

Abstract
TNF is an important mediator in numerous inflammatory diseases, e.g., in inflammatory bowel diseases (IBDs). In IBD, acute increases in TNF production can lead to disease flares. Glucocorticoids (GCs), which are steroids that bind and activate the glucocorticoid receptor (GR), are able to protect animals and humans against acute TNF-induced inflammatory symptoms. Mice with a poor transcriptional response of GR dimer–dependent target genes were studied in a model of TNF-induced lethal inflammation. In contrast to the GRWT/WT mice, these GRdim/dim mice displayed a substantial increase in TNF sensitivity and a lack of protection by the GC dexamethasone (DEX). Unchallenged GRdim/dim mice had a strong IFN-stimulated gene (ISG) signature, along with STAT1 upregulation and phosphorylation. This ISG signature was gut specific and, based on our studies with antibiotics, depended on the gut microbiota. GR dimers directly bound to short DNA sequences in the STAT1 promoter known as inverted repeat negative GRE (IR-nGRE) elements. Poor control of STAT1 in GRdim/dim mice led to failure to repress ISG genes, resulting in excessive necroptosis induction by TNF. Our findings support a critical interplay among gut microbiota, IFNs, necroptosis, and GR in both the basal response to acute inflammatory challenges and pharmacological intervention by GCs.
Keywords: Endocrinology, Immunology
Keywords: Cytokines, Inflammatory bowel disease, Innate immunity

Introduction
The cytokine TNF is of great interest in biomedical research. Low but chronic expression levels of TNF have been shown to be sufficient for the development of diseases that have a large economic and social impact, e.g., rheumatoid arthritis and inflammatory bowel diseases (IBDs), such as Crohn disease. For the treatment of these and other diseases, TNF-inhibiting biological drugs have been shown to be a good option for patients not responding to first-line treatments, for example, with methotrexate (1). Although anti-TNF treatment is often successful, there are a number of problems associated with anti-TNF therapy, such as side effects, nonresponsiveness, and high cost (1). Interestingly, in such chronic diseases, acute exacerbations leading to inflammation flares are observed that may be caused by several molecules, including damage-associated molecular patterns (DAMPS) and cytokines and even an acute increase in TNF production (2). Therefore, understanding the impact, mechanism, and therapeutic inhibition of acute TNF-induced toxicity is essential.
Although there is debate about their underlying mechanism of action, glucocorticoids (GCs) are used in the management of several acute inflammatory conditions. TNF-induced acute lethal inflammation represents an interesting mouse model, in which intestinal epithelial cells (IECs) have been described as the main target cells of TNF (3–5). In mice, at least 2 gut-associated mechanisms contribute to loss of IEC barrier function, which is a crucial step in TNF-induced lethality: (a) loss of mucus in goblet cells and loss of antibacterial granules in Paneth cells and (b) damage and cell death in the IEC layer (5, 6).
GCs protect against TNF-induced lethal shock and TNF-induced intestinal permeability (5). GCs, such as dexamethasone (DEX), bind and activate the glucocorticoid receptor (GR) (7). They are used to treat a variety of inflammatory disorders, such as rheumatoid arthritis and IBDs (8). After binding, GR moves to the nucleus and regulates gene transcription as a monomer or as a homodimer. For a long time, the antiinflammatory effects of GCs were believed to result mainly from tethering (i.e., protein-protein interactions) of monomeric GR to inflammatory transcription factors, such as NF-κB and AP-1. However, more recent research indicates that GR homodimers, which form a DNA-binding transcription factor, are essential for mediating the antiinflammatory properties of GCs in acute inflammatory settings, such as systemic inflammatory response syndrome (SIRS) (9, 10). Most of these data are based on findings in GRdim/dim mice. These mice express a mutant version of GR, carrying a missense point mutation (A458T) that leads to reduced GR dimerization and DNA binding, yet maintains an intact monomer profile (11, 12). Upon injection of synthetic GCs, GRdim/dim mice show significantly reduced expression of GC response element (GRE) responsive target genes (9, 13, 14). The GRdim/dim mutant protein binds much less strongly to the classical type imperfect palindrome GRE (AGAACA(N)3TGTTCT) (12). As described by Surjit et al., GR dimers additionally downregulate several hundreds of genes by interaction with a short inverted repeat negative GRE (IR-nGRE), defined as (CTCC(N)0-2GGAGA) (15). GRdim/dim mice are extremely sensitive to contact hypersensitivity, endotoxemia, sepsis, and TNF-induced SIRS (9, 10). Antiinflammatory pathways induced by GR dimers are hampered in GRdim/dim mice. For example, a crucial role for the GC-inducible and GR dimer–dependent gene Dusp1, encoding MKP1, was described in the TNF model and for the Il1ra gene encoding the IL1 receptor antagonist in the endotoxemia model (9, 10).
Here, we studied the mechanism by which GR dimers control TNF-mediated pathological changes at the level of the IECs. We measured genome-wide mRNA expression profiles of PBS-, TNF-, and DEX-stimulated IECs of GRWT/WT and GRdim/dim mice. In nonstimulated conditions, we found an IEC-specific IFN-stimulated gene (ISG) signature in GRdim/dim mice, likely driven by STAT1 and strongly reduced by antibiotics treatment. Our data suggest a tight interplay among gut microbiota, induction of ISGs, and repression by locally produced GCs and GR dimers. They also explain why GRdim/dim mice are extremely sensitive to SIRS and why exogenous GCs, such as DEX, fail to protect GRdim/dim mice against TNF-induced intestinal permeability and lethality. Among the ISGs induced by TNF, but poorly controlled in GRdim/dim mice, are genes essential in necroptosis. Ripk3, Zbp1, and Mlkl were identified, suggesting that necroptosis is a process promoted by the microbiome and IFNs and controlled by dimeric GR. Therefore, we conclude that under physiological conditions, the microbiota sustain a local GC production in order to prevent a detrimental ISG expression signature induced by the very same microbiota in a GR dimer–dependent way. Failure of this protective loop leads to extreme sensitization for cell death in IECs and TNF-induced gut permeability and subsequent lethality.
Results
Intestinal epithelial GR and GR dimers play an essential role in protection against TNF-induced lethality, gut permeability, and cell death.
We previously showed that GRdim/dim mice display a significant increase in sensitivity toward TNF-induced lethality and that TNF lethality is linked with apparent loss of goblet cells and Paneth cells and permeability of the IECs (5, 9). Hence, we decided to study the role of GR dimers in the intestinal epithelium during TNF-induced SIRS. GRVillinKO mice, which selectively lack a functional GR-coding gene (Nr3c1) in IECs, were used (16). GRVillinKO and control GRfl/fl mice were injected with a single dose of 35 μg TNF, and survival was monitored. GRVillinKO mice were significantly more sensitive compared with the control GRfl/fl mice (Figure 1A). It has been known for a long time that injection of GCs protects against TNF-induced lethal shock (7). Interestingly, DEX could not protect GRVillinKO mice (Figure 1A), strongly indicating that protection by DEX requires GR expression in IECs.
Figure 1. IEC GR and GR dimers play an essential role in protection against TNF-induced lethality, gut permeability, damage, and cell death.

(A) GRfl/fl (n = 14 per group, black) and GRVillinKO mice (n = 7 per group, white) were pretreated with PBS (squares) or with 10 mg/kg DEX (triangles). Thirty minutes later, mice were injected with 35 μg TNF, and lethality was monitored. P values for survival curves were analyzed with a log-rank test (combined data of 2 independent experiments). (B) TNF dose-response curves of GRWT/WT (black) and GRdim/dim (white) mice pretreated with PBS (squares) or with 10 mg/kg DEX (triangles) for 30 minutes (n = 8–10 per group; combined data of 2 independent experiments). (C) LD50 values of TNF are depicted on top of each bar for each group. 95% confidence intervals were calculated for the LD50 of each group. (D–F) GRWT/WT and GRdim/dim mice were injected with PBS or 12.5 μg TNF (n = 10 per group; combined data of 2 independent experiments). Asterisks refer to significant differences compared with PBS control, unless indicated otherwise. (D) Relative permeability, 8 hours after TNF challenge, is based upon systemic appearance of orally gavaged FITC-dextran in plasma samples. Standard H&E (E) and TUNEL staining (F) on ileum samples 8 hours after TNF were scored in order to calculate bowel damage and TUNEL scores, respectively. Notice a perfect appearance of GRWT/WT villi, but villi damage, shortening, cell death, and loss of goblet cells in GRdim/dim as well as TUNEL signals at villi tops and crypts. Scale bars: 50 μm. Bars represent mean ± SEM. P values were calculated using 2-way ANOVA. ****P < 0.0001 ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05.
