

Abstract
Disruptive mutations in chromatin remodeler CHD8 cause autism spectrum disorders, exhibiting widespread white matter abnormalities; however, the underlying mechanisms remain elusive. We show that cell-type specific Chd8 deletion in oligodendrocyte progenitors, but not in neurons, results in myelination defects, revealing a cell-intrinsic dependence on CHD8 for oligodendrocyte lineage development, myelination and post-injury re-myelination. CHD8 activates expression of BRG1-associated SWI/SNF complexes that in turn activate CHD7, thus initiating a successive chromatin remodeling cascade that orchestrates oligodendrocyte lineage progression. Genomic occupancy analyses reveal that CHD8 establishes an accessible chromatin landscape, and recruits MLL/KMT2 histone methyltransferase complexes distinctively around proximal promoters to promote oligodendrocyte differentiation. Inhibition of histone demethylase activity partially rescues myelination defects of CHD8-deficient mutants. Our data indicate that CHD8 exhibits a dual function through inducing a cascade of chromatin reprogramming and recruiting H3K4 histone methyltransferases to establish oligodendrocyte identity, suggesting potential strategies of therapeutic intervention for CHD8-associated white matter defects.
eTOC blurb
Mutations in chromatin regulator CHD8 are associated with autism and white matter abnormalities. Zhao et al. show that CHD8 functions in oligodendrocyte progenitors to promote oligodendrocyte lineage development, myelination, and post-injury re-myelination by establishing an open chromatin landscape for a cascade of chromatin reprogramming events and recruiting KMT2 histone methyltransferase.
Introduction
Recent studies indicate that mutations in CHD8, encoding a member of chromodomain helicase DNA-binding (CHD) family of SNF2H-like ATP-dependent chromatin remodelers, manifest in and define a specific subtype of autism spectrum disorders (ASD) (Bernier et al., 2014; Cotney et al., 2015; Stolerman et al., 2016). ASDs are etiologically heterogeneous disorders with significant genotypic and phenotypic complexity. CHD8 represents one of the most high-risk susceptibility genes in ASD (Bernier et al., 2014; Cotney et al., 2015). Disruptive mutations in CHD8 result in a wide spectrum of congenital anomalies encompassing growth retardation and intellectual disability. Intriguingly, individuals with ASD often develop white matter abnormalities in the brain, such as deficits in myelin content and compaction (Boddaert et al., 2009; Casanova, 2006; Deoni et al., 2015; Hardan et al., 2016). These observations raise a possibility that CHD8 may be required for myelinating cell development and myelination in the central nervous system (CNS).
Myelination by oligodendrocytes is required for saltatory nerve impulse conduction and proper CNS function (Bercury and Macklin, 2015). Defects in myelination and remyelination impair saltatory nerve conduction and functional connectivity leading to cognitive, behavioral, and motor deficits in neurological disorders including multiple sclerosis and leukodystrophies (Franklin and Goldman, 2015; McKenzie et al., 2014; Trapp et al., 1998). Oligodendrocyte precursor cells (OPCs), which develop from neural progenitors in the CNS, proliferate and differentiate to form mature oligodendrocytes through a stepwise process (Kessaris et al., 2006; Rowitch, 2004; Wegner and Stolt, 2005). Chromatin modification and remodeling is critical for the oligodendrocyte differentiation process, and their dysregulation has been implicated in neurological diseases (Emery and Lu, 2015; Hota and Bruneau, 2016; Kuspert and Wegner, 2016).
ATP-dependent chromatin remodeling enzymes use ATP as an energy source to gate the accessibility of chromatin to transcriptional modulators, and regulate various biological processes including cell growth, differentiation, and regeneration (Hota and Bruneau, 2016; Wu et al., 2009). There are at least four chromatin remodeler families depending on their catalytic ATPase subunit: CHD, INO80, ISWI, and SWI/SNF families (Hota and Bruneau, 2016). The BRG1-containing SWI/SNF-related BAF complex is required for the developmental transition from Olig1+ lineage progenitors or neural progenitors to oligodendrocytes (Bischof et al., 2015; Matsumoto et al., 2016; Yu et al., 2013). In addition, BRG1, together with the lineage transcription factor Olig2, activates a downstream chromatin remodeler CHD7 to control OPC differentiation and the timing of myelination in the CNS (He et al., 2016). At present, whether and how these chromatin remodelers act in concert with temporal or spatial specificity for CNS myelinogenesis and regeneration remains elusive. Additionally, how chromatin remodeling enzymes coordinate with the histone modifying machinery to regulate the transcriptional program for oligodendrocyte myelination has not been fully defined.
Although CHD8 is expressed broadly in the developing brain including neurons and glial cells and has been implicated in neuronal development, functions and animal behaviors by analyzing heterozygous Chd8+/− mutant animals or in utero knockdown experiments (Cotney et al., 2015; Durak et al., 2016; Gompers et al., 2017; Katayama et al., 2016; Platt et al., 2017), the important questions regarding the cell-type-specific function of CHD8 are not resolved by previous studies. Since ASD patients with CHD8 mutations exhibit severe white matter abnormalities, whether the myelination defect of ASD brains is caused by cell-autonomous and cell non-autonomous actions of CHD8 mutations remains unanswered. Defining the lineage-specific molecular and cellular mechanisms of how Chd8 mutations cause white matter abnormalities will provide a better understanding of and treatment strategies for this complex disease.
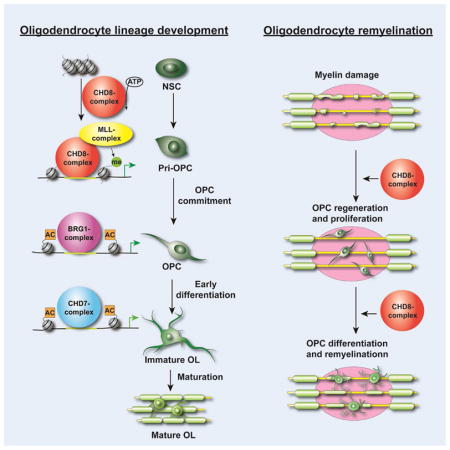
By generating conditional Chd8 knockout mice lacking Chd8 in different neural cell types, we identify a critical, previously unrecognized cell-intrinsic function of CHD8 in oligodendrocyte lineage progression with distinct specificity across CNS regions, as well as in remyelination after demyelinating injury. Chromatin occupancy profiling reveals that CHD8 establishes an accessible chromatin landscape and initiates a successive chromatin remodeling cascade to activate lineage progression. We further show that CHD8 recruits KMT2 histone methyltransferase complexes to promote transcriptional programs necessary for oligodendrocyte differentiation. Elevation of H3K4 trimethylation levels by inhibition of KDM5 histone demethylases partially restores the dysmyelinating phenotype in Chd8-mutant mice. Collectively, our studies provide evidence that a successive cascade of chromatin remodeling events through a CHD8-BRG1-CHD7 axis establishes an accessible chromatin landscape to orchestrate sequential oligodendrocyte lineage progression and enforces developmental checkpoints for establishing oligodendrocyte identity. Our findings point to the potential benefit of modulating CHD8 complex activity to enhance (re)myelination programs in neurological disorders such as ASD or after demyelination.
