

Abstract
Enterotoxigenic Escherichia coli (ETEC) infections are highly prevalent in developing countries, where clinical presentations range from asymptomatic colonization to severe cholera-like illness. The molecular basis for these varied presentations, which may involve strain-specific virulence features as well as host factors, has not been elucidated. We demonstrate that, when challenged with ETEC strain H10407, originally isolated from a case of cholera-like illness, blood group A human volunteers developed severe diarrhea more frequently than individuals from other blood groups. Interestingly, a diverse population of ETEC strains, including H10407, secrete the EtpA adhesin molecule. As many bacterial adhesins also agglutinate red blood cells, we combined the use of glycan arrays, biolayer inferometry, and noncanonical amino acid labeling with hemagglutination studies to demonstrate that EtpA is a dominant ETEC blood group A–specific lectin/hemagglutinin. Importantly, we have also shown that EtpA interacts specifically with glycans expressed on intestinal epithelial cells from blood group A individuals and that EtpA-mediated bacterial-host interactions accelerate bacterial adhesion and effective delivery of both the heat-labile and heat-stable toxins of ETEC. Collectively, these data provide additional insight into the complex molecular basis of severe ETEC diarrheal illness that may inform rational design of vaccines to protect those at highest risk.
Keywords: Infectious disease, Vaccines
Keywords: Bacterial infections, Bacterial vaccines, Glycobiology

Introduction
Enterotoxigenic Escherichia coli (ETEC) is a major cause of infectious diarrhea in the developing world (1, 2). These organisms are estimated to be responsible for millions of infections and hundreds of thousands of deaths, particularly of young children (3). Likewise, these pathogens are a principal cause of diarrhea in travelers to endemic areas and an occasional cause of illness in developed countries (4–7). Despite significant declines in deaths from acute diarrhea (8), enteric infections caused by ETEC and other pathogens remain inextricably associated with malnutrition, stunted growth, and impaired intellectual development (9), poorly understood sequelae that appear to result from gut dysfunction that follows repeated episodes of diarrhea in childhood (10). The ultimate impact of these illnesses is further amplified by poor responses to vaccines, increased mortality from other infections, and emerging evidence that early gut dysfunction may be reflected in the subsequent development of obesity and its attendant metabolic complications (11, 12). Consequently, these infections continue to exact an extraordinary toll on global health.
The ETEC pathovar is defined by the production of plasmid-encoded heat-labile (LT) and/or heat-stable (ST) enterotoxins (13). Effective delivery of these toxins to cognate receptors on the surface of intestinal epithelial cells is ultimately required to drive net salt and water efflux into the lumen of the small intestine, which results in diarrhea. Although ETEC was originally identified in individuals suffering from severe cholera-like diarrhea (14), the manifestations of infection may range from asymptomatic colonization to fulminant illness clinically indistinguishable from that caused by Vibrio cholerae (15–17). The molecular basis for these varied clinical presentations is presently unclear.
In the classic paradigm for ETEC virulence, these organisms adhere to small intestinal epithelia via plasmid-encoded fimbrial colonization factors to effect toxin delivery. However, recent studies suggest that the molecular pathogenesis of this diverse group of pathogens may involve a repertoire of additional virulence features (18–20), including a variety of different adhesins to interact with the intestinal epithelium (21).
Because of their impact on global health, these pathogens are a principal target for vaccine development (22). Although there is currently no vaccine based on the classical virulence paradigm that affords significant broad-based protection against ETEC (23), more recently discovered virulence factors and an improved understanding of ETEC microbial pathogenesis could offer additional avenues to protect those at highest risk for severe disease and uncover novel molecular targets for future vaccine development (24).
Among the more recently described putative virulence molecules not targeted in the current generation of ETEC vaccines is EtpA, a high–molecular weight secreted adhesin (25) bridge between ETEC and intestinal epithelia (26). EtpA appears to be conserved among a phylogenetically and geographically diverse population of ETEC strains (27–29) and has shown promise as a protective antigen in preclinical vaccine studies (30–33). Nevertheless, many questions remain to be answered about the contribution of this antigen and other molecules to disease pathogenesis and their potential use in vaccines relative to canonical ETEC pathovar–specific adhesins.
One approach that has been used to explore ETEC pathogen biology is the controlled human infection model, in which healthy immunologically naive volunteers are challenged by ingestion of enterotoxigenic E. coli. Notably, this model was used initially to establish these toxin-producing E. coli as a cause of diarrheal illness in humans (34) and subsequently to examine virulence features of ETEC (35) and immune responses to experimental infection (36) and to conduct vaccine studies (37–40). The majority of ETEC human-challenge studies conducted to date have involved H10407, a prototype strain originally isolated from an adult with severe, cholera-like illness in Bangladesh (41). While diarrheal illness following H10407 challenge tends to be significantly more severe than that associated with other ETEC strains (42, 43), the severity of diarrhea can vary substantially among volunteers within the same cohort, implicating the involvement of one or more host factors in disease outcome.
Here, we demonstrate that, when challenged with H10407, naive human volunteers with blood group A are at substantially increased risk for severe diarrhea relative to other blood groups. In addition, we found that the EtpA adhesin is a lectin that selectively promotes interaction of ETEC with human small intestinal epithelial cells expressing blood group A glycans. These EtpA-dependent ETEC-host interactions potentially provide a molecular basis for selection and an accelerated course of illness in blood group A–positive hosts.
Results
Blood group A volunteers challenged with ETEC have more severe disease.
Because pathogen interactions with human blood group antigens can influence the outcome of infection by other gastrointestinal pathogens (44, 45), we questioned whether diarrheal severity following ETEC infection was determined by blood group. To examine the impact of ABO blood groups on the development of diarrheal illness, we analyzed samples from 106 adult human volunteers who had participated in any 1 of 4 earlier controlled human infection model studies in which they were challenged with ETEC strain H10407 at the Johns Hopkins Center for Immunization Research (Baltimore, Maryland, USA) and for whom prospectively collected metadata on diarrheal severity were available. Each volunteer was challenged with 1 to 2 × 107 colony-forming units of ETEC H10407 and was ETEC-naive prior to experimental challenge. All subjects were treated with antibiotics upon meeting early treatment criteria (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI97659DS1) or at 120 hours after inoculation to clear their infections.
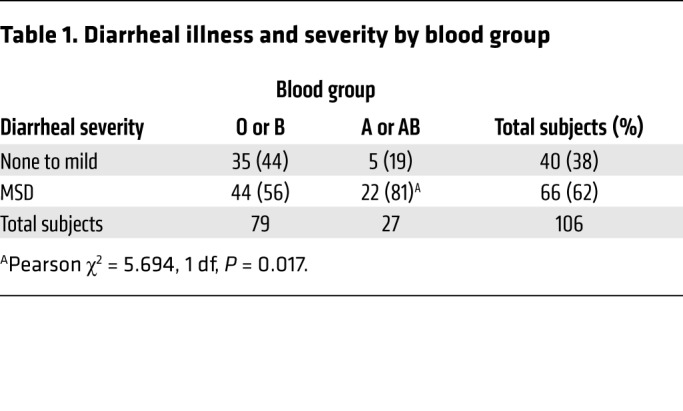
Interestingly, we found that development of severe diarrheal illness was significantly (P = 0.015) accelerated in individuals with blood group A relative to those in non-A blood groups (Figure 1A), with volunteers in the A blood group requiring early initiation of antibiotic therapy more frequently (72%) than those in blood group B or O (43%). Despite earlier antibiotic administration, maximal diarrheal volumes over 24 hours, total diarrheal volume prior to treatment, and the frequency of diarrheal episodes tended to be higher in A versus non-A individuals (Supplemental Figure 1, A–D). Notably, individuals with blood groups B or O were considerably more likely to remain free of diarrhea or have mild diarrhea (44%) following challenge than volunteers expressing the A blood group (19%) (Table 1). Conversely, of the 106 subjects, 81% of those expressing the A blood group antigen (type A or AB) had moderate-to-severe diarrhea (MSD) compared with 56% of individuals with non-A (B or O) blood types (P = 0.017), and the overall relative risk of MSD among individuals with the A blood group compared with the non-A blood groups was 1.44 (95% CI = 1.1–1.9, P = 0.009) (Figure 1B and Supplemental Table 2). These data therefore suggest that, upon challenge with H10407, blood group A volunteers (Supplemental Tables 3 and 4) are markedly more likely to manifest the severe, cholera-like spectrum of illness originally associated with this isolate.
Figure 1. Diarrheal severity is increased in blood group A–positive volunteers relative to those in the non-A blood groups.

(A) Development of severe diarrheal illness is accelerated in A blood group hosts. Kaplan-Meier curves indicate the proportion of subjects treated following the development of severe diarrheal illness in volunteers belonging to A and non-A blood groups. Time-to-treatment data were available for 79 of the 106 subjects (18 subjects in the A blood group and 61 subjects in the non-A group). Data were censored at 120 hours, when all remaining untreated individuals received antibiotics to clear their infection. P = 0.015, log-rank (Mantel-Cox) testing. (B) Individuals of A blood group are more likely to develop MSD. Shown are relative risk data from 4 independent controlled human infection model challenge studies of H10407 involving a total of 106 volunteers. Study numbers at left refer to ClinicalTrials.gov designations. Box size indicates relative study size, and lines represent 95% CIs for the relative risk of severe diarrhea. The width of the diamond indicates the 95% CIs for the pooled effect. The relative risk (pooled fixed effects) of MSD illness dichotomized by A (blood groups A and AB) vs. non-A (O and B) was 1.441 (95% CI, 1.097 to 1.893; P = 0.009). Cochran’s Q test of heterogeneity was insignificant (P = 0.667, Q = 1.5682, df = 3).
Table 1. Diarrheal illness and severity by blood group.
EtpA interacts specifically with blood group A glycans.
