

Abstract
Mutations in superoxide dismutase 1 (SOD1) are responsible for 20% of familial ALS. Given the gain of toxic function in this dominantly inherited disease, lowering SOD1 mRNA and protein is predicted to provide therapeutic benefit. An early generation antisense oligonucleotide (ASO) targeting SOD1 was identified and tested in a phase I human clinical trial, based on modest protection in animal models of SOD1 ALS. Although the clinical trial provided encouraging safety data, the drug was not advanced because there was progress in designing other, more potent ASOs for CNS application. We have developed next-generation SOD1 ASOs that more potently reduce SOD1 mRNA and protein and extend survival by more than 50 days in SOD1G93A rats and by almost 40 days in SOD1G93A mice. We demonstrated that the initial loss of compound muscle action potential in SOD1G93A mice is reversed after a single dose of SOD1 ASO. Furthermore, increases in serum phospho-neurofilament heavy chain levels, a promising biomarker for ALS, are stopped by SOD1 ASO therapy. These results define a highly potent, new SOD1 ASO ready for human clinical trial and suggest that at least some components of muscle response can be reversed by therapy.
Keywords: Neuroscience, Therapeutics
Keywords: ALS, Genetic diseases, Neurodegeneration
Introduction
Amyotrophic lateral sclerosis (ALS) is caused by the loss and dysfunction of neurons in motor pathways and leads to severe weakness, stiffness, and ultimately death in 3–5 years. Approximately 15%–20% of ALS can be associated with a genetic cause. Among the genetically defined ALS cases, about 20% are associated with mutations in the superoxide dismutase 1 (SOD1) gene. Although the exact toxicity of SOD1 mutations remains incompletely understood, there is a consensus that there is a toxic gain of function, and some key data support this idea. First, overexpression of mutant SOD1 in mice or rats recapitulates important aspects of human ALS, including loss of neuromuscular junction innervation and motor neuron death (1, 2). Second, loss of SOD1, while resulting in eventual motor neuron dysfunction, does not result in motor neuron death (3, 4). Third, in humans, individual disease mutations are associated with varying levels of SOD1 activity, yet there is no correlation between disease severity and SOD1 activity (5–7). Given the toxic gain-of-function role of SOD1, lowering levels of SOD1 is predicted to be therapeutic.
One method of lowering SOD1 is to target the SOD1 mRNA using antisense oligonucleotides (ASOs) that bind to the SOD1 mRNA by Watson-Crick base pairing. The ASO activates RNase H1 that destroys the targeted RNA. Direct delivery of ASOs to cerebral spinal fluid (CSF) affords widespread distribution of ASOs throughout the brain and spinal cord in rodents and nonhuman primates (8–13). ASOs for CNS disorders have advanced to clinical trial for Huntington’s disease (registered at ClinicalTrials.gov, NCT02519036), SOD1-associated ALS (NCT02623699), and Alzheimer’s disease (NCT03186989). Most impressively, a splicing-modifying ASO has recently been approved by the FDA (14) for spinal muscular atrophy (SMA) (15).
An SOD1 ASO was used both in the first demonstration of the utility of ASOs in animal models of CNS disease (8, 16) and the first human clinical trial (9). Although there was tremendous enthusiasm for the early SOD1 ASO (ASO 333611), effects in the animal models were modest and advances in ASO technology identified other, more effective ASO designs for CNS disease, such that it became likely that a more potent ASO could be developed. We describe here the recent development of more potent ASOs with substantial prolongation of survival in SOD1 rodent models. The increased efficacy of these ASOs allowed us to test how changes in disease markers may be halted or reversed in those models, a key question for application of this therapy in human clinical trials. Suppression of SOD1 after disease onset reversed the disease course, indicating that intervention in symptomatic patients may not only halt disease but also reverse some aspects of it. The work described here provided the foundation for advancement of this compound to the ongoing human clinical trial in SOD1 ALS patients (NCT02623699).
Results
Next-generation SOD1 ASOs more potently reduce mRNA and protein in rodent models compared with previous ASOs.
Based on an in vitro screen of over 2,000 ASOs targeting the human SOD1 gene and subsequent optimization, 2 particularly potent ASOs targeting the 3′ UTR were selected for further testing (Figure 1A). In a human neuroblastoma cell line (SH-SY5Y), these new ASOs (ASO 1 and ASO 2) potently lowered SOD1 mRNA in a dose-dependent manner (Figure 1B). The new ASOs were more efficacious than the previous SOD1 ASO, 333611 (Figure 1B and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI99081DS1). To determine the potency of these compounds in vivo, ASOs were delivered to the CSF of mice and rats expressing human SOD1G93A via intraventricular (mice) or intrathecal (rats) bolus injection. CSF administration distributes ASOs throughout the brain and spinal cord (8–13). Human SOD1 mRNA was reduced in spinal cords of SOD1G93A mice in a dose-dependent manner (Figure 1C) with approximately 75% maximum reduction of SOD1 following a single injection of ASO 1 or ASO 2. Again, ASO 1 and ASO 2 were substantially more active and efficacious than the previous ASO 333611. Similar dose-dependent reductions in human SOD1 mRNA were achieved in SOD1G93A rats (Figure 1D), with ASO 1 and ASO 2 exhibiting greater activity than 333611 (Figure 1D). Potency of the lead ASO 1 was indistinguishable in SOD1G93A mice and rats, with a lumbar cord EC50 of 0.9 μg/g and 1.4 μg/g tissue, respectively (Figure 1E). ASO 1 and ASO 2 also exhibit prolonged duration of action, with nearly 10 weeks of sustained SOD1 mRNA lowering following a single intrathecal bolus injection in SOD1G93A rats (Figure 1F). Similarly, misfolded SOD1 also exhibits sustained reduction after a single bolus injection (Figure 1G). Consistent with our previous findings, native SOD1 mRNA (Supplemental Figure 1B) and protein (Figure 1H) are also reduced in spinal cord tissue following ASO treatment.
Figure 1. New SOD1 ASOs reduce mRNA and protein in vitro and in vivo, and are more potent than the previous SOD1 lead.