Results
CHD8 exhibits unique targeting specificity at different stages of the oligodendrocyte lineage and activates expression of BRG1-associated SWI/SNF complex
To explore the potential function of CHD8 in CNS myelination, we analyzed a cohort of ASD patients carrying disruptive CHD8 mutations. These patients exhibited severe defects in the cerebral white matter and volumetric loss compared to the age-matched normal brains (Figure 1A). We then examined murine white matter tracts by immunolabeling of PDGFRα and CC1, which mark OPC and differentiated oligodendrocytes, respectively (Figure 1B). The majority of CHD8+ cells were CC1+ differentiated oligodendrocytes in the corpus callosum, optic nerve, and spinal white matter at P14 (Figure 1B, C). By contrast, CHD8 expression was hardly detectable in GFAP+ astrocytes in the corpus callosum (Figure 1D). These data indicated that CHD8 is highly expressed in oligodendrocyte lineage cells in the developing white matter. CHD8 expression appeared more robust in A2B5+ OPCs than in CNP+ differentiating oligodendrocytes and MBP+ mature rat oligodendrocytes in culture (Figure 1E). Moreover, CHD8 expression was detected in the white matter of normal human cerebellum and co-labeled with an oligodendrocyte marker Sox10 (Figure 1F). Consistent with immunostaining results in vitro, CHD8 protein amounts were higher in OPCs than in mature oligodendrocytes (mOLs), which were differentiated from OPCs after three days of triiodothyronine (T3) exposure (Figure 1G). In contrast, CHD7 was expressed at higher levels in mOLs than A2B5+ OPCs (Figure 1G).
Figure 1. CHD8 is enriched in oligodendrocyte lineage cells and targets SWI/SNF complex genes.

(A) Representative T2-weighted magnetic resonance imaging (MRI) scans of cortices in normal subject (2 year old) and ASD individuals carrying CHD8 disruptive mutations (patient #1, c.5391 A>T, splicing site mutation (2 year old); patient #2, missense mutation c.3485 T>C (p.L1162P); 5 year old). Arrows indicate the corpus callosum.
(B) Co-immunolabeling of CHD8, PDGFRα, and CC1 on the CNS regions of wild-type mice at P14. Yellow and white arrowheads indicate co-labeled PDGFRα+ and CC1+cells, respectively. Scale bar, 10 μm.
(C) CC1+ or PDGFRα+ cell numbers among total CHD8+ cells in the optic nerve, spinal cord, and corpus callosum.
(D) Immunostaining for CHD8 and GFAP in the P14 GFAP-GFP mouse corpus callosum. Arrow indicates GFAP+ astrocytes. Scale bar, 50 μm.
(E) Immunolabeling of CHD8, A2B5, CNP, and MBP in cultured rat oligodendrocytes. Scale bar, 50 μm.
(F) CHD8 and Sox10 immunostaining in oligodendrocytes (arrows) of the human cerebellar white matter. Scale bar, 50 μm.
(G) Immunoblotting of CHD8 and CHD7 in cultured OPCs and differentiating oligodendrocytes at day 1 (d1) and day 3 (d3) after T3 treatment.
(H, I) ChIP-seq density heatmaps for CHD8 (H) and CHD7 (I) within ± 1 kb of the CHD8 peak center in OPCs and mOLs.
(J) Histogram showing the distribution of CHD8, CHD7, and BRG1 binding peaks in the genome of OPCs.
(K) The signal density of CHD8 and CHD7 peaks plotted relative to TSS sites and H3K27Ac peaks.
(L, M) Venn diagrams showing overlap of CHD8 and CHD7 peaks in (L) OPCs and (M) mOLs. (N) CHD8 or CHD7 binding on SWI/SNF complex genes.
(O) Relative expression of SWI/SNF component mRNAs in CG4 cells transfected with control or Chd8 overexpression vectors (n = 3 independent experiments; * p < 0.05 and ** p < 0.01, two-tailed unpaired Student’s t test).
See also Figure S1.
Since mutations in CHD8 and CHD7 results in distinct disease states in humans (ASD and CHARGE syndrome, respectively), we compared the targets of CHD8 and CHD7 by characterizing their genomic occupancy using chromatin-immunoprecipitation and sequencing (ChIP-seq) in A2B5+ OPCs and mOLs. The signals for CHD8 occupancy were much greater in OPCs than in mOLs (Figure 1H). In contrast, CHD7 signals were enriched in mOLs (Figure 1I). A previous study indicated that BRG1 occupancy is more enriched in differentiating immature oligodendrocytes (iOL) (Yu et al., 2013). Furthermore, unlike BRG1 and CHD7 occupancy, CHD8 peaks were overrepresented in the promoter regions (Figure 1J). CHD8 occupancy signals in OPCs were highly enriched around transcriptional start sites (TSS) and in proximal enhancer regions with activating H3K27ac deposition compared with BRG1 and CHD7 signals (Figure 1J, K). Further, a large majority of CHD8 binding regions were not targeted by CHD7 in A2B5+ OPCs or in mOLs (Figure 1L, M), indicating that CHD8 harbors unique targeting specificity during oligodendrocyte development even though CHD8 and CHD7 are within the same CHD subfamily.
The SWI/SNF complex component BRG1 activates downstream CHD7 expression during OPC differentiation (He et al., 2016; Yu et al., 2013). Strikingly, we found that CHD8 binds to the promoters of genes encoding key components of the BRG1-associated SWI/SNF complex (BAF) including Brg1, Baf45b, Baf45d, Baf60a, and Baf250a (Figure 1N). In contrast, CHD7 did not substantially target genes encoding BAF components. Furthermore, overexpression of CHD8 in an oligodendrocyte cell line CG4, induced the expression of these BAF complex genes but not a related remodeler gene Brm (a.k.a. Smarca2) (Figure 1O). To further examine the genetic hierarchy between CHD8 and BRG1, we examined CHD8 expression in the spinal white matter of Brg1-cKO (Brg1flox/flox;Olig1-cre+/−) animals at P7. We found that CHD8 expression was retained in BRG1-depleted cells, suggesting that CHD8 acts as an upstream regulator of Brg1 in the oligodendrocyte lineage (Figure S1). Together, these data suggest that CHD8 can initiate a cascade of BRG1-CHD7 mediated chromatin remodeling events to activate oligodendrocyte differentiation programs.
Mice lacking Chd8 in the oligodendrocyte lineage exhibit myelination deficits
To investigate the in vivo function of CHD8 specifically in CNS myelination, we first generated a Chd8 conditional mouse line carrying a floxed exon4 using CRISPR-Cas9 technology to cause premature termination of CHD8 after Cre recombination (Figure S2). We ablated Chd8 in oligodendrocyte lineage cells by breeding mice with the floxed Chd8 allele with an oligodendrocyte-lineage expressing Olig1-Cre line that commences in primitive OPCs (Pri-OPC) prior to PDGFRα+ OPCs (Xin et al., 2005) (Figure 2A). We compared conditional Chd8 knockout mice (Chd8flox/flox;Olig1-Cre+/−, hereafter referred to as Chd8 cKO) with their heterozygous Chd8flox/+;Olig1-Cre+/− littermates (control mice). CHD8 was substantially reduced in the white matter of the spinal cord and corpus callosum in Chd8 cKO mice at P14 (Figure 2B; Figure S3A), while CHD8 was present at normal expression levels in neurons, astrocytes and microglia in the Chd8 cKO brain (Figure S3B–D), suggesting that CHD8 was effectively depleted in the oligodendrocyte lineage but not in other neural cell types in the developing CNS of Chd8 cKO mice. All Chd8 cKO mice developed severe dysmyelinating phenotypes and exhibited hindlimb clasping, generalized tremors and tonic-clonic seizures beginning around postnatal week 3 (Figure 2C, D, Movies S1 and S2), and died around postnatal day 21 (Figure 2E). The heterozygous control mice were phenotypically normal compared with wildtype or Chd8flox/flox mice. The myelinating optic nerve tract from Chd8 cKO mice at P14 appeared translucent (Figure 2F), indicating a myelin deficit in this tract. Consistently, expression of myelin genes Mbp and Plp1 was diminished in the CNS regions of mutant mice including the spinal cord, cerebral cortex, and cerebellum at P1 and P14 (Figure 2G–I). Notably, myelin gene expression was essentially absent in the spinal cord and cerebellum relative to the cortex at P14, indicating a regional difference in dysmyelinating severity (Figure 2G–I). In light of these findings, we further examined the ultrastructure of myelin sheaths in the optic nerves, spinal cord, and corpus callosum and found severely compromised axonal ensheathment in the CNS of Chd8 cKO animals at P14 (Figure 2J, K).