Pathogens use adhesins possessing glycan-specific lectin activity (46) to engage blood group antigens expressed on mucosal surfaces, including the gastrointestinal tract (47). The apical surface of intestinal epithelial cells is heavily decorated with a variety of glycoproteins that form the glycocalyx (48), and glycosylation patterns of these proteins are in part determined by blood group (47, 49). Because earlier studies had suggested that EtpA, a secreted adhesin molecule discovered in H10407 (25), engages the surface of gastrointestinal epithelia, we conducted glycan-array screening to further elucidate the nature of EtpA-mediated bacterial adhesion. Using recombinant EtpA (rEtpA) to probe arrays containing more than 400 unique glycan species, we found that rEtpA interacted almost exclusively with glycans related to blood group A. In contrast, we observed little or no binding to other glycans, including those of blood group B or blood group O (Figure 2A and Supplemental Data Set 1). Next, in biolayer interferometry experiments, we found that EtpA interacted with blood group A–related glycans with high affinity when the glycan was presented as a multivalent trisaccharide conjugated to polyacrylamide (PAA): GalNAcα1-3 (Fucα1,2)Galβ-PAA, where Gal indicates galactose; kD, ~9 × 10–8 M (Figure 2B). Similarly, we demonstrated that EtpA can engage N-acetylgalactosamine (GalNAc) (kD, ~1.6 × 10–8 M), the terminal sugar in blood group A, with higher affinity than Gal (kD, ~1.9 × 10–7), the terminal sugar of blood group B (Figure 2C). Collectively, these studies suggest that the EtpA adhesin is a blood group A–specific lectin.
Figure 2. EtpA interacts preferentially with blood group A glycans.

(A) Glycan-array data (top row) demonstrating EtpA binding predominately to blood group A glycans. The top 100 of 411 glycans are ranked in order of diminishing binding activity from left to right. BSA (middle row) is shown as a control. Values represent background-corrected median data from 2 experimental replicates. Distribution of blood group (bg) binding is shown in the bottom row of the figure. Heatmap key (lower right) shows relative fluorescence units (rfu) and coding for blood group antigens. Names for glycans correspond to Supplemental Data Set 1. (B) EtpA binding to blood group A glycans assessed by Bio-Layer Interferometery (Pall ForteBio Corp.) assays. Data are representative of 3 independent experiments. Blood group A3 refers to the biotinylated trisaccharide-PAA conjugate GalNAcα1-3 (Fucα1,2)Galβ-PAA-biotin; blood group A2 refers to the disaccharide conjugate GalNAcα1-3Galβ-PAA-biotin. (C) EtpA binding to terminal GalNAc residues relative to Gal schematic (right) shows structures of blood groups A and B terminating in GalNAc and Gal sugars, respectively. Blood group O (core H) lacks either terminal sugar residue.
Many bacterial adhesins possess the ability to agglutinate RBCs. Included among these adhesins is filamentous hemagglutinin from Bordetella pertussis (50), a component of acellular pertussis vaccines (51), which, like EtpA, belongs to the 2-partner family of bacterial exoproteins (25). We found that EtpA could decorate the surface of human RBCs belonging to blood group A1 (Figure 3A) and, to a lesser extent, cells from those individuals from blood group A2 who exhibit lower levels of the glycosyltransferase enzyme activity required for addition of terminal GalNAc residues that determine the A blood group (52); however, only a small percentage of blood group B or O erythrocytes had detectable EtpA on the surface. Similarly, in RBC pull-down assays, more rEtpA remained associated with erythrocytes from the A1 and A2 blood groups (Figure 3B) than those from the B or O blood groups. In addition, incubation of EtpA with RBCs from blood group A1 resulted in agglutination, but yielded poor agglutination activity with other RBCs (Figure 3C). In addition, pretreatment of target A1 cells with α-N-acetylgalactosaminidase completely abrogated the hemagglutinin activity of EtpA (Figure 3D) as did addition of excess GalNAc, but not GlcNAc or Gal, the terminal sugar of blood group B (Supplemental Figure 2). We also demonstrated that latex microparticles coated with EtpA specifically agglutinated A1 RBCs (Figure 3E). EtpA therefore belongs to a large family of bacterial adhesins that possess lectin/hemagglutinin activity (46), and this protein preferentially targets GalNac residues presented in the context of the A blood group.
Figure 3. EtpA is a blood group A–specific hemagglutinin.

(A) Binding of rEtpA to the surface of group A1 and A2 RBC ghosts relative to B and O RBCs is shown in FACS data and a graph. (B) RBC pull-down of rEtpA with different blood group ghost erythrocytes. Shown in the immunoblot is rEtpA-myc-his identified by anti-myc antibodies. (C) EtpA agglutinates A1 RBCs, but exhibits minimal or no hemagglutination activity with RBCs from those with A2, B, or O blood groups. The column on the right includes anti–A blood group antibodies as a positive control. (D) EtpA-mediated hemagglutination is dependent on terminal GalNac residues. A1 RBCS pretreated with α-N-acetylgalactosaminidase (top row) fail to agglutinate in the presence of rEtpA. Shown (row 2, left) are positive controls (no enzyme pretreatment) and blood group O–negative controls (bottom rows). (E) EtpA-coated latex microspheres (top row) agglutinate RBCs from blood group A, but not B or O. Bottom row shows BSA-coated particles as controls. Each of the images are representative of 3 experimental replicates. Original magnification, ×40.
EtpA is a predominant blood group A–binding partner in ETEC.
Next, to determine whether EtpA is the predominant blood group A–binding partner in ETEC, we examined interaction of A blood group glycans with subcellular protein fractions isolated from H10407. Blood group A glycans interacted only with a high molecular weight species (in excess of 150 kD) in culture supernatants, with no detectable interaction with preparations from the inner or outer membranes (Figure 4A). Because the EtpA adhesin is the only known ETEC-specific virulence molecule in the ETEC secretome of this size (53), we next compared binding of blood group A glycans to secreted proteins from the WT H10407 strain and isogenic etpA mutants. Binding occurred only in supernatants prepared from WT ETEC or the complemented etpA mutant strain, but not the etpA isogenic deletion mutant (Figure 4, B–D). Interestingly, blood group A erythrocytes have been traditionally used to screen for the presence of CFA/I fimbriae in ETEC (54). However, we found that CFA/I mediated nonspecific hemagglutination of RBCs regardless of blood type, a result that is consistent with the idea that CFA/I interactions are mediated via sialic acid (55) (Supplemental Figure 3). To further confirm the role of EtpA in mediating interaction with the A blood group, we next tagged the ETEC proteome using the noncanonical amino acid azidonorleucine (ANL) (56) and incubated tagged supernatant and outer membrane protein (OMP) fractions with erythrocytes. Again, we found that a single band from tagged WT supernatant, corresponding to EtpA, bound appreciably only to A1 erythrocytes (Figure 4E). However, testing of ANL-labeled OMPs purified from H10407 failed to detect any specific interactions of ETEC OMPs with RBCs of any major blood group (Supplemental Figure 4), further suggesting that the secreted EtpA molecule is the dominant blood group A–specific lectin expressed on the surface of ETEC. We also demonstrated that EtpA expression by a geographically diverse group of ETEC conferred the ability to bind blood group A, suggesting that these findings are not limited to the H10407 prototype strain (Supplemental Figure 5, A–D).
Figure 4. EtpA is a dominant blood group A–binding partner of ETEC H10407.

(A) Blood group A antigen far-Western blot with subcellular fractions of ETEC H10407 including supernatant (S), outer membrane (OM), and inner membrane (IM) demonstrates binding of biotinylated blood group A to high molecular weight protein in concentrated culture supernatant. (B) Coomassie-stained gel of supernatant proteins from H10407 (WT), etpA mutant jf1668, etpA mutant complemented with the etpBAC locus plasmid pJY019 (etpA(P+); etpA(P–)) equals mutant complemented with cloning vector alone. (C) EtpA immunoblot confirming the presence of EtpA in culture supernatants from H10407 and the complemented mutant. (D) Far-Western blot of culture supernatants shown in B and C shows binding of biotinylated blood group A only in the presence of EtpA. (E) EtpA is the dominant A blood group–specific interacting partner among ETEC proteins from ANL-labeled bacteria. Shown is a fluorescence image of ANL-labeled proteins from ETEC H10407 that interact with erythrocyte ghosts from A1, A2, B, and O blood groups (middle, RBCs/ANL). RBC ghosts alone are shown at left, and input protein (ANL) from H10407 and the etpA deletion mutant (jf1668) are shown at right. The migration of EtpA is indicated by the arrow, and TAMRA-labeled rEtpA is shown at far right as a positive control. Each image is representative of 3 experimental replicates.
EtpA directs ETEC interaction with blood group A on host intestinal epithelial cells.
We questioned whether blood group A–related glycans on the surface of intestinal enterocytes might therefore direct EtpA-mediated interactions required for effective bacterial adhesion and toxin delivery. Using HT-29 intestinal cells derived from a blood group A–positive host, we found that EtpA bound to the surface of these cells and colocalized with blood group A glycans (Figure 5A). Conversely, rEtpA bound very poorly to the surface of HT-29A–/– cells, an HT-29–derived line that was CRISPR-Cas9 engineered to eliminate the gene encoding the glycosyltransferase responsible for the addition of terminal GalNac residues to the core H (blood group O) antigen (45). Likewise, we found that EtpA from ANL-tagged WT ETEC interacted preferentially with HT-29 parental cells compared with the mutant HT-29A–/– cells (Figure 5B). Interestingly, EtpA-expressing ETEC localized to regions of blood group A expression on the surface of HT-29 cells (Figure 5C), and EtpA-expressing ETEC adhered more readily to parental cells expressing blood group A (Figure 5, D and E, and Supplemental Figure 6A). Importantly, we found that optimal delivery of LT toxin required both EtpA (Supplemental Figure 6B) and the presence of the A blood group antigen (Figure 5F). Similarly, we noted that addition of exogenous GalNAc impaired both EtpA-mediated adhesion and toxin delivery to HT29 parental cells (Supplemental Figure 7, A and B), suggesting that this terminal glycan on blood group A is important for EtpA pathogen–epithelial cell interactions. Notably, while purified LT was significantly less potent than cholera toxin (CT) in activating cAMP in target cells, direct toxin-mediated activation of this cyclic nucleotide was actually enhanced in cells lacking the A blood group (Supplemental Figure 8), recapitulating recent observations with CT (45). Collectively, these data suggest that the presence of blood group A effectively promotes bacterial adhesion and ETEC-host interactions that culminate in enhanced toxin delivery.