To investigate the importance of GR dimers in protection against TNF lethality by exogenous GCs, we pretreated GRWT/WT and GRdim/dim mice with 10 mg/kg DEX or PBS, followed by increasing doses of TNF. Survival was monitored (Figure 1B), and an LD50 was defined for all mouse groups (Figure 1C). GRdim/dim mice displayed a higher sensitivity to TNF compared with GRWT/WT mice, with LD50 values of, respectively, 10 μg and 30.5 μg per mouse. In GRWT/WT mice, a single injection of DEX increased the LD50 of TNF over 3 times (from 30.5 μg to 96.8 μg per mouse), but in GRdim/dim mice, DEX had no significant protective effect and increased the LD50 from 10 μg to 11.9 μg per mouse. Our data suggest an essential role for IEC GR and, in particular, an optimal dimerization function in resistance against TNF-induced lethal shock, in both the absence and presence of exogenous GCs.
Next, we investigated whether GR dimers in IECs protect against TNF by reducing intestinal permeability previously shown to be strongly linked with TNF lethality (5). We studied the intestinal permeability and cell death of IECs in the ileum of GRWT/WT and GRdim/dim mice after TNF challenge. In both groups of mice, we used 12.5 μg TNF, which is a lethal dose in GRdim/dim mice, but is not lethal in GRWT/WT mice. In contrast to GRWT/WT mice, GRdim/dim mice displayed significant increased intestinal permeability determined via FITC-dextran leakage into the blood upon oral gavage (Figure 1D) and bowel damage as judged by H&E staining after TNF challenge (Figure 1E). GRdim/dim mice also displayed TNF-induced cell death of IECs, while this was virtually absent in GRWT/WT mice, as quantified by TUNEL stainings. Positive red signals were particularly prominent at the villi tops and less so in the crypts (Figure 1F).
GR dimers are essential for suppressing STAT1 expression and activity specifically in the gut.
In order to investigate why GRdim/dim mice are sensitive to these TNF-induced effects on IECs, we performed an RNA-sequencing (RNA-seq) analysis of IECs of GRWT/WT and GRdim/dim mice 8 hours after injection with PBS or with TNF. In control PBS-injected mice, 460 and 240 genes showed significant up- and downregulation, respectively, in GRdim/dim compared with GRWT/WT IECs (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI96636DS1). Known motif analysis of the differentially expressed (DE) genes by HOMER (http://homer.ucsd.edu/homer/ngs/analyzeRNA.html) revealed a significant enrichment of ISRE, IRF1, and IRF2 motif–containing genes in IECs of GRdim/dim compared with GRWT/WT mice (Figure 2A). For example, upregulation of Ifit1, Stat1, Irf8, and Irf1 in IECs of GRdim/dim mice was found (Figure 2B). Additionally, Supplemental Figure 1 displays a more extensive heat map of ISGs, and Supplemental Table 2 lists all ISGs that are expressed at higher and lower levels in GRdim/dim compared with GRWT/WT IECs. These data were confirmed via quantitative PCR (qPCR) (Figure 2C). Out of 460 genes expressed significantly higher in IECs of GRdim/dim mice, 228 were genes defined as belonging to the Interferome database (17), which is a much higher frequency than expected by chance (hypergeometric test P = 1.61 × 10–66).
Figure 2. ISGs are expressed in the IECs of naive GRdim/dim mice.

(A–C) RNA-seq of IECs of GRWT/WT (black) and GRdim/dim (white) mice (n = 3 per group). (A) HOMER motif analysis of DE genes in GRdim/dim compared with GRWT/WT mice. Motifs with highest rank and their P values and q values are displayed. (B) Heat map of DE genes containing an ISRE and/or IRF-1 element. (C) Confirmation of RNA-seq data with qPCR on independent new samples (n = 3 per group). Ifit1, Irf8, Irf1, and Stat1 mRNA expression are shown as mean ± SEM. P values were calculated using Student’s t test. (D–F) GRWT/WT and GRdim/dim mice received drinking water with or without antibiotics for 3 weeks, after which IEC samples were taken (n = 3 per group). (D) STAT1 and p-STAT1 protein levels were analyzed via Western blot using actin as a loading control. (E) Relative STAT1 and p-STAT1 signal intensities were quantified and normalized to ACTIN and STAT1 levels respectively. P values were calculated using 2-way ANOVA. (F) Stat1 and Ifit1 mRNA expression were determined in IECs of GRfl/fl (black) and GRVillinKO mice (white) via qPCR and are shown as mean ± SEM (n = 5 per group). P values were calculated using Student’s t test. (G) GR recruitment to 2 IR-nGRE sites in the STAT1 promoter (IR-nGRE1 and IR-nGRE2). GRWT/WT (black) and GRdim/dim (white) mice were treated with PBS or 10 mg/kg DEX for 2 hours (n = 5 per group; combined data of 3 independent experiments). ChIP on IEC samples was performed against GR using an H300 antibody. Data were normalized to input for each sample and expressed as fold change of H300 to IgG control. P values were calculated using 2-way ANOVA. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
The induction of ISGs, such as Ifit1, is under the control of the transcription factor complex IFN-stimulated gene factor 3 (ISGF3), which consists of STAT1, STAT2, and IRF9 (18). In agreement with the fact that many of the STAT1-responsive ISG genes are upregulated in GRdim/dim mice, suggesting an active ISGF3 complex, we found that STAT1 protein levels were increased in GRdim/dim mice compared with GRWT/WT mice and that STAT1 was only phosphorylated in the IECs of GRdim/dim mice (Figure 2, D and E). Furthermore, an ISG signature was detected in the IECs of GRVillinKO mice (Figure 2F), suggesting that the ISG signature is mediated by a lack of local (dimeric) GR signaling.
To confirm that the ISG signature in IECs of GRdim/dim mice is also observed at the protein level, we performed a proteome-wide mass spectrometry (liquid chromatography–tandem mass spectrometry [LC-MS/MS]) experiment on ileum samples of GRWT/WT and GRdim/dim mice. Differential protein concentrations with a log-fold change (LFC) of 1.0 and an adjusted P value of 0.05 are listed in Supplemental Figure 2. We found 32 proteins upregulated in GRdim/dim, including 17 known as IFN stimulated (18) (17/32 significant enrichment of IFN proteins, P = 0.003), including STAT1.
A conceivable interpretation of our data is that GR mediates repression of ISGs via the control of STAT1 expression in a GR dimer–dependent way. To repress gene expression in a GR dimer–dependent way, the gene should contain one or more negative or inhibitory GREs in its promoter. By motif analysis, we identified 2 so-called IR-nGRE elements, as previously defined (15), in the promoter of STAT1: IR-nGRE1 with sequence CACCTGGAGA and IR-nGRE2 with sequence CTCCAGGACA at positions –2841 and –757 relative to the transcription start site, respectively. We studied GR binding to these elements via ChIP on ileum tissue extracts (Figure 2G). In the graph, data are represented as the fold change of the GR-specific H300 antibody to the IgG control, thus showing specific recruitment of GR to both IR-nGRE elements in the STAT1 promoter (ratio GR to IgG control). In basal conditions, GR was recruited to the IR-nGRE elements in the STAT1 promoter in both GRWT/WT mice and GRdim/dim mice, as shown by a 2-fold change. Interestingly, GR was substantial more recruited to these elements upon DEX treatment in GRWT/WT mice compared with DEX-treated GRdim/dim mice. These data potentially validate the IR-nGRE elements in the STAT1 promoter as GR dimer–bound elements that might be involved in the GR dimer–dependent repression of Stat1 gene expression.
The IEC-specific ISG signature and corticosterone production in GRdim/dim mice are modulated by the gut microbiome.
Interestingly, the increased expression of Stat1 mRNA observed in GRdim/dim mice was only observed in the intestinal epithelium and not in other organs, such as liver, macrophages, spleen, and adrenal glands (Figure 3A). The gut is a unique organ in which local GCs are produced and the gut microbiota are present (19). Microbiota are known to trigger signaling pathways, leading to the production of IFNs and activation of their signaling cascades (20–22). We hypothesize that, under basal GR WT conditions, IFN signaling in IECs is stimulated by the microbiota, but that this IFN response is controlled by local GC production that leads to low but important GR dimer–repressor activity.
