

Abstract
The present study aimed to investigate the effects of colony-stimulating factor-1 receptor (CSF-1R) on proliferation, migration and invasion in the human nasopharyngeal carcinoma (NPC) 6–10B cell line, and to investigate the possible underlying mechanisms. Using a lentiviral transfection method, a virus carrying the CSF-1R gene was transfected into 6–10B cells. The expression of CSF-1R was then detected by reverse transcription-quantitative polymerase chain reaction and western blot analysis, and was revealed to be markedly enhanced in 6–10B cells. Subsequently, an MTT assay was performed to assess cell proliferative ability, and flow cytometric analysis was utilized to measure the apoptotic rate of the cells. Wound healing and Transwell assays were also performed to observe cell migration and invasion capabilities. Additionally, western blot analysis was used to detect the protein expression of the proliferation and apoptosis signaling factors cyclin D1, B-cell lymphoma 2, Bcl-2-associated X protein, and phosphorylated and total extracellular protein kinase B (Akt/PKB) in 6–10B cells. The results showed that CSF-1R overexpression promoted the proliferation, migration and invasion of the 6–10B cells. The corresponding mechanism may be associated with activation of the phosphoinositide 3-kinase/Akt pathway, which promotes cell survival and proliferation. These results indicated a potential molecular target for the treatment of NPC.
Keywords: colony-stimulating factor-1 receptor, nasopharyngeal carcinoma, proliferation, migration, invasion, cyclin D1, B-cell lymphoma 2, Bcl-2-associated X protein, phosphoinositide 3-kinase/Akt pathway
Introduction
Nasopharyngeal carcinoma (NPC), which originates in the nasopharynx, is the most common type of head and neck malignancy in Southern China and certain regions of Southeast Asia. Currently, the standard treatment for NPC consists of concurrent chemo-radiotherapy, followed by adjuvant chemotherapy (1). Despite marked improvements in clinical treatments for NPC, neck lymph node metastasis occurs in up to 75% of NPC patients, which represents an unfavorable prognostic factor for the disease (2). Radiotherapy resistance is a cause of local recurrences and distant metastases (3). A previous study revealed that colony-stimulating factor-1 receptor (CSF-1R) expression was 4.1-times higher in NPC patients who were resistant to radiation than in those who were sensitive to radiation (4). Furthermore, the expression of CSF-1R in NPC tissues was markedly higher than that in the nasopharyngitis tissues, and patients with moderate or strong-intensity of CSF-1R expression were more likely to develop metastasis and recurrence (5). Therefore, a greater understanding of the biological features of NPC is urgently required.
CSF-1R, a member of the receptor tyrosine kinase (RTK) family, is one of the main regulatory factors in the immune system, and is encoded by the proto-oncogene c-fms. Ligand binding activates CSF-1R through a process of oligomerization and trans-phosphorylation, to ultimately activate tyrosine kinase signaling pathways, which may result in tumor cell proliferation (6). Growing evidence has indicated that CSF-1R, together with its ligand colony-stimulating factor 1 (CSF-1/M-CSF), serves an important role in the development of cancer and may be involved in the process of carcinogenesis, and tumor cell proliferation and metastasis (7,8). Thus far, CSF-1R has been revealed to be upregulated in breast cancer (9), ovarian cancer (10) and head and neck malignancies (11). The expression of CSF-1R in the blood has been identified as a biomarker in numerous malignant tumors (12). A previous study demonstrated that CSF-1R amplification in breast cancer increased cell proliferation (13). In addition, another study reported that the CSF-1R inhibitor significantly reduced the volume of microglia and suppressed the proliferative and invasive abilities of the cells (14). However, the effects of CSF-1R on NPC and the possible underlying mechanisms of this have not yet been evaluated. Therefore, the present study investigated the potential promotional effects of CSF-1R on the proliferation, migration and invasion of the 6–10B NPC cell line, and the possible molecular mechanisms underlying CSF-1R-induced cell proliferation and metastasis in NPC.
Materials and methods
Cell culture
The human NPC 6–10B cell line was obtained from Sun Yat-Sen University Cancer Centre (State Key Laboratory of Oncology in South China, Guangzhou, China). The cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) in a humidity-controlled 37°C incubator with 5% CO2. The medium was refreshed every other day.
