation. Although we were not able to test Case II-1 due to a lack of available samples, her phenotypes were very similar to those of the other two affected siblings who carried the mutation.
Discussion and conclusions
GAMOS is a heterogeneous disease, both phenotypically and genetically. Phenotypically, in addition to major findings of early-onset nephrotic syndrome and microcephaly, most patients have facial dysmorphia including hypertelorism, ear abnormalities, and micrognathia. Some patients may present arachnodactyly, visual impairment, or other manifestations [9].
Colin et al. [3] identified 2 different truncating mutations in the WDR73 gene in 3 patients from 2 unrelated families with GAMOS. The authors suggested that WDR73 plays a role in regulating the microtubule network during the cell cycle, and loss of its function leads to impaired neuronal growth and brain development, as well as impaired podocyte growth and maintenance in the kidneys. After the investigation by Colin et al. [3], several studies have reported on additional cases of GAMOS in association with WDR73 mutations [4–8]. In one of those studies [6], WDR73 mutations were found in 3 (5.9%) of the 51 unrelated patients with cerebellar atrophy and variable brain anomalies, and in 2 (5%) of the 40 unrelated patients with a clinical diagnosis of GAMOS. Rosti et al. [9] detected NUP107 mutations in 5 Turkish patients with GAMOS-like phenotypes from two consanguineous families. However, in other reports [14, 15], NUP107 mutations were associated with isolated nephrotic syndrome. Therefore, there may be an allele-specific critical role for NUP107 in regulating brain growth and GAMOS-like presentation [9].
Recently, a large-scale study [11] identified novel causative mutations in one of the four genes encoding KEOPS subunits, OSGEP, TP53RK, TPRKB and LAGE3, in 37 individuals (from 32 families) out of 91 individuals with GAMOS. In that study, OSGEP mutations were the most common, and were detected in 28 individuals from 24 families, while mutations in TP53RK were detected in only 4 individuals from 3 families. Almost half of the families carrying OSGEP mutations were of Taiwanese or East Asian descent, while the Korean family described in the present case study carried a TP53RK mutation. In addition, an independent study [12] reported on a familial case of GAMOS with OSGEP mutations. Currently, the five genetic subtypes of GAMOS are listed in the Online Mendelian Inheritance in Men (https://www.ncbi.nlm.nih.gov/omim/) database. GAMOS1–5 are associated with mutations in WDR73, LAGE3, OSGEP, TP53RK, and TPRKB, respectively. All subtypes exhibit autosomal recessive inheritance, except for GAMOS2, which exhibits X-linked recessive inheritance.
The TP53RK mutation identified in the present study [i.e., c.194A > T (p.Lys65Met)] is novel. Co-segregation of the recessive mutation with the phenotype in the family members, including both parents, was confirmed. Interestingly, a known variation involving the same Lys65 [i.e., c.194A > G (p.Lys65Arg)] has a reported allele frequency of 0.00009747 (ExAC Browser, http://exac.broadinstitute.org/). Although we did not perform functional studies, both variations are predicted to be pathogenic according to the web-based program MutationTaster (http://www.mutationtaster.org/). Whole exome sequencing performed for Case II-2 revealed no pathogenic variants in other known candidate genes of GAMOS, namely WDR73, NUP107, WHAMM, and other genes encoding KEOPS subunits. In our whole exome sequencing pipeline, common nucleotide variants (minor allele frequency > 1%) were removed in the middle of the analysis. The only variant in other known proteinuria/focal segmental glomerulosclerosis-associated genes, which was detected by the whole exome sequencing, was a heterozygous c.449–26C > CT (rs35640989) in the NUP107 gene. Although a mutational study for Case II-1 was not performed owing to a lack of samples, all the three affected babies included in this study showed common manifestations of very early-onset nephrotic syndrome, microcephaly, dysmorphic faces, and early fatal outcome. Case II-1 had an additional finding of a hiatal hernia with gastric volvulus. A hiatal hernia was one of the features observed in the original cases as reported by Galloway and Mowat [1]. However, there have been only few case reports of GAMOS associated with hiatal hernias, including one patient with WDR73 mutations [6] and a familial case with OSGEP mutations [11]. Case II-2 in the present study displayed additional skeletal deformities, including arachnodactyly and clasp thumb. Skeletal deformities are more commonly reported than hiatal hernias in patients with GAMOS [11, 16–18]. In a large study conducted by Braun et al. [11], most of the Taiwanese patients had arachnodactyly and carried OSGEP mutations. All three patients in the present study had an early fatal outcome; therefore, we could not evaluate long-term clinical changes in growth and development. To date, only four patients with GAMOS from three families have been reported in association with TP53RK mutations [11]. Among those four patients, two sibling cases showed similar phenotypes to those of our patients, i.e., early-onset proteinuria at 2 months of age in both cases, early fatal outcome at 2.5 months and 11 months of age, respectively, and facial dysmorphism with skeletal abnormalities in one case. The other two patients showed relatively later onset at 1 year and 10 months of age, respectively, and later fatal outcomes at 2.5 years and 3 years of age, respectively.