Figure 3. The gut microbiota determine the gut-specific ISG signature in GRdim/dim mice and local GC production.

(A) RNA was isolated from ileum, liver, bone marrow–derived macrophages (BMDM), spleen, and adrenal glands dissected from GRWT/WT (black) and GRdim/dim (white) mice. Stat1 mRNA expression was analyzed via qPCR (n = 4 per group). For each organ, different housekeeping genes were used for normalization. Expression in GRWT/WT mice was set as 1. P values were calculated using Student’s t test. (B–E) Effects of commensal bacteria depletion on ISG expression, p-STAT1 levels, TNF-induced lethality, and GC production. GRWT/WT and GRdim/dim mice were treated with antibiotics (+AB) for 3 weeks. IECs were isolated, and Stat1 (B) and Ifit1 (C) mRNA expression were determined via qPCR (n = 8 per group, combined data of 2 independent experiments). P values were calculated using 2-way ANOVA. (D) TNF dose-response curves of GRWT/WT and GRdim/dim mice treated with (triangles) or without (squares) antibiotics (n = 4–6 per dose). (E–G) Effects of antibiotics on GC production. GRWT/WT and GRdim/dim mice were treated with antibiotics for 3 weeks. Cyp11a1 (E) and Cyp11b1 (F) mRNA expression in IECs were determined via qPCR (n = 4 per group). P values were calculated using 2-way ANOVA. (G) GC production in supernatant of ileum explants of GRWT/WT mice treated without (–AB) or with antibiotics (+AB) (n = 5 per group). P values were calculated by Student’s t test. All bars represent mean ± SEM. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
To investigate this hypothesis, we studied the effect of the microbiome on basal gene expression in the IECs of GRWT/WT and GRdim/dim mice. Both groups of mice were treated with an antibiotic cocktail (+AB) for 3 weeks to deplete the bacteria, which was confirmed by plating out stool samples. In both groups of mice, antibiotic treatment reduced Stat1 and Ifit1 expression in IECs (Figure 3, B and C). Following mRNA expression, total STAT1 protein levels were reduced in both groups of mice after antibiotic treatment. In addition, a downward trend in the levels of phosphorylated STAT1 (p-STAT1) was observed in GRdim/dim mice only (P = 0.07), as there are no p-STAT1 levels measurable in GRWT/WT mice (Figure 2, D and E). Interestingly, in GRdim/dim mice, the levels of Stat1 and Ifit1 mRNA after antibiotic treatment remained higher than in untreated GRWT/WT mice, suggesting not only that the triggering of TLRs or IFN-β production by the antibiotic-sensitive microbiota induces the ISG signature in GRdim/dim mice, but also that additional factors, e.g., antibiotic-resistant bacteria, viruses, and fungi, are involved. The reduction in the ISG signature by antibiotics was somewhat reflected in the sensitivity to TNF-induced lethality because antibiotic-treated GRWT/WT and GRdim/dim mice were found to be partly protected (Figure 3D).
Next to adrenal cortical cells, IECs are known to produce GCs (19). Therefore, local stimulation of corticosterone (CS) production by microbiota components could be an additional factor needed to control the ISG signature in the IECs. To address this, we first studied the impact of antibiotics on the expression of the Cyp11a1 and Cyp11b1 genes, which encode critical steroidogenic enzymes involved in CS synthesis, in IECs of GRWT/WT and GRdim/dim mice. In both mouse groups, the expression of these genes was reduced in IECs of mice subjected to antibiotics, supporting our hypothesis (Figure 3, E and F). Interestingly, the expression of Cyp11a1 and Cyp11b1 genes was higher in GRdim/dim mice compared with GRWT/WT mice. This correlates with the fact that both genes are known ISG genes and are found in the Interferome database (17). Next, the local production of CS in IECs in the absence of microbiota was determined. Interestingly, ex vivo CS production by ileum explants was significantly reduced by antibiotic treatment in mice (Figure 3G). Serum CS levels, which are higher in GRdim/dim than GRWT/WT mice (23), were also reduced by about half by antibiotics (data not shown). Together, these data support our hypothesis that microbiota stimulate local CS production and activate GR dimers, which are essential in controlling the microbiota-stimulated ISG signature in the gut.
STAT1 and its responsive genes are master regulators of sensitivity to TNF.
STAT1-deficient (STAT1–/–) mice exhibit strong IFN-signaling defects and do not display any of the physiological responses associated with IFNs (24–26). In contrast to STAT1+/+ mice, full STAT1–/– mice fail to express Ifit1, Ifit2, or other ISGs (including Cyp11a1 and Cyp11b1) in IECs in unstimulated conditions (Figure 4A). STAT1–/– mice were found to resist a lethal dose of TNF (Figure 4B) as well as TNF-induced intestinal permeability or cell death (Figure 4, C and D). These data support the idea that STAT1 is a crucial regulator of TNF-induced intestinal permeability, IEC cell death, and subsequent lethality. Unfortunately, homozygous STAT1–/– GRdim/dim double-mutant mice were not viable. To investigate whether the upregulated and phosphorylated intestinal STAT1 is a master regulator of GRdim/dim hypersensitivity to TNF, we administered the JAK/STAT inhibitor tofacitinib (27). Tofacitinib (100 mg/kg) or vehicle was given orally to GRdim/dim mice. Mice received tofacitinib twice a day for 2 days before challenge with 20 μg TNF and 2 times (1 hour before and 8 hours after) on the day of the challenge. Compared with treatment with vehicle, tofacitinib treatment significantly protected GRdim/dim mice against TNF-induced lethality (Figure 4E).
Figure 4. STAT1 is a master regulator of TNF sensitivity.

(A) RNA was isolated from IECs of STAT1+/+ (black bars) and STAT1–/– mice (white bars). Ifit1 and Ifit2 mRNA expression levels were determined via qPCR (n = 4 per group). P values were calculated by Student’s t test. (B–D) STAT1 mediates TNF-induced lethality, intestinal permeability, and cell death. (B) STAT1+/+ (black) and STAT1–/– (white) mice were injected with 35 μg TNF, and survival was monitored (n = 7 per group). Survival curves were analyzed with a log-rank test. (C) As a measure for intestinal permeability, systemic appearance of orally gavaged FITC-dextran in plasma samples was determined 8 hours after TNF injection in STAT1+/+ mice (PBS, n = 7; TNF, n = 14) and in STAT1–/– mice (PBS, n = 7; TNF, n = 15). (D) TUNEL staining was performed on ileum tissue sections and quantified (combined data of 2 independent experiments). P values were calculated using 2-way ANOVA. (E) STAT1 is an important regulator of GRdim/dim TNF sensitivity. Tofacitinib (100 mg/kg) or vehicle was given orally to GRdim/dim mice (n = 10 per group; combined data of 2 independent experiments). Mice received tofacitinib twice a day for 2 days before challenge (20 μg TNF) and 2 times (1 hour before and 8 hours after) on the day of challenge. Survival was monitored and analyzed with a log-rank test. (F) LFCs of TNF-induced ISGs, detected by RNA-seq, in IECs of GRWT/WT mice (black) or GRdim/dim mice (white) injected with 12.5 μg TNF (n = 4 per group). (G) Ifit1 and Ifit2 mRNA expression, measured by qPCR, in IECs of GRWT/WT and GRdim/dim, 8 hours after PBS or 12.5 μg TNF (n = 4 per group). P values were calculated using 2-way ANOVA. All bars represent mean ± SEM. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
To further explore how the gut ISG signature might be connected to the increased sensitivity of GRdim/dim mice to TNF, we studied gene-expression profiles in IECs of GRWT/WT and GRdim/dim mice challenged with different doses of TNF. Mice were injected with 12.5 μg TNF, which is a sublethal dose in GRWT/WT mice (referred to as WT12.5), but is a lethal dose in GRdim/dim mice (DIM12.5). GRWT/WT mice were also challenged with a lethal dose (50 μg) of TNF (WT50). A comparison between the response to different doses of TNF is discussed in Supplemental Figure 3. Ingenuity Pathway Analysis (IPA) revealed that the major pathway activated by lethal doses of TNF in GRWT/WT and GRdim/dim mice is the IFN-signaling pathway. When focusing on all 2,029 genes that are significantly induced by TNF (across the 3 groups), 736 of these genes were identified as ISGs by the Interferome database (17). In both GRWT/WT and GRdim/dim mice, a dose of 12.5 μg TNF induced an almost identical set of ISGs, but TNF induction of these genes was significantly higher in GRdim/dim than in GRWT/WT mice (Figure 4F, log y axis). This is also illustrated by the expression pattern of Ifit1 and Ifit2 (Figure 4G). Together, these results suggest a poorly controllable ISG induction in GRdim/dim mice upon TNF challenge. Based on the data with STAT1-deficient mice (Figure 4C) and our previous work using IFN- and IFN-receptor-1–deficient mice (3), the insufficient control of ISG induction in GRdim/dim mice is likely to form the basis of their hypersensitivity to TNF-induced lethality.