(A) Oligonucleotides containing phosphorothioate backbone modifications (unmodified phosphodiester linkages noted with red o) and 2′-O-methoxyethylribose (MOE; orange) and (S)-2′,4′-constrained 2′-O-ethyl (cEt; blue) groups in the 5′ and 3′ wings were targeted to the 3′ UTR of SOD1 mRNA (ASO 1 and ASO 2), exon 1 of SOD1 mRNA (333611), or nothing in the human or rodent genome (inactive ASO). The sequences evaluated and location of chemical modifications are provided. (B) SHSY5Y cells were treated with SOD1 ASOs by electroporation. After 24 hours, SOD1 mRNA was measured. New SOD1 ASOs are more potent than the previous lead ASO (n = 2 per concentration, average ± range). (C and D) Candidate ASOs for human SOD1 were screened in mice (C) and rats (D) expressing human SOD1. ASOs were injected i.c.v. (mice) and intrathecally (rats) and mRNA was measured in lumbar spinal cord 2 weeks after dosing. Potent ASOs were identified that lowered SOD1 mRNA in both species with ED50 between 50 μg and 70 μg (n = 3 per dose, individual animals). (E) Tissue concentrations of ASO were measured and correlated with SOD1 mRNA lowering, demonstrating an EC50 of 0.9 μg/g in mice and 1.4 μg/g in rats (n = 24). (F) A single intrathecal bolus of 500 μg ASO 1 or ASO 2 was given to SOD1G93A rats. SOD1 mRNA levels were assessed in the spinal cord at 2, 4, 8, and 16 weeks after bolus. ASO 1 and ASO 2 suppressed SOD1 mRNA levels for more than 8 weeks (n = 2–7 per time point, average ± SEM). (G) A single intraventricular bolus of ASO 1 or ASO 2 was given to SOD1G93A rats. Misfolded SOD1 protein from spinal cord was assessed at 1, 2, 4, and 8 weeks after bolus (n = 7 per time point, average ± SEM). (H) Six weeks after a single intraventricular bolus of ASO 1, SOD1 protein was quantified in the lumbar spinal cord of SOD1G93A rats. *** P = 0.0005.
Treatment with SOD1-lowering ASO significantly delays disease onset and extends survival in SOD1G93A mice and rats.
Expression of mutant SOD1 in SOD1G93A rodent models is known to cause severe atrophy of the limbs and trunk that leads to loss of motor function and eventually death (17). To investigate whether a SOD1-lowering ASO strategy could delay disease parameters, SOD1G93A mice were injected intraventricularly with bolus doses of 300 μg ASO 1 at 50 and 94 days of age. Weight and performance on rotarod were tested weekly. Mice that received ASO 1 maintained weight 26 days longer and performed better on rotarod than mice injected with a control inactive ASO (inactive ASO) at similar concentrations (Figure 2, A and B). Median survival for ASO 1–treated mice was 37 days longer compared with inactive ASO, representing a 22% extension of survival in the mouse model (Figure 2C).
Figure 2. SOD1 ASOs prolong onset, survival, and motor performance of SOD1G93A mutant animals.

(A–C) Mice were dosed i.c.v. twice, at day 50 and again at day 94, each time with 300 μg ASO 1 (n = 20 per treatment group, all females). (A) Onset of disease was scored as percentage of animals losing 10% of peak body weight. Median onset for mice treated with the control ASO was 140 days, whereas treatment with ASO 1 increased median onset to 183 days (P < 0.0001, log-rank Mantel-Cox). (B) Motor performance was tested through the rotarod test: mice were tested once a week starting at 80 days of age until they could not stay on the rod for at least 30 seconds. ASO 1 treatment significantly increased rotarod performance: in the control group, the median age at which 50% decrease in performance was reached was 147 days, whereas in the ASO 1–treated animals, the median increased significantly to 182 days (P < 0.0001, 2-way ANOVA). (C) The median survival of mice treated with the control ASO was 168 days, whereas the treatment with ASO 1 increased median survival to 205 days (P < 0.0001, log-rank Mantel-Cox). (D and E) Rats were injected intrathecally with a 1,000 μg single bolus dose of inactive control ASO (n = 16), aCSF vehicle control (n = 19), ASO 333611 (n = 17), ASO 1 (n = 19), or ASO 2 (n = 18). (D) Rats treated with ASO 1 or ASO 2 maintained weight 70 days (P < 0.0001) and 67 days (P < 0.001) longer, respectively, than rats treated with aCSF. 333611 delayed onset of weight loss modestly (median 139 days, compared with aCSF median 121 days). (E) Survival was markedly prolonged in the ASO 1 and ASO 2 treatment groups by 53 days (P < 0.0001) and 64 days (P < 0.0001), respectively, compared with aCSF control treatment in which rats survived to a median age of 166 days. This represents a 32% (ASO 1) and 39% (ASO 2) extension of survival. P values were determined by log-rank (Mantel-Cox) test.
SOD1G93A transgenic rats were injected intrathecally with a single bolus dose of artificial CSF (aCSF) or 1,000 μg ASO vehicle, ASO 333611, ASO 1, ASO 2, or inactive ASO. ASO 333611 is the previous SOD1 ASO as described in reference 8 and used in a phase I study in SOD1 ALS participants (9). Rats assigned to the lead ASO treatment groups performed better in all categories when compared with the vehicle treatment group. SOD1G93A rats treated with ASO 1 or ASO 2 maintained weight 70 days (P < 0.0001) and 67 days (P < 0.001) longer, respectively, than rats treated with aCSF (Figure 2D). In contrast, the intrathecal bolus of ASO 333611 or inactive ASO did not significantly delay onset when compared with the aCSF group (delayed by 17.5 days and 7 days, respectively). Survival was markedly prolonged in the ASO 1 and ASO 2 treatment groups by 53 days (P < 0.0001) and 64 days (P < 0.0001), respectively (Figure 2E), compared with aCSF control treatment in which rats survived to a median age of 166 days. This represents a 32% (ASO 1) and 39% (ASO 2) extension of survival. The effect was similar for both male and female rats (Supplemental Figure 2). This result is substantially improved compared with published results using ASO 333611 infused intraventricularly (10 days, ref. 8) or compared with ASO 333611 delivered intrathecally in this study (195-day median survival, 29-day prolongation) (Figure 2E).