Transfection
Lentiviral vectors were constructed by Shanghai GeneChem Co., Ltd. (Shanghai, China). The viruses carried the enhanced green fluorescent protein (eGFP) gene. CSF-1R (NM 005211; GeneChem Co., Ltd., Shanghai, China) was transformed into GV358 vector (GeneChem Co., Ltd., Shanghai, China) using ClonExpressTM II One Step Cloning kit (Vazyme Biotech Co., Ltd., Nanjing, China). The constructs were then transfected into 293T cells (GeneChem Co., Ltd., Shanghai, China) with lentiviral packaging vectors using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.). Lentivirus was collected 48 h after transfection and used to infect the 6-10B cells. The viruses were diluted in sterile phosphate-buffered saline (PBS) to a final titer of 5E+8 TU/ml. Lentiviruses carrying the CSF-1R gene (ID, LV-CSF1R) or a control vector were transfected into 6-10B cells respectively when cell confluence reached 20-30%. Expression of the eGFP protein was assessed 3 days following transfection to determine transfection efficiency using an inverted fluorescence microscope at magnification, ×100. CSF-1R expression in the 6-10B cells treated with control vector (control group) or CSF-1R overexpression vector (transfection group) was then verified using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analysis.
RT-qPCR
Total RNA was extracted from the 6-10B cells using TRIzol® reagent (Ambion; Thermo Fisher Scientific, Inc.). cDNA was synthesized using a FastQuant RT kit (Tiangen Biotech Co., Ltd., Beijing, China) according to the manufacturer's protocol with the following temperature protocol: 42°C for 15 min, followed by 95°C for 3 min and then cooling on ice at 4°C for 10 min. Subsequently, 100 ng total cDNA was added per 25-µl reaction with CSF-1R or GAPDH primers, and quantitative PCR analysis was performed using a SuperReal PreMix Plus SYBR Green PCR kit (Tiangen Biotech Co., Ltd.), according to the manufacturer's protocols. The reactions are typically performed on a PCR machine, and the qPCR thermo cycling conditions were as follows: 95°C for 15 min, 40 cycles of 95°C for 10 sec and 60°C for 32 sec. Finally, the data were analyzed using the 2−ΔΔCq method (15) with GAPDH expression as the endogenous control. All mRNA primers were designed with Primer Premier 5.0 software (Premier Tech Co, Ltd., Quebec, Canada) and synthesized by Thermo Fisher Scientific, Inc. The qPCR primers used to amplify the CSF-1R and GAPDH genes are as follows: CSF-1R forward, 5′-TCTGGTCCTATGGCATCCTC-3′ and reverse, 5′-GATGCCAGGGTAGGGATTC-3′; and GAPDH forward, 5′-AGCCACATCGCTCAGACAC-3′ and reverse, 5′-GCCCAATACGACCAAATCC-3′.
Western blot analysis
Cells were harvested and lysed in radioimmunoprecipitation assay cell lysis buffer (Beyotime Institute of Biotechnology, Haimen, China), mixed with protease inhibitor cocktail (100:1), on ice for ~30 min. The cells were then pelleted by centrifugation at 4°C for 20 min at 12,000 × g. Total protein was extracted from the supernatant and the concentration was determined using a bicinchoninic acid assay kit (Beyotime Institute of Biotechnology). Equal amounts of protein (40 µg) were separated on a 12% SDS-PAGE gel and were transferred onto polyvinylidene difluoride membranes. The membranes were subsequently blocked with 3% bovine serum albumin for 1 h at room temperature, prior to being incubated overnight at 4°C with the following antibodies: Polyclonal rabbit anti-CSF-1R (dilution, 1:1,000; catalog no. 3152; Cell Signaling Technology, Inc., Danvers, MA, USA), monoclonal rabbit anti-cyclin D1 (dilution, 1:1,000; catalog no. ab134175; Abcam, Cambridge, UK), monoclonal rabbit anti-Bcl-2 (dilution, 1:1,000; catalog no. 4223; Cell Signaling Technology, Inc.), monoclonal rabbit anti-Bax (dilution, 1:2,000; catalog no. ab32124; Abcam, Cambridge, UK), monoclonal rabbit anti-Akt (dilution, 1:1,000; catalog no. 9272; Cell Signaling Technology, Inc.), monoclonal rabbit anti-phosphorylated (p)-Akt (dilution, 1:2,000; catalog no. 4060; Cell Signaling Technology, Inc.) and monoclonal rabbit anti-GAPDH (dilution, 1:1,000; catalog no. ab181602; Abcam). Membranes were subsequently incubated with a goat anti-rabbit IgG secondary antibody (dilution, 1:3,000; catalog no. 7074; Cell Signaling Technology, Inc.) for 1.5 h at 37°C. Following washing three times with TBST, signals were detected with an enhanced chemiluminescence detection system (Pierce; Thermo Fisher Scientific, Inc.).