Lack of protective effects of exogenous GCs against TNF-induced changes in the gut of GRdim/dim mice.
To study the effect of exogenous GCs on TNF-induced intestinal permeability, we pretreated mice with vehicle or DEX (minus 30 minutes) and challenged GRWT/WT and GRdim/dim mice with a lethal dose of TNF, 50 μg and 12.5 μg, respectively. Eight hours after TNF challenge, the TNF-induced intestinal permeability was significantly decreased by DEX in GRWT/WT mice, but not in GRdim/dim mice (Figure 5A). To determine the damage of the intestine, ileum was sectioned 2 hours and 8 hours after TNF challenge and damage was quantified visually using the validated protocol (28), including scores for villus length, villus erosion, cell death, and loss of goblet cells (Figure 5B). TNF caused massive damage to the gut in all samples. DEX was able to protect partially against the TNF-induced damage to the villi in GRWT/WT mice, but not in GRdim/dim mice. TUNEL staining on ileum sections 8 hours after TNF challenge revealed more TUNEL activity induced by TNF in GRdim/dim compared with GRWT/WT mice. Moreover, DEX had a significant protective effect in GRWT/WT mice, but not in GRdim/dim mice (Figure 5C).
Figure 5. Lack of protective effects of DEX against TNF-induced changes in the gut of GRdim/dim mice.

(A–C) GRWT/WT (black) and GRdim/dim (white) mice were pretreated with vehicle or 10 mg/kg DEX for 30 minutes, followed by challenge with 50 μg or 12.5 μg TNF, respectively. (A) Eight hours after TNF challenge, systemic concentrations of FITC-dextran in plasma were determined after oral gavage and reflect relative intestinal permeability (n = 12–20 per group; combined data of 3 independent experiments). (B) Ileum was sampled 2 hours and 8 hours after TNF injection (n = 5 per group), then stained with H&E and bowel damage scores determined. (C) Sections were stained for TUNEL and quantified. All bars represent mean ± SEM. P values were calculated using 2-way ANOVA. (D–F) RNA-seq results of IECs of GRWT/WT and GRdim/dim mice pretreated with 10 mg/kg DEX or PBS, followed 30 minutes later by a lethal dose of TNF (50 μg for GRWT/WT and 12.5 μg for GRdim/dim mice) or PBS. (D) Overview of the experimental protocol. For all groups, n = 3 per group. (E) Venn diagram of DE genes upon DEX treatment compared with PBS-stimulated conditions. (F) Overview of the most significant transcription factor–binding motifs found enriched in the DEX-induced (left panel) and DEX-reduced (right panel) genes in GRWT/WT mice, using HOMER. Motifs with the highest rank and their P values and q values. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
GR dimer–specific repression of ISGs, including necroptosis master switches Ripk3, Zbp1, and Mlkl.
To identify the biological pathway behind the observed GR dimer–dependent protective effects in Figure 5, A–C, we studied the effects of DEX or PBS pretreatment (minus 30 minutes) on gene expression by RNA-seq of GRWT/WT and GRdim/dim IECs 8 hours after TNF or PBS. An overview of the numbers of DE genes (up and down) under DEX conditions, relative to noninduced (PBS injected) levels is provided in Figure 5E. No specifically enriched motifs were found by HOMER in the GRdim/dim-specific regulated genes. As expected, one of the prominent enriched motifs found in the family of GRWT/WT-specific DEX-induced genes is the classical GRE. Interestingly, the major motifs found in GRWT/WT-specific DEX downregulated genes are ISRE and IRF motifs (Figure 5F).
A dimer-specific repression of ISGs seems a plausible mechanism underlying the molecular basis of the GR dimer–specific protection of DEX against TNF-induced gut permeability and lethality. To investigate whether DEX represses TNF-induced ISRE genes more prominently in GRWT/WT compared with GRdim/dim mice, we studied Stat1 and Ifit1 gene-expression profiles in both mouse groups (Figure 6, A and B). Although DEX is able to repress TNF-induced expression of these genes, the process is less efficient in GRdim/dim mice compared with GRWT/WT mice. As mentioned before, we found DEX-stimulated GR recruitment to both IR-nGRE elements in the STAT1 promoter upon DEX treatment, but only in GRWT/WT mice (Figure 2G). When evaluating the impact of DEX on transcriptional levels of TNF-induced ISGs specifically in GRWT/WT and GRdim/dim mice, we focused on 736 ISGs (induced by TNF and identified by the Interferome database; ref. 17) and studied the impact of DEX on the log-fold induction of these genes. As shown in Figure 6C, the reduced expression by DEX was found to be stronger in GRWT/WT mice compared with GRdim/dim mice (35% and 28%, respectively), suggesting poorer repression of ISGs by DEX in GRdim/dim mice. Next, we questioned whether DEX was able to significantly repress TNF-induced ISGs in GRWT/WT mice, but not in GRdim/dim mice, and found a list of 93 genes following this pattern (Figure 6D and Supplemental Table 3). Interestingly, this list contains genes coding for proteins important in the induction of necroptotic cell death, namely Ripk3, Zbp1, and Mlkl.
Figure 6. Exogenous GC treatment represses ISG signature in a GR dimer–dependent way.

(A–E) Analysis of the DEX repression on TNF-induced ISGs based on RNA-seq data. GRWT/WT (black) and GRdim/dim (white) mice were pretreated with vehicle or 10 mg/kg DEX for 30 minutes, followed by challenge with 50 μg or 12.5 μg TNF, respectively (n = 3 per group). RNA was isolated and Stat1 (A) and Ifit1 (B) mRNA expression were analyzed via qPCR. (C) 736 ISGs were induced by TNF in GRWT/WT and GRdim/dim mice. The impact (LFC) of DEX pretreatment is depicted as a percentage of reduction. (D) In total, across GRWT/WT and GRdim/dim mice, 2,029 genes were induced by TNF, and 93 genes were only significantly inhibited by DEX in GRWT/WT mice. (E) Average normalized counts determined by RNA-seq for Ripk3, which is one of these 93 genes. All bars represent mean ± SEM, and statistical analysis was with 2-way ANOVA. (F and G) GRdim/dim mice have an increased necroptosis signature in IECs. GRWT/WT (black) and GRdim/dim mice (white) were injected with PBS or 12.5 μg TNF (n = 5 per group). After 2 hours, Ripk3 was detected on ileum samples by IHC (F; white arrows; representative images are shown). (G) GRdim/dim mice were pretreated with vehicle (black) or 250 μg Nec1s (white). Thirty minutes later, mice were challenged with 12.5 μg TNF, and survival was monitored (n = 5 per group). P values for survival curves were analyzed with a log-rank test. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05. (H) General overview of the interaction between the microbiota and local CS production in controlling the ISG signature in IECs. PRR, pattern recognition receptor; TNFR, TNF receptor.
The expression levels of these 3 genes, as determined by RNA-seq, are depicted in Figure 6E (Ripk3) and Supplemental Table 3 (Zbp1 and Mlkl). Based on the data in Figure 6E, it is clear that Ripk3 is significantly more highly expressed in GRdim/dim mice compared with GRWT/WT mice in basal conditions and after stimulation with an equal dose of TNF (12.5 μg/mouse) in both groups. Moreover, expression of Ripk3 mRNA is less repressed by DEX pretreatment in these mice. The function of these genes is strictly linked to necroptotic cell death (29). The induction of RIPK3 protein was measured by IHC on ileum sections (Figure 6F) and appeared more pronounced in the gut of GRdim/dim compared with GRWT/WT mice. The staining was particularly strong in crypts. Supporting a role for necroptosis, the extreme sensitivity for TNF-induced lethal SIRS of GRdim/dim mice was reverted by pretreatment with Nec1s (29), a specific necroptosis inhibitor (Figure 6G). These data combined suggest that necroptosis is a contributing pathway that is more strongly induced by TNF in GRdim/dim mice, potentially because the Ripk3, Zbp1, and Mlkl genes are more strongly induced in these mice.