To assess if timing of administration impacts disease onset or survival, SOD1G93A mice were dosed with either ASO 1 or inactive control by i.c.v. bolus at a single time point, either day 80 or 110. Disease onset was delayed and survival markedly extended regardless of the time point of injection (Supplemental Figure 3). There was no significant difference in the onset or survival between the 80-day active ASO treatment and the 110-day active ASO treatment.
SOD1 ASO treatment preserves compound muscle action potential, maintains neuromuscular innervation, and results in lower levels of phospho-neurofilament in SOD1G93A mice.
To test whether SOD1 reduction by ASO could affect markers of disease, SOD1G93A mice were injected intraventricularly once at 5 weeks of age with 100 μg ASO 1 and evaluated for changes in compound muscle action potential (CMAP), neuromuscular junction innervation, and serum phospho-neurofilament heavy chain (pNFH) levels. In SOD1G93A mice, CMAP declines over time, preceding the loss of motor neurons (18, 19). SOD1G93A mice treated with ASO 1 at 5 weeks maintained CMAP over the next 12 weeks, whereas the control-treated animals’ CMAP was reduced by more than half over the same time period (Figure 3A). Consistent with CMAP electrophysiological demonstration of preserved muscle function, ASO 1–treated mice maintained innervation of the tibialis anterior muscles in the hind limbs, whereas control-treated animals showed evidence of denervation of more than 75% of muscle endplates (Figure 3B). pNFH increases in CSF and serum in ALS rodent models, human patients with ALS, and other neurodegenerative diseases and it has been proposed as a potential pharmacodynamics marker (20–22). SOD1 mice treated with ASO 1 showed lower levels of pNFH compared with SOD1 mice treated with control ASO (Figure 3C).
Figure 3. One single i.c.v. injection of SOD1 ASO extensively preserves neuromuscular synapses and neuronal loss in SOD1G93A mutant mice.

(A–C) Mice were injected once at 5 weeks of age with a single dose of 300 μg ASO 1. Compound muscle action potential (CMAP), neuromuscular junctions (NMJ), and phospho-neurofilament heavy chain (pNFH) were measured at multiple time points (n = 12 per group; average ± SEM). (A) Results validated a significant effect of ASO 1 as compared with the inactive ASO (significant at weeks 7–17). CMAP values recorded from ASO 1–treated animals were not significantly different from those recorded from WT animals except at weeks 15 and 17 (P < 0.001 and P < 0.0001, respectively, 2-way ANOVA). (B) ASO 1 significantly protected SOD1 mice from NMJ loss at the tibialis anterior muscle. Although there was a significant difference in NMJ number between WT animals and SOD1 animals injected with a control ASO at all ages analyzed (data not shown), ASO 1–treated mice did not show significant differences compared with WT mice (P < 0.0001). (C) Blood was collected at baseline (5 weeks) before i.c.v. injection, and then again at 8 and 10 weeks of age. pNFH serum levels were significantly decreased at 8 and 10 weeks of age by ASO 1 treatment (P < 0.001, 2-way ANOVA).
Reversal of SOD1-mediated neuronal dysfunction may be achieved by treatment with SOD1 ASO and is measurable by CMAP and pNFH levels.
Two major questions regarding markers of disease in neurodegenerative diseases are: to what extent therapeutics may be able reverse disease and whether this reversal can be measured. SOD1G93A mice were treated at 9 weeks of age when CMAP had clearly begun to decline and pNFH had begun to rise (Figure 4, A and B). Treatment with ASO 3, an SOD1 ASO with dose-dependent reductions in SOD1 mRNA similar to those in ASO 1 and ASO 2 (Supplemental Figure 4), demonstrated a sustained increase in CMAP whereas control ASO–treated mice showed a continued steady decline (Figure 4A). Similarly, pNFH, which at time of injection (9 weeks) had already begun to rise in SOD1G93A mice (Figure 3C), was still significantly lower 8 weeks after treatment with ASO 3 (inactive ASO–treated group, 22 ng/ml ± 1.9 ng/ml; ASO 3–treated group, 12 ng/ml ± 1.2 ng/ml; mean ± SEM, P < 0.001) (Figure 4B). We also found that both pNFH serum and CSF levels were responsive to SOD1 ASO treatment in ALS rats (Supplemental Figure 5). Furthermore, it was recently demonstrated that motor neuron–enriched miR-218 is temporally increased in ALS rat model CSF and its levels correlate with motor neuron loss (23). Here, we found that miR-218 CSF levels were also responsive to SOD1 ASO therapy (Supplemental Figure 5). These results suggest that SOD1-mediated neuronal dysfunction is, at least in part, reversible with a potent therapeutic.
Figure 4. SOD1 ASO injected at 9 weeks of age is able to reverse CMAP amplitudes and lower serum pNFH levels in SOD1G93A mutant mice.

(A and B) Mice were injected once at 9 weeks of age with either a control ASO or with ASO 3 (100 μg). (A) CMAP amplitudes at the tibialis anterior muscles were recorded at baseline (5 weeks) and then every other week thereafter (n = 12 per group, average ± SEM). On the week of dosing (9 weeks), CMAP was recorded prior to i.c.v. injection. Typically, SOD1 mice show a steady decline in the CMAP amplitudes recorded at the tibialis anterior; however, one single dose of ASO 3 at 9 weeks of age was able to reverse the trend and by 15 weeks the ASO 3–treated mice had CMAP amplitudes significantly higher than mice treated with a control ASO (P < 0.001, 2-way ANOVA). (B) Blood was collected from each animal at 9, 11, 13, 15, and 17 weeks of age and pNFH levels were quantified. pNFH serum levels of control mice showed a steady increase whereas those of ASO 3–treated mice did not. Levels at 15 weeks: inactive ASO, 19.9 ng/ml ± 5.1 ng/ml; ASO 3, 10.6 ng/ml ± 2.5 ng/ml. Levels at 17 weeks: inactive ASO, 33.96 ng/ml ± 9.3 ng/ml; ASO 3, 16.5 ng/ml ± 4.3 ng/ml. P < 0.0001, 2-way ANOVA, n = 12 per group, average ± SEM.