MTT assay
6-10B cells transfected with the control vector (control group) or the CSF-1R vector (transfection group) were seeded onto 96-well plates at a density of 1,000 cells/well. Following incubation for 24, 48, 72, 96 or 120 h, 20 µl MTT reagent was added (at a final concentration of 0.5 mg/ml). The cells were then cultured for another 4 h and 150 µl dimethyl sulfoxide was added to each well to dissolve the purple formazan. The cells were subjected to an absorbance reading at 490 nm using a 96-well microplate reader. All experiments were performed in triplicate. Cell proliferation was evaluated at 24, 48, 72, 96 or 120 h.
Flow cytometric apoptosis assay
The two groups of 6-10B cells were seeded onto 6-well plates and trypsinized when the cells reached 70-80% confluence. Annexin V and propidium iodide (PI) staining was performed using an Annexin V-APC/7-AAD Apoptosis Detection kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China), according to the manufacturer's protocols. Following incubation for ~15 min in the dark, apoptotic cells were immediately analyzed by a flow cytometer (fluorescence-activated cell sorting; BD Biosciences, Franklin Lakes, NJ, USA). Using Annexin V-APC and 7-AAD staining enables different stages of cell apoptosis to be distinguished. For example, APC-positive and 7-AAD-negative staining indicates early apoptotic cells, while 7-AAD-positive and APC-positive staining indicates late apoptotic cells. FlowJo 7.6 software (Tree Star Inc., Ashland, OR, USA) was used to calculate rate of apoptosis.
Wound healing assay
The two groups of 6-10B cells were cultivated in 6-well plates (5×105 cells/well). When cell confluence reached 70–80%, a 200 µl pipette tip was used to scrape a line-shaped wound in the cell monolayer on each plate. Cells were subsequently cultured for 24 h in serum-free medium. Cell migration was evaluated at 0, 12 and 24 h by a light microscope at magnification, ×100 (Olympus Corporation, Tokyo, Japan).
Transwell invasion assay
An invasion assay was performed in a 24-well Transwell chamber (Corning Incorporated, Corning, NY, USA). A total of 5×104 cells were seeded onto filters pre-coated with Matrigel with 150 µl serum-free RPMI-1640 medium. The lower chamber was supplied with 500 µl medium containing 10% FBS. After culturing for 24 h, all the medium was removed. The filters were fixed with 95% methanol for 15–20 min at room temperature and stained with 0.05% crystal violet for 20 min at 25°C, prior to being washed three times with PBS. Finally, the number of cells adhered to the bottom of the filters was counted using a light microscope at magnification, ×100.
Statistical analysis
Data are presented as the mean ± standard deviation. Student's t-test was used to compare the diversity between the two groups, using SPSS statistical software version 22.0 (IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Overexpression of CSF-1R in 6–10B cells
Following the transfection of a CSF-1R overexpression vector into 6–10B cells, the transfection efficiency was analyzed by inverted fluorescence microscopy (Fig. 1A). Subsequently, RT-qPCR analysis revealed that the mRNA expression of CSF-1R was significantly upregulated in the transfection group, but was downregulated in the control group (Fig. 1B; P<0.01). Similarly, the protein expression of CSF-1R was markedly increased in the transfection group (Fig. 1C; P<0.01), which indicated that CSF-1R was successfully overexpressed in the 6–10B cells.
Figure 1.

Overexpression of CSF-1R in 6–10B cells. (A) Transfection efficiency was detected using an inverted fluorescence microscope (magnification, ×100). (B) The mRNA expression level of CSF-1R in human nasopharyngeal carcinoma 6–10B cells following transfection with a CSF-1R lentiviral vector. (C) The protein expression of CSF-1R in human nasopharyngeal carcinoma 6–10B cells following lentiviral transfection. (D) The relative protein expression levels of CSF-1R in the transfection group and the control group were assessed. GADPH was used as the loading control for the western blot analyses. **P<0.01 vs. control group. CSF-1R, colony-stimulating factor 1 receptor.