Discussion
GCs are powerful in protecting against the TNF-induced lethal shock model (7), but the mechanism of protection is not clear. Here, we show that DEX confers protection against TNF in GRWT/WT mice, yet fails to do so in GRdim/dim mice. Both GRdim/dim and GRVillinKO mice exhibit a stronger response to TNF with gut barrier leakiness. DEX failed to provide protection against TNF in both these mutant mice. GRdim/dim mice exhibited more structural damage and cell death of IECs in response to TNF.
RNA-seq analysis on IECs of GRWT/WT and GRdim/dim mice under basal conditions revealed an unexpected and strong IFN signature in GRdim/dim mice. The phosphorylation state of STAT1 in GRdim/dim mice implied that STAT1 is more transcriptionally active. The enhanced expression of Stat1 and ISGs depends on the GR, since the same ISG signature could be seen in IECs of GRVillinKO mice and, importantly, appeared to be IEC specific. Interestingly, the ISG signature in IECs of GRdim/dim mice was confirmed by a proteome-wide MS experiment: 32 proteins were identified as substantially upregulated, including 17 IFN-stimulated proteins, such as STAT1.
The RNA-seq analysis showed that, under basal conditions, 664 genes were DE in GRdim/dim mice; of these, 460 were upregulated and 204 downregulated. This result is in contrast with the genome-wide expression profiling by Frijters et al., in which no DE genes were found in liver samples of GRWT/WT and GRdim/dim mice. Based on their findings, the authors suggested that the GR dim mutation itself does not cause differential gene regulation (13). The discrepancy between their liver and our IEC results might be explained by the fact that local GC production in the gut could lead to GR dimer formation even in the absence of exogenous ligand. It is important to note that recent studies in unchallenged mice suggest that, in the absence of synthetic ligand, the DNA-bound GR almost exclusively resides in its monomer form and that virtually the complete population of GR forms GR dimers upon injection of pharmacological doses of ligand (14). Hence, it is plausible that, in organs where GCs are produced locally, CS levels rise to a level sufficient to support GR dimerization, leading to transactivation or repression of particular GR dimer–sensitive target genes. This mechanism may play a role in the IECs, as these are known to produce CS (19).
As an additional explanation for the gut specificity of this ISG signature, the role of the gut microbiota was investigated. Elimination of this factor by antibiotics strongly (but not completely) reduced expression of Stat1 and other ISGs and led to some, albeit incomplete, protection against TNF-induced lethality in GRdim/dim mice. These results suggest a link between ISG signature and TNF sensitivity, which is in line with earlier data using IFN receptor–KO mice (3). The fact that antibiotics only partly protect against TNF and do not completely revert the GRdim/dim sensitivity for TNF suggests that either the elimination of gut flora is incomplete, e.g., leaving certain resistant bacteria, viruses, and fungi intact, or that the contribution of ISGs is only partial. STAT1, however, is a central player in TNF-induced lethal SIRS as well as gut permeability, as we show using STAT1–/– mice. Unfortunately GRdim/dimSTAT1–/– mice appeared not viable due to an unknown cause. Tofacitinib, a JAK/STAT1 inhibitor, like antibiotics, led to a marked yet incomplete reversal of the TNF sensitivity of GRdim/dim mice.
Our data suggest that endogenous GCs are (a) induced in a gut flora–dependent way as a result of the ISG signature, which includes the steroidogenic Cyp11a1 and Cyp11b1 genes, but (b) also repress ISGs via GR dimers. Since STAT1 is a central core node regulator of ISGs, it is conceivable that GR dimers block ISGs at the level of STAT1. Activated STAT1/STAT1 homodimers or STAT1/STAT2 heterodimers translocate to the nucleus, where they participate in the regulation of the expression of ISGs (26). GCs are known to modulate IFN signaling at different levels. First, GCs repress type I IFN production, leading to reduced expression of ISGs (30, 31). Second, GCs inhibit STAT1 activation at therapeutic, but also physiological, levels by repressing its mRNA and protein expression (32). Third, STAT1 phosphorylation and occupancy of ISRE sequences in the DNA are actively inhibited by GC treatment (33–35). Inhibition of the STAT1 system, including expression, phosphorylation, and occupancy of ISREs, might be an important part of the antiinflammatory capacities of GCs (34). However, we found no such evidence in GRdim/dim mice. Fourth, increased Stat1 expression could result from binding of GR dimers to GRE elements with nGRE activity, leading to downregulation of gene expression. Several GRE elements have recently been described in the mouse Stat1 gene (36). Fifth, and of interest here, GR dimers inhibit STAT1 transcription via so-called IR-nGREs (consensus sequence: CTCC(N)0-2GGAGA), first described by Surjit et al., and present in the promoter of many genes, leading to transcriptional repression via binding of GR dimers (15). In silico analysis of the STAT1 promoter predicted 2 IR-nGRE elements (IR-nGRE1 and IR-nGRE2). Although we found no difference in GR occupancy on these elements between naive GRWT/WT and GRdim/dim mice, we clearly found GR recruitment after DEX injection in GRWT/WT mice, but not in GRdim/dim mice, suggesting a dimer-specific interaction. It is also important to note that, although Surjit et al. clearly showed that DEX-induced IR-nGRE–mediated repression was abolished in GRdim/dim mice, it is suggested that GR dimers are formed in a structurally alternative way on these elements (37). Sixth, since the genes that are downregulated by DEX in IECs in a GR dimer–dependent way appear to be strongly enriched in IRF1 and ISRE sequences, GR dimers may bind to IRF1 on the one hand or STAT1, STAT2, or IRF9 on the other hand and form a transcriptionally inhibitory complex.
The genes coding for key enzymes of CS synthesis were found to be upregulated in naive GRdim/dim mice, which is compatible with the fact that they are known in the Interferome database as ISGs (17). Antibiotics downregulate expression of these genes and also downregulate IEC-specific production of CS. These data are in line with the hypothesis that IECs are stimulated by gut flora to produce ISGs, including genes leading to local CS production, which triggers GR dimers to suppress this ISG signature at the level of STAT1. Larsson et al. showed dramatic transcriptional responses to microbiota in all segments of the gut by comparing the gene-expression profile of germ-free and conventionally raised WT mice (38). Similarly to what was shown in other studies (39), Larsson et al. observed a significant microbial induction of genes related to immunity. Moreover, sensing of microbial products by various receptors triggers ISG expression via IFN-β and IFN-α production (22).
IFNs are known to prime cells for subsequent inflammatory triggers (40). By RNA-seq in IECs of GRWT/WT and GRdim/dim mice, we found that TNF induces ISGs and that an equal dose in both groups of mice led to higher LFCs of ISG expression in GRdim/dim mice. IFN signaling and activation of IRF were the 2 most prominent pathways predicted by IPA in GRdim/dim mice injected with a lethal dose of TNF compared with GRWT/WT mice injected with a lethal TNF dose. These data suggest that the increased sensitivity for TNF in GRdim/dim mice is based on an uncontrolled induction of ISGs, not on a priming sensu stricto.
Finally, to understand why GRdim/dim mice are not protected against TNF-induced permeability and lethality by exogenous GCs, we studied the impact of DEX on TNF-induced changes in IECs and performed genome-wide expression profiling via RNA-seq in IECs of DEX- and DEX/TNF-treated GRWT/WT and GRdim/dim mice. DEX inhibits several TNF-induced physiological changes in a dimer-dependent way, namely gut permeability, gut damage, and cell death. The transcription factor–binding motifs found in the genes repressed by DEX in a dimer-specific way were ISRE and IRF elements only. This suggests a poor repression of ISGs in GRdim/dim mice, which was confirmed by the lower impact of DEX on the repression on TNF-induced ISGs in GRdim/dim compared with GRWT/WT mice. Whether GR dimers limit the expression of ISGs through inhibition of STAT1 or at the level of each individual ISG via their IRF or ISRE element is so far unclear. We identified 93 genes induced of which induction by TNF, was prevented by DEX pretreatment in a GRWT/WT-specific way. In this group, we found Ripk3, Zbp1, and Mlkl, which are important mediators of necroptosis, a form of cell death recently associated with SIRS (29, 41). Zbp1, also known as DAI, is a molecule known to be induced by microbial/viral stimuli and is able to lead to necroptosis, in combination with Ripk3, in the absence of Ripk1 (42–44). Mlkl is the major target of the Ripk1/Ripk3 heterodimer or Zbp1/Ripk3 heterodimer, and cleavage of Mlkl eventually leads to necroptotic cell death. Clearly, these molecules behave like ISGs and are more highly expressed in and less repressed by DEX in GRdim/dim mice. Therefore, a more outspoken induction of necroptosis by TNF as well as a lack of repression of cell death by DEX in the IECs of GRdim/dim mice might form an attractive explanation of the phenotypes discussed in this paper.