Figure 2. The chromatin remodeler CHD8 is required for proper CNS myelination.

(A) Diagram depicting generation of Chd8 cKO mice.
(B) Immunolabeling of CHD8 in the ventral white matter of control and Chd8 cKO spinal cords at P14. Scale bar, 50 μm.
(C, D) Representative images of control and Chd8 cKO mice at P14 (C) and P18 (D). The cKO mouse is suffering from a seizure (D).
(E) Survival curves of control and Chd8 cKO mice (n ≥ 32).
(F) Representative images of optic nerves from control and Chd8 cKO mice at P14.
(G–I) Expression of Mbp and Plp1 in spinal cord (G), cortex (H), and cerebellum (I) of control and Chd8 cKO mice at P1 and P14. Scale bars, 300 μm.
(J) Electron micrographs of indicated CNS regions from P14 control and Chd8 cKO mice. Scale bars, 2 μm (left and right panels), 4 μm (middle panel).
(K) Myelinated axon numbers (per mm2) in the optic nerve (ON), spinal cord (SC), and corpus callosum (CC) of control and Chd8 cKO at P14 (n = 4 controls and 4 mutant animals; *** p < 0.001, two-tailed unpaired Student’s t test).
See also Figure S2.
Region-dependent requirement for CHD8 in OPC proliferation and survival in the CNS
To investigate whether the dysmyelinating phenotype in Chd8 cKO mice is due to a defect in OPC generation, we assessed OPC development by examining expression of the OPC marker Pdgfra by in situ hybridization. Pdgfra+ OPCs were observed in the brain of Chd8 cKO mice, and their numbers were comparable to those in controls in the developing cortex at P1 and P14 (Figure 3A, B). Further, we found similar rates of OPC proliferation (as judged by percentage of BrdU+ proliferative OPCs) in control and Chd8 cKO brains (Figure 3C, D).
Figure 3. CHD8 is required for OPC generation and survival in the spinal cord but not in the cortex.

(A) In situ hybridization for Pdgfra in the cortices at P1 and P14. Scale bar, 300 μm.
(B) Quantification of Pdgfra+ OPCs in the cortex (n = 3 animals).
(C) Immunostaining for BrdU and Olig2 in the corpus callosum from P1 mice. Scale bar, 50 μm.
(D) Percentage of BrdU+ cells relative to all Olig2+ OPCs in the corpus callosum of P1 mice. (n = 3 animals).
(E,F) In situ hybridization for Pdgfra in the spinal cord of control and Chd8 cKO mice at E14.5, P1, P14. Scale bar, 300 μm. Panel F, quantification of Pdgfra+ OPCs.
(G,H) Olig2 immunostaining in the spinal cord of control and Chd8 cKO mice at P1. Scale bar, 200 μm. Panel H, quantification of Olig2+ cells.
(I) Olig2 and Ki67 immunostaining in the spinal cord of control and Chd8 cKO mice at P1. Scale bar, 100 μm.
(J) Quantification of Ki67+ relative to total Olig2+ OPCs.
(K) Primary OPCs from control and Chd8 cKO mice cultured in Sato medium without PDGFAA and NT3 for 24 hr were immune-stained with cleaved caspase 3 and Olig2. Scale bar, 100 μm.
(L) Percentage of cl-caspase3+ among total Olig2+ cells from above control and Chd8 cKO OPCs.
n = 3 animals/genotype; * p < 0.05 and ** p < 0.05, two-tailed unpaired Student’s t test in F, H, J, L.
See also Figure S3.
In contrast, in the spinal cord, OPC numbers were substantially reduced relative to control levels over the course of Chd8 cKO development from E14.5 to P14 (Figure 3E, F) despite their initial formation at early embryonic stages. Pdgfra+ OPCs were hardly detectable at P14 (Figure 3E, F). In the P1 spinal cord, the number of Olig2+ OPCs was significantly reduced in Chd8 cKO (Figure 3G, H) as were numbers of proliferating Ki67+/Olig2+ OPCs (Figure 3I, J), indicating a significantly lower degree of proliferation of Chd8-cKO OPC populations in the spinal cord. The percentage of cleaved caspase 3+ apoptotic cells among Olig2+ OPCs was not significantly higher in Chd8 cKO spinal cord at P7 (Figure S3E), although we could not exclude that cell death might occur at specific time-windows during development. However, we observed a significant increase in cell death among OPCs isolated from Chd8 cKO cortices compared with controls when they are maintained in the spontaneous differentiation medium after mitogen PDGFAA and NT3 withdrawal (Figure 3K, L), suggesting an increase of apoptosis upon OPC differentiation. Chd8 inactivation in oligodendrocyte lineage cells did not change either overall numbers of astrocytes (GFAP+), neurons (NeuN+), or microglia (Iba1+), or the CHD8 expression pattern in these cells in Chd8 cKO cortices (Figure S3B–D). Together, these observations demonstrate a regionally specific requirement for CHD8 in OPC development and proliferation in the CNS.
Deletion of Chd8 in oligodendrocyte progenitors, but not in neurons, leads to defects in myelination in the developing brain
To determine if defects in oligodendrocyte differentiation are a cell-autonomous effect of Chd8 ablation, we purified OPCs from the neonatal cortex of Chd8flox/flox pups, and then transduced them with adenoviral vectors to express either eGFP as a control (Ade-GFP) or Cre-eGFP (Ade-Cre-GFP) to excise Chd8flox/flox alleles in culture. After transduction, mitogens PDGFAA and NT3 were withdrawn to examine differentiation capacity (Figure 4A). Chd8flox/flox OPCs transduced with Ade-GFP and wild-type OPCs transduced with Ade-Cre-GFP were able to differentiate into mature MBP+ oligodendrocytes with a complex morphology. In contrast, OPCs transduced with Ade-Cre-GFP failed to differentiate into MBP+ oligodendrocytes (Figure 4A, B), suggesting that Chd8-depleted OPCs are intrinsically defective in maturation.
Figure 4. CHD8 deletion impairs oligodendrocyte differentiation in a cell-autonomous manner.

(A) MBP immunostaining of Chd8f/f OPCs 4 days after transduction with GFP control or Cre-GFP expressing adenovirus vectors in differentiation medium. Arrows: transduced cells. Scale bar, 100 μm.
(B) Percentage of MBP+ oligodendrocytes among total GFP+ cells after 4 days of differentiation.
(C) Diagram showing tamoxifen (TAM) administration to induce Chd8 deletion.
(D, E) Immunostaining of CHD8 (D), CC1 and MBP (E) on the corpus callosum of control and Chd8 OPC-iKO mice. Scale bars, D, 50 μm; E,100 μm.
(F) Quantification of CC1+ oligodendrocyte cell numbers in the corpus callosum of control and Chd8 OPC-iKO mice at P14.
(G) Percentage of MBP+ area in the corpus callosum of control and Chd8 OPC-iKO mice at P14.
(H, I) Immunolabeling of PDGFRα on the corpus callosum of control and Chd8 OPC-iKO mice. Scale bar, 50 μm. Panel I, quantification of PDGFRα+ OPCs.
n = 4 animals/genotype; *** p < 0.001, two-tailed unpaired Student’s t test in B, F, G, I.
See also Figure S4 and S5.