CSF-1R overexpression induces the proliferation of 6–10B cells
6-10B cell proliferation was measured via an MTT assay. As demonstrated in Fig. 2, the number of 6–10B cells in the transfection group significantly increased from day 4, compared with that in the control group (P<0.05), which suggested that CSF-1R overexpression promoted 6–10B cell proliferation.
Figure 2.
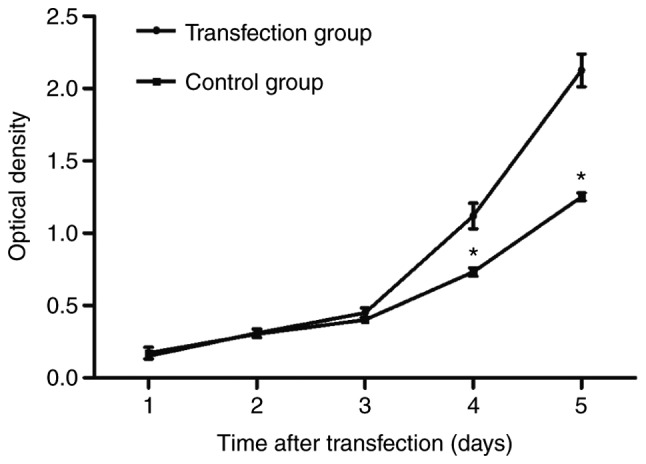
Cell proliferation increased significantly in the colony-stimulating factor 1 receptor transfection group from day 4. *P<0.05 compared with the transfection group.
CSF-1R overexpression reduces the apoptosis of 6–10B cells
In order to assess whether CSF-1R was able to inhibit the apoptosis of 6–10B cells, staining with Annexin V-APC and 7-AAD was performed. As demonstrated in Fig. 3, a lower rate of cell apoptosis was observed in the transfection group compared with that in the control group, with regards to early-stage and late-stage apoptosis (10.82±0.75 vs. 17.11±0.46%; P<0.05). This result revealed that CSF-1R overexpression significantly inhibited the apoptosis of 6–10B cells.
Figure 3.

CSF-1R reduced the cell death rate of human nasopharyngeal carcinoma 6–10B cells. (A) Cell apoptosis was detected by fluorescence-activated cell sorting following staining. (B) The percentage of apoptotic cells was significantly lower in the transfection group than in the control group. *P<0.05 compared with the control group. CSF-1R, colony-stimulating factor 1 receptor.
CSF-1R overexpression promotes 6–10B cell migration and invasion
In order to demonstrate the effects of CSF-1R on cell invasion, an invasion assay was performed, in which cells passing through the polycarbonate membrane of a Transwell invasion chamber were counted. Five visual fields were randomly selected to calculate the average number of migrated cells in each group, which was determined to be 180.400±9.633 per field in the transfection group and 19.200±5.541 per field in the control group (magnification, ×100; Fig. 4A). Analysis of these results indicated that the invasive ability was significantly increased in the cells overexpressing CSF-1R compared with that in the control cells (Fig. 4B; P<0.01).
Figure 4.

Effect of CSF-1R on cell invasion was assessed using a Transwell assay. (A) The number of cells that invaded to the underside of the chamber in the transfection group was significantly higher than that in the control group. (B) Statistical analysis of the number of migrated cells. **P<0.01 compared with the control group. CSF-1R, colony-stimulating factor 1 receptor.
In order to prove that CSF-1R regulates cell migration, the migration of cells was assessed in a wound healing assay. As demonstrated in Fig. 5, CSF-1R expression markedly accelerated cell migration in the transfection group. Overall, these results suggested that CSF-1R may induce 6–10B cell invasion and migration.
Figure 5.

Effect of CSF-1R on cell migration assessed by a wound healing assay. Overexpression of CSF-1R markedly stimulated cell migration compared with control group. CSF-1R, colony-stimulating factor 1 receptor; Akt, protein kinase B; p-Akt, phosphorylated Akt; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
CSF-1R overexpression enhances the activity of PI3K/Akt in 6–10B cells
To determine whether CSF-1R may promote cell proliferation by activating the PI3K/Akt pathway, western blotting was performed to analyze the protein expression of cyclin D1, Bcl-2, Bax, Akt and p-Akt. Notably, the expression of p-Akt was increased in the transfection group (Fig. 6A). Accordingly, the expression of cyclin D1 (Fig. 6B) and Bcl-2 (Fig. 6C) was also enhanced. By contrast, Bax, as a factor that inhibits tumor growth, was downregulated (Fig. 6C). These results indicated that the activation of PI3K/Akt signaling may mediate the increased proliferative ability of 6–10B cells overexpressing CSF-1R.