SOD1 ASOs lower SOD1 mRNA and protein in nonhuman primates.
To evaluate the distribution and pharmacokinetic properties of the new SOD1 ASO in a larger brain, ASO 1 was delivered via intrathecal injection to cynomolgus monkeys. ASO 1 was optimized for targeting human SOD1, but it is complementary to nonhuman primate (NHP) SOD1 with a 1-base mismatch, and it is active in NHP cells (Supplemental Figure 6). ASO 1 was evaluated in NHPs at doses of 4 mg, 12 mg, and 35 mg delivered intrathecally, with each monkey receiving 5 doses. One week after the final dose, SOD1 mRNA was lowered broadly in the CNS in a dose-dependent manner (Figure 5A). Quantification of drug levels in various tissues confirmed a pharmacokinetic/pharmacodynamic relationship, with an estimated composite EC50 of 20 μg/g tissue (Figure 5B). This is consistent across all tissues assessed (Supplemental Figure 7). The decrease in potency in NHP SOD1 compared with human SOD1 in transgenic rodents (Figure 1E, EC50 0.9–1.4 μg/g) is likely attributable, in part, to the single mismatch of ASO 1 to the NHP SOD1 transcript. SOD1 protein levels in the tissue were lowered by approximately 50% (Figure 5C). A corresponding dose-dependent decrease in CSF SOD1 levels was also observed, confirming CSF SOD1 levels reflect target reduction in the CNS in the larger NHP brain (Figure 5D). CSF SOD1 protein remains reduced approximately 100 days after the last administration (Figure 5E). These data in NHPs demonstrate effectiveness of the SOD1 ASO in a larger animal and are an important step toward understanding the use of the compound in human clinical trials.
Figure 5. SOD1 ASO lowers SOD1 in nonhuman primate CNS and CSF.

(A–C) The safety of the ASO was assessed in cynomolgus monkeys, which also provided an opportunity to understand the on-target activity of the ASO and the utility of SOD1 in the cerebrospinal fluid as a marker of target suppression in the tissue. Monkeys were dosed with artificial CSF or 4, 12, or 35 mg ASO 1, with each monkey receiving 5 doses. One week after the final dose, SOD1 mRNA and protein were quantified and associated with ASO concentration across brain regions. (A) A dose-dependent lowering of mRNA across CNS regions, including the lumbar spinal cord, was observed (n = 6, average ± SEM). L, lumbar; Th, thoracic; Cerv, cervical; Fr, frontal cortex; Motor, motor cortex; Hip, hippocampus; Pons, pons; Cb, cerebellum. (B) A pharmacokinetic/pharmacodynamic relationship could be observed, with an estimated IC50 of 20 μg/g tissue (n = 126, each point representing one tissue). (C) A dose-dependent lowering of SOD1 protein across CNS regions, including the lumbar spinal cord, was observed (n = 6, average ± SEM). aCSF, artificial CSF. (D) A dose-dependent lowering of SOD1 protein in CSF as compared with the vehicle-treated group was observed (n = 6; line, average; 4 mg, P = 0.0019; 12 mg, P = 0.0006; 35 mg, P = 0.0003; 1-way ANOVA). **P < 0.005; ***P < 0.001. (E) CSF SOD1 protein remains significantly reduced as compared with day 0, roughly 100 days following the last intrathecal ASO administration (n = 6, average ± SEM; ****P = 0.0001; 1-way ANOVA).
Discussion
ASOs targeting SOD1 were previously used to demonstrate widespread distribution of ASOs in the CNS following CSF delivery, proof of principle in animal models, and important early safety in a first-in-humans clinical study (8, 9, 24). Benefiting from advancements in ASO technology, here we describe other, more potent ASOs that demonstrate a prolonged survival benefit in both mouse and rat SOD1 models, the longest to date using a relevant therapeutic. With this added ASO potency, we were able to test whether treating after the disease progressed would also provide benefit, and we found a notable reversal of the CMAP. These data establish a novel therapeutic for SOD1-mediated ALS, which is currently under clinical investigation (NCT02623699).
For many genetically linked neurodegenerative diseases, patients remain asymptomatic for more than 5 decades but when they develop signs of disease, the progression is relatively rapid over a period of about 5 years. Understandably, there is concern that once the disease starts, it may be difficult or impossible to slow or reverse the neurodegenerative process. We have attempted to address the importance of timing in 2 approaches. The first was to limit the intervention to a single time point, ranging from early in the mouse’s life to well after some disease processes, such as denervation of the tibialis, have occurred. Using mouse clinical measures such as disease onset or survival, timing was irrelevant to the magnitude of benefit. Potentially of greater interest are the data in Figure 4A showing reversal of a specific, highly quantifiable endpoint in this very rapid model of ALS. CMAP is the summation of the amount of functional innervation to a given muscle. The fact that this measure increases in mice in the ASO treatment setting demonstrates a remarkable ability for the motor system to recover. Concomitant with this, there was decreased denervation at the neuromuscular junction, reinforcing the functional recovery (Figure 4B). We also demonstrated that serum and CSF pNFH levels, which are increased in rodent and human ALS, were decreased by this therapy. This defines, for the first time, serum and CSF pNFH levels as a pharmacodynamic marker for ASO therapy. Perhaps even more importantly, these neurofilament data suggest that any particularly effective therapy in humans may reverse the rise in serum/CSF pNFH. Taken together, these data show reversal of neurodegeneration and define electrophysiologic and serum pharmacodynamic markers that can be directly translated to human clinical studies. miR-218, a motor neuron–specific microRNA, also shows changes in CSF with SOD1 ASO treatment in SOD1G93A rats and may be another outstanding pharmacodynamic marker for motor neuron–targeted therapeutics (23). Measuring miR-218 in human CSF will be a critical next step.