To further examine the effects of Chd8 inactivation on OPC differentiation during postnatal development in a time-controlled manner, we generated OPC-inducible Chd8 mutants by breeding Chd8flox/flox mice with PDGFRα-CreERT mice (Kang et al., 2010), which carry an OPC-specific tamoxifen-inducible Cre and a Rosa26:ccGFP reporter (Nakamura et al., 2006). Tamoxifen administration in PDGFRα-CreERT;Chd8flox/flox mutants (referred to as Chd8 OPC-iKO) pups from P3 to P7 induced effective depletion of CHD8 (Figure 4C, D). Loss of Chd8 in OPCs led to a dramatic (~87%) reduction in CC1+ differentiating oligodendrocytes in the cortices of Chd8 OPC-iKO mice compared to control littermates (PDGFRα-CreERT:Chd8flox/+) (Figure 4E, F). The expression of MBP in oligodendrocytes from Chd8 OPC-iKO brains was significantly reduced compared to the robust expression observed in controls (Figure 4E, G), while we did not detect a difference in the number of OPCs in the cortices of iKO mice (Figure 4H, I). Similarly, in the spinal cord, Chd8 depletion in OPCs impaired OPC differentiation (Figure S4A, B). Although the OPC number and their proliferation rate were not altered (Figure S4C, D), we detected an increase in cleaved caspase 3+ apoptotic cells among Chd8-deleted OPCs (Figure S4E). Thus, these observations indicate that Chd8-deletion in OPCs not only inhibits OPC differentiation but also impairs OPC survival in the developing spinal cord.
To examine the potential non-cell-autonomous impact of Chd8 ablation on myelination, we deleted Chd8 in developing neurons with synapsin1-Cre. In this line, the neuron-specific Cre activity commences at the early embryonic stage E12.5 (Zhu et al., 2001). We did not observe substantial differences in expression of MBP or in the number of CC1+ oligodendrocytes in the developing cortex between controls and mice with neuron-specific Chd8 deletion at P14 (Figure S5), suggesting a cell-autonomous role of CHD8 in the control of OPC differentiation.
CHD8 is critical for OPC proliferation and remyelination after demyelination
Given the essential role of CHD8 in oligodendrocyte development, we hypothesized that CHD8 is also required for remyelination after demyelination in the adult CNS. We induced demyelinating lesions in the ventral white matter of the spinal cord by lysolecithin (LPC) treatment (Franklin, 2002; Zhao et al., 2016). In this model, a spontaneous myelin regeneration process begins with an OPC recruitment phase approximately 7 days post-LPC-lesion-inducement (dpl) and a remyelinating phase begins around dpl 14 (Franklin, 2002). CHD8 expression was barely detectable in oligodendrocytes in the adult intact spinal cord (Figure 5A); however, CHD8+ cell numbers were increased substantially in the lesions during remyelination but not in non-lesion regions of spinal white matter at dpl 7 (Figure 5A). CHD8 expression was detected in PDGFRα+ OPCs and CC1+ oligodendrocytes within the lesion (Figure 5B, C). Similarly, CHD8 expression was detected in the remyelinating lesions in multiple sclerosis brains (Figure S6).
Figure 5. CHD8 is required for CNS remyelination.

(A) CHD8 and CC1 immunostaining in non-lesion control and LPC lesion spinal cords at dpl 7. Dashed line indicates the border between white (WM) and gray matter (GM). Scale bar, 200 μm.
(B, C) CHD8 and PDGFRα immunostaining (B) and CC1 (C) at LPC-lesion sites. Scale bar, 50 μm.
(D) Diagram showing TAM administration and LPC injection schedule.
(E) In situ hybridization for Pdgfra, Mbp, and Plp1 in spinal LPC lesions of control and Chd8 OPC-iKO mutants at dpl 14 and 28. Scale bar, 100 μm.
(F) Quantification of Plp1+ oligodendrocytes in LPC lesion sites at dpl 14 and 28. Data are presented as means ± s.e.m.
(G, H) Immunostaining for CC1 in LPC lesions from control and Chd8 OPC-iKO spinal cords at dpl 14. Scale bar, 100 μm. H, quantification of CC1+ oligodendrocytes in LPC lesion.
(I, J) In situ hybridization for Pdgfra in spinal LPC lesions of control and Chd8 OPC-iKO mutants at dpl 14. Scale bar, 100 μm. J, quantification of Pdgfra+ OPCs in LPC lesion sites
(K) Immunostaining for Ki67 and Olig2 in LPC lesions from control and Chd8 OPC-iKO spinal cords at dpl 14. Scale bar, 100 μm.
(L) Quantification of Ki67+ cells among total Olig2+ OPCs in LPC lesion sites at dpl 14.
(M) Electron microscopy of LPC lesions from control and Chd8 OPC-iKO spinal cords at dpl 14. Scale bar, 2 μm.
(N) The percentage of remyelinated axons in LPC-induced lesions of control and Chd8 OPC-iKO spinal cords at dpl 14.
(O) The myelin g-ratio in LPC-induced lesions of control and Chd8 OPC-iKO mutants at dpl 14 (n > 200 axons from 3 animals; p < 0.001, two-tailed unpaired Student’s t test).
* p < 0.01 and ** p < 0.001, two-tailed unpaired Student’s t test in F, H, J, L, N (n = 4 animals/genotype).
See also Figure S6.
To determine the potential role of CHD8 in oligodendrocyte regeneration, we administered tamoxifen three days prior to LPC injection to delete Chd8 in 8-week-old Chd8 OPC-iKO with heterozygous mice (PDGFRα-CreERT:Chd8flox/+) as controls (Figure 5D). LPC was then injected into the ventral white matter of the spinal cord, which was followed by five additional days of tamoxifen administration (Figure 5D), and the lesioned spinal cords were harvested at dpl 14 and 28. To determine the extent of remyelination, we examined the expression of the OPC marker PDGFRa and myelin genes Mbp and Plp1 using in situ hybridization. There were significantly fewer Mbp- and Plp1-expressing oligodendrocytes in the spinal cord of Chd8 OPC-iKO mice at dpl 14 and 28 than in control spinal cords (Figure 5E, F). Consistent with these findings, fewer CC1+-differentiating oligodendrocytes were detected in the lesions of Chd8 OPC-iKO spinal cords than controls at 14 dpl (Figure 5G, H). In addition, the number of PDGFRα-expressing OPCs was lower in Chd8 OPC-iKO mice at 14 dpl (Figure 5I, J). The proliferation rate of OPCs was significantly reduced in Chd8 OPC-iKO relative to control animals (Figure 5K, L). Ultrastructural analysis by electron microscopy further indicated that the number of myelinated axons was substantially lower in the lesions of Chd8 OPC-iKO mice at dpl 14 (Figure 5M, N), and newly generated myelin sheaths around axons, as assessed by g-ratios, were significantly thinner in Chd8 OPC-iKO mice compared to controls (Figure 5O). These data suggest that CHD8 regulates OPC proliferation and regeneration during the repair process, and therefore is essential for remyelination after demyelinating injury.
CHD8 regulates the transcriptional program for oligodendrocyte-lineage differentiation
To investigate the potential mechanisms of dysmyelination in Chd8-deficient animals, we performed transcriptome profiling and analyzed global gene expression patterns of OPCs acutely isolated from control and Chd8 cKO cortices by fluorescence activated cell sorting (FACS) with PDGFRα and O4 antibodies (Figure S7A) (Robinson et al., 2014). Expression of ~2,342 genes was altered in Chd8 cKO compared with control OPCs (fold change ≥ 1.6, p < 0.05; Figure 6A). The control and Chd8 cKO OPCs had distinct gene expression profiles as shown by principal component analysis (Figure 6B). Gene Set Enrichment Analysis (GSEA) revealed that the genes downregulated in Chd8-deficient cells were enriched in those associated with oligodendrocyte differentiation and cholesterol biosynthesis (Figure 6C, D), consistent with the dysmyelination phenotype in Chd8 cKO mice. In addition, differentiation-inhibitory genes such as those encoding hedgehog- and p53-signaling pathways as well as Wnt/β-catenin signaling effectors Id4 and Sp5 were up-regulated in the Chd8-deficient OPCs (Figure 6A, C). Quantitative real-time PCR analysis confirmed a reduction in myelination-associated genes Myrf, Plp1, Cnp, Mag, and Mbp, alongside an increase in differentiation inhibitory genes Sp5 and Id4 (Figure 6E).
Figure 6. CHD8 controls the core regulatory network for oligodendrocyte differentiation.

