

Abstract
We present here an integrated nanotechnology/biology strategy for cancer immunotherapy that uses arginine nanoparticles (ArgNPs) to deliver CRISPR-Cas9 gene editing machinery into cells to generate SIRP-α knockout macrophages. The NP system efficiently co-delivers single guide RNA (sgRNA) and Cas9 protein required for editing to knock out the “don’t eat me signal” in macrophages that prevents phagocytosis of cancer cells. Turning off this signal increased the innate phagocytic capabilities of the macrophages by 4-fold. This improved attack and elimination of cancer cells makes this strategy promising for the creation of ‘weaponized’ macrophages for cancer immunotherapy.
Table of Contents
INTRODUCTION
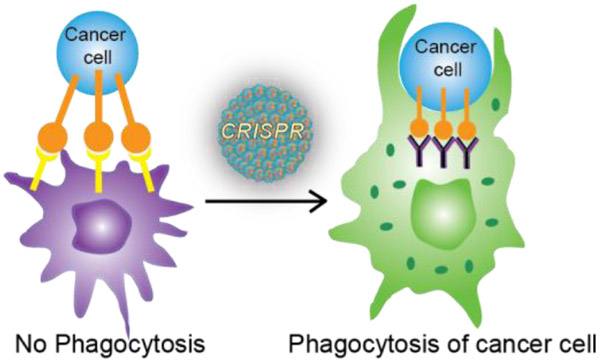
The immune system plays a critical role in preventing tumor initiation and growth; evasion of this system is required for cancer progression.1 One such mechanism is the generation of “don’t eat me” signals by the cancer cells, preventing phagocytosis by macrophages 2 The avoidance signal of perhaps greatest interest is CD47,3 a cell surface protein overexpressed by most cancer cells. Interaction between cancer cell CD47 receptors and macrophage signal regulatory protein-α (SIRP-α) is sufficient to bypass phagocytosis even if phagocytic signals are present (Figure 1). CD47:SIRP-α binding leads to activation of SIRP-α via phosphorylation of its immune-receptor tyrosine-based inhibition motifs on the cytoplasmic tail,4 resulting in binding and activation of Src homology phosphatase-1 (SHP-1) and SHP-2.5 As a result, phagocytosis is blocked by preventing accumulation of myosin-IIA at the phagocytic synapse. This inhibitory mechanism of CD47:SIRP-α binding is evident in a wide range of cancer-initiated malignancies making it a promising therapeutic target.6
Figure 1.

(a) Prevention of cancer cell phagocytosis by CD47:SIRP-α interaction (b) Genomic editing using CRISPR-Cas9 machinery (c) Cell-based immunotherapy through elimination of CD47:SIRP-α interaction by knocking out SIRP-α using nanoparticle-mediated delivery of CRISPR-Cas9/sgRNA and resulting phagocytosis of cancer cell by SIRP-α- macrophage
Recently, strategies have been developed to block the interaction of CD47 with SIRP-α. Use of an anti-CD47 monoclonal antibody has shown efficacy in preclinical studies with different human cancers both in vitro and in mouse xenotransplantation models.2,7–8,9,10,11 An engineered SIRP-α variant, CV1,12 has been used as an antibody adjuvant and shown to facilitate macrophage-mediated phagocytosis in tumor models with increased tumor penetration and low toxicity. However, overexpression of CD47 by cancer cells leads to a large antigen sink in the employment of antibody-based strategies that both reduces bioavailability and increases the potential for toxicity to normal cells.7,13 An alternative to blocking CD47 is the targeting of SIRP-α. Studies have shown that anti-SIRP-α antibodies significantly enhanced antibody-mediated killing of tumor cells by phagocytes in vitro. Nevertheless, due to their large size, penetration of the antibodies into solid tumors was a major limitation for therapeutic efficacy. SIRP-α-targeting agents must have sufficient tumor penetration to interact with and block tumor infiltrating macrophages,14,15 to be a viable therapeutic approach. 16
Recently, we have developed nanomaterial platforms for the delivery of biologics. These scaffolds can simultaneously transport proteins and nucleic acids directly to the cytosol through a membrane fusion mechanism. In our approach, we have delivered the complete CRISPR/Cas9 machinery (Figure 1b) by engineering Cas9 protein to facilitate association with cationic arginine-coated gold nanoparticles (ArgNPs).17 This strategy has demonstrated ~90% delivery efficiency along with ~30% gene editing efficiency. Here, we use the same system to knock out SIRP-α in macrophages to turn off this “don’t eat me” signal and enable phagocytosis of cancer cells (Figure 1c), thus providing a strategy for cancer immunotherapy.18,19
RESULTS AND DISCUSSION
Delivery of CRISPR-Cas9 protein and subsequent knockout of SIRP-α gene.