Figure 5. A blood group–dependent interactions of EtpA and ETEC with intestinal epithelia.

(A) EtpA binds to regions of blood group A expression on the surface of HT-29 epithelial cells. Shown in columns from left to right are blood group A (green, antibodies against blood group A and fluorescent conjugate αbgA/AF488); nuclei (blue, DAPI); biotinylated EtpA (red, SA-coated Qdots594); and merged images. Middle row: no EtpA control. Bottom row: no EtpA binding to the surface of HT29A–/– cells engineered to remove the α1→3 GalNac glycosyltransferase required for A antigen expression. Original magnification, ×100. (B) EtpA preferentially engages cells expressing A blood group. Key: upper left indicates the target (bait) cell lines used to attract ANL-labeled bacterial (prey) proteins from H10407 (WT) or the etpA mutant. Labeled rEtpA and the arrow at left are shown to indicate the predicted migration of EtpA. Lanes at far right show input proteins from the EtpA-expressing H10407 WT and the etpA mutant. (C) Localization of EtpA-expressing ETEC to areas of blood group A expression on the surface of HT-29 cells. Original magnification, ×63. Images in A–C represent 1 of 3 experimental replicates. (D) EtpA and blood group A are required for optimal adhesion. Data are representative of 4 independent experiments. HT-29A–/– data include results from 2 independently generated engineered blood group A glycosyl transferase mutant lines (1e6, circular symbols; and 1g10, square symbols; n = 8 technical replicates in total). *P = 0.03, ANOVA, Kruskal-Wallis. (E) Blood group A accelerates adhesion of ETEC to target intestinal epithelial cells (n = 8 technical replicates for HT-29 cells and n = 6 technical replicates for HT-29–/– cells; representative of 2 independent experiments); symbols represent mean ± SEM. **P = 0.003; ***P = 0.0003, Mann-Whitney U test, 2-tailed comparison. (F) Presence of A blood group and EtpA are required for optimal delivery of heat-labile toxin by ETEC (n = 5 technical replicates representative of 3 independent experiments). **P < 0.01, ANOVA, Kruskal-Wallis.
HT-29 cells are derived from a metastatic colorectal carcinoma, and glycan distribution can vary significantly with location within the gastrointestinal tract (57). In addition, ETEC are thought to preferentially target the small intestine, where the bulk of fluid secretion occurs following colonization, rather than the colon.
We sought, therefore, to confirm these interactions using small intestinal enteroids (58, 59) derived from human stem cells obtained from individuals belonging to each of the major ABO blood groups. Using ileal enteroids, we found that, while EtpA clearly bound to cells from group A individuals, the purified adhesin bound very poorly to cells expressing either B or O blood groups (Figure 6, A and B). We also found EtpA-expressing bacteria within areas of blood group A expression on the epithelial surface of polarized enteroids (Figure 6C), and interestingly, we also observed apparent rearrangement or “footprints” of the A blood group at sites of bacterial attachment, suggesting an intimate interaction of ETEC with these cell-surface glycans (Figure 6D). Finally, we found that ETEC adhered much more readily to polarized intestinal monolayers derived from individuals with blood group A compared with other blood groups (Figure 6E) and that adhesion of WT ETEC to A blood group enteroids was significantly accelerated relative to that of bacteria lacking etpA (Figure 6F), culminating in enhanced toxin delivery by the EtpA-expressing WT (Figure 6, G and H). EtpA therefore appears to be a dominant pathovar-specific lectin uniquely capable of preferentially engaging A blood group glycans expressed on small intestinal epithelial surfaces to accelerate effective ETEC pathogen-host interactions.
Figure 6. EtpA and EtpA-expressing ETEC interact preferentially with the surface of blood group A enteroids.

(A) Confocal imaging of EtpA binding to surface of polarized small intestinal enteroid monolayers from a blood group A subject. Differential interference contrast (DIC) image at bottom shows architecture of polarized enteroids on Transwell filter. Original magnification, ×63. (B) Kinetic ELISA quantification of biotinylated EtpA binding to surface of small intestinal enteroid cells from blood groups A, B, and O (n = 4 technical replicates representative of 3 separate experiments). *P < 0.05; **P < 0.01, ANOVA, Kruskal-Wallis. (C) Association of EtpA-expressing ETEC H10407 with A blood group on surface of small intestinal cells. Images represent volocity-processed confocal laser-scanning microscopy (CLSM) data of ETEC H10407 (red) to blood group A (green) and nuclei (blue). Lower panel shows polarized orientation of cells with bacteria adherent to the apical surfaces of enterocytes. Original magnification, ×63. (D) Top panel: CLSM of ETEC (green) and A blood group (red) on jejunal enteroids. Bottom panel: A blood group “footprints” (arrows) at sites of bacterial attachment. Data shown in C and D are representative of 3 experimental replicates. Original magnification, ×60. (E) Bacterial density on surface of small intestinal enteroids determined by CLSM quantification of H10407 (serotype O78). Quantitation based on imaging 10 fields per blood group, at ×20 magnification. *P = 0.039; #P < 0.0001, ANOVA, Kruskal-Wallis. (F) Adhesion of WT H10407 and etpA mutant bacteria to blood group A small intestinal enteroids (n = 5 technical replicates; representative of 3 biological replicates). Bars represent geometric mean values. (G) Production of cAMP by blood group A target enteroids following infection by WT (H10407) or the etpA mutant. Data represent total of 5 technical replicates from 2 separate experiments. (H) Production of cGMP by blood group A small intestinal enteroids following infection with WT (n = 6) or the etpA mutant (n = 4). Data are representative of 3 independent biological replicates. Inset graph indicates relative (mean ± SD, n = 6 technical replicates) production of heat stable toxin (ST) by the WT (blue bars) vs. the mutant (gray bars). **P < 0.01, Mann-Whitney U test, 2 tailed nonparametric testing.
Discussion
Blood group–dependent microbial-host interactions can dictate clinical outcomes of infection with important gastrointestinal pathogens. In the case of Helicobacter pylori, the BabA adhesin facilitates attachment to the gastric mucosae by engaging ABO (60) and Leb, blood group antigens potentiating the delivery of other secreted virulence factors that lead to development of gastric ulcers and cancer (44, 60, 61). Similarly, the clear epidemiologic associations between severe V. cholerae infections and the O blood group (62–66) are reflected in recent observations that CT preferentially activates enterocytes of blood group O lineage (45). Both CT and the closely related LT toxin of enterotoxigenic E. coli appear to share in a differential binding of blood group antigens that favors intoxication of blood group O enterocytes (67, 68). Interestingly, because of the molecular similarity between CT and LT, some earlier field studies examined ETEC outcomes relative to blood group with the implicit assumption that LT-producing ETEC would lead to more severe disease following infection of O blood group hosts; however, these studies yielded conflicting results (69, 70).
In contrast, from the human volunteer ETEC challenge studies, a surprising, but clear, relationship between the A blood group and disease severity began to emerge. The ETEC controlled human infection model has a number of potential advantages over prior field studies (3, 62, 69, 70) in defining a molecular basis for disease. First, the present studies were confined to ETEC-naive adults challenged with a defined inoculum of a single strain (H10407) isolated from an individual with severe cholera-like illness (41). The controlled human infection model may have therefore permitted us to unmask somewhat subtle molecular associations that might have otherwise been hidden by confounding variables common to field studies of enteric infections in developing countries, including malnutrition, breast feeding, altered microbiota, and coinfection with other enteric pathogens as well as the genetic variability inherent in the infecting ETEC strains.
The studies presented here provide an expanded view of ETEC pathogen-host interactions beyond a classical paradigm in which these organisms engage the intestinal surface via fimbrial colonization factors and deliver LT and/or ST enterotoxins. The present studies of ETEC paint a more complex picture in which A blood group expression potentially facilitates EtpA-mediated lectin-specific bacterial interactions with intestinal epithelia that culminate in effective toxin delivery and symptomatic diarrheal illness.
Our studies suggest that the many ETEC strains that produce the EtpA adhesin (27, 29) may be particularly well equipped to preferentially infect hosts who express the A blood group antigen on mucosal surfaces. When infected with H10407, an EtpA-producing strain, immunologically naive subjects with blood group A appear to be at significantly increased risk for diarrheal illness, and the ensuing diarrhea tends to be more severe relative to the other major blood groups. Intriguingly, the ETEC H10407 strain, isolated from a case of severe cholera-like diarrheal illness more than 40 years ago, is typically among the most virulent strains to be tested in human-challenge studies (43, 71).
It is likely however, that while EtpA leads to enhanced virulence in blood group A hosts, other putative virulence factors identified in H10407, including the mucin-degrading EatA protease (19), collectively augment virulence by accelerating toxin delivery that culminates in severe diarrhea.