(A) Differentially expressed transcripts (highlighted in color; FDR < 0.05) between OPCs isolated from control and Chd8 cKO mouse brains at P7.
(B) Principal components analysis (PCA) of RNA-seq data from control and Chd8 cKO OPCs.
(C) GSEA terms that are significantly different in the Chd8 cKO OPC cells compared to controls. NES: net enrichment score.
(D) GSEA plot shows oligodendrocyte signature genes are downregulated in the Chd8 cKO cells.
(E) qRT-PCR analysis of myelination-associated genes in control and Chd8 cKO OPCs. Data are presented as means ± s.e.m. (n = 3 experiments; * p < 0.05, two-tailed unpaired Student’s t test).
(F, G) GSEA plots show KMT2/MLL target genes (F) and H3K4me3 (G) targeted genes are downregulated in Chd8 cKO cells.
(H) Percentage of binding peaks of CHD8, CHD7, and BRG1 in the genome of OPCs.
(I) Enriched motifs in the CHD8-bound regions.
(J) The signal density of Olig2 and Sox10 peaks plotted relative to CHD8 peaks.
(K) Venn diagram showing the overlap between CHD8-bound genes and differentially expressed genes in control and Chd8 cKO OPCs. The overlapping genes are direct CHD8 target genes.
(L) Enrichment analysis of pathway terms overrepresented in CHD8 target genes.
(M) Heatmaps for ATAC-seq peaks from control and Chd8 cKO OPCs showing ± 1 kb around the ATAC-seq peak center.
(N, O) Tag enrichments of accessible genomic regions relative to ATAC-seq peaks (N) and Olig2 binding peaks (O) in control and Chd8 cKO OPCs.
(P) Heatmaps for H3K4me3 ChIP-seq peaks from control and Chd8 cKO OPCs showing ± 1 kb around the peak center.
(Q, R) Representative ATAC-seq and H3K4me3 ChIP-seq tracks of OPC specification regulatory genes (Q), myelination-related genes (R) in the control and Chd8 cKO OPCs.
(S) Representative ATAC-seq tracks of neuronal, astrocytic, microglial, and endothelial genes in the control and Chd8 cKO OPCs.
See also Figure S7.
Strikingly, histone methyltransferase KMT2/MLL target genes and H3K4me3-marked genes were also significantly downregulated in Chd8-deficient cells (Figure 6F, G). KMT2/MLL catalyzes histone 3 lysine 4 trimethylation and creates a H3K4me3 mark, which is associated with actively transcribed and poised gene promoters in mammals (Zhang et al., 2015). Given that the H3K4me3 mark is associated with promoter activity, these data suggest that CHD8 regulates a transcriptional activation program for OPC differentiation.
ChIP-seq analysis revealed that CHD8 targeted approximately 23,153 sites with a large majority of binding sites detected at putative promoter or proximal enhancer regions at 5′ UTR compared with BRG1 and CHD7 in OPCs (Figure 6H). The sites occupied by CHD8 showed considerable enrichment for consensus binding motifs for the transcriptional regulators including Olig2 and Sox10 (Figure 6I, J). To identify the direct CHD8 target genes, we intersected the CHD8-occupied genes with those genes differentially expressed in Chd8-mutant OPCs and identified approximately 1,656 genes as candidate CHD8-regulated targets (Figure 6K). GO analysis of pathway terms for potential CHD8 targets were overrepresented by those involved in neurogenesis, gliogenesis, oligodendrocyte differentiation, Wnt signaling and regulation of cell death (Figure 6L).
CHD8 establishes an accessible chromatin landscape to activate lineage-specific transcriptional programs
Since CHD8, a chromatin remodeling factor, is critical for oligodendroglial differentiation, we hypothesized that CHD8 might regulate transcriptional activation by facilitating nucleosome depletion across an ensemble of enhancers on the loci of oligodendrocyte lineage regulators. To test this hypothesis, we performed an assay for transposase-accessible chromatin (ATAC-seq) to capture open chromatin sites (Buenrostro et al., 2015) in OPCs isolated from control and Chd8 cKO brains. Signals due to chromatin accessibility were dramatically reduced in Chd8-mutant OPCs (Figure 6M), suggesting that CHD8 is required for maintenance of an open chromatin structure. Next, we plotted ATAC-Seq tag counts as a function of genomic separation from TSS and Olig2-bound sites, and found that TSS and regions associated with Olig2 occupancy were more accessible to transposase activity in control cells than Chd8 mutant OPCs (Figure 6N, O). Since H3K4me3 is associated with transcriptionally active promoter elements, we investigated the status of H3K4me3 enrichment in the gene loci in OPCs in the absence of CHD8 by ChIP-seq. H3K4me3-enriched regions were substantially diminished in the corresponding elements in Chd8-depleted OPCs (Figure 6P).
Compared with control OPCs, chromatin accessibility in the promoters and proximal enhancers of oligodendrocyte specification genes Olig2 and Ascl1 (Nakatani et al., 2013), and differentiation-related genes Olig1/2, Sox10, Tcf7l2, Zeb2, Nkx2.2, Nkx6.2, Myrf, Zfp488, Zfp365, Mbp, Ugt8a, Caspg4, LncOL1, and Fyn (Emery and Lu, 2015; He et al., 2017) were drastically decreased in Chd8 cKO OPCs (Figure 6Q, R). The abundance of RNA transcripts encoding these oligodendrocyte lineage-specific transcription factors was downregulated by depletion of CHD8 expression in OPCs (Figure 6A), suggesting that CHD8 promotes accessibility of the regulatory elements of genes to activate OPC differentiation programs.
Consistent with chromatin accessibility changes, the H3K4me3 marks in the promoter and proximal enhancer regions of oligodendrocyte lineage regulatory genes Olig1/2, Ascl1, Sox10, Tcf7l2, Zeb2, Nkx2.2, Nkx6.2, Myrf, Zfp488, and Zfp365 and myelination-associated genes Mbp, Ugt8a, Caspg4, LncOL1, and Fyn were markedly reduced in Chd8-mutant OPCs compared to controls (Figure 6Q, R). In contrast, the chromatin accessibility assessed by ATAC-seq at loci of neuron-enriched genes e.g. Neurod6, astrocyte gene GFAP, microglial gene Rgs2, and endothelial genes e.g. Mfsd7c were unaltered in Chd8 mutant OPCs (Figure 6S), suggesting a selective CHD8 function in OPCs. Therefore, these data suggest that CHD8 is required for establishment of accessible chromatin landscape and H3K4me3 enrichment that promote the transcriptional program necessary for oligodendrocyte lineage progression.
CHD8 recruits the KMT2 methyltransferase complex to the promoter regions of oligodendrocyte lineage genes
Since CHD8 controls chromatin accessibility and H3K4me3 enrichment in OPC regulatory gene loci, we hypothesized that CHD8 recruits a histone methyltransferase complex to induce H3K4me3 deposition on these promoter elements. KMT2/MLL methyltransferase complexes including core subunits ASH2L, DPY30, RBBP5, and WDR5, are required for the catalytic activity of mono-, di-, and trimethylation at lysine 4 of histone H3 (Dou et al., 2006; Takahashi et al., 2011). A co-immunoprecipitation assay revealed that CHD8 forms a complex with KMT2 components ASH2L and WDR5 (Figure 7A). Furthermore, in Chd8-deficient OPCs, the enrichment of ASH2L and WDR5 in the promoter regions of oligodendrocyte lineage genes Olig2, Pdgfra, Zfp365 and Sox10 was significantly reduced (Figure 7B, C). Inhibition of Ash2L and Wdr5 expression in rat OPC cultures resulted in a decrease in expression of oligodendrocyte differentiation-associated genes Plp1, Cnp, Myrf, Mag, and Mbp, but not in expression of differentiation inhibitors such as Hes5 (Figure 7D, E). These data suggest that CHD8 recruits a KMT2/MLL histone methyltransferase complex to activate the transcriptional program necessary for OPC differentiation.