Figure 6.

CSF-1R overexpression enhances the activity of PI3K/Akt in 6–10B cells. (A) CSF-1R overexpression activates the PI3K/Akt signaling pathway in 6–10B cells, confirmed by an enhanced signal for p-Akt in western blot analysis. (B) The expression of cyclin D1 was increased compared with the control group. (C) Expression of Bcl-2 was increased, while the expression of Bax was reduced in the transfection group, compared with the control group. (D) The relative expression levels in the transfection group and the control group were assessed. GADPH was used as the loading control for the western blot analyses. **P<0.01 compared with the control group. CSF-1R, colony-stimulating factor 1 receptor; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; p-Akt, phosphorylated Akt; Bcl-2, B-cell lymphoma 2.
Discussion
Although substantial progress has been achieved in the treatment of human NPC via radiotherapy, the 5-year local recurrence rate of patients remains at 30, and 30–60% of patients experience metastasis (16). Radiotherapy resistance is a cause of local recurrence and distant metastases. The involvement of CSF-1R in NPC has recently been a focus of research. CSF-1R is established as a member of the receptor tyrosine kinase (RTK) family, and is involved in regulating the immune system (17,18). To date, it has been demonstrated that the expression of CSF-1R and its ligand in neoplastic epithelial cells is correlated with metastasis, angiogenesis and cell invasion, resulting in poor prognosis and recurrence (19). Accumulating evidence has proven that PI3K/Akt pathways are involved in RTK-induced progression and migration in human cancer cell lines (20). However, few studies have focused on the effects of CSF-1R on NPC cells.
Therefore, the present study overexpressed CSF-1R in the human NPC 6–10B cell line by lentiviral transfection (Fig. 1A), and measured its expression level by RT-qPCR (Fig. 1B) and western blot analysis (Fig. 1C). Subsequently, a series of tests were performed to compare the proliferation, migration and invasion abilities of the transfection group with those of a control group (Figs. 2–5). The results demonstrated that cell proliferation, migration and invasion were increased in the transfection group when compared with their levels in the control group. Furthermore, p-Akt protein abundance was increased following CSF-1R overexpression. As a consequence, the protein abundance of cyclin D1 and Bcl-2 was also increased, whereas that of Bax was decreased (Fig. 6), which suggested that the CSF-1R-induced proliferation of 6–10B cells involved p-Akt activation. Therefore, it may be hypothesized that CSF-1R elicits the PI3K/Akt pathway, involving Akt phosphorylation, to induce cyclin D1 and Bcl-2 expression and reduce Bax expression, and ultimately promote cell proliferation.
It is well known that cyclin D1 belongs to the highly conserved cyclin family and primarily functions as a factor that promotes cell proliferation. Namely, it regulates the transition of the G1 phase into the S phase by dimerizing with CDK4/6. Any mutation, amplification or overexpression of this gene may alter cell cycle progression, which may contribute to tumorigenesis (21). Cyclin D1 overexpression contributes to increased chemotherapeutic resistance and protection from apoptosis, resulting in tumor progression. Furthermore, cyclin D has been widely confirmed as a biomarker in breast carcinoma (22), with its overexpression leading to tumorigenesis and malignancy. To date, cyclin D1 expression has been detected at very high levels in numerous types of malignant tumor, including head and neck squamous cell carcinoma (23), breast carcinoma (24), ovarian cancer (25), colorectal cancer (26) and gastric cancer (27).
Members of the Bcl-2 family are among the most established regulators of apoptosis. The Bcl-2 family proteins are divided into two groups according to their function: One group includes Bcl-2, B-cell lymphoma-XL and Bcl-2-like protein 2, all of which promote apoptosis, while the other group includes Bax, Bcl-2 homologous antagonist killer and phorbol-12-myristate-13-acetate-induced protein 1, which inhibit apoptosis. These proteins control apoptosis by regulating outer mitochondrial membrane permeability and the release of cytochrome C (28). Bcl-2 is generally considered to be an anti-apoptotic protein that serves an important role in promoting cellular survival and inhibiting the actions of pro-apoptotic proteins (29). By contrast, Bax is identified as a pro-apoptotic member of the Bcl-2 protein family that stimulates apoptosis by heterodimerizing with Bcl-2 to inhibit its anti-apoptotic function (30). To date, it has been widely reported that the Bcl-2/Bax ratio is a key factor that reflects the degree of cell apoptosis resistance (31). The present study demonstrated that CSF-1R overexpression was able to increase the expression of Bax and reduce the expression of Cyclin D1 and Bcl-2. From these results, it may be concluded that CSF-1R overexpression promotes proliferation and inhibits apoptosis in NPC 6–10B cells.