Clinical trials in a rare population such as SOD1-related ALS may be challenging given the relatively small number of patients affected by the disease, but much of the groundwork for such a trial has been established. Natural history studies of SOD1 ALS have defined survival times in this patient group. For people with A4V mutations in SOD1, which represent 50% of the SOD1-associated ALS population in the United States, survival is 1.2 years. If we anticipate a change of 50% in survival in patients, as few as 52 participants would be needed to determine if an ASO changed survival (25). An even more important first step in clinical trials is to determine if SOD1 in the CSF is lowered by the ASO. The NHP CSF data included here demonstrate that an intrathecally administered ASO can reduce CSF target protein concentrations, confirming CSF protein as a translational pharmacodynamic end point. Similar clinical measures of CSF SOD1 protein are crucial to demonstrate drug action and pharmacodynamic activity for ongoing clinical trials (24). Recent SOD1 protein kinetics (26) have determined the half-life of CSF SOD1 in humans to be 25 ± 7.4 days.
One concern for this approach is the safety associated with loss of SOD1 expression. While safety will remain an important consideration in human clinical studies, for SOD1 ALS there are several data points that provide some reassurance. As noted above, extensive evidence supports the primary toxicity of SOD1 mutations as resulting from gaining a novel toxic property. Overexpression of mutant SOD1 in mice or rats mimics important aspects of ALS in humans, including loss of neuromuscular junction innervation and motor neuron death (1, 2). Loss of SOD1 results in motor neuron dysfunction but not motor neuron death (3, 4), and neither loss nor increase of normal SOD1 activity in mouse models of SOD1 ALS alters survival (27). In humans, individual disease mutations are associated with varying levels of SOD1 activity, and some patients with disease-causing mutations even have apparently normal SOD1 activity (7). Lastly, SOD1 ASOs have been found to be safe in humans (9). A clinical trial based on this prior work and the work presented here is already underway in SOD1-related ALS (NCT02623699) using ASO 1.
For many neurodegenerative diseases, the proteins implicated by genetic forms of the disease are also implicated in the pathways or pathology of a broad number of cases. Based on this experience, it was anticipated that SOD1 would be involved broadly in ALS. The data regarding this issue are not yet clear. Using some staining protocols, misfolded SOD1 may be found in pathological samples from non-SOD1 genetic ALS (28). However, other studies have not replicated these findings (29). The simple explanation of using different reagents and samples does not fully explain the discrepancies. Perhaps most intriguing are data showing that lowering SOD1 in human astrocytes or oligodendrocytes decreases the toxicity of these cells to cultured motor neurons (30, 31). Overall, while these data are suggestive that SOD1 may be involved more broadly in ALS than the 1%–2% of the ALS population with genetic SOD1 mutations, the data are not yet strong enough to push forward a SOD1-lowering therapy in all ALS. As further data emerge on SOD1 in sporadic ALS and as we learn more about safety and efficacy of ASOs in the SOD1 genetic population, SOD1 ASOs for ALS without known SOD1 mutations will likely need to be reconsidered.
ASO therapeutics for neurologic diseases are in clinical trial or are anticipated shortly for clinical trial for a number of disorders. Phase III trials for a splicing ASO for spinal muscular atrophy (SMA) were stopped early because of success and led to recent FDA approval of these drugs for clinical use (14). A splicing ASO recently received conditional approval for Duchenne muscular dystrophy (32). ASOs for Huntington’s disease are currently in phase I trial in Europe. ASO lowering of tau showed impressive efficacy in mouse preclinical studies (12) and is in early clinical trials (NCT03186989). ASOs targeting C9ORF72 expansions that cause ALS/FTD showed efficacy in stem cell–derived neurons (33, 34) and mouse models (35) and are likely to be in human clinical trial in the near future. Thus, the approach outlined here for targeting mRNA in the brain and spinal cord with ASOs is increasingly being used for neurological disorders and hopefully will become part of a clinical treatment paradigm, as in SMA.
While we are enthusiastic and encouraged by the demonstration of reversal in these studies, we recognize that these results may or may not directly translate to humans. Subsequent data from human clinical studies compared with the data presented here may be the best opportunity to fully evaluate the translational potential of SOD1 models. There has been much prior criticism of SOD1 models and their failure to predict efficacy in humans, but for the vast majority of these failed clinical studies, 2 important components were often missing: first, a demonstration that the pathway targeted by the drug was involved in the participants being treated; and second, a pharmacodynamic marker to demonstrate that the drug worked as designed in humans. The ASO SOD1 trial in humans with SOD1 ALS is directly on target and the pharmacodynamic marker, SOD1 in the CSF, has been clearly defined (26).
There are other methods to target SOD1 mRNA. Indeed, recent data in animal models using virally driven RNAi showed promising results (36, 37). While there may be some additional issues surrounding delivery of virus, these strategies are plausible. Antibodies that target SOD1 are currently being developed (38–41). In addition, small molecules targeting SOD1 have been considered. Pyrimethamine has been tested in phase I/II trial and showed an approximately 10%–15% lowering of SOD1 in CSF (42). Arimoclomol, a heat shock protein activator, was tested in rapidly progressive ALS. Patients treated with arimoclomol showed a trend toward longer survival (43). It is possible that some of these approaches could be used in conjunction with ASOs. All, including ASOs, require ongoing studies in human SOD1 ALS to determine success.
In conclusion, ASOs targeting SOD1 show extension of survival in SOD1 mice and rat models and reversal of CMAP. These ASOs are also effective in NHPs. Given the growing experience with ASOs in clinical trials, the new SOD1 ASOs are an exciting therapeutic for human SOD1 ALS.
Methods
Antisense oligonucleotides.