Figure 7. CHD8 regulates H3K4me3 deposition at the promoters of oligodendrocyte genes through interaction with ASH2L and WDR5.

(A) CHD8 co-immunoprecipitated with ASH2L and WDR5.
(B) Levels of Chd8 mRNA in rat OPCs treated with Chd8 siRNA (siChd8) or control siRNA (Ctrl).
(C) Enrichment of ASH2L, WDR5, and H3K4me3 in promoter regions of oligodendrocyte genes in control and siChd8-treated OPCs.
(D, E) Ash2L and Wdr5 (D) and myelin gene (E) mRNA expression in cultured OPCs treated with siRNAs targeting Ash2L or Wdr5. Data are presented as means ± s.e.m. (n = 3 experiments; ** p < 0.001, two-tailed unpaired Student’s t test).
(F) CHD8 immunostaining in OPCs from control and Chd8 cKO cortices. Scale bar, 100 μm.
(G, H) H3K4me3 immunostaining in control and Chd8 cKO OPCs treated with vehicle or CPI-455. Scale bar, 100 μm. Panel H, percentage of H3K4me3+ oligodendrocytes among total cells.
(I, J) MBP and Olig2 immunostaining in control and Chd8 cKO OPCs treated with vehicle or CPI-455. Scale bar, 100 μm. Panel J, percentage of MBP+ oligodendrocytes among total Olig2+ cells.
(K, L) CC1 and MBP immunostaining of spinal cords from control and Chd8 cKO littermates treated with vehicle or CPI-455 and harvested at P14. Scale bar, 100 μm. Panel L, quantification of CC1+ oligodendrocytes.
(M) A schematic model showing that CHD8 executes a dual function by establishing an accessible chromatin landscape and recruiting KMT2/MLL histone methyltransferase to activate the transcriptional program for oligodendrocyte lineage progression.
Inhibition of the KDM5 subfamily of histone demethylases partially rescues differentiation defects in Chd8-deficient OPCs
Since CHD8 interacts with histone methyltransferase complexes essential for histone H3K4 trimethylation, we hypothesized that restoring H3K4 trimethylation levels indirectly via histone demethylase inhibition might rescue differentiation defects in Chd8 mutant OPCs. The KDM5 subfamily of histone demethylases remove tri- and di-methylations of lysine 4 from histone H3 (Cloos et al., 2008). CPI-455 is a potent and selective inhibitor of the KDM5 subfamily (Vinogradova et al., 2016), and exhibits a blood-brain barrier permeability potential (Banelli et al., 2017). The H3K4me3 mark is lost in the majority of Chd8-ablated OPCs isolated from Chd8 cKO animals (Figure 7F–H). Strikingly, CPI-455 treatment significantly increased levels of H3K4me3 in Chd8-depleted OPCs (Figure 7G, H). Further, we detected a significant higher percentage of MBP-expressing mature oligodendrocytes in Chd8 cKO OPCs treated with CPI-455 (Figure 7I, J), suggesting that inhibition of the KDM5 histone demethylase activity also partially restores differentiation capacity of Chd8-deficient OPCs.
To determine whether CPI-455 treatment could rescue the oligodendrocyte differentiation defect in Chd8-cKO animals, we treated both control and Chd8 cKO pups with CPI-455 via intraperitoneal injection. The pups were injected with either CPI-455 or vehicle every other day starting from P1 to P7, and spinal cords were harvested at P14 for analysis. KDM5 inhibitor administration did not alter the number of CC1+ oligodendrocytes in the spinal cords of control animals, however, treatment substantially increased the number of CC1+ oligodendrocytes and expression of MBP in Chd8 cKO spinal cord (Figure 7K, L). These data indicate that inhibition of the KDM5 histone demethylase activity increases H3K4me3 levels and enhances differentiation capacity of Chd8-deficient OPCs.
Discussion
A critical role of autism-related CHD8 for oligodendrocyte development and survival with regional specificity
The myelination defects caused by CHD8 mutations could lead to impaired brain connectivity and function in an ASD cohort (Bernier et al., 2014; Platt et al., 2017). Recent studies have been focusing on CHD8 functions in neuronal development and functions (Cotney et al., 2015; Durak et al., 2016; Gompers et al., 2017; Platt et al., 2017), the mechanisms underlying the white matter defects in the autistic individuals are poorly defined. By generating cell-type specific Chd8 mutant mice, we demonstrate for the first time that CHD8 is required cell-autonomously for OPC development and differentiation.
The Chd8 cKO mice die around P21, the peak period of myelination. This may be due to the complete absence of myelin sheaths in the Chd8 cKO spinal cord, leading to severe tremors and tonic seizures, and animal mortality eventually. This phenotype resembles other mutants that cause an unmyelinated phenotype accompanied by mortality at the time of peak myelinogenesis (Weng et al., 2012; Yu et al., 2013). Alternatively, the potential alteration of functions of other neural cell types in Chd8 cKO mutants may also contribute to early lethality of these mice. However, deletion of Chd8 in neuronal cells of the developing brain starting at the early embryonic stage did not alter the number of oligodendroglia and myelin protein expression (Figure S5), suggesting that Chd8 loss-of-function in postmitotic neurons has a minimal impact on myelination and white matter development. These observations indicate that myelination defects in ASD individuals carrying CHD8 mutations are likely caused by the cell-intrinsic loss-of-function of CHD8 in the oligodendrocyte lineage.
Our data suggest that the extent of CHD8 regulation in OPC maintenance and differentiation is region-specific across the CNS. Chd8 deletion blocks the differentiation from PDGFRα+ OPCs to oligodendrocytes but not the initial formation of OPCs. In contrast to the brain, however, in the developing spinal cord, Chd8 deletion not only inhibits the OPC-to-oligodendrocyte transition but also impairs OPC proliferation and survival, resulting in severe deficits of OPC populations and essentially absence of oligodendrocytes over the course of development. The underlying mechanisms for the regionally specific function of CHD8 remain to be determined. Since Chd8 deletion efficiency is comparable between the spinal cord and brain in Chd8 cKO mutants (Figures 2B and S3A), it suggests that oligodendrocyte development and survival in the spinal cord and cortex may have an intrinsic difference in the degree to which Chd8 is required.
The reduction of OPCs in the Chd8 mutant spinal cord might reflect a reduced OPC proliferation rate and/or an increase in OPC cell death upon differentiation, leading to a failure in maintaining OPC pools. Our data indicate that Chd8 loss-of-function leads to an upregulation of the p53 signaling pathway in OPCs (Figure 6C), which is consistent with CHD8 regulation of p53-target genes (Feng et al., 2017). Given that p53 induces cell apoptosis (Aubrey et al., 2018; Ding and Fisher, 1998), upregulation of p53 signaling in Chd8-dificient mutants could impair OPC survival. This suggests that CHD8 is critical for keeping balance between OPC proliferation, differentiation and survival. In the developing brain, there may be a compensatory effect from other chromatin remodelers such as CHD7, which can interact with CHD8 (Batsukh et al., 2010) and has a limited but substantial temporal overlap in expression, whereas CHD8 could act as the primary driver of OPC proliferation and differentiation in the developing spinal cord.
CHD8 initiates a chromatin remodeling cascade to drive oligodendrocyte differentiation
During oligodendrocyte development, transcriptional regulators act together to activate stage-specific genes. How the transcriptional machinery establishes promoter or enhancer repertoires to initiate lineage-specific gene expression programs is poorly understood. Our studies revealed that stage- and target-specific chromatin landscape remodeling can be mediated by a cascade of chromatin remodeling events; such mechanisms have not been previously defined in oligodendrocyte development.