The PI3K/Akt pathway is an intracellular signaling pathway that is associated, not only with tumor proliferation and survival, but also with invasion and metastasis (32). Only class I PI3K has been shown to regulate this pathway and is associated with tumor cell behavior. By interacting with growth factors or linked proteins that possess phosphorylated tyrosine residues, PI3K is activated, resulting in the generation of the second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 then combines with Akt and phosphoinositide-dependent kinase-1 (PDK1), thereby stimulating PDK1 to phosphorylate Thr(308) on the central kinase catalytic domain of Akt. Subsequently, Akt is activated and phosphorylates specific downstream proteins, including the pro-apoptotic proteins of the Bcl-2 family and glycogen synthase kinase 3 (GSK-3), thereby preventing the phosphorylation and degradation of cyclin D1 (33). Several RTKs regulate the activation of the PI3K/Akt pathway (34). It has been reported that, by inhibiting the activity of PI3K, cell proliferation may be significantly reduced and cell cycle progression prohibited; however, these effects may be prevented when activated Akt (p-Akt) is expressed (32).
The present study further revealed that one of the RTK family members, CSF-1R, may promote cell proliferation and inhibit cell apoptosis via the PI3K/Akt pathway. It was revealed that CSF-1R activated PI3K-dependent Akt phosphorylation, resulting in the upregulation of Cyclin D1 and Bcl-2 and the downregulation of Bax. Consequently, there was an increase in cell cycle progression and a reduction in cell apoptosis. However, these observations were based on one cell line, and future studies involving additional cell lines and more clinical data are required to confirm these effects in patients with NPC.
In conclusion, the present study demonstrated that CSF-1R promoted the proliferation, migration and invasion of the 6–10B NPC cell line. The possible underlying mechanisms may involve activation of the PI3K/Akt pathway. These findings provide further understanding of the processes underlying NPC development, and indicate that a CSF-1R inhibitor may be an effective treatment for NPC.
Acknowledgements
The authors would like to thank Dr Musheng Zeng and his colleagues at Sun Yat-Sen University Cancer Center for providing the 6–10B cell line.
Glossary
Abbreviations
- CSF-1R
colony stimulating factor-1 receptor
- CSF-1/M-CSF
colony stimulating factor 1
- NPC
nasopharyngeal carcinoma
- RTK
receptor tyrosine kinase
- Bcl-2
B-cell lymphoma 2
- BAX
Bcl-2-associated X protein
- PI3K
phosphoinositide 3-kinase
- Akt/PKB
protein kinase B
- PIP3
phosphatidylinositol-3,4,5-triphosphate
- PDK1
phosphoinositide-dependent kinase-1
- CAT
central kinase catalytic domain
- GSK-3
glycogen synthase kinase 3
Funding
The present study was supported by Guangxi Natural Science Fund (grant no. 2010037) and the National Natural Science Fund (grant no. 81260348).
Availability of data and resources
The datasets used or analyzed during the current study are available from the corresponding authors on reasonable request.
Authors' contributions
JYC performed experiments and was a major contributor in writing the manuscript. YH contributed to the conception and design of the present study. JXC also contributed to the conception and design of the present study. LH conducted cell transfection and RT-qPCR. WA conducted western blotting and the flow cytometric apoptosis assay. JY analyzed and interpreted the data. WL collected clinical data. LL conducted IHC. JH followed up the patients and analyzed the clinical data. ZC and XH performed radiotherapy. WG evaluated the NPC patients during the treatment in the preliminary study.
Ethics statement and consent to participate
Not applicable.