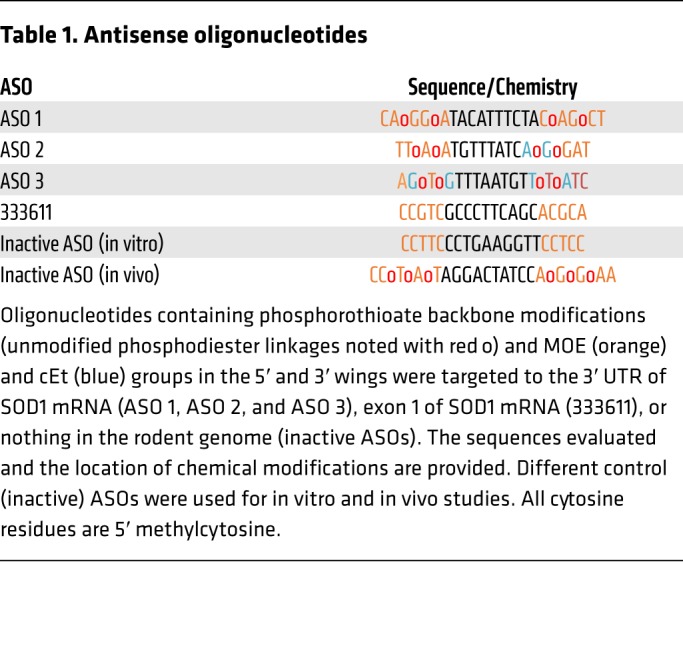
The antisense oligonucleotides used in the present study are described in Table 1 and were manufactured and provided by Ionis Pharmaceuticals.
Table 1. Antisense oligonucleotides.
Intrathecal bolus injection in the rat.
SOD1G93A transgenic rats received a 30-μl intrathecal bolus injection via polyethylene catheter placed between the L3 and L5 vertebrae of the lumber spinal cord, as previously described (44). For proof-of-concept studies, 65-day-old rats were given 1000 μg ASO 1, ASO 2, ASO 3, ASO 333611, inactive ASO, or an artificial cerebrospinal fluid (aCSF) vehicle control. All ASOs were provided by Ionis Pharmaceuticals and diluted in aCSF. All treatment groups were sexual phenotype– and litter-matched to control for drift in copy count or sexual phenotype–induced variances. In addition, researchers were blinded to the identity of the ASO at the time of treatment and throughout the duration of the experiment. For pharmacokinetic and pharmacodynamic studies, adult animals were dosed and tissue was collected 2 weeks after dosing for RNA quantification, unless otherwise noted.
Dosing of mice.
Intracerebroventricular (i.c.v.) infusions were performed as previously described (45). Surgeries were conducted according to Biogen Guidance for Aseptic Rodent Surgery. Animals were anesthetized by isoflurane inhalation. The eyes of the animal were treated with ocular lubricant. The fur of the scalps and anterior backs were clipped and the animal was placed in a stereotaxic apparatus. Buprenorphine was administered subcutaneously prior to incision (1.0 mg/kg). An 11.5-cm, slightly off-center incision was made in the scalp. A 25-gauge needle attached to a Hamilton syringe was placed at bregma level and the needle was then moved to the appropriate anterior/posterior and medial/lateral coordinates (0.2 mm anterior/posterior and 1 mm to the right medial/lateral). The proper amount of injection solution was injected at rates of approximately 1 μl/second for a total of 10 μl. This flow rate has been shown to deliver sufficient compound with consistency and no side effect to the animal. The incision was sutured closed using 1 horizontal mattress stitch with 5O Ethilon suture. Depending on the experiment, mice were injected either once (at day 35) or twice (at day 50 and day 94) in order to obtain sustained mRNA lowering of SOD1.
Motor assessments in the rat.
Prior to surgery, rats were assessed for baseline weight and general motor capabilities. Following surgery, rats were monitored weekly for changes in these values. Weight loss was calculated from the time of surgery to identify disease onset (peak weight) (46). When an animal began to show paralysis in any portion of the body, a HydroGel pack (Clear H2O) was placed on the bottom of the home cage to limit dehydration. Monitoring continued until the rats reached end-stage, defined as loss of the ability to right itself after being placed on its side, or inability to ambulate from a given location. The date of end-stage and subsequent euthanasia was recorded for survival analyses.
Euthanasia and tissue collection.
For proof-of-concept studies, upon reaching end-stage, rats were exsanguinated by transcardial perfusion with heparinized (0.03%) PBS. Sections of the brain and spinal cord were flash frozen in liquid nitrogen for RNA and protein analysis. For pharmacodynamic and pharmacokinetic studies, following in-life completion, animals were euthanized by CO2 asphyxiation as approved by IACUC protocol. For RNA analysis, 2-mm sections of lumbar spinal cord were collected. Adjacent to the RNA sampling, pieces of lumbar cord were also collected for ASO quantification. Each dissected region was then frozen at –80°C until analysis.
Rotarod in mice.
Mice were given 1 day to become familiar with the rotarod apparatus before the test by placing them on a rod slowly rotating at 2 rpm for 5 minutes. To determine motor function deficiency, mice were put on the rod at increasing speeds from 2 rpm to 40 rpm in 180 seconds and at 40 rpm for an additional 60 seconds. Tests were given 2 times in a day. Mice were tested once a week starting at 80 days of age until they could not stay on the rod for at least 30 seconds.
Compound muscle action potential.
CMAP recordings were performed at the indicated time points under isoflurane anesthesia. Disposable monopolar 28-gauge needle electrodes were used for stimulation and recording. The sciatic nerve was stimulated near the sciatic notch with constant-current monophasic square-wave pulses (0.1 millisecond duration, 2-second intervals). For CMAP recordings, the recording electrode was placed intramuscularly 1 mm deep in the tibialis anterior. Stimulation current was increased until a maximum CMAP was obtained, and recordings were performed using a current level 0.5 mA above this value (supramaximal stimulation). The amplitude represents the peak-to-peak amplitude of the biphasic CMAP waveform (averaged over 4 stimuli). For each animal, right and left leg amplitudes were averaged.
NMJ staining and counting.
Tibialis anterior muscle samples were postfixed in 4% paraformaldehyde (PFA) for 15 minutes after collection and then incubated into 20% sucrose for 24 hours for cryoprotection. Samples were shipped to Clarapath Inc. for analysis. All samples were sectioned using the Clarapath Inc. tape-transfer system at 20 μm in a cryostat. Sections were collected directly onto glass slides. Slides were stained with a rabbit antivesicular acetylcholine transporter (VAchT; 1:11,000); the antibody was provided by Target ALS through Covance. The secondary antibody used was Alexa Fluor 488 donkey anti-rabbit (Life Technologies). All slides were counterstained with TMR-α–bungarotoxin (Life Technologies) at a concentration of 1:750. All stained slides were imaged using a ×20 objective (0.75 NA) at a resolution of 0.45 μm/pixel. A single focal plane was collected for all samples. A multiband fluorescence filter set was used to simultaneously image GFP and RFP. Both automated and manual counting methods for TMR-α–bungarotoxin and VAchT were performed.
pNFH quantification.
pNFH serum levels were measured through the ELLA microfluidic ELISA platform (Protein Simple) according to the manufacturer’s instructions. Briefly, a maximum of 100 μl blood was collected through facial vein puncture at the indicated time points. Serum samples were prepared by centrifugation of BD Vacutainer SST tubes (BD Diagnostics) and stored at –80°C until used.