The BRG1-containing BAF remodeling complex initiates the differentiation process from Olig1+ progenitors (Yu et al., 2013). Here we showed that CHD8 binds to the promoters of genes encoding key components of the BAF complex and induces their expression. In contrast, CHD8 did not induce expression of Brm/Smarca2, which encodes a BRG1-related remodeler, demonstrating specificity. Additionally, characterization of CHD8, BRG1, and CHD7 genomic occupancy showed that these chromatin remodelers have distinct preferences for regulatory elements. The majority of the sites targeted by CHD8 are in promoter or proximal enhancer regions, and are distinct from those bound by BRG1 and CHD7. In A2B5+ OPCs, CHD8 binds near transcription start sites, whereas CHD7 and BRG1 preferentially target distal enhancers, suggesting that distinct chromatin remodelers exert unique functions during OPC development. Like CHD7, CHD8 also targets distal enhancers of myelination-associated genes in maturing oligodendrocyte to regulate their terminal differentiation (Figure S7C). Thus, CHD8 regulates the chromatin landscape in both promoter/proximal and distal enhancers during oligodendrocyte development. The mechanisms of dynamic CHD8 recruitment to distinct promoters remain to be further defined, nonetheless, our data reveal that a cascade of chromatin remodeler action initiated by CHD8, which promotes Brg1 expression, which in turn initiates Chd7 expression (He et al., 2016). The successive chromatin remodeling events regulated by a CHD8-BRG1-CHD7 cascade likely establish the checkpoints to ensure oligodendrocyte lineage progression in a timely and orderly manner and prevent stage-inappropriate gene program activation.
CHD8 establishes an accessible chromatin landscape and recruits histone methyltransferase complexes to promote oligodendrocyte differentiation
Using transcriptome profiling and stage-specific ChIP-seq analyses, we created an unbiased genome-wide occupancy map showing that CHD8 selectively targets OPC differentiation and myelinogenic factors at different stages of oligodendrocyte development. Evaluation of chromatin accessibility by ATAC-seq indicated that loss of CHD8 resulted in closed chromatin conformations in the promoter regulatory regions of differentiation-associated gene loci, suggesting that CHD8 is essential for increasing accessibility near lineage-specific regulatory genes to promote the OPC differentiation program.
In eukaryotes, H3K4me3 is a major chromatin modification associated with actively transcribed promoters (Santos-Rosa et al., 2002; Strahl and Allis, 2000). We found that H3K4me3 marks were substantially reduced in Chd8-mutant OPCs, suggesting that CHD8 induces deposition of H3K4me3 marks over promoter regions of the genes involved in OPC development. The chromodomain in CHD family chromatin remodelers has been reported to bind the methylated histone H3 tail (Flanagan et al., 2005; Saha et al., 2006). CHD8 may recognize the H3K4me3 mark to further promote an open chromatin structure.
KMT2/MLL histone methyltransferase complexes, which belong to the trithorax group protein family (Kingston and Tamkun, 2014), specifically methylate histone H3 to produce the H3K4me3 mark. The H3K4me3 may form a pre-initiation complex by interacting with other transcriptional regulators to activate gene expression (Cano-Rodriguez et al., 2016). Our data demonstrate that CHD8 forms a complex with KMT2 components in OPCs and that the loss of CHD8 causes a failure of the KMT2 complex to target the promoter regions of oligodendrocyte lineage genes. Therefore, CHD8-mediated chromatin remodeling activity has a dual function in both establishing an open chromatin landscape and recruiting histone methyltransferase to enhance H3K4me3 deposition in lineage regulatory genes for promoting OPC proliferation and differentiation programs (Figure 7M).
Broad relevance of CHD8 to human disease
CHD8 mutations are observed in subjects with a specific cohort of autism spectrum disorders, which often exhibit white matter defects (Barnard et al., 2015; Bernier et al., 2014; Deoni et al., 2015; Hardan et al., 2016). Consistently, Chd8 cKO mice develop myelination deficits. These mutant mice also exhibit tonic-clonic seizures, which are commonly seen in autistic patients (Steffenburg et al., 2003; Volkmar and Nelson, 1990; Wakeford et al., 2015), suggesting that myelination defects caused by CHD8 mutations contribute to seizures and epilepsy in autism disorders. In addition, we detected robust CHD8 expression in active remyelinating lesions in the brains of multiple sclerosis patients. The role of CHD8 in remyelination in the brain remains to be determined, however, previous studies indicate that the defects in developmental myelination likely generalize to remyelination (Zhang et al., 2018; Zhang et al., 2009). Nonetheless, we show that CHD8 is critical for OPC replenishment and remyelination in demyelinating lesions after injury in the spinal cord, suggesting a previously unrecognized role of CHD8 in white matter pathogenesis and myelin repair.
Although CHD7 and CHD8 have similar domain structures, deficiencies in the individual enzymes cause dissimilar human diseases. In contrast to the essential role of CHD8 in OPC development and differentiation, CHD7 mutation mainly causes a delay in oligodendrocyte myelination (He et al., 2016). This may be due to their distinct substrate specificities and remodeling activities (Manning and Yusufzai, 2017). In addition, a differential sensitivity to gene dosage across species or dominant-negative effects of human mutant proteins might contribute to the phenotypic variation between the haploinsufficient humans and mouse mutants. Intriguingly, our genome-wide profiling study revealed that CHD8 also targets a number of genes previously linked to ASD (Liu et al., 2014; Willsey et al., 2013) in OPCs (Figure S7D), in addition to those involved in oligodendrocyte development. It remains to be defined whether CHD8-targeted ASD-associated genes regulate CNS myelination.
Our data indicate that CHD8 interacts with and recruits histone methyltransferase complex components to activate the transcriptional program for oligodendrocyte lineage commitment. Mutations in these KMT2/MLL complex proteins are also found in individuals with white matter abnormalities (Berdasco and Esteller, 2013; Matsumoto and Niikawa, 2003; Rangasamy et al., 2013), suggesting a shared molecular network of CHD8 and histone methyltransferase complexes underlies white matter malformations in various human disorders. Importantly, selective inhibition of the histone demethylase KDM5, which enhanced H3K4me3 deposition, partially restored myelination defects in Chd8 cKO mice. Thus, our observations revealed that CHD8 cooperates with KMT2 histone methyltransferases and acts as a key molecular nexus of the regulatory network that activates the oligodendrocyte lineage transcriptional program. It is conceivable that enhancing CHD8-KMT2/MLL complex activity might potentiate and promote oligodendrocyte differentiation, resulting in induction of remyelination and ameliorating demyelinating symptoms of neurological disorders such as ASD and multiple sclerosis.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CHD8 antibody | Abcam | Cat# ab84527, RRID:AB_1860139 |