Consent for publication
No applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Razak AR, Siu LL, Liu FF, Ito E, O'Sullivan B, Chan K. Nasopharyngeal carcinoma: The next challenges. Eur J Cancer. 2010;46:1967–1978. doi: 10.1016/j.ejca.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Chua DT, Sham JS, Wei WI, Ho WK, Au GK. The predictive value of the 1997 American Joint Committee on cancer stage classification in determining failure patterns in nasopharyngeal carcinoma. Cancer. 2001;11:2845–2855. doi: 10.1002/1097-0142(20011201)92:11<2845::AID-CNCR10133>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Guo Y, Zhu XD, Qu S, Li L, Su F, Li Y, Huang ST, Li DR. Identification of genes involved in radioresistance of nasopharyngeal carcinoma by integrating gene ontology and protein-protein interaction networks. Int J Oncol. 2012;40:85–92. doi: 10.3892/ijo.2011.1172. [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Chen J, Guo Y, Lin H, Zhang Z, Feng G, Hao Y, Cheng J, Liang P, Chen K, et al. Identification of prognostic biomarkers for response to radiotherapy by DNA microarray in nasopharyngeal carcinoma patients. Int J Oncol. 2012;40:1590–1600. doi: 10.3892/ijo.2012.1341. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Xu X, Hao Y, Chen J, Li L, Cheng J, Chen Z, Liang W, Yang J, Ao W. Overexpression of CSF-1R in nasopharyngeal carcinoma. Rom J Morphol Embryol. 2015;56:1279–1283. [PubMed] [Google Scholar]
- 6.Yu W, Chen J, Xiong Y, Pixley FJ, Dai XM, Yeung YG, Stanley ER. CSF-1 receptor structure/function in MacCsflr-/-macrophages: Regulation of proliferation, defferetiation, and morphology. J Leukoc Biol. 2008;84:852–863. doi: 10.1189/jlb.0308171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, West RB. The macrophage colony stimulating factor-1 response signature in breast carcinoma. Clin Cancer Res. 2009;15:778–787. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pixley FJ. Macrophage migration and its regulation by CSF-1. Int J Biochem Cell B. 2012;2012:501962. doi: 10.1155/2012/501962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, Hamilton JA. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. 2014;2:765–776. doi: 10.1158/2326-6066.CIR-13-0190. [DOI] [PubMed] [Google Scholar]
- 10.Gruessner C, Gruessner A, Glaser K, Abushahin N, Laughren C, Zheng W, Chambers SK. Biomarkers and endosalpingiosis in the ovarian and tubal microenvironment of women at high-risk for pelvic serous carcinoma. Am J Cancer Res. 2014;4:61–72. [PMC free article] [PubMed] [Google Scholar]
- 11.Kuropkat C, Dünne AA, Plehn S, Ossendorf M, Herz U, Renz H, Werner JA. Macrophage colony-stimulating factor as a tumor marker for squamous cell carcinoma of the head and neck. Tumour Biol. 2003;24:236–240. doi: 10.1159/000076138. [DOI] [PubMed] [Google Scholar]
- 12.Gazdar AF, Minna JD. Deregulated EGFR signaling during lung cancer progression: Mutations, amplicons, and autocrine loops. Cancer Prev Res (Phila) 2008;1:156–160. doi: 10.1158/1940-6207.CAPR-08-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirma N, Luthra R, Jones J, Liu YG, Nair HB, Mandava U, Tekmal RR. Overexpression of the colony stimulating factor (CSF-1) and/or its receptor c-fms in mammary glands of transgenic mice results in hyperplasia and tumor formation. Cancer Res. 2004;64:4162–4170. doi: 10.1158/0008-5472.CAN-03-2971. [DOI] [PubMed] [Google Scholar]
- 14.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anticancer therapy: Promises and perils. EMBO Mol Med. 2011;3:623–636. doi: 10.1002/emmm.201100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 17.Kogan M, Haine V, Ke Y, Wigdahl B, Fischer-Smith T, Rappaport J. Macrophage colony stimulating factor regulation by nuclear factor kappa B: A relevant pathway in human immunodeficiency virus type 1 infected macrophages. DNA Cell Biol. 2012;31:280–289. doi: 10.1089/dna.2011.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Xu GZ, Jiang CH, Tian J. Macrophage colony-stimulating factor and its receptor signaling augment glycated albumin-induced retinal microglial inflammation in vitro. BMC Cell Biol. 2011;12:5. doi: 10.1186/1471-2121-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. 2016;44:333–341. doi: 10.1042/BST20150245. [DOI] [PubMed] [Google Scholar]