Recently, the CRISPR-Cas9 nuclease system has emerged as a powerful tool for genome editing.20 It is a two-component system consisting of sgRNA and Cas9 nuclease (generally derived from Streptococcus pyogenes, or SpCas9) for generating sequence-specific targeted mutations in the genome.21 This targeted modification of the genome is permanent and can be passaged to offspring cells. In our previous research,17,22 we have demonstrated CRISPR/Cas9 mediated gene editing by engineering Cas9 protein to facilitate association with cationic ArgNPs. We inserted a peptide tag containing glutamic acids (E-tags) at the N-terminus of Cas9 protein derived from S. pyogenes (SpCas9) and appended a nuclear localization signal (NLS) tag23 at the C terminus to enhance nuclear accumulation (Figure 2a-d). When E-tagged SpCas9 was mixed with ArgNPs, self-assembled superstructures were generated via carbo ylate-guanidinium binding. We found that E20-tag provided the most efficient delivery of SpCas9 into the cytosol and nucleus in multiple cell lines, including the RAW 264.7 model macrophage cell line. Therefore, we engineered SpCas9, first, by introducing an E-20 tag at the N-terminus of the protein so that the protein could self-assemble with cationic ArgNPs. After engineering and purifying CasE20 protein, we fabricated nanoassemblies with CasE20-sgRNA (Cas9-ribonucleoprotein, hereafter, referred to as Cas9-RNP) and ArgNPs. These nanoassemblies were incubated with RAW264.7 cells in cell culture media. Delivery efficiency was monitored by using Cas9E20 labelled with fluorescein isothiocyanate (FITC), and imaging via confocal laser scanning microscopy (CLSM) after 3 h of incubation. Cas9E20 was readily delivered to cytosol via membrane fusion mechanism 24 and accumulated in the nucleus, stained with Hoechst 33342 (Figure 2f, Supporting Information Video S1), a prerequisite for gene editing. We further demonstrated the intracellular release dynamics of Cas9E20-RNP by performing time lapse video imaging. We recorded images at 30 s intervals for 1h following addition of the nanoassemblies into the RAW264.7 cell culture media (Supporting Information Video S2). Real time tracking of a delivery event showed almost instantaneous release of Cas9E20 in the cytosol with subsequent transport of the protein to the nucleus, presumably due to active nuclear transport of NLS-tagged Cas9E20. This remarkably fast intracellular delivery is consistent with prior studies showing that the protein payload was directly released from the cell membrane without being trapped in the endosomes. After efficient intracellular delivery of Cas9E20 was demonstrated, we identified the target gene sequence and generated the appropriate sgRNA 25 for the transmembrane region of SIRP-α in RAW264.7 cells (Figure 2e). We subsequently assembled Cas9E20-RNP with ArgNPs and delivered these nanoassemblies into RAW264.7 cells. The nanoassemblies were incubated with the RAW264.7 cells for 3 h in serum-free media. Genome editing efficiency was evaluated after48 h, using indel analysis (Figure 2g). We observed 27% indel efficiency, fully competitive with other gene delivery-based editing approaches. Cells treated with only Cas9E20-RNP and untreated cells did not show any gene editing. Once the RAW264.7 cells were edited using Cas9E20-RNP and ArgNP nanoassemblies, the knockouts were isolated by dilution (single cell isolation).