Several additional features of our studies deserve comment. We were not able to examine “secretor status,” i.e., expression of the FUT2 α1,2 fucosyltransferase required to synthesize ABO blood group antigens on intestinal epithelia (52). Because approximately 20% of the population are nonsecretors due to defects in FUT2 (72), it is likely that some of the A blood group–positive individuals did not express the A antigen on their gastrointestinal epithelia. Theoretically, this might account for some individuals in the A blood group who failed to become ill following challenge. Conversely, while the A blood group is a risk factor in this model, the development of severe diarrheal illness among non-A volunteers demonstrates that expression of A glycans is not an absolute requirement for disease. V. cholerae infections provide an important analogous paradigm for severity of illness related to blood group. While severe cholera is significantly associated with the O blood group, it is not exclusively observed in blood group O individuals (66). We should also note that the ETEC-challenge studies were done with a relatively high inoculum of bacteria (~107) designed to assure a reasonable attack rate (73), a feature that may not be reflected in naturally occurring infections in young children where the A blood group also appears to be a risk for development of symptomatic infections (3). It is also worth noting that volunteers in these studies were treated with antibiotics to clear the infection once they met the definition of severe diarrheal illness, a feature that might also have affected the ultimate outcome.
The secreted EtpA lectin likely acts in concert (21) with at least 2 additional glycan-binding tip adhesins of ETEC fimbriae with different substrate specificities. These include FimH of the chromosomally encoded type 1 pili, which binds to mannosylated surface proteins on epithelial cells (74), and the CfaE adhesin located at the tips of canonical plasmid-encoded CFA/I pili, which putatively binds to sialylated glycoproteins (55, 75). The latter is consistent with the observation that ETEC expressing CFA/I mediates mannose-resistant (54) and neuraminidase-sensitive (75, 76) hemagglutination in all major blood groups and across species to include chicken and bovine erythrocytes. Therefore, it appears that EtpA is the only blood group A–specific lectin identified in ETEC to date. Nevertheless, we cannot rule out the possibility that our studies failed to identify another blood group A lectin that was not efficiently expressed under laboratory culture conditions.
Interestingly, findings presented here are in keeping with earlier birth-cohort studies of children under the age of 2 years in Bangladesh who also demonstrated an association between the A blood group and symptomatic ETEC infection (3). Therefore, while our recent studies have potentially important clinical and vaccinology implications, further study of the relationship among blood group, disease severity, and antigen expression could guide and inform use of these antigens in vaccines.
These additional molecular details regarding the nature of ETEC pathogen–host interactions could promote an improved understanding of the virulence elements and host determinants related to the most severe forms of illness that are clinically indistinguishable from severe cholera (15–17). Consequently, identification of EtpA as blood group A lectin could focus antigen development and inform vaccine design to specifically protect individuals that are at highest risk for severe cholera-like diarrheal illness.
Methods
ETEC controlled human infection volunteer challenge studies.
Samples for blood group analysis were obtained from a convenience sample of 106 human volunteers from 4 prior independent controlled human infection model ETEC challenge studies conducted at the Johns Hopkins Center for Immunization Research (ClinicalTrials.gov NCT01739231, NCT01060748, NCT00844493, and NCT01922856). All volunteers were experimentally infected with 1 to 2 × 107 colony-forming units of the EtpA-expressing ETEC strain H10407. Subjects in these studies were treated with antibiotics once they met previously defined endpoint criteria (summarized in Supplemental Table 1) or at 120 hours after inoculation to clear their infections. Additional details of the of the study design are included in the Supplemental Methods.
Glycan-array screening.
Glycan arrays containing 411 separate features were fabricated as previously reported, except for the addition of a washable fluorescent dye, the free acid of DyLight 649 (0.7 μg/ml, Thermo Scientific), to the print buffer as an indicator of successful liquid deposition and spot morphology. The array format and assay have been described previously (77) along with analysis of reproducibility (78) and validation with numerous antibodies and lectins (79, 80). Slides were blocked overnight at 4°C with 3% BSA (w/v; 200 μl/well) in PBS and then washed 6 times with PBST (PBS with 0.05% Tween 20; 200 μl/well). Polyhistidine-tagged rEtpA-6His was diluted to final concentrations of 20 and 200 μg/ml in PBST buffer containing 3% BSA (MilliporeSigma) and 1% HSA (MilliporeSigma), and then 50 μl of each sample was added into 2 different wells on different slides. Incubation buffer alone served as the negative control for samples analyzed in duplicate. After incubation with rEtpA for 2 hours at 37°C in an orbital shaker (100 rpm), arrays were washed and then probed with mouse monoclonal IgG anti-His5 antibody (QIAGEN, 34660) diluted 1:500 to a final concentration of 0.4 μg/ml (1 hour at 37°C; 100 rpm), followed by DyLight 549–conjugated affinity purified goat anti-mouse IgG (Jackson ImmunoResearch, 115-505-071; 1 hour at 37°C; 100 rpm). After washing 7 times with PBST (200 μl/well), slides were immersed in wash buffer for 5 minutes and centrifuged at 200 g for 5 minutes. Slides were scanned at 10 μm resolution with a GenePix 4000B Microarray Scanner (Molecular Devices Corp.) with excitation/emission wavelengths of 532/575 nm and analyzed with GenePix Pro 6.0 software, as previously reported (77). Heatmaps were assembled in R version 3.2.2 (The R Foundation for Statistical Computing), using the gplots and RColorBrewer packages made available through the Comprehensive R Archive Network (CRAN; http://cran.wustl.edu/). Additional details of the glycan-array screening can be found in Supplemental Methods.
Bacterial strains and culture conditions.
ETEC strain H10407 was provided from GMP stock produced at the Walter Reed Army Institute of Research (Silver Spring, Maryland, USA). The etpA mutant H10407 (jf1668) and complemented (jf1697) strains were generated in previous studies. ETEC strains were routinely grown in Luria broth (LB) or LB agar (2% agar) at 37°C from glycerol stocks preserved at –80°C. Bacterial strains and plasmids used in this study are listed in Supplemental Table 5. Strains 100137, 200145, 30051, 400643, 500632, 601142, and 700241 were provided by David Rasko (Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, Maryland, USA). Strain 12005000131-1 was provided by Elizabeth Cebelinski (Molecular Epidemiology Section of the Infectious Diseases Laboratory, Minnesota Department of Health, St. Paul, Minnesota, USA).
Protein expression and purification.
The 2-partner secreted EtpA glycoprotein was purified from culture supernatants of E. coli Top10 coexpressing plasmids pJL017 and pJL030 (81). Briefly, E. coli Top10 carrying pJL017 and pJL030 (Supplemental Table 5) was grown in LB with ampicillin (100 μg/ml) and chloramphenicol (15 μg/ml) at 37°C, in a shaking incubator at 230 rpm, until OD 600 nm reached approximately 0.5–0.6. Recombinant protein expression was then induced with 0.0002% arabinose for 6 hours at 37°C. Culture supernatant was concentrated using a 100 kD Molecular Weight Cutoff (MWCO) filter (Millipore), and rEtpA-myc-6His protein was purified using immobilized metal affinity chromatography as described previously (81).
ETEC subcellular fractionation and far-Western analysis.
ETEC H10407 was grown overnight in 10 ml cultures of LB at 37°C as described above, diluted into 500 ml cultures the next morning, and grown for 3 hours at 37°C. Cells were harvested by centrifugation at 5000 g for 10 minutes at 4°C and the pellet resuspended in 8 ml 50 mM Tris buffer pH 7.8 containing 1 mM EDTA and 1 mg DNAse. Subcellular fractions were then prepared as previously described (82). Briefly, cells were disrupted using a French press, followed by the addition of 2 mM MgCl2 to E. coli lysate and centrifugation at 5000 g for 10 minutes at 4°C to remove large debris/intact cells. Supernatant was collected, supplemented with PMSF to a final concentration of 1 mM, and ultracentrifuged at 100,000 g for 1 hour at 4°C. Supernatant from this step was saved as a cytosolic fraction. The remaining cell pellet was resuspended in ice-cold 10 mM HEPES pH 7.4, layered over a gradient of 69%, 52.8%, and 26.4% sucrose in HEPES pH 7.4 and ultracentrifuged at 113,000 g for 16 hours at 4°C. The visible upper layer containing inner membranes and the lower high-density layer with outer membranes were collected. Membrane proteins were reconstituted in ice-cold HEPES pH 7.4 and centrifuged for 1 hour at 100,000 g, 4°C. Finally, the protein pellets from the inner and outer membranes were resuspended in 0.25 and 0.5 ml dH2O, respectively.
Far-Western blotting was used to test blood group A glycan interaction with ETEC proteins. Briefly, approximately 10 μg of ETEC proteins were resolved by SDS-PAGE on NuPAGE 4%–20% Tris-Glycine Gel (Life Technologies) and transferred to nitrocellulose membranes. Membranes were blocked with 5% BSA in TBS-T (Tris[hydroxymethyl]aminomethane–buffered saline, 0.01% Tween-20) for 1 hour and then incubated with a final concentration of 5 μg/ml of biotinylated multivalent blood group A trisaccharide (Glycotech; 01-032) overnight at 4°C. Biotinylated glycans were detected using streptavidin-HRP (SA-HRP) conjugate (1:20,000), and EtpA was detected by immunoblotting using anti-EtpA rabbit polyclonal antibody.
Hemagglutination studies.
We examined the ability of EtpA to agglutinate human erythrocytes belonging to blood groups A1, A2, B, and O (Immucor, catalog 0002338). Briefly, 2-fold dilutions of rEtpA in PBS in a final volume of 10 μl were mixed with 40 μl of approximately 1% cell suspensions (in PBS) of RBCs expressing the A1, A2, B, and O antigens in 96-well round-bottom plates, then incubated for 30 minutes at 4°C. Plates were then imaged to show agglutinated versus nonagglutinated (settled) RBCs. In parallel experiments, recombinant polyhistidine-tagged EtpA was immobilized onto cobalt-impregnated super-paramagnetic polystyrene beads (Dynabeads TALON, Thermo Fisher, catalog 101.01D) that were then incubated with RBCs.