Gyrolab assay for misfolded SOD1 protein.
Rat spinal cord samples were homogenized in 50 mM Tris pH 7.4, 100 mM NaCL, 10% glycerol, 1% Triton X-100 and then diluted at 1:30. Misfolded SOD1 protein was measured with the following antibodies: capture antibody D3H5 (40) (working concentration: 100 μg/ml); and detection antibody B8H10 (working concentration, 50 nM; Medimabs MM-0070-P). Concentrations were measured on a Gyrolab XP Workstation based on a standard curve of denatured recombinant human SOD1 protein.
Cynomolgus monkey SOD1 protein quantification.
Protein levels were measured using an ELISA method specifically qualified to quantify the amount of SOD1 in cynomolgus monkey brain tissue and CSF. Briefly, microtiter plates were coated with SD-G6 antibody diluted in 0.2 M carbonate-bicarbonate buffer and blocked with SuperBlock-PBS (Thermo Fisher Scientific) for 2 hours at ambient temperature. The SOD1 reference standard was generated at Biogen and was used to prepare standards and quality controls (QCs) to final concentrations in SuperBlock-PBS. Cynomolgus brain tissue, CSF samples, and the endogenous QC (EQC) were diluted 1:64 with SuperBlock-PBS. Plates were incubated for 1 hour with sample, washed, and then incubated with 2.5 μg/ml SOD-100 antibody diluted in SuperBlock-PBS for 1 hour, then washed and incubated for 1 hour with 1:32,000 dilution goat anti-rabbit IgG HRP-conjugated antibody. After washing, TMB substrate was added.
Study approval and experimental animals.
SOD1G93A rats (Taconic Biosciences, model 2148) were bred in-house on a Sprague-Dawley background. All rats were kept on a 12-hour light/dark cycle. SOD1G93A mice (B6.CgTg(SOD1*G93A)Gur/J) were bred and maintained at the Jackson Laboratories breeding facility. Food and water were provided ad libitum. Breeding and experimental procedures were completed in accordance with an animal use protocol approved by the Animal Studies Committee at Washington University School of Medicine, Biogen, and Ionis Pharmaceuticals. All mouse studies were conducted using female animals only.
The NHP study was performed at Covance Laboratories GmbH. Covance Laboratories GmbH test facility is fully accredited by the AAALAC. All procedures in the study plan were in compliance with the German Animal Welfare Act and were approved by the local IACUC. Briefly, 30 healthy cynomolgus monkeys (Macaca fascicularis) approximately 2–5 years of age and between 2–6 kg in weight at study start were used for testing. NHPs were administered, by intrathecal bolus injection, 5 doses of ASO or aCSF on days 1, 14, 28, 56, and 84 with 1.0 ml dosing volume using a 35 mg/ml dosing solution. NHPs were euthanized on day 91. CSF collection was performed predose at day 1 (baseline) and at day 91, just before necropsy. To determine the time frame for SOD1 protein recovery, CSF was collected from a separate cohort of animals predose on day 1 (baseline), and predose on days 56, 91, 112, and 119. NHPs received neurological examinations predose on days 1, 14, 28, 56, 84, and 91 and at least once every 2 weeks between days 28 and 91.
Statistics.
P < 0.05 was considered significant for all studies. For survival studies for mice and rats, a log-rank Mantel-Cox test was used. One-way ANOVA followed by Tukey’s post hoc test was performed for Figure 5, D and E. IC50 values were calculated with the formula log (inhibitor) vs. response – variable slope (4 parameters), using Prism6 software formula log (agonist) vs. response - find ECanything, with the following constraints: bottom > 0, top = 100, F = 50 for ED50 and EC50, and F = 80 for EC80. For all other comparisons, a 2-way ANOVA was used.
Author contributions
AM, TC, AJW, CFB, HBK, EES, DLG, MC, and TMM designed the study. TC, AJW, GST, AS, BJF, KMS, MLH, MS, LS, YL, MZ, ST, DLG, and DWS collected data. AM, TC, AW, GST, CFB, HK, ES, DLG, and TMM analyzed data. NC, JA, and BW designed and generated the electrophysiology data in Figures 2 and 3. AM, TC, GST, CFB, HBK, EES, DLG, and TMM wrote the manuscript.
Supplementary Material
Acknowledgments
Funding was provided by the NIH, National Institute of Neurological Disorders and Stroke (NINDS) (R01NS078398 and U01NS084970 to TMM). We would like to thank the preclinical development team, the oligo synthesis group, and the vivarium staff and histology core staff at Ionis Pharmaceuticals and Biogen for their logistical and technical support. We thank Toby Ferguson (Biogen) for comments regarding these data and the manuscript.
Version 1. 07/16/2018
Electronic publication
Version 2. 08/01/2018
Print issue publication
Version 3. 11/01/2018
Added authors Nicole Comfort, Bin Wang, and Jessica Amacker.
Footnotes
Conflict of interest: AM, GST, BJF, LS, YL, MZ, ST, DLG, and DWS are employees of Biogen. TC, CFB, HBK, and EES are employees of Ionis Pharmaceuticals. Ionis Pharmaceuticals provides antisense oligonucleotides to TMM. Biogen provides support to TMM for clinical studies. TMM has participated in a medical advisory board for Biogen and is a consultant for Cytokinetics. TMM and Washington University have licensed technology regarding SOD1 protein kinetic measurements to C2N. MC is a consultant for Cytokinetics, Lilly, Orion, Biohaven, Wave and Biogen.
Reference information: J Clin Invest. 2018;128(8):3558–3567.https://doi.org/10.1172/JCI99081.