Figure 2.

(a) Engineering Cas9 by introducing an E20-tag at the N-terminus and nuclear localization signal (NLS) at the C-terminus (b) Chemical structure of ArgNPs (c) Schematic showing formation of nanoassembly by Cas9E20-RNP and ArgNPs (d) Delivery of Cas9E20 via membrane fusion mechanism. Fusion of nanoassemblies to the cell membrane facilitates direct release of the protein payload into cytoplasm, bypassing endosomes (e) Endogenous SIRP-α gene locus showing the CRISPR-Cas9 target site (f) Cytoplasmic/nuclear delivery of FITC- labelled SpCas9E20 into RAW264.7 cells; (Cell nuclei stained with Hoechst 33342)(g) Delivery of Cas9E20-RNP to target SIRP-α gene in RAW264.7 cells resulted in efficient gene editing, as determined by indel (insertion and deletion) assay: Lane 1: Cas9E20-RNP:ArgNPs, Lane 2: cells only and Lane 3: Marker (the bottom one is 200 bp and each increase in fragment size is 100 bp). Indel efficiency is given in percentages. (h) Fluorescence histogram from flow cytometry analysis on SIRP-α- RAW264.7 cells and RAW264.7 cells; RAW264.7 cells: green histogram; SIRP-α- RAW264.7 cells purple histogram
To further assess the expression level of SIRP-α in SIRP-α- RAW264.7 cells (after single cell isolation), we labelled both RAW264.7 and SIRP-α- RAW264.7 cells with APC anti mouse SIRP-α- antibody. Labelling with APC anti-mouse SIRP-α antibody caused an increase in fluorescence intensity of the RAW264.7 cells as shown by a flow cytometry data (Figure 2h). The fluorescence intensity of the RAW264.7 cells shifted to the right (green histogram) compared to that of the SIRP-α- RAW264.7 cells (purple histogram) due to the increased level of SIRP-α expression in RAW264.7 cells compared to SIRP-α- RAW264.7 cells. This result clearly proved that ArgNPs facilitated delivery of CRISPR-Cas9 in RAW264.7 cells has indeed knock-out SIRP-α.
SIRP-α knockout RAW264.7 macrophages promote phagocytosis of cancer cells.
Next the phagocytic ability of the SIRP-α knockout RAW264.7 (SIRP-α- RAW264.7) cells was tested against cancer cells. Here, we chose human osteosarcoma cells expressing and not expressing GFP, (U2OS-GFP+ and U2OS-GPF-, respectively), as a cancer cell model. We separately co-cultured RAW264.7 and SIRP-α- RAW264.7 cells (both labeled with PE anti mouse F4/80 antibody) with U2OS-GFP+ cells for 4 h at 370C. SIRP-α- RAW264.7 cells showed a 4-fold increase in phagocytosis of U2OS-GFP+ cells compared to non-edited RAW264.7 cells, as measured by flow cytometry (Figure 3a, b). This demonstrates that the U2OS cells were being recognized and engulfed by the SIRP-α- RAW264.7 cells (Supporting Information Video S3) at a higher rate, revealing the efficacy of the strategy.
Figure 3.