The KEOPS complex contains five subunits: LAGE3, OSGEP, TP53RK, TPRKB, and the recently identified C14orf142 [19]. Braun et al. [10] identified and meticulously studied multiple mutations in the genes encoding the four KEOPS subunits, including OSGEP, TP53RK, TPRKB, and LAGE3. The authors conducted short hairpin RNA (shRNA)-mediated knockdown of KEOPS subunits and showed that mutations in OSGEP, TP53RK, and TPRKB could not rescue proliferation defects in human podocytes, suggesting that these alleles impaired protein functionality. Co-immunoprecipitation experiments revealed that the p.Lys60Serfs*61 and p.Thr81Arg mutations in TP53RK detected in a family abrogated the interaction between TPRKB and TP53RK [11]. However, other mutations in any of the four genes did not abrogate intermolecular interactions. The canonical function of the KEOPS complex is to mediate N6-threonylcarbamoyladenosine modifications, known as a t6A modification, at position 37 of all tRNAs that recognize codons that start with adenosine, i.e., ‘ANN codons’ [20–23]. Braun et al. [11] demonstrated that knocking down OSGEP, TP53RK, or TPRKB using shRNA in human podocytes resulted in various effects, including decreased t6A levels, inhibition of nascent protein synthesis, decreased cell proliferation, activation of the unfolded protein response with ER stress, upregulation of the ER-associated proteasomal degradation system, and increased apoptosis associated with activation of the DDR. Knockdown of one of these three components of the KEOPS complex also disrupts the formation of the sublamellar actin network in human podocytes and decreases podocyte migration [11]. Therefore, the authors concluded that mutations in the KEOPS complex impair both the canonical and noncanonical functions of the KEOPS complex, which would play a major pathogenic role in the development of GAMOS [11].
In conclusion, we report a familial case of GAMOS associated with a novel homozygous TP53RK mutation (p.Lys65Met). Three of the four children in the family were affected and displayed similar phenotypes, including very early-onset nephrotic syndrome, microcephaly, facial dysmorphia, and early fatality. In addition, renal biopsy revealed focal segmental glomerulosclerosis in one patient.
Acknowledgements
We are grateful to the patients and their parents for their permission to publish this case report.
Funding
This study was supported by a grant (HI12C0014) of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DDR
DNA damage response
- ER
Endoplasmic reticulum
- GAMOS
Galloway–Mowat syndrome
- KEOPS
Kinase, endopeptidase and other proteins of small size
- MRI
Magnetic resonance imaging
Authors’ contributions
HIC, HGK, and HT designed and organized the study. HSH, SHK, MHC, and EP cared for the patients, acquired the clinical data, and prepared the samples from the family members. HSH, EP, and SHK wrote the manuscript that was edited by all other authors. MHC, EP, and HGK performed Sanger sequencing of the TP53RK gene. NMi, NMa, and HT performed WES, interpreted the WES data, and drafted the genetic diagnostic report. HSL, a renal pathologist, performed and interpreted the renal histological examination and drafted the pathologic report. HIC obtained funding. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Seoul National University Hospital’s Institutional Review Board (IRB No. 0812–002-264). The parents of the patients provided informed consent to participate in this study.
Consent for publication
The parents of the patients provided informed consent to publish this case report, including case description, medical data, and images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hye Sun Hyun, Email: inthy@naver.com.
Seong Heon Kim, Email: pedksh@gmail.com.
Eujin Park, Email: ukishiro@hanmail.net.
Myung Hyun Cho, Email: dong82dong82@naver.com.
Hee Gyung Kang, Email: kanghg@snu.ac.kr.
Hyun Soon Lee, Email: Hyunsoonlee3@gmail.com.
Noriko Miyake, Email: nmiyake@yokohama-cu.ac.jp.
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Hiroyasu Tsukaguchi, Email: tsukaguh@otokoyam.kmu.ac.jp.
Hae Il Cheong, Phone: 82-2-2072-2810, Email: cheonghi@snu.ac.kr.