In conclusion (Figure 6H), we reveal a hitherto unknown physiological pathway in the epithelial cells of the gut involving microbiota-stimulated ISG expression via STAT1. The family of ISGs contains Cyp11a1 and Cyp11b1, which code for essential enzymes involved in local CS production. CS subsequently stimulates GR dimer formation and inhibits Stat1 and other ISG gene expressions. Interestingly, genes coding for proteins involved in cell death also belong to the family of ISGs, namely Ripk3, Zbp1, and Mlkl. Expression of these genes, as well as cell death, is inhibited by CS pretreatment. TNF stimulates STAT1 and ISGs, including these cell death–promoting genes. In the absence of GCs or GR dimers, the induction of ISGs by microbiota and TNF is no longer properly controlled, leading to excessive TNF-induced cell death, permeability, and lethality. Our work suggests that GCs protect against acute TNF-induced inflammation, predominantly in IECs, by a dimer-dependent repression of microbiota- and TNF-induced ISGs, including the master regulator STAT1.
Which microbes are involved in this pathway and whether this is relevant in mammals without coprophagy behavior, or only in mice, are not known. Moreover, since the role of TNF in real human pathologies may be different than in a mouse model of TNF-induced acute inflammation, and given the fact that, based on alternative splicing and alternative start codon use (as elegantly shown by Cidlowski and his team), several different GR proteins with different functions are expressed in different tissues during diseases, the impact of our findings in other mouse models and in human patients will need further investigation (45).
Methods
Mice.
GRdim/dim mice were generated by H.M. Reichardt et al. and kept on an FVB/N background (11). Heterozygous GRdim/WT mice were intercrossed to generate GRWT/WT and GRdim/dim homozygous mutant mice. All offspring were genotyped by PCR on genomic DNA isolated from toe biopsies. GRfl/fl mice (46) were crossed with Villin-Cre transgenic mice (47) and the offspring intercrossed to generate GRVillinKO and GRfl/fl mice, all on a C57BL6/J background. C57BL6/J WT mice and STAT1–/– mice were purchased from JANVIER LABS and the Jackson Laboratory, respectively. Mice were kept in individually ventilated cages under a 12-hour dark/12-hour light cycle in a conventional and specific pathogen–free (SPF) animal facility and received food and water ad libitum. All mice were used at 8 to 12 weeks of age.
Reagents.
Recombinant mouse TNF was produced in E. coli and purified to homogeneity in our laboratories. TNF had a specific activity of 1.2 × 108 IU/mg and no detectable endotoxin contamination. DEX (D-4902) and FD4 FITC-dextran (FD4-1G, Mw 3,000–5,000 Da) were purchased from Sigma-Aldrich NV. Recombinant TNF, DEX, and FITC-dextran were diluted in pyrogen-free PBS. Tofacitinib (CP-690550) citrate (S5001; Shelleckchem Inc.) was diluted in 2% Tween 80 with 0.5% methylcellulose diluted in PBS. Nec1s, a necroptosis inhibitor, was diluted in DMSO and was used as recently described (48).
Injections and sampling.
All injections were given intraperitoneally except in Nec1s experiments, for which intravenous injections were used. Injection volumes were always adapted to the body weight of the mice. In lethality experiments, lethal response was followed until no further deaths occurred. FITC-dextran and tofacitinib were administered via oral gavage. Blood was taken via cardiac puncture after sedation of the mice with a ketamine/xylazine solution (Sigma-Aldrich NV). To obtain mouse serum, samples were allowed to clot overnight at 4°C. The next day, the cloth was removed and samples were centrifuged at 20,000 g for 3 minutes. Serum samples were stored at –20°C. For sampling of different organs and IECs, the mice were killed by cervical dislocation at indicated time points. IEC samples for RNA isolations were prepared as follows: ileum was dissected, extensively flushed with PBS, and incubated with lysis buffer (732 6802; Bio-Rad) supplemented with 2-mercaptoethanol on ice for 5 minutes. Samples were then snap-frozen in liquid nitrogen. IEC samples for Western blot analysis were also snap-frozen. For histology, a piece of ileum was fixed in 4% paraformaldehyde and embedded in paraffin by a standard protocol.
Intestinal permeability.
An in vivo permeability assay was performed using FITC-dextran as described previously (4). Three hours after TNF challenge, 100 μl FITC-dextran (25 mg/ml in PBS) was administered by oral gavage. Five hours later, blood was collected in an EDTA-coated tube and centrifuged at 500 g for 10 minutes. Plasma was collected and fluorescence was measured as follows: λexc/λem = 488/520 nm.
Tissue sections, histology, and immunohistochemistry.
Tissue sections of 4 μm were cut and stained with H&E using standard protocols. Pictures were taken with an Olympus Bx51 Light microscope (×40). Tissue damage was quantified by 4 neutral observers using the necrotizing enterocolitis scoring system published and validated by Halpern et al. (28). The mean values of all estimations were used. Cell death was identified by TUNEL (Fluorescein In Situ Cell Death Detection Kit; Roche) according to the manufacturer’s protocol.
Immunohistochemistry of RIPK3.
For immunochemistry, tissue sections were dewaxed, incubated in Dako Antigen Retrieval Solution (Agilent) at a boiling temperature for 20 minutes in a Pick cell cooking unit, and cooled down for 2.5 hours. Blocking buffer (5% goat serum in PBT, i.e., PBS, 0.1% BSA, and 1% Triton X-100) was added to the slides for 30 minutes at room temperature. Primary antibody against RIPK3 (ADI-905-242-100; Enzo Life Sciences) was diluted 1:200 in PBT and incubated overnight at 4°C. Slides were then incubated with secondary antibody (goat anti-rabbit–labeled E0432, Dako, Agilent; 1:500 in blocking buffer) and streptavidin–Alexa Fluor 568 (S11223, 1:500 in blocking buffer). Counterstaining was done with Hoechst reagent (Sigma-Aldrich NV, 1:1,000 in PBS). Fluorescence microscopy was performed using a Zeiss Axioscan Z.1.
qPCR analysis.
RNA was isolated with the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. RNA concentration was measured with the Nanodrop 1000 (Thermo Fisher Scientific), and 300–1,000 ng RNA was used to prepare cDNA with Superscript II (Invitrogen, Thermo Fisher Scientific). qPCR was performed using the Roche LightCycler 480 System (Applied Biosystems). The best performing housekeeping genes were determined by geNorm (49). Results are given as relative expression values normalized to the geometric mean of the housekeeping genes. Primers used for qPCR are depicted in Table 1.
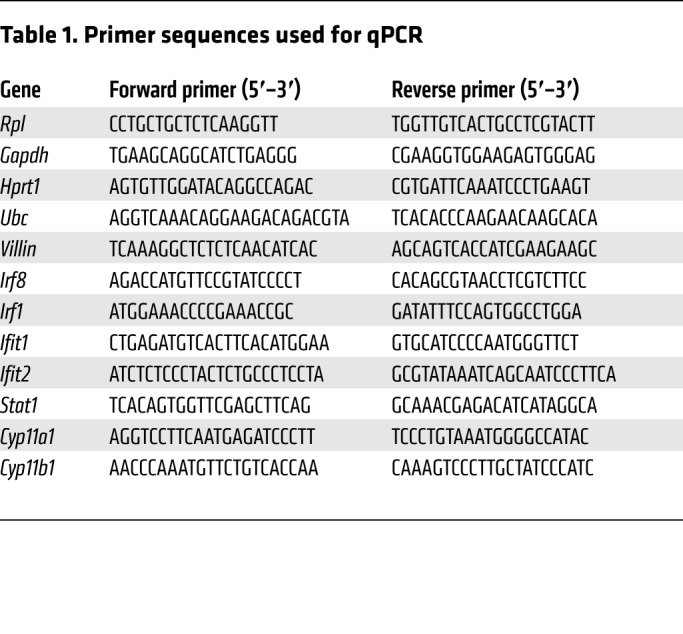
Table 1. Primer sequences used for qPCR.