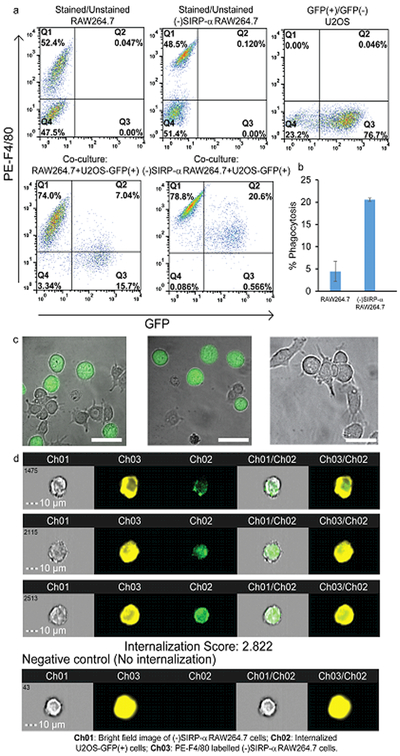
(a) Flow cytometry plots of PE anti-mouse F4/80 antibody-stained and unstained RAW264.7 cells, PE anti-mouse F4/80 antibody-stained and unstained SIRP-α- RAW264.7cells, GFP+/GFP- U2OS cells, co-culture of PE anti-mouse F4/80 antibody-stained RAW264.7 cells and U2OS-GFP+ cells and co-culture of PE anti-mouse F4/80 antibody-stained SIRP-α- RAW264.7 cells and U2OS-GFP+ cells (b) Percentage of phagocytosis of U2OS-GFP+ by SIRP-α- RAW264.7 cells and RAW264.7 cells (c) Confocal images showing phagocytosis or no phagocytosis of U2OS cells; U2OS cells (green) labelled with pHrodo Green AM Intracellular pH Indicator. Scale bar: 20 µm (d) Representative images U2OS-GFP+ cells internalized by PE-F4/80 labelled SIRP-α- RAW264.7 cells measured by Imaging flow cytometry. The negative control (no internalization) is a representative PE-F4/80 labelled SIRP-α- RAW264.7 cell without internalized U2OS-GFP+ cells. Scale bar: 10 µm.
To confirm the internalization of the U2OS cells within the macrophages, we labeled U2OS-GFP- cells with pHrodo Green AM Intracellular pH Indicator.26 This dye is non-fluorescent at neutral pH, however it becomes fluorescent as phagolysosomes are formed in acidic environments. The brightly fluorescent cells (green) confirmed the internalization of the U2OS cells by the modified macrophages as well as formation of phagolysosomes (Figure 3c). Fluorescent cells are not seen when U2OS cells are not internalized by the macrophages (Figure 3c, right image). To provide further confirmation, we used an imaging flow-cytometer that provided an image of each cell that passed through the system. Using this method, we observed the internalization of the U2OS cells by SIRP-α- RAW264.7 cells with an internalization score of 2.822 (Figure 3d). Unmodified RAW264.7 cells, on the other hand, had an internalization score of 1.134 (Supporting Information Figure S1). The images clearly exhibited that the PE-F4/80 stained SIRP-α- RAW264.7 cells contained U2OS-GFP+ cells. No cancer cells bound to, but not internalized by, macrophages were detected with this imaging technology due to the setting of gates on single cells. These results collectively demonstrated the efficiency of our method.
In summary, we have created an integrated nanotechnology/biology approach to engineer macrophages in vitro into therapeutic tools to fight cancer. We have utilized a protein based approach to deliver CRISPR-Cas9 into macrophages to knock out the SIRP-α gene that governs macrophage interactions with CD47 on cancer cells to prevent phagocytosis. Knocking out SIRP-α weaponizes the macrophages, greatly enhancing their ability to phagocytose tumor cells, providing a new immunotherapeutic strategy for cancer therapy.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Dr. James Chambers for helping with confocal microscopy. Special thanks to Ankita Mitra and Ilker Emrah Ozay for assistance in flow cytometry and Amnis data analysis.
Funding Sources
This research was supported by the NIH (GM077173 and EB022641 (VR)) and the Chemistry-Biology Interface Training grant (JH, 5T32GM008515).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details and additional figures and videos. This material is available free of charge on the ACS Publications website at http://pubs.acs.org
Notes
The authors declare no competing financial interests.