Contributor Information
Alex McCampbell, Email: alexander.mccampbell@biogen.com.
Tracy Cole, Email: TCole@ionisph.com.
Amy Setnicka, Email: asetnicka@dei-central.com.
Brandon J. Farley, Email: Brandon.j.farley@gmail.com.
Mark Shabsovich, Email: shabsovichmark@wustl.edu.
Yi Luo, Email: Yi.Luo@biogen.com.
Mingdi Zhang, Email: mingdi.zhang@biogen.com.
Nicole Comfort, Email: nicole.comfort@columbia.edu.
Bin Wang, Email: bin.wang@biogen.com.
Sai Thankamony, Email: Sai.Thankamony@biogen.com.
David W. Salzman, Email: david.salzman@srnalytics.com.
Merit Cudkowicz, Email: Cudkowicz.Merit@mgh.harvard.edu.
C. Frank Bennett, Email: FBennett@ionisph.com.
References
- 1.Bruijn LI, Cleveland DW. Mechanisms of selective motor neuron death in ALS: insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol. 1996;22(5):373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 2.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 3.Fischer LR, Li Y, Asress SA, Jones DP, Glass JD. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp Neurol. 2012;233(1):163–171. doi: 10.1016/j.expneurol.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reaume AG, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 5.Andersen PM, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120(Pt 10):1723–1737. doi: 10.1093/brain/120.10.1723. [DOI] [PubMed] [Google Scholar]
- 6.Ratovitski T, et al. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet. 1999;8(8):1451–1460. doi: 10.1093/hmg/8.8.1451. [DOI] [PubMed] [Google Scholar]
- 7.Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136(Pt 8):2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith RA, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller TM, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kordasiewicz HB, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeVos SL, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33(31):12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVos SL, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481. doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoch KM, et al. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. 2016;90(5):941–947. doi: 10.1016/j.neuron.2016.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. FDA approves first drug for spinal muscular atrophy. FDA. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm534611.htm. Published December 23, 2016. Updated March 28, 2018. Accessed June 1, 2018.
- 15.Finkel RS, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 16.Miller TM, Smith RA, Kordasiewicz H, Kaspar BK. Gene-targeted therapies for the central nervous system. Arch Neurol. 2008;65(4):447–451. doi: 10.1001/archneur.65.4.nnr70007. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto A, et al. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res. 2006;83(1):119–133. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- 18.Mancuso R, Osta R, Navarro X. Presymptomatic electrophysiological tests predict clinical onset and survival in SOD1(G93A) ALS mice. Muscle Nerve. 2014;50(6):943–949. doi: 10.1002/mus.24237. [DOI] [PubMed] [Google Scholar]
- 19.Mancuso R, Santos-Nogueira E, Osta R, Navarro X. Electrophysiological analysis of a murine model of motoneuron disease. Clin Neurophysiol. 2011;122(8):1660–1670. doi: 10.1016/j.clinph.2011.01.045. [DOI] [PubMed] [Google Scholar]
- 20.Lu CH, Petzold A, Kalmar B, Dick J, Malaspina A, Greensmith L. Plasma neurofilament heavy chain levels correlate to markers of late stage disease progression and treatment response in SOD1(G93A) mice that model ALS. PLoS One. 2012;7(7):e40998. doi: 10.1371/journal.pone.0040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCombe PA, Pfluger C, Singh P, Lim CY, Airey C, Henderson RD. Serial measurements of phosphorylated neurofilament-heavy in the serum of subjects with amyotrophic lateral sclerosis. J Neurol Sci. 2015;353(1-2):122–129. doi: 10.1016/j.jns.2015.04.032. [DOI] [PubMed] [Google Scholar]
- 22.Steinacker P, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry. 2016;87(1):12–20. doi: 10.1136/jnnp-2015-311387. [DOI] [PubMed] [Google Scholar]
- 23.Hoye ML, et al. MicroRNA profiling reveals marker of motor neuron disease in ALS models. J Neurosci. 2017;37(22):5574–5586. doi: 10.1523/JNEUROSCI.3582-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winer L, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol. 2013;70(2):201–207. doi: 10.1001/jamaneurol.2013.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bali T, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88(2):99–105. doi: 10.1136/jnnp-2016-313521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crisp MJ, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest. 2015;125(7):2772–2780. doi: 10.1172/JCI80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruijn LI, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281(5384):1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 28.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Da Cruz S, et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017;134(1):97–111. doi: 10.1007/s00401-017-1688-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 31.Ferraiuolo L, et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A. 2016;113(42):E6496–E6505. doi: 10.1073/pnas.1607496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm. Published September 19, 2016. Updated September 26, 2018. Accessed June 1, 2018.
- 33.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang J, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foust KD, et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther. 2013;21(12):2148–2159. doi: 10.1038/mt.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoica L, et al. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann Neurol. 2016;79(4):687–700. doi: 10.1002/ana.24618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu HN, et al. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci. 2012;32(26):8791–8799. doi: 10.1523/JNEUROSCI.5053-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104(7):2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gros-Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113(5):1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- 41.Rakhit R, et al. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13(6):754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- 42.Lange DJ, Andersen PM, Remanan R, Marklund S, Benjamin D. Pyrimethamine decreases levels of SOD1 in leukocytes and cerebrospinal fluid of ALS patients: a phase I pilot study. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(3):199–204. doi: 10.3109/17482968.2012.724074. [DOI] [PubMed] [Google Scholar]
- 43.Benatar M, et al. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology. 2018;90(7):e565–e574. doi: 10.1212/WNL.0000000000004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mazur C, Fitzsimmons B, Kamme F, Nichols B, Powers B, Wancewicz E. Development of a simple, rapid, and robust intrathecal catheterization method in the rat. J Neurosci Methods. 2017;280:36–46. doi: 10.1016/j.jneumeth.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Hua Y, Krainer AR. Antisense-mediated exon inclusion. Methods Mol Biol. 2012;867:307–323. doi: 10.1007/978-1-61779-767-5_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomsen GM, et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci. 2014;34(47):15587–15600. doi: 10.1523/JNEUROSCI.2037-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.