To examine the specificity of EtpA-induced agglutination of RBCs expressing the A antigen, agglutination assays were performed in the presence of excess terminal sugars specific to individual blood types: GalNAc (blood group A), Gal (blood group B), or control GlcNAc at 50 mM final concentration. α-N-acetylgalactosaminidase (P0734S; New England Biolabs) was used to remove terminal GalNAc from the A blood group antigen by incubating 50 μl of a 1% RBC suspension with 20 U α-N-acetylgalactosaminidase for 4 hours at 37°C.
To examine the potential contribution of the CFA/I colonization factor to hemagglutination, we followed a protocol similar to that previously described by Evans (54). Briefly, bacteria were grown overnight on CYE agar (1% casamino acids; BD), 0.15% yeast extract (BD), 2% agar, 0.005% MgSO4, and 0.0005% MnCl2. Bacteria were resuspended in PBS (A600 3.0) and diluted 4-fold prior to use. Human erythrocytes (Immucor, 2%–4% suspension) were diluted 1:4 in PBS, and 25 μl each of bacterial and RBC suspension were added to a flat-bottomed 96-well plate and rotated at 1,000 g at RT for 15 minutes, after which agglutination was visualized. Control anti–A IgM antibodies (Immucor) were used at 1:32 dilution.
FACS.
RBC ghosts were prepared at 4°C. 800 μl of a 2%–4% suspension of RBCs was washed once in PBS, resuspended in 1.5 ml of ghost buffer (50 mM NaPO4, pH 8.0), and incubated on ice for 1 minute. RBCs were pelleted at 25,000 g for 10 minutes, then washed twice in ghost buffer. Ghosts were resuspended in 400 μl PBS.
For FACS analysis, 15 μg biotinylated EtpA or BSA was added to 190 μl of RBC ghosts and rotated at RT for 1.5 hours. Cells were washed twice in PBS and pelleted by spinning at 25,000 g for 5 minutes, rotated for 30 minutes with a 1:200 dilution of SA-conjugate nanocrystals (Qdot 525, Q10141MP, Thermo Fisher), and then washed twice in PBS and resuspended in 200 μl of PBS. Fluorescence was measured using FACSCalibur (BD Biosciences), and data were analyzed using FlowJo software, v10.
Molecular cloning of mutant Met-RS.
To permit noncanonical amino acid labeling of ETEC proteins with azidonorlucine (Anl), a mutant form of the methionyl-tRNA synthetase (NLL-MetRS) gene from pAM1 (56) was cloned (In-Fusion, Clontech) into the NcoI and EcoRI sites of pBAD/Myc-His B (Supplemental Table 5) using primers jf091715.1 and jf091715.2 (Supplemental Table 6). The resulting plasmid pBAD-MetRS was transformed into WT H10407 or the jf1668 etpA mutant to generate jf3422 and jf3904, respectively.
Protein interaction studies using noncanonical amino acid labeling of ETEC.
Proteomes of strains jf3422 and jf3904 bearing the MetRS expression plasmid were labeled by growth with azidonorleucine as previously described. Briefly, strains were grown overnight in LB containing 100 μg/ml ampicillin, 0.2% arabinose, and 1 mM azidonorleucine (HAA1625, Iris Biotech GmbH). Cell-free supernatants were concentrated 10-fold using 10 kD MWCO centrifugal concentrators and dialyzed against PBS prior to use.
OMPs were prepared from ANL-tagged bacteria grown in the same fashion using an established protocol (83). Briefly, labeled bacteria were resuspended in 20 ml ice-cold lysis buffer (50 mM Tris, 1 mM EDTA, pH 7.8) and lysed by sonication (4 × 20-second bursts). Approximately 1 mg of DNase I (MilliporeSigma) was added to each tube and the extracts adjusted to 2 mM MgCl2 and 100 μM PMSF. Intact cells and large debris were removed by low-speed centrifugation at 3,000 g for 10 minutes, and membranes were pelleted from clarified lysates by ultracentrifugation for 1 hour at 100,000 g. Membrane pellets were then resuspended in 2 ml 10 mM HEPES pH 7.4 and layered onto discontinuous sucrose gradients consisting of a top layer of 0.77 M sucrose, a middle layer of 1.44 M sucrose, and a bottom layer of 2.02 M sucrose. After centrifugation at 113,000 g for 16 hours at 4°C, the OMPs were recovered from the interface between the 1.44 and 2.02 M layers. OMP pellets were washed by resuspension in 14 ml 10 mM HEPES pH 7.4, collected by centrifugation at 100,000 g for 1 hour, and resuspended in 0.5 ml PBS.
Erythrocyte membranes were prepared using the method previously described by Steck et al. (84). Briefly, suspensions of RBCs were sedimented at 2,300 g for 10 minutes at 4°C, the pellet recovered, and then resuspended in 5 volumes of cold PBS, pH 8.0. After centrifugation, packed RBCs were hemolyzed by mixing with 5 mM sodium phosphate, pH 8.0, and incubating on ice for 1 minute. The resulting membranes were then harvested by centrifugation at 22,000 g for 10 minutes and the supernatant removed by aspiration.
HT-29 membranes or membranes from the HT-29A–/– line were prepared as follows. Cells grown in monolayers on a T25 flask were washed with PBS and the supernatant decanted. After addition of 4 ml fresh PBS, cells were recovered by scraping and the suspension centrifuged for 5 minutes at 4,200 g. Cell pellets were resuspended in 1 ml of ice-cold lysis buffer (5 mM PO4, pH 8, 0.5 mg/ml DNase I, containing Pierce Protease Inhibitor; Thermo Fisher, 88665) and transferred to a microfuge tube spun for 5 minutes at 3,400 g. Supernatant was removed and replaced with 1 ml fresh lysis buffer. Cells were then incubated on ice for 30 minutes, flash-frozen in an ethanol-dry ice bath, and thawed at 37°C. After repeating the freeze-thaw cycles twice, the extract was passed through a 26-gauge needle 4 to 5 times to sheer DNA and spun at 25,000 g for 10 minutes at 4°C. The resulting translucent membrane pellets were then resuspended in PBS.
Erythrocyte ghosts or HT-29 membrane preparations were incubated with ANL-labeled bacterial proteins at 4°C overnight, pelleted by centrifugation, washed in 1 ml of ice-cold PBS, then resuspended and boiled in click buffer (50 mM HEPES, 0.5% [w/v] SDS, pH 7.5). ANL-containing proteins were labeled for 1 hour at 37°C in the dark with 5 μM tetramethylrhodamine-alkyne (TAMRA-alkyne) (Molecular Probes) in the presence of 25 mM ascorbate, 1.5 mM CuSO4, and 2.5 mM Tris (3-hydroxypropyltriazolylmethyl)amine. Labeled proteins were separated by SDS-PAGE in the dark and the gel scanned on a Typhoon FLA 9500 phosphorimager (GE Healthcare Life Sciences) using excitation at 532 nm and a 575 nm long-pass filter. Positive control TAMRA-labeled rEtpA was produced by reacting EtpA with 120 μM azidobutyric acid NHS ester (Lumiprobe). The unreacted label was then removed by dialysis, and the azide-labeled EtpA reacted with TAMRA-alkyne, as before.
Bio-layer interferometry.
Bio-layer interferometry was used to determine affinity of EtpA–blood group binding using Octet Red96 (Pall ForteBio Corp.). Briefly, biotinylated blood group A antigens conjugated to PAA (either as the disaccharide GalNAcα1-3Galβ-PAA-biotin; GlycoTech, catalog 01-017; or the trisaccharide GalNAcα1-3 [Fucα1,2]Galβ-PAA-biotin, GlycoTech 01-032) or related target sugars (α-d-Gal-PAA-biotin, GlycoTech 01-003; GalNAcα1,3GalNAcβ-PAA-biotin, GlycoTech 01-026; αGalNAc-PAA-biotin, GlycoTech 08-010) were immobilized onto the SA biosensors (18-5019; Pall ForteBio Corp.). These were then incubated with 2-fold serial dilutions (2000–500 nM) of purified rEtpA or control BSA (2000 nM) prepared in 1× PBS as analyte. Affinity constant (kD) values were calculated using Octet software version 8.1.
Growth and differentiation of human enteroids from different blood groups.
Enteroid experiments were conducted at either John Hopkins University School of Medicine or at Washington University School of Medicine, and detailed experimental protocols for both institutions are included in the Supplemental Methods. In brief, biopsy samples were obtained from adult patients undergoing routine endoscopy at each institution. Small intestinal epithelial (enteroid) cell lines were then generated from dissociated crypts as previously described (85–87), stored, and then accessed from the Washington University Digestive Diseases Research Core Center Biospecimens Core or the respective aligned program at John Hopkins University School of Medicine. Purified cell lines of each of the described ABO types were thawed and resuspended in Matrigel (BD Biosciences) and then propagated at 37°C, 5% CO2 with advanced DEM/F12 media (Invitrogen) supplemented with Wnt3a, R-spondin 1, and Noggin (WRN). Protocols for cellular differentiation and growth of polarized epithelial monolayers were performed as described in detail in the Supplemental Methods. Blood typing by PCR and immunofluorescence microscopy was performed as previously described.
α1-3-N-acetylgalactosaminyltransferase–deficient cell line.
Cells lacking α1-3-N-acetylgalactosaminyltransferase (GI 55773627; GenBank D82843.2) were constructed from HT-29 cells (ATCC HTB-38) by CRISPR/Cas9 genome editing in the Genome Engineering and iPSC Center at Washington University (http://geic.wustl.edu) as previously reported (45). Both parental HT-29 cells and the resulting HT-29A–/– line were propagated in McCoy’s-5A medium (Gibco, Life Technologies) supplemented with 10% FBS.
ETEC adhesion and toxin delivery assays.