Institute for Protein Design, School of Medicine, University of Washington, Seattle, Washington 98195, USA
Laboratory for Biomaterials and Drug Delivery, Division of Critical Care Medicine, Boston Children’s Hospital, Harvard Medical School, 300 Longwood Avenue, Boston, Massachusetts 02115, USA
REFERENCES
- 1.Chao MP, Weissman IL, and Majeti R (2012) The CD47–SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol 24, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD et al. (2012) The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U.S.A 109, 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fehervari Z (2015) Don’t eat me, activate me. Nat. Immunol 16, 1113–1113. [Google Scholar]
- 4.Slee J, Christian A, Levy R, and Stachelek S (2014) Addressing the inflammatory response to clinically relevant polymers by manipulating the host response using ITIM domain-containing receptors. Polymers 6, 2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inagaki K, Yamao T, Noguchi T, Matozaki T, Fukunaga K, Takada T, Hosooka T, Akira S, and Kasuga M (2000) SHPS-1 regulates integrin-mediated cytoskeletal reorganization and cell motility. EMBO J. 19, 6721–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barclay AN, and Van den Berg TK (2014) The interaction between signal regulatory protein alpha (SIRP-α) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol 32, 25–50. [DOI] [PubMed] [Google Scholar]
- 7.Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, Finetti P, Van Egmond M, Matozaki T, Kraal G et al. (2011) CD47–signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. U.S.A 108, 18342–18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, Park CY, Weissman IL, and Majeti R (2011) Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 71, 1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT et al. (2010) Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-hodgkin lymphoma. Cell 142, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, and Weissman IL (2011) Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therap. Blood 118, 4890–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edris B, Weiskopf K, Volkmer AK, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Liu J, Majeti R, West RB, Fletcher JA et al. (2012) Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. U.S.A 109, 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiskopf K, Ring AM, Ho CCM, Volkmer J-P, Levin AM, Volkmer AK, Özkan E, Fernhoff NB, van de Rijn M, Weissman IL, and Garcia KC (2013) Engineered SIRP-α variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho CCM, Guo N, Sockolosky JT, Ring AM, Weiskopf K, Özkan E, Mori Y, Weissman IL, and Garcia KC (2015) “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein α (SIRP-α) antagonists that enhance antibody-dependent cellular phagocytosis. J. Biol. Chem 290, 12650–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panni RZ, Linehan DC, and DeNardo DG (2013) Targeting tumor-infiltrating macrophages to combat cancer. Immunother 5 DOI: 10.2217/imt.2213.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D et al. (2013) Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Response. Cancer Res 73, 1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chames P, Van Regenmortel M, Weiss E, and Baty D (2009) Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol 157, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mout R, Ray M, Yesilbag Tonga G, Lee Y-W, Tay T, Sasaki K, and Rotello VM (2017) Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano 11, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varela-Rohena A, Carpenito C, Perez EE, Richardson M, Parry RV, Milone M, Scholler J, Hao X, Mexas A, Carroll RG, June CH, and Riley JL (2008) Genetic engineering of T cells for adoptive immunotherapy. Immunol. Res 42, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossig C, and Brenner MK (2004) Genetic modification of T lymphocytes for adoptive immunotherapy. Mol. Ther 10, 5–18. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F (2015) CRISPR-Cas9: prospects and challenges. Hum. Gene Ther 26, 409–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mout R, Ray M, Lee Y-W, Scaletti F, and Rotello VM (2017) In Vivo Delivery of CRISPR/Cas9 for therapeutic gene editing: progress and challenges. Bioconjugate Chem 28, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray M, Tang R, Jiang Z, and Rotello VM (2015) Quantitative tracking of protein trafficking to the nucleus using cytosolic protein delivery by nanoparticle-stabilized nanocapsules. Bioconjugate Chem 26, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang R, Kim CS, Solfiell DJ, Rana S, Mout R, Velázquez-Delgado EM, Chompoosor A, Jeong Y, Yan B, Zhu Z-J, Kim C, Hardy JA, and Rotello VM (2013) Direct delivery of functional proteins and enzymes to the cytosol using nanoparticle-stabilized nanocapsules. ACS Nano 7, 6667–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sano S. i., Ohnishi H, and Kubota M (1999) Gene structure of mouse BIT/SHPS-1. Biochem. J 344, 667–675. [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Zhong M-C, Guo H, Davidson D, Mishel S, Lu Y, Rhee I, Pérez-Quintero L-A, Zhang S, Cruz-Munoz M-E, Wu et al. (2017) SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integri. Nature 544, 493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.