RNA-seq analysis of IEC samples.
Total RNA was isolated with the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Biological triplicates were used for every condition. RNA concentration was measured, and RNA quality was checked with the Agilent RNA 6000 Pico Kit (Agilent Technologies) and sequenced on a Illumina Genome Analyzer IIx. Data were mapped to the mouse (mm10) reference genome transcriptome with tophat2 (50). Only uniquely mapped reads were retained. Gene-level read counts were obtained with the HTSeq python package. Differential gene expression was assessed with the DESeq2 package, and the FDR was set at the 1% level. Functional analysis of DE gene lists was performed via IPA. Part of the comparison between different TNF conditions was also done using IPA. Motif finding for multiple motifs or de novo motif finding was performed using HOMER software. We used the promoter region (start offset: –500; end offset: default) to search for known motifs and de novo motifs of length 8, 10, 12, 14, and 16. Searching for specific motifs in a set of sequences was done with the meme suite. Visualizations were made using R software. All original microarray data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE113691).
Proteomics sample preparation, LC-MS/MS analysis, and data analysis.
See Supplemental Methods.
Western blot analysis.
For the detection of STAT1 and p-STAT1, protein was isolated out of IECs (lysis buffer with 0.1% SDS, 150 mM NaCl, 50 mM Tris pH 8, 1% NP-40, 0.5% deoxycholate, 0.5 mM EDTA supplemented with protease and phosphatase inhibitor cocktails from Roche). Protein samples containing 50 μg protein were separated by electrophoresis in a 10% gradient SDS polyacrylamide gel and transferred to nitrocellulose membranes (pore size, 0.45 μm). After blocking the membranes with 1:2 dilution of Starting Block/PBST 0.1% (Thermo Fisher Scientific), membranes were incubated overnight at 4°C with primary antibodies against p-STAT1 (1:1,000, catalog 9167S; Cell Signaling), STAT1 (1:1,000, catalog D1K9Y; Cell Signaling), and actin (1:5,000, catalog 691002; Bio-Connect). Blots were washed with PBST 0.1% and then incubated for 1 hour at room temperature with anti-rabbit antibody (1:10,000, catalog 926-32211; LI-COR) and anti-mouse antibody (1:10,000, catalog 926-32220; LI-COR). Immunoreactive bands were detected using the Odyssey Infrared Imager (LI-COR) and quantified using Image Studio Lite Version 4.0.
Isolation of mouse IECs and ChIP.
IECs were isolated as previously described (51). Briefly, whole small intestines were cut into pieces and incubated at 37°C in DMEM containing 5% FCS and 1 mM DTT for 30 minutes. The remaining tissue was incubated in PBS containing 1.5 mM EDTA for an additional 10 minutes. Supernatant was filtered using a 70 μm cell strainer and centrifuged for 5 minutes at 400 g; the cell pellet was resuspended in Opti-MEM (Gibco, Thermo Fisher Scientific). The ChIP assay protocol was based on that of Schmidt et al. (52). Mouse IECs diluted in Opti-MEM were immediately fixed in 1% formaldehyde for 10 minutes at room temperature while gently shaking. The reaction was stopped by adding 125 mM glycine for 10 minutes with shaking. Cells were washed with PBS, and pellets were lysed in lysis buffer supplemented with protease inhibitors (0.1% SDS, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA and 20 mM Tris pH 8). Lysates were sonicated using a Bioruptor instrument (Diagenode) for 30 minutes using 30 seconds on/30 seconds off intervals. Supernatant was taken and used as input and for immunoprecipitation. nProtein sepharose beads (GE Healthcare) were washed in incubation buffer (0.15% SDS, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 20 mM HEPES) and incubated with 1 μg BSA for 2 hours. Samples were first incubated with 5 μg anti-GR H300 antibody (catalog sc-8992; Santa Cruz Biotechnology Inc.) or IgG control (catalog 10500C; Invitrogen, Thermo Fisher Scientific) and then added to the blocked beads overnight. All incubations were performed at 4°C while rotating. The next day, samples were extensively washed using spin columns with 0.35 μm pore size filters (MoBiTec) and eluted with elution buffer (1% SDS and 0.1 M NaHCO3) for 20 minutes. Next, supernatant was incubated with 200 mM NaCl, 20 μg/ml RNase A (QIAGEN) and 1 mg/ml proteinase K (VWR International) for 2 hours at 55°C and for 5 hours at 65°C. DNA was purified with the QIAquick PCR Purification Kit according to the manufacturer’s protocol (QIAGEN). qPCR was performed using the Roche LightCycler 480 System (Applied Biosystems). The following primers for the STAT1 promoter were used: IR-nGRE1 (forward 5′-CCTGCTGCTCTCAAGGTT-3′; reverse 5′-TGGTTGTCACTGCCTCGTACTT-3′) and IR-nGRE2 (forward 5′-TGAAGCAGGCATCTGAGGG-3′; reverse 5′-CGAAGGTGGAAGAGTGGGAG-3′). Normalization of the ChIP data was done based on a method described by Haring et al., as follows (53): for each sample, data with GR-specific H300 antibody or IgG control were normalized to an input sample. This input sample is indicative of the presence and amount of chromatin used in the ChIP reaction. This DNA sample yields a PCR product with all primer sets used.
Antibiotic-mediated depletion of commensal bacteria.
Mice were kept in individual cages and treated with a dilution of a 100 mg/l ciprofloxacin (Sigma-Aldrich NV), 500 mg/l ampicillin (Sigma-Aldrich NV), 500 mg/l metronidazole (Sigma-Aldrich NV), and 250 mg/l vancomycin (Duchefa Biochemie) solution in their drinking water for 3 weeks. After 2 weeks, the presence of microflora was determined by culturing fecal samples in brain-heart infusion (BD) and thioglycollate medium (Sigma-Aldrich NV).
Measuring CS produced by IECs using ileum explants.
Mice were anesthetized with a ketamine/xylazine solution and perfused via the vena cava inferior with PBS supplemented with heparin to remove contamination of peripheral tissues by adrenal-produced CS. After perfusion, ileum samples of equal size were collected and flushed with PBS in order to remove remaining feces. Next, ileum samples were cut longitudinally and into small pieces that were incubated in Opti-MEM at 37°C. Supernatant of ileum explants was collected after 5 hours, and CS levels were determined with the Corticosterone EIA Kit (Arbor Assays) and according to the supplier’s standard protocol.
Statistics.
For dose curve experiments, 95% confidence intervals were calculated for the LD50 of each group by GenStat, 15th edition. Statistics on survival curves were performed using the log-rank test, but a χ2 test was used in order to determine differences between final outcomes. Statistical significance of differences between groups was determined by 2-tailed Student’s t tests, 1-way ANOVA, or 2-way ANOVA tests with 95% confidence intervals. All data, except RNA-seq data, are expressed as mean ± SEM and analyzed using GraphPad Prism software. P < 0.05 was considered significant.
Study approval.
All experiments described in this paper were approved by the ethical committee of the Faculty of Sciences, Ghent University.
Author contributions
MB, KVL, ST, ME, SV, FT, KD, JS, JV, LVW, RDR, NT, FI, and REV performed and guided experimental work. CL supervised the design and performance of the experiments. ST performed the analysis of RNA-seq data and made graphical overviews for the manuscript. ME, KD, and JS provided technical assistance. HMR and JT provided the GRdim/dim and GRfl/fl mice, respectively. RB, PV, FI, and KDB provided other essential tools and useful comments and suggestions. MB and CL wrote the manuscript.
Supplementary Material
Acknowledgments
The authors wish to thank Joke Vanden Berghe, Sara Van Ryckeghem, and the staff of the animal care facilities for animal care. We also want to thank Lien Dejager and Filip Van Hauwermeiren for their help with isolation in intestinal permeability experiments. We acknowledge the VIB Nucleomics Core for RNA-seq analysis and Marnik Vuylsteke for statistical assistance. We are grateful to Kelly Lemeire for performing TUNEL staining and IHC on tissue sections. Research in these laboratories was funded by the Agency for Innovation of Science and Technology in Flanders (IWT), the Research Council of Ghent University (GOA program), the Research Foundation Flanders (FWO Vlaanderen), COST action BM1402, and the Interuniversity Attraction Poles Program of the Belgian Science Policy (IAP-VI-18). Funding was also provided by the DFG–ANR Tu 220/13-1 (to J.T.) and the Collaborative Research Center 1149, C02/ INST 40/492-1 (to J.T.).