To examine delivery of the LT toxin, cultures of HT-29 or HT-29A–/– were differentiated by growing cultures at confluence for 1 week prior to use in 96-well plates. Where indicated, phosphodiesterase (PDE) inhibitors vardenafil HCL·3H20 (MilliporeSigma, Y0001647), cilostazol (MilliporeSigma, PHR1503), and rolipram (MilliporeSigma, R6520) were added to cell cultures 1 hour prior to infections at a final concentration of 6.125 μM (88). ETEC (H10407) or the etpA mutant (jf1668) was grown overnight in 1 ml of LB media. The following morning, bacteria were diluted 1:100 for 90 minutes to mid-log growth phase. HT-29 and HT-29A–/– were infected with bacteria at an MOI of approximately 100. MOI was determined by plating serial dilutions of bacterial suspensions and counting HT-29 cells on a hemocytometer after trypsinization of adherent cells. Infection of HT-29 cells by H10407 or the etpA mutant proceeded for 2 hours at 37°C and 5% CO2 in media containing PDE inhibitors. After 2 hours, the cells were washed once in prewarmed media and the infection continued for an additional hour in media with PDE inhibitors. Cellular cAMP (Arbor Assays) levels were used to assess the efficiency of LT delivery.
Adhesion assays were performed as previously described using mid-log phase bacterial cultures. After 30 minutes, the monolayers were washed 3 times with prewarmed media, then treated with 0.1% Triton X-100 in PBS for 5 minutes. Ten-fold dilutions of the Triton X-100 lysates were plated on Luria agar, and bacterial adherence was calculated as the percentage of organisms recovered per colony-forming unit of inoculum. Adherence assays for enteroids were performed in the same fashion, but washed at different time points, as indicated in the figure legends.
Confocal laser scanning microscopy.
HT-29 cells were seeded onto glass coverslips pretreated with poly-l-lysine in 24-well tissue culture plates. Enteroids grown on Transwell filters were processed by removing the filter from the well and then treated as below. Cells were blocked with 1% BSA in PBS and incubated with biotinylated EtpA (50 μg/ml final concentration) for 2 hours at 37°C or overnight at 4°C. After washing with PBS 3 times, EtpA was detected using Qdot 605 SA conjugate (Life Technologies). A blood group antigen was detected using mouse monoclonal antibody (IgM; Santa Cruz Biotechnology Inc., sc-69951) against human blood group A, followed by Alexa Fluor 647–conjugated goat anti-mouse IgM heavy chain (Molecular Probes, A21238). CellMask deep red plasma membrane stain (Thermo Fisher Scientific, C10046) (1:2,000) and DAPI (1:6,000) were used to stain cellular membranes and nuclei, respectively. ETEC H10407 (serotype O78:H11) was visualized with polyclonal rabbit antibodies against O78 obtained from the Penn State University E. coli Reference Center, followed by detection with goat anti-rabbit IgG (H&L) cross-absorbed Alexa Fluor (488 or 594) secondary antibody conjugates (Invitrogen, A11070, A11072).
Confocal microscopy images were captured using a Zeiss LSM 510 Meta Confocal Laser Scanning Microscope. Image files were saved in LSM format and processed using Fiji (89), Image J v 2.0.0 (NIH), and Volocity 3D analysis software (version 6.3, PerkinElmer).
Blood group typing.
Coded, deidentified samples forwarded to Washington University were used in a blinded fashion to type for the major ABO blood group antigens. Plasma or serum was used in reverse immunotyping for antibodies directed at the ABO antigens, which was performed using standard methods. Briefly 2 μl RBCs (Immucor) from each blood group were incubated with 4 μl of patient sera or plasma at RT for 5 minutes in a 96-well plate, then examined microscopically for agglutination. For available saliva, DNA was extracted from 50 μl of sample (QIAGEN 69504), and genotyping by PCR was performed as previously described (90). Salivary genotyping results were confirmed by dot immunoblotting. Briefly, 0.5 μl saliva diluted 1:4 in PBS was dotted onto nitrocellulose membranes that were then incubated in a solution of H2O2 (3%) for 5 minutes. Membranes were then incubated with 1:20 dilutions of anti-A, anti-B, or anti-H antibodies (MilliporeSigma) for 30 minutes, washed 3 times in PBS with 0.05% Tween, and probed with goat anti-mouse IgM HRP-conjugate (1:5,000 dilution; Santa Cruz Biotechnology Inc., sc-2064) and developed using the ECL Western Blotting Substrate (Thermo Fisher).
ABO determinations were then matched to diarrheal outcomes in which the severity of illness was dichotomized into none (no or mild disease) and MSD using established criteria (71).
Statistics.
We used log-rank (Mantel-Cox) testing to compare time-to-treatment outcomes, with P < 0.05 considered significant. Meta-analysis (pooled fixed effects) was used to compare the relative risk of moderated to severe diarrheal illness. Pearson χ2 testing was used to compare diarrheal illness and severity by blood group. Mann-Whitney U testing was used to compare nonparametric data. More than 2 groups of data were analyzed using Kruskal-Wallis testing for multiple comparisons of nonparametric data.
Study approval.
Institutional Review Board approval was obtained at each participating study site (Washington University, the Naval Medical Research Center, and Johns Hopkins University) prior to sample interrogation and analysis. Written, informed consent was received from all participants prior to inclusion in the studies.
Author contributions
JMF, MAC, and MD were involved in study design, data curation, manuscript preparation, and project administration. TJV, AP, and PK conducted cloning of MetRS and ANL-labeling experiments. SC, ALB, DAS, BD, CDH, RLG, CKP, MGP, and MSR designed and conducted ETEC controlled human infection model studies in volunteers and processed the data and samples. WSW and JCG conducted glycan-array experiments. PK FMK, JFA, and BT were involved in protein interaction, hemagglutination, microscopy, adhesion, and toxin delivery studies. AS and MK conducted flow cytometry experiments. MAC, SS, JFA, MD, MK, and BT acquired, maintained, and propagated small intestinal stem cell banks or intestinal epithelial cells. PK and TJV were responsible for recombinant protein production.
Supplementary Material
Acknowledgments
The authors wish to thank Margaret A. Olsen (Center for Administrative Data Research at Washington University) for her expert help and advice in analysis of clinical metadata associated with these studies, and David A. Rasko (University of Maryland) and Elizabeth Cebelinski (Minnesota Department of Health) for sending additional ETEC isolates. This work was supported by funding from The Enteric Vaccine Initiative of PATH; The Department of Veterans Affairs (VA) (5I01BX001469-05); NIH grants R01AI89894, R01AI126887, K23 AI130389-01, and P01AI125181; Clinical and Translational Sciences Award UL1TR000448 from the NIH; the Digestive Diseases Research Core Center at Washington University School of Medicine; and grant P30 DK52574 from the National Institute of Diabetes and Digestive and Kidney Diseases. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the PATH, the VA, the National Institute of Allergy and Infectious Diseases (NIAID), the NIH, the Department of the Navy, the Department of Defense, or the US Government. Some of the authors are military service members (RLG, MGP, MSR) or employees of the US government (CKP). This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States government.” Title 17 U.S.C. §101 defines a US government work as a work prepared by a military service member or employee of the US government as part of that person’s official duties.
Version 1. 05/17/2018
In-Press Preview
Version 2. 06/25/2018
Electronic publication
Version 3. 08/01/2018
Print issue publication
Footnotes
Conflict of interest: JMF is listed as the inventor on US patent 12/079,304, involving the EtpA adhesin.
Reference information: J Clin Invest. 2018;128(8):3298–3311.https://doi.org/10.1172/JCI97659.
Contributor Information
Pardeep Kumar, Email: pradeepkumar.19@gmail.com.
F. Matthew Kuhlmann, Email: FKUHLMAN@wustl.edu.
A. Louis Bourgeois, Email: lbourgeois@path.org.
Jennifer Foulke-Abel, Email: jfoulke@jhmi.edu.
Brunda Tumala, Email: tumalabr@wustl.edu.
Tim J. Vickers, Email: tjvickers@wustl.edu.
David A. Sack, Email: dsack@jhsph.edu.
Barbara DeNearing, Email: bdenear1@jhu.edu.
Clayton D. Harro, Email: charro@jhsph.edu.
W. Shea Wright, Email: wsheawright@gmail.com.
Jeffrey C. Gildersleeve, Email: gildersj@mail.nih.gov.
Matthew A. Ciorba, Email: mciorba@wustl.edu.
Srikanth Santhanam, Email: srikanth@wustl.edu.
Chad K. Porter, Email: chad.k.porter2.civ@mail.mil.
Ramiro L. Gutierrez, Email: ramiro.l.gutierrez.mil@mail.mil.
Michael G. Prouty, Email: michael.g.prouty2.mil@mail.mil.
Mark S. Riddle, Email: mark.riddle@usuhs.edu.
Alexander Polino, Email: ajpolino@wustl.edu.
Alaullah Sheikh, Email: asheikh@wustl.edu.
Mark Donowitz, Email: mdonowit@jhmi.edu.
James M. Fleckenstein, Email: jflecken@wustl.edu.