- 20.Foster P, Yamaguchi K, Hsu PP, Qian F, Du X, Wu J, Won KA, Yu P, Jaeger CT, Zhang W, et al. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther. 2015;14:931–940. doi: 10.1158/1535-7163.MCT-14-0833. [DOI] [PubMed] [Google Scholar]
- 21.Li XR, Liu M, Zhang YJ, Wang JD, Zheng YQ, Li J, Ma B, Song X. Evaluation of ER, PgR, HER-2, Ki-67, cyclin D1, and nm23-H1 as predictors of pathological complete response to neoadjuvant chemotherapy for locally advanced breast cancer. Med Oncol. 2011;28(Suppl 1):S31–S38. doi: 10.1007/s12032-010-9676-z. [DOI] [PubMed] [Google Scholar]
- 22.He Y, Liu Z, Qiao C, Xu M, Yu J, Li G. Expression and significance of Wnt signaling components and their target genes in breast carcinoma. Mol Med Rep. 2013;9:137–143. doi: 10.3892/mmr.2013.1774. [DOI] [PubMed] [Google Scholar]
- 23.Kanazawa T, Misawa K, Misawa Y, Uehara T, Fukushima H, Kusaka G, Maruta M, Carey TE. G-protein-coupled receptors: Next generation therapeutic targets in head and neck cancer? Toxins (Basel) 2015;7:2959–2984. doi: 10.3390/toxins7082959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czapiewski P, Wełnicka-Jaśkiewicz M, Seroczyńska B, Skokowski J, Sejda A, Szade J, Wiewiora C, Biernat W, Żaczek A. CD99 correlates with low cyclin D1, high topoisomerase 2 status and triple negative molecular phenotype but is prognostically irrelevant in breast carcinoma. Pol J Pathol. 2015;66:269–275. doi: 10.5114/pjp.2015.54961. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Chen X, Zhang X, Zhan J, Chen J. Knockdown of TMEM14A expression by RNAi inhibits the proliferation and invasion of human ovarian cancer cells. Biosci Rep. 2016;36:e00298. doi: 10.1042/BSR20150258. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Chen Y, Jiang T, Shi L, He K. hcrcn81 promotes cell proliferation through Wnt signaling pathway in colorectal cancer. Med Oncol. 2016;33:3. doi: 10.1007/s12032-015-0713-9. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Z, Wang L, Song W, Cui H, Chen G, Qiao F, Hu J, Zhou R, Fan H. Reduced miR-29a-3p expression is linked to the cell proliferation and cell migration in gastric cancer. World J Surg Oncol. 2015;13:101. doi: 10.1186/s12957-015-0513-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MS, Ha JH, Yoon HS, Lee CK, Chi SW. Structural basis for the conserved binding mechanism of MDM2-inhibiting peptides and anti-apoptotic Bcl-2 family proteins. Biochem Biophys Res Commun. 2014;445:120–125. doi: 10.1016/j.bbrc.2014.01.130. [DOI] [PubMed] [Google Scholar]
- 29.Edison N, Curtz Y, Paland N, Mamriev D, Chorubczyk N, Haviv-Reingewertz T, Kfir N, Morgenstern D, Kupervaser M, Kagan J, et al. Degradation of Bcl-2 by XIAP and ARTS promotes apoptosis. Cell Rep. 2017;21:442–454. doi: 10.1016/j.celrep.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou L, Gao R, Wang Y, Zhou M, Ding Z. Loss of BAX by miR-365 promotes cutaneous squamous cell carcinoma progression by suppressing apoptosis. Int J Mol Sci. 2017;18(pii):E1157. doi: 10.3390/ijms18061157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song S, Jacobson KN, McDermott KM, Reddy SP, Cress AE, Tang H, Dudek SM, Black SM, Garcia JG, Makino A, Yuan JX. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am J Physiol Cell Physiol. 2016;310:C99–C114. doi: 10.1152/ajpcell.00092.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh SS, Yap WN, Arfuso F, Kar S, Wang C, Cai W, Dharmarajan AM, Sethi G, Kumar AP. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J Gastroenterol. 2015;21:12261–12273. doi: 10.3748/wjg.v21.i43.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahajan K, Mahajam NP. PI3K-Independent AKT activation in cancers: A Treasure trove for novel therapeutics. J Cell Physiol. 2012;227:3178–3184. doi: 10.1002/jcp.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan AC, Vyse S, Huang PH. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov Today. 2017;22:72–84. doi: 10.1016/j.drudis.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]