Version 1. 05/10/2018
In-Press Preview
Version 2. 06/25/2018
Electronic publication
Version 3. 08/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(8):3265–3279.https://doi.org/10.1172/JCI96636.
Contributor Information
Marlies Ballegeer, Email: marliesballegeer@hotmail.com.
Kelly Van Looveren, Email: Kelly.Vanlooveren@IRC.VIB-UGent.be.
Steven Timmermans, Email: steven.timmermans@dmbr.ugent.be.
Melanie Eggermont, Email: melanie.eggermont@irc.ugent.be.
Sofie Vandevyver, Email: Sofie.Vandevyver@gmail.com.
Fabien Thery, Email: fabien.thery@vib-ugent.be.
Karen Dendoncker, Email: Karen.Dendoncker@IRC.VIB-UGent.be.
Jolien Souffriau, Email: Jolien.Souffriau@IRC.VIB-UGent.be.
Jolien Vandewalle, Email: jolien.vandewalle@irc.ugent.be.
Lise Van Wyngene, Email: Lise.Vanwyngene@irc.vib-ugent.be.
Riet De Rycke, Email: Riet.DeRycke@dmbr.vib-ugent.be.
Nozomi Takahashi, Email: nozomi.takahashi@dmbr.vib-ugent.be.
Peter Vandenabeele, Email: Peter.Vandenabeele@irc.vib-Ugent.be.
Jan Tuckermann, Email: jan.tuckermann@uni-ulm.de.
Holger M. Reichardt, Email: hreichardt@med.uni-goettingen.de.
Francis Impens, Email: francis.impens@vib-ugent.be.
Rudi Beyaert, Email: Rudi.Beyaert@dmbr.vib-UGent.be.
Karolien De Bosscher, Email: karolien.debosscher@vib-ugent.be.
Roosmarijn E. Vandenbroucke, Email: Roosmarijn.Vandenbroucke@irc.VIB-UGent.be.
Claude Libert, Email: claude.libert@irc.vib-ugent.be.
References
- 1.Van Hauwermeiren F, Vandenbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011;22(5-6):311–319. doi: 10.1016/j.cytogfr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Billiet T, Rutgeerts P, Ferrante M, Van Assche G, Vermeire S. Targeting TNF-α for the treatment of inflammatory bowel disease. Expert Opin Biol Ther. 2014;14(1):75–101. doi: 10.1517/14712598.2014.858695. [DOI] [PubMed] [Google Scholar]
- 3.Huys L, et al. Type I interferon drives tumor necrosis factor-induced lethal shock. J Exp Med. 2009;206(9):1873–1882. doi: 10.1084/jem.20090213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Hauwermeiren F, et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J Clin Invest. 2013;123(6):2590–2603. doi: 10.1172/JCI65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Hauwermeiren F, et al. TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol. 2015;8(4):828–840. doi: 10.1038/mi.2014.112. [DOI] [PubMed] [Google Scholar]
- 6.Vereecke L, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. 2010;207(7):1513–1523. doi: 10.1084/jem.20092474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libert C, Van Bladel S, Brouckaert P, Fiers W. The influence of modulating substances on tumor necrosis factor and interleukin-6 levels after injection of murine tumor necrosis factor or lipopolysaccharide in mice. J Immunother. 1991;10(4):227–235. doi: 10.1097/00002371-199108000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Sacta MA, Chinenov Y, Rogatsky I. Glucocorticoid signaling: an update from a genomic perspective. Annu Rev Physiol. 2016;78:155–180. doi: 10.1146/annurev-physiol-021115-105323. [DOI] [PubMed] [Google Scholar]
- 9.Vandevyver S, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. 2012;122(6):2130–2140. doi: 10.1172/JCI60006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleiman A, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2012;26(2):722–729. doi: 10.1096/fj.11-192112. [DOI] [PubMed] [Google Scholar]
- 11.Reichardt HM, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93(4):531–541. doi: 10.1016/S0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 12.Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20(7):876–883. doi: 10.1038/nsmb.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frijters R, et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. 2010;11:359. doi: 10.1186/1471-2164-11-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim HW, et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. 2015;25(6):836–844. doi: 10.1101/gr.188581.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Surjit M, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145(2):224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Reichardt SD, et al. Glucocorticoids enhance intestinal glucose uptake via the dimerized glucocorticoid receptor in enterocytes. Endocrinology. 2012;153(4):1783–1794. doi: 10.1210/en.2011-1747. [DOI] [PubMed] [Google Scholar]
- 17.Rusinova I, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41(Database issue):D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cima I, et al. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med. 2004;200(12):1635–1646. doi: 10.1084/jem.20031958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abt MC, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37(1):158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganal SC, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37(1):171–186. doi: 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Kawashima T, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity. 2013;38(6):1187–1197. doi: 10.1016/j.immuni.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Oitzl MS, Reichardt HM, Joëls M, de Kloet ER. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci USA. 2001;98(22):12790–12795. doi: 10.1073/pnas.231313998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meraz MA, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84(3):431–442. doi: 10.1016/S0092-8674(00)81288-X. [DOI] [PubMed] [Google Scholar]
- 25.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84(3):443–450. doi: 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 26.Akira S. Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells. 1999;17(3):138–146. doi: 10.1002/stem.170138. [DOI] [PubMed] [Google Scholar]
- 27.Cada DJ, Demaris K, Levien TL, Baker DE. Tofacitinib. Hosp Pharm. 2013;48(5):413–424. doi: 10.1310/hpj4805-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halpern MD, et al. Hepatic inflammatory mediators contribute to intestinal damage in necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2003;284(4):G695–G702. doi: 10.1152/ajpgi.00353.2002. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 30.Dupont E, Huygen K, Schandené L, Vandercruys M, Palfliet K, Wybran J. Influence of in vivo immunosuppressive drugs on production of lymphokines. Transplantation. 1985;39(2):143–147. doi: 10.1097/00007890-198502000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Norbiato G, Bevilacqua M, Vago T, Clerici M. Glucocorticoids and interferon-alpha in the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1996;81(7):2601–2606. doi: 10.1210/jcem.81.7.8675584. [DOI] [PubMed] [Google Scholar]
- 32.Hu X, Li WP, Meng C, Ivashkiv LB. Inhibition of IFN-gamma signaling by glucocorticoids. J Immunol. 2003;170(9):4833–4839. doi: 10.4049/jimmunol.170.9.4833. [DOI] [PubMed] [Google Scholar]
- 33.Flammer JR, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol. 2010;30(19):4564–4574. doi: 10.1128/MCB.00146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber S, et al. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut. 2002;51(3):379–385. doi: 10.1136/gut.51.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc Natl Acad Sci U S A. 2011;108(23):9554–9559. doi: 10.1073/pnas.1017296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human glucocorticoid receptor β regulates gluconeogenesis and inflammation in mouse liver. Mol Cell Biol. 2015;36(5):714–730. doi: 10.1128/MCB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. 2013;20(1):53–58. doi: 10.1038/nsmb.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsson E, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61(8):1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10(3):159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 40.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9(4):378–387. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 41.Duprez L, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 42.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016;26(10):721–732. doi: 10.1016/j.tcb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Newton K, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540(7631):129–133. doi: 10.1038/nature20559. [DOI] [PubMed] [Google Scholar]
- 44.Lin J, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540(7631):124–128. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gross KL, Cidlowski JA. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 2008;19(9):331–339. doi: 10.1016/j.tem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tronche F, Kellendonk C, Reichardt HM, Schütz G. Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev. 1998;8(5):532–538. doi: 10.1016/S0959-437X(98)80007-5. [DOI] [PubMed] [Google Scholar]
- 47.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi N, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-beta/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic Enterococcus faecalis. J Immunol. 2005;174(5):2990–2999. doi: 10.4049/jimmunol.174.5.2990. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48(3):240–248. doi: 10.1016/j.ymeth.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haring M, Offermann S, Danker T, Horst I, Peterhansel C, Stam M. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007;3:11. doi: 10.1186/1746-4811-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.