References
- 1.Kotloff KL, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382(9888):209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, et al. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study. Lancet. 2016;388(10051):1291–1301. doi: 10.1016/S0140-6736(16)31529-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm AM. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect Immun. 2007;75(8):3961–3968. doi: 10.1128/IAI.00459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roels TH, Proctor ME, Robinson LC, Hulbert K, Bopp CA, Davis JP. Clinical features of infections due to Escherichia coli producing heat-stable toxin during an outbreak in Wisconsin: a rarely suspected cause of diarrhea in the United States. Clin Infect Dis. 1998;26(4):898–902. doi: 10.1086/513923. [DOI] [PubMed] [Google Scholar]
- 5.Beatty ME, et al. Epidemic diarrhea due to enterotoxigenic Escherichia coli. Clin Infect Dis. 2006;42(3):329–334. doi: 10.1086/499246. [DOI] [PubMed] [Google Scholar]
- 6.Jain S, et al. An outbreak of enterotoxigenic Escherichia coli associated with sushi restaurants in Nevada, 2004. Clin Infect Dis. 2008;47(1):1–7. doi: 10.1086/588666. [DOI] [PubMed] [Google Scholar]
- 7.Medus C, et al. Long-term sentinel surveillance for enterotoxigenic escherichia coli and non-o157 shiga toxin-producing E. coli in Minnesota. Open Forum Infect Dis. 2016;3(1):ofw003. doi: 10.1093/ofid/ofw003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker CL, et al. Global burden of childhood pneumonia and diarrhoea. Lancet. 2013;381(9875):1405–1416. doi: 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MAL-ED Network Investigators The MAL-ED study: a multinational and multidisciplinary approach to understand the relationship between enteric pathogens, malnutrition, gut physiology, physical growth, cognitive development, and immune responses in infants and children up to 2 years of age in resource-poor environments. Clin Infect Dis. 2014;59(Suppl 4):S193–S206. doi: 10.1093/cid/ciu653. [DOI] [PubMed] [Google Scholar]
- 10.Keusch GT, et al. Environmental enteric dysfunction: pathogenesis, diagnosis, and clinical consequences. Clin Infect Dis. 2014;59(Suppl 4):S207–S212. doi: 10.1093/cid/ciu485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerrant RL, DeBoer MD, Moore SR, Scharf RJ, Lima AA. The impoverished gut--a triple burden of diarrhoea, stunting and chronic disease. Nat Rev Gastroenterol Hepatol. 2013;10(4):220–229. doi: 10.1038/nrgastro.2012.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colombara DV, et al. Chronic health consequences of acute enteric infections in the developing world. Am J Gastroenterol Suppl. 2016;3(2):4–11. doi: 10.1038/ajgsup.2016.9. [DOI] [Google Scholar]
- 13.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010;12(2):89–98. doi: 10.1016/j.micinf.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sack RB. The discovery of cholera - like enterotoxins produced by Escherichia coli causing secretory diarrhoea in humans. Indian J Med Res. 2011;133:171–180. [PMC free article] [PubMed] [Google Scholar]
- 15.Sack RB, Gorbach SL, Banwell JG, Jacobs B, Chatterjee BD, Mitra RC. Enterotoxigenic Escherichia coli isolated from patients with severe cholera-like disease. J Infect Dis. 1971;123(4):378–385. doi: 10.1093/infdis/123.4.378. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein RA, Vasil ML, Jones JR, Anderson RA, Barnard T. Clinical cholera caused by enterotoxigenic Escherichia coli. J Clin Microbiol. 1976;3(3):382–384. doi: 10.1128/jcm.3.3.382-384.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vicente AC, et al. Outbreaks of cholera-like diarrhoea caused by enterotoxigenic Escherichia coli in the Brazilian Amazon rainforest. Trans R Soc Trop Med Hyg. 2005;99(9):669–674. doi: 10.1016/j.trstmh.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Dorsey FC, Fischer JF, Fleckenstein JM. Directed delivery of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Cell Microbiol. 2006;8(9):1516–1527. doi: 10.1111/j.1462-5822.2006.00736.x. [DOI] [PubMed] [Google Scholar]
- 19.Kumar P, Luo Q, Vickers TJ, Sheikh A, Lewis WG, Fleckenstein JM. EatA, an immunogenic protective antigen of enterotoxigenic Escherichia coli, degrades intestinal mucin. Infect Immun. 2014;82(2):500–508. doi: 10.1128/IAI.01078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Q, et al. Enterotoxigenic Escherichia coli secretes a highly conserved mucin-degrading metalloprotease to effectively engage intestinal epithelial cells. Infect Immun. 2014;82(2):509–521. doi: 10.1128/IAI.01106-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kansal R, et al. Transcriptional modulation of enterotoxigenic Escherichia coli virulence genes in response to epithelial cell interactions. Infect Immun. 2013;81(1):259–270. doi: 10.1128/IAI.00919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourgeois AL, Wierzba TF, Walker RI. Status of vaccine research and development for enterotoxigenic Escherichia coli. Vaccine. 2016;34(26):2880–2886. doi: 10.1016/j.vaccine.2016.02.076. [DOI] [PubMed] [Google Scholar]
- 23.Svennerholm AM, Lundgren A. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev Vaccines. 2012;11(4):495–507. doi: 10.1586/erv.12.12. [DOI] [PubMed] [Google Scholar]
- 24.Fleckenstein J, Sheikh A, Qadri F. Novel antigens for enterotoxigenic Escherichia coli vaccines. Expert Rev Vaccines. 2014;13(5):631–639. doi: 10.1586/14760584.2014.905745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleckenstein JM, Roy K, Fischer JF, Burkitt M. Identification of a two-partner secretion locus of enterotoxigenic Escherichia coli. Infect Immun. 2006;74(4):2245–2258. doi: 10.1128/IAI.74.4.2245-2258.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy K, Hilliard GM, Hamilton DJ, Luo J, Ostmann MM, Fleckenstein JM. Enterotoxigenic Escherichia coli EtpA mediates adhesion between flagella and host cells. Nature. 2009;457(7229):594–598. doi: 10.1038/nature07568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo Q, Qadri F, Kansal R, Rasko DA, Sheikh A, Fleckenstein JM. Conservation and immunogenicity of novel antigens in diverse isolates of enterotoxigenic Escherichia coli. PLoS Negl Trop Dis. 2015;9(1):e0003446. doi: 10.1371/journal.pntd.0003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahl JW, et al. A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infect Immun. 2011;79(2):950–960. doi: 10.1128/IAI.00932-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Canto F, et al. Distribution of classical and nonclassical virulence genes in enterotoxigenic Escherichia coli isolates from Chilean children and tRNA gene screening for putative insertion sites for genomic islands. J Clin Microbiol. 2011;49(9):3198–3203. doi: 10.1128/JCM.02473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo Q, Vickers TJ, Fleckenstein JM. Immunogenicity and protective efficacy against enterotoxigenic Escherichia coli colonization following intradermal, sublingual, or oral vaccination with EtpA adhesin. Clin Vaccine Immunol. 2016;23(7):628–637. doi: 10.1128/CVI.00248-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy K, Hamilton D, Allen KP, Randolph MP, Fleckenstein JM. The EtpA exoprotein of enterotoxigenic Escherichia coli promotes intestinal colonization and is a protective antigen in an experimental model of murine infection. Infect Immun. 2008;76(5):2106–2112. doi: 10.1128/IAI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy K, Hamilton D, Ostmann MM, Fleckenstein JM. Vaccination with EtpA glycoprotein or flagellin protects against colonization with enterotoxigenic Escherichia coli in a murine model. Vaccine. 2009;27(34):4601–4608. doi: 10.1016/j.vaccine.2009.05.076. [DOI] [PubMed] [Google Scholar]
- 33.Roy K, Hamilton DJ, Fleckenstein JM. Cooperative role of antibodies against heat-labile toxin and the EtpA Adhesin in preventing toxin delivery and intestinal colonization by enterotoxigenic Escherichia coli. Clin Vaccine Immunol. 2012;19(10):1603–1608. doi: 10.1128/CVI.00351-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DuPont HL, et al. Pathogenesis of Escherichia coli diarrhea. N Engl J Med. 1971;285(1):1–9. doi: 10.1056/NEJM197107012850101. [DOI] [PubMed] [Google Scholar]
- 35.Satterwhite TK, Evans DG, DuPont HL, Evans DJ. Role of Escherichia coli colonisation factor antigen in acute diarrhoea. Lancet. 1978;2(8082):181–184. doi: 10.1016/s0140-6736(78)91921-9. [DOI] [PubMed] [Google Scholar]
- 36.Skrede S, et al. Experimental infection of healthy volunteers with enterotoxigenic Escherichia coli wild-type strain TW10598 in a hospital ward. BMC Infect Dis. 2014;14:482. doi: 10.1186/1471-2334-14-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harro C, et al. A combination vaccine consisting of three live attenuated enterotoxigenic Escherichia coli strains expressing a range of colonization factors and heat-labile toxin subunit B is well tolerated and immunogenic in a placebo-controlled double-blind phase I trial in healthy adults. Clin Vaccine Immunol. 2011;18(12):2118–2127. doi: 10.1128/CVI.05342-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tacket CO, et al. Protection by milk immunoglobulin concentrate against oral challenge with enterotoxigenic Escherichia coli. N Engl J Med. 1988;318(19):1240–1243. doi: 10.1056/NEJM198805123181904. [DOI] [PubMed] [Google Scholar]
- 39.Freedman DJ, Tacket CO, Delehanty A, Maneval DR, Nataro J, Crabb JH. Milk immunoglobulin with specific activity against purified colonization factor antigens can protect against oral challenge with enterotoxigenic Escherichia coli. J Infect Dis. 1998;177(3):662–667. doi: 10.1086/514227. [DOI] [PubMed] [Google Scholar]