References
- 1.Galloway WH, Mowat AP. Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs. J Med Genet. 1968;5(4):319–321. doi: 10.1136/jmg.5.4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen AH, Turner MC. Kidney in Galloway-Mowat syndrome: clinical spectrum with description of pathology. Kidney Int. 1994;45(5):1407–1415. doi: 10.1038/ki.1994.184. [DOI] [PubMed] [Google Scholar]
- 3.Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet. 2014;95(6):637–648. doi: 10.1016/j.ajhg.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Omran T, Fahiminiya S, Sorfazlian N, Almuriekhi M, Nawaz Z, Nadaf J, et al. Nonsense mutation in the WDR73 gene is associated with Galloway-Mowat syndrome. J Med Genet. 2015;52(6):381–390. doi: 10.1136/jmedgenet-2014-102707. [DOI] [PubMed] [Google Scholar]
- 5.Jinks RN, Puffenberger EG, Baple E, Harding B, Crino P, Fogo AB, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain. 2015;138(Pt8):2173–2190. doi: 10.1093/brain/awv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vodopiutz J, Seidl R, Prayer D, Khan MI, Mayr JA, Streubel B, et al. WDR73 mutations cause infantile neurodegeneration and variable glomerular kidney disease. Hum Mutat. 2015;36(11):1021–1028. doi: 10.1002/humu.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosti RO, Dikoglu E, Zaki MS, Abdel-Salam G, Makhseed N, Sese JC, et al. Extending the mutation spectrum for Galloway-Mowat syndrome to include homozygous missense mutations in the WDR73 gene. Am J Med Genet A. 2016;170A(4):992–998. doi: 10.1002/ajmg.a.37533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang C, Gai N, Zou Y, Zheng Y, Ma R, Wei X, et al. WDR73 missense mutation causes infantile onset intellectual disability and cerebellar hypoplasia in a consanguineous family. Clin Chim Acta. 2017;464:24–29. doi: 10.1016/j.cca.2016.10.029. [DOI] [PubMed] [Google Scholar]
- 9.Rosti RO, Sotak BN, Bielas SL, Bhat G, Silhavy JL, Aslanger AD, et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J Med Genet. 2017;54(6):399–403. doi: 10.1136/jmedgenet-2016-104237. [DOI] [PubMed] [Google Scholar]
- 10.Mathiowetz AJ, Baple E, Russo AJ, Coulter AM, Carrano E, Brown JD, et al. An Amish founder mutation disrupts a PI(3)P-WHAMM-Arp2/3 complex-driven autophagosomal remodeling pathway. Mol Biol Cell. 2017;28(19):2492–2507. doi: 10.1091/mbc.e17-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun DA, Rao J, Mollet G, Schapiro D, Daugeron MC, Tan W, et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat Genet. 2017;49(10):1529–1538. doi: 10.1038/ng.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edvardson S, Prunetti L, Arraf A, Haas D, Bacusmo JM, Hu JF, et al. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur J Hum Genet. 2017;25(5):545–551. doi: 10.1038/ejhg.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Latypova X, Matsumoto N, Vinceslas-Muller C, Bézieau S, Isidor B, Miyake N. Novel KCNB1 mutation associated with non-syndromic intellectual disability. J Hum Genet. 2017;62(5):569–573. doi: 10.1038/jhg.2016.154. [DOI] [PubMed] [Google Scholar]
- 14.Miyake N, Tsukaguchi H, Koshimizu E, Shono A, Matsunaga S, Shiina M, et al. Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am J Hum Genet. 2015;97(4):555–566. doi: 10.1016/j.ajhg.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park E, Ahn YH, Kang HG, Miyake N, Tsukaguchi H, Cheong HI. NUP107 mutations in children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant. 2017;32(6):1013–1017. doi: 10.1093/ndt/gfw103. [DOI] [PubMed] [Google Scholar]
- 16.Hou JW, Wang TR. Galloway-Mowat syndrome in Taiwan. Am J Med Genet. 1995;58(3):245–248. doi: 10.1002/ajmg.1320580310. [DOI] [PubMed] [Google Scholar]
- 17.Chen CP, Chang TY, Lin SP, Huang JK, Tsai JD, Chiu NC, et al. Prenatal magnetic resonance imaging of Galloway-Mowat syndrome. Prenat Diagn. 2005;25(6):441–445. doi: 10.1002/pd.1183. [DOI] [PubMed] [Google Scholar]
- 18.Chen CP, Lin SP, Tsai JD, Huang JK, Yen JL, Tseng CC, et al. Perinatal imaging findings of Galloway-Mowat syndrome. Genet Couns. 2007;18(3):353–355. [PubMed] [Google Scholar]
- 19.Wan LC, Maisonneuve P, Szilard RK, Lambert JP, Ng TF, Manczyk N, et al. Proteomic analysis of the human KEOPS complex identifies C14ORF142 as a core subunit homologous to yeast Gon7. Nucleic Acids Res. 2017;45(2):805–817. doi: 10.1093/nar/gkw1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugeron MC, Lenstra TL, Frizzarin M, El Yacoubi B, Liu X, Baudin-Baillieu A, et al. Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res. 2011;39(14):6148–6160. doi: 10.1093/nar/gkr178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Yacoubi B, Hatin I, Deutsch C, Kahveci T, Rousset JP, Iwata-Reuyl D, et al. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011;30(5):882–893. doi: 10.1038/emboj.2010.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan M, Mehta P, Yu Y, Prugar E, Koonin EV, Karzai AW, et al. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011;30(5):873–881. doi: 10.1038/emboj.2010.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yarian C, Townsend H, Czestkowski W, Sochacka E, Malkiewicz AJ, Guenther R, et al. Accurate translation of the genetic code depends on tRNA modified nucleosides. J Biol Chem. 2002;277(19):16391–16395. doi: 10.1074/jbc.M200253200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.