- 40.Darsley MJ, et al. The oral, live attenuated enterotoxigenic Escherichia coli vaccine ACE527 reduces the incidence and severity of diarrhea in a human challenge model of diarrheal disease. Clin Vaccine Immunol. 2012;19(12):1921–1931. doi: 10.1128/CVI.00364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans DJ, Evans DG. Three characteristics associated with enterotoxigenic Escherichia coli isolated from man. Infect Immun. 1973;8(3):322–328. doi: 10.1128/iai.8.3.322-328.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter CK, et al. A systematic review of experimental infections with enterotoxigenic Escherichia coli (ETEC) Vaccine. 2011;29(35):5869–5885. doi: 10.1016/j.vaccine.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 43.Coster TS, et al. Immune response, ciprofloxacin activity, and gender differences after human experimental challenge by two strains of enterotoxigenic Escherichia coli. Infect Immun. 2007;75(1):252–259. doi: 10.1128/IAI.01131-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262(5141):1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 45.Kuhlmann FM, Santhanam S, Kumar P, Luo Q, Ciorba MA, Fleckenstein JM. Blood group O-dependent cellular responses to cholera toxin: parallel clinical and epidemiological links to severe cholera. Am J Trop Med Hyg. 2016;95(2):440–443. doi: 10.4269/ajtmh.16-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Esko JD, Sharon N. Microbial lectins: hemagglutinins, adhesins, and toxins. In: Varki A, et al. eds. Essentials of Glycobiology. Cold Spring Harbor, NY; Cold Spring Harbor Laboratory Press; 2009:1–13. [PubMed] [Google Scholar]
- 47.Bergstrom KS, Xia L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology. 2013;23(9):1026–1037. doi: 10.1093/glycob/cwt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9(4):265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 50.Locht C, Bertin P, Menozzi FD, Renauld G. The filamentous haemagglutinin, a multifaceted adhesion produced by virulent Bordetella spp. Mol Microbiol. 1993;9(4):653–660. doi: 10.1111/j.1365-2958.1993.tb01725.x. [DOI] [PubMed] [Google Scholar]
- 51.Greco D, et al. A controlled trial of two acellular vaccines and one whole-cell vaccine against pertussis. Progetto Pertosse Working Group. N Engl J Med. 1996;334(6):341–348. doi: 10.1056/NEJM199602083340601. [DOI] [PubMed] [Google Scholar]
- 52.Cooling L. Blood groups in infection and host susceptibility. Clin Microbiol Rev. 2015;28(3):801–870. doi: 10.1128/CMR.00109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy K, Bartels S, Qadri F, Fleckenstein JM. Enterotoxigenic Escherichia coli elicits immune responses to multiple surface proteins. Infect Immun. 2010;78(7):3027–3035. doi: 10.1128/IAI.00264-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans DG, Evans DJ, Tjoa W. Hemagglutination of human group A erythrocytes by enterotoxigenic Escherichia coli isolated from adults with diarrhea: correlation with colonization factor. Infect Immun. 1977;18(2):330–337. doi: 10.1128/iai.18.2.330-337.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartus H, Actor P, Snipes E, Sedlock D, Zajac I. Indications that the erythrocyte receptor involved in enterotoxigenic Escherichia coli attachment is a sialoglycoconjugate. J Clin Microbiol. 1985;21(6):951–954. doi: 10.1128/jcm.21.6.951-954.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahdavi A, et al. Identification of secreted bacterial proteins by noncanonical amino acid tagging. Proc Natl Acad Sci U S A. 2014;111(1):433–438. doi: 10.1073/pnas.1301740111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbe C, et al. Evidence of regio-specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J Biol Chem. 2003;278(47):46337–46348. doi: 10.1074/jbc.M302529200. [DOI] [PubMed] [Google Scholar]
- 58.In JG, Foulke-Abel J, Estes MK, Zachos NC, Kovbasnjuk O, Donowitz M. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol. 2016;13(11):633–642. doi: 10.1038/nrgastro.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 60.Aspholm-Hurtig M, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305(5683):519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- 61.Moonens K, et al. Structural insights into polymorphic ABO glycan binding by Helicobacter pylori. Cell Host Microbe. 2016;19(1):55–66. doi: 10.1016/j.chom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glass RI, et al. Predisposition for cholera of individuals with O blood group. Possible evolutionary significance. Am J Epidemiol. 1985;121(6):791–796. doi: 10.1093/oxfordjournals.aje.a114050. [DOI] [PubMed] [Google Scholar]
- 63.Barua D, Paguio AS. ABO blood groups and cholera. Ann Hum Biol. 1977;4(5):489–492. doi: 10.1080/03014467700002481. [DOI] [PubMed] [Google Scholar]
- 64.Clemens JD, et al. ABO blood groups and cholera: new observations on specificity of risk and modification of vaccine efficacy. J Infect Dis. 1989;159(4):770–773. doi: 10.1093/infdis/159.4.770. [DOI] [PubMed] [Google Scholar]
- 65.Swerdlow DL, et al. Severe life-threatening cholera associated with blood group O in Peru: implications for the Latin American epidemic. J Infect Dis. 1994;170(2):468–472. doi: 10.1093/infdis/170.2.468. [DOI] [PubMed] [Google Scholar]
- 66.Harris JB, et al. Blood group, immunity, and risk of infection with Vibrio cholerae in an area of endemicity. Infect Immun. 2005;73(11):7422–7427. doi: 10.1128/IAI.73.11.7422-7427.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heggelund JE, Burschowsky D, Bjørnestad VA, Hodnik V, Anderluh G, Krengel U. High-resolution crystal structures elucidate the molecular basis of cholera blood group dependence. PLoS Pathog. 2016;12(4):e1005567. doi: 10.1371/journal.ppat.1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holmner A, Askarieh G, Okvist M, Krengel U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol. 2007;371(3):754–764. doi: 10.1016/j.jmb.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 69.van Loon FP, et al. ABO blood groups and the risk of diarrhea due to enterotoxigenic Escherichia coli. J Infect Dis. 1991;163(6):1243–1246. doi: 10.1093/infdis/163.6.1243. [DOI] [PubMed] [Google Scholar]
- 70.Black RE, Levine MM, Clements ML, Hughes T, O’Donnell S. Association between O blood group and occurrence and severity of diarrhoea due to Escherichia coli. Trans R Soc Trop Med Hyg. 1987;81(1):120–123. doi: 10.1016/0035-9203(87)90302-6. [DOI] [PubMed] [Google Scholar]
- 71.Porter CK, et al. An evidenced-based scale of disease severity following human challenge with enteroxigenic Escherichia coli. PLoS ONE. 2016;11(3):e0149358. doi: 10.1371/journal.pone.0149358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kelly RJ, Rouquier S, Giorgi D, Lennon GG, Lowe JB. Sequence and expression of a candidate for the human Secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J Biol Chem. 1995;270(9):4640–4649. doi: 10.1074/jbc.270.9.4640. [DOI] [PubMed] [Google Scholar]
- 73.Harro C, et al. Refinement of a human challenge model for evaluation of enterotoxigenic Escherichia coli vaccines. Clin Vaccine Immunol. 2011;18(10):1719–1727. doi: 10.1128/CVI.05194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheikh A, et al. Highly conserved type 1 pili promote enterotoxigenic E. coli pathogen-host interactions. PLoS Negl Trop Dis. 2017;11(5):e0005586. doi: 10.1371/journal.pntd.0005586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li YF, Poole S, Rasulova F, McVeigh AL, Savarino SJ, Xia D. A receptor-binding site as revealed by the crystal structure of CfaE, the colonization factor antigen I fimbrial adhesin of enterotoxigenic Escherichia coli. J Biol Chem. 2007;282(33):23970–23980. doi: 10.1074/jbc.M700921200. [DOI] [PubMed] [Google Scholar]
- 76.Evans DG, Evans DJ, Clegg S, Pauley JA. Purification and characterization of the CFA/I antigen of enterotoxigenic Escherichia coli. Infect Immun. 1979;25(2):738–748. doi: 10.1128/iai.25.2.738-748.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campbell CT, Zhang Y, Gildersleeve JC. Construction and use of glycan microarrays. Curr Protoc Chem Biol. 2010;2(1):37–53. doi: 10.1002/9780470559277.ch090228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oyelaran O, McShane LM, Dodd L, Gildersleeve JC. Profiling human serum antibodies with a carbohydrate antigen microarray. J Proteome Res. 2009;8(9):4301–4310. doi: 10.1021/pr900515y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manimala JC, Roach TA, Li Z, Gildersleeve JC. High-throughput carbohydrate microarray profiling of 27 antibodies demonstrates widespread specificity problems. Glycobiology. 2007;17(8):17C–23C. doi: 10.1093/glycob/cwm047. [DOI] [PubMed] [Google Scholar]
- 80.Manimala JC, Roach TA, Li Z, Gildersleeve JC. High-throughput carbohydrate microarray analysis of 24 lectins. Angew Chem Int Ed Engl. 2006;45(22):3607–3610. doi: 10.1002/anie.200600591. [DOI] [PubMed] [Google Scholar]
- 81.Fleckenstein JM, Roy K. Purification of recombinant high molecular weight two-partner secretion proteins from Escherichia coli. Nat Protoc. 2009;4(7):1083–1092. doi: 10.1038/nprot.2009.87. [DOI] [PubMed] [Google Scholar]
- 82.Fleckenstein JM, Kopecko DJ, Warren RL, Elsinghorst EA. Molecular characterization of the tia invasion locus from enterotoxigenic Escherichia coli. Infect Immun. 1996;64(6):2256–2265. doi: 10.1128/iai.64.6.2256-2265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schnaitman CA. Protein composition of the cell wall and cytoplasmic membrane of Escherichia coli. J Bacteriol. 1970;104(2):890–901. doi: 10.1128/jb.104.2.890-901.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Steck TL, Kant JA. Preparation of impermeable ghosts and inside-out vesicles from human erythrocyte membranes. Meth Enzymol. 1974;31:172–180. doi: 10.1016/0076-6879(74)31019-1. [DOI] [PubMed] [Google Scholar]
- 85.VanDussen KL, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64(6):911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foulke-Abel J, et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014;239(9):1124–1134. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.In J, et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol. 2016;2(1):48–62.e3. doi: 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Read LT, Hahn RW, Thompson CC, Bauer DL, Norton EB, Clements JD. Simultaneous exposure to Escherichia coli heat-labile and heat-stable enterotoxins increases fluid secretion and alters cyclic nucleotide and cytokine production by intestinal epithelial cells. Infect Immun. 2014;82(12):5308–5316. doi: 10.1128/IAI.02496-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taki T, Kibayashi K. A simple ABO genotyping by PCR using sequence-specific primers with mismatched nucleotides. Leg Med (Tokyo) 2014;16(3):168–172. doi: 10.1016/j.legalmed.2014.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.