| Rabbit anti-CHD8 antibody | Bethyl Laboratories | Cat# A301-225A, RRID:AB_890577 |
| Rabbit anti-CHD7 mAb (D3F5) antibody | Cell Signaling Technology | Cat# 6505 RRID:AB_11220431 |
| Mouse anti-HA-Tag (6E2) antibody | Cell Signaling Technology | Cat# 2367S, RRID:AB_10691311 |
| Mouse anti-c-Myc Antibody (9E10) | Santa Cruz biotechnology | Cat# sc-40, RRID:AB_627268 |
| Rabbit anti-DYKDDDDK Tag (D6W5B) antibody | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| Sheep Anti-Digoxigenin Fab fragments Antibody, AP Conjugated | Roche | Cat# 11093274910, RRID:AB_514497 |
| Goat anti-Sox10 | R&D systems | Cat# AF2864, RRID:AB_442208 |
| Rabbit anti-Nogo A | Millipore | Cat# AB5888, RRID:AB_1587321 |
| Chicken anti-MBP | Millipore | Cat# AB9348, RRID:AB_11213157 |
| Rabbit anti-Olig2 | Millipore | Cat#AB9610; RRID:AB_10141047 |
| Rat anti-PDGFRα | BD Bioscience | Cat# 558774; RRID:AB_397117 |
| Mouse anti-APC (CC1) | Oncogene Research | Cat#OP80; RRID:AB_2057371 |
| Goat anti-MBP | Santa Cruz biotechnology | Cat#sc-13914; RRID:AB_648798 |
| Rabbit anti-GFAP | Invitrogen | Cat# 13-0300, RRID:AB_2532994 |
| Rabbit anti-cleaved caspase 3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Rabbit anti-GFP | Molecular Probes | Cat# A-11122, RRID:AB_221569 |
| Rabbit Monoclonal anti-Sox10 | Abcam | Cat# ab180862, RRID:AB_2721184 |
| Mouse anti-NeuN | Millipore | Cat# MAB377, RRID:AB_2298772 |
| Rabbit anti-CHD8 | Abcam | Cat# ab114126, RRID:AB_10859797 |
| H3K4me3 antibody | Millipore | Cat# 07-473, RRID:AB_1977252 |
| H3K27Ac antibody | Active motif | Cat# 39135, RRID:AB_2614979 |
| Mouse anti-O4 | Millipore | Cat# MAB345; RRID:AB_94872 |
| Cy™2 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno Research Laboratories | Cat# 715-225-151, RRID:AB_2340827 |
| Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno Research Laboratories | Cat# 711-165-152, RRID:AB_2307443 |
| Cy™5 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immuno Research Laboratories | Cat# 705-175-147, RRID:AB_2340415 |
| Bacterial and Virus Strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | NEB | C2987 |
| Ad-GFP Adenovirus | Vector biolabs | 1060 |
| Ad-Cre-GFP Adenovirus | Vector biolabs | 1700 |
| Biological Samples | ||
| Mouse Optic Nerve, cortex, spinal cord | This study | N/A |
| Human cerebellum white matter tissue | CCHMC pathology core | N/A |
| Human Multiple Sclerosis patient brain tissue | United Kingdom MS tissue bank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CPI-455 | Selleckchem | Cat# S8287 |
| Tamoxifen | Sigma | T5648 |
| Sunflower oil | Sigma | S5007 |
| Lysophosphatidylcholine | Sigma | L4129 |
| PolyJet | SignaGen Laboratories | SL100688 |
| MinElute PCR Purification Kit | Qiagen | ID: 28004 |
| Pierce™ Protein A/G Magnetic Beads | Thermofisher | 88802 |
| Human PDGF AA | Shenandoah Biotechnology | 100-16 |
| Human FGF-basic 154 aa | Shenandoah Biotechnology | 100-146 |
| Insulin from bovine pancreas | Sigma-Aldrich | I6634 |
| iScript™ cDNA Synthesis Kit, 100 × 20 μl rxns | Bio-Rad | #1708891 |
| Agencourt AMPure XP | Beckman coulter | A64880 |
| NAiMAX | ThermoFisher Scientific | Cat#13778030 |
| DIG RNA Labeling Mix | Roche | Cat# 11 277 073 910 |
| T3 RNA polymerase | Promega | P2083 |
| T7 RNA polymerase | Promega | P207B |
| BCIP®/NBT Alkaline Phosphatase Substrate | Sigma | B5655 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| TRIzol reagent | ThermoFisher Scientific | Cat#15596018 |
| Critical Commercial Assays | ||
| NEBNext ChIP-seq Library Prep Master Mix Set for Illumina | NEB | cat # E6240L |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1024 |
| Nucleofector Kits | Lonza | Cat# VPI-1006 |
| Deposited Data | ||
| ATAC-seq | This paper | GEO: GSE107919 |
| H3K4me3 ChIP-seq | This paper | GEO: GSE107919 |
| RNA-seq | This paper | GEO: GSE107919 |
| CHD8 ChIP-seq | This paper | GEO: GSE107919 |
| BRG1 ChIP-seq | (Yu et al. 2013) | GEO: GSE42454 |
| CHD7 ChIP-seq | (He et al. 2016) | GEO: GSE72727 |
| Experimental Models: Cell Lines | ||
| Rat OPC primary culture | This study | N/A |
| Mouse OPC | This study | N/A |
| Rat: CG4 cell line | Dr. Jean-Claude Louis, University of California | RRID: CVCL_0210 |
| Human: HEK 293 cell | ATCC | CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Chd8 flox/flox | This paper | N/A |
| Mouse: Olig1 Cre/+ | (Xin et al., 2005) | N/A |
| Sprague Dawley® Rats | Charles River Laboratories | N/A |
| Mouse: PDGFRα-CreER | (Kang et al. 2010) | N/A |
| Mouse: Synapsin1-Cre | (Zhu et al. 2001) | N/A |
| Mouse: Brg1 flox/flox | (Yu et al. 2013) | N/A |
| Oligonucleotides | ||
| Chd8 flox Mouse genotyping F: 5′-AGTACACTGCGGGATAAAGTGAC-3′ | This paper | N/A |
| Chd8 flox Mouse genotyping R: 5′-ACCTAAGATTGAAACAAGAACCTATC-3′ | This paper | N/A |
| Mouse Gapdh qPCR Forward: TGCCAAATATGATGACATCAAGAA | This paper | N/A |
| Mouse Gapdh qPCR Reverse: GGAGTGGGTGTCGCTGTTG | This paper | N/A |
| Mouse Sox10 qPCR Forward: GTTGGTACTTGTAGTCCGGATG | This paper | N/A |
| Mouse Sox10 qPCR Reverse: GTACCCTCACCTCCACAATG | This paper | N/A |
| Mouse Plp1 qPCR Forward: TGCTCGGCTGTACCTGTGTACATT | This paper | N/A |
| Mouse Plp1 qPCR Reverse: TACATTCTGGCATCAGCGCAGAGA | This paper | N/A |
| Mouse Cnp qPCR Forward: TCCACGAGTGCAAGACGCTATTCA | This paper | N/A |
| Mouse Cnp qPCR Reverse: TGTAAGCATCAGCGGACACCATCT | This paper | N/A |
| Mouse Mbp qPCR Forward: TCACAGAAGAGACCCTCACA | This paper | N/A |
| Mouse Mbp qPCR Reverse: GCCGTAGTGGGTAGTTCTTG | This paper | N/A |
| Mouse Mag qPCR Forward: ACAGCGTCCTGGACATCGTCAACA | This paper | N/A |
| Mouse Mag qPCR Reverse: CATACAACTGACCTCCACTTCCGT | This paper | N/A |
| Mouse Myrf qPCR Forward: CAGACCCAGGTGCTACAC | This paper | N/A |
| Mouse Myrf qPCR Reverse: TCCTGCTTGATCATTCCGTTC | This paper | N/A |
| Mouse Hes5 qPCR Forward: AGCTACCTGAAACACAGCAAAGCC | This paper | N/A |
| Mouse Hes5 qPCR Reverse: TAAAGCAGCTTCATCTGCGTGTCG | This paper | N/A |
| Mouse Id4 qPCR Forward: CTGTGCCTGCAGTGCGATATGAA | This paper | N/A |
| Mouse Id4 qPCR Reverse: TGCAGGATCTCCACTTTGCTGACT | This paper | N/A |
| Mouse Sp5 qPCR Forward: TTCGTGTGCAACTGGCTCTT | This paper | N/A |
| Mouse Sp5 qPCR Reverse: AGGTGATCGCTTCGCATGAA | This paper | N/A |
| Mouse Chd8 qPCR Forward: GTGAAGACGAGAAGGAAGAGAAG | This paper | N/A |
| Mouse Chd8 qPCR Reverse: GGGAATCCATCTTGGGACATAG | This paper | N/A |
| Rat Gapdh qPCR Forward: TCCAGTATGACTCTACCCACG | This paper | N/A |
| Rat Gapdh qPCR Reverse: CACGACATACTCAGCACCAG | This paper | N/A |
| Rat Mbp qPCR Forward: TTGACTCCATCGGGCGCTTCTTTA | This paper | N/A |
| Rat Mbp qPCR Reverse: TTCATCTTGGGTCCTCTGCGACTT | This paper | N/A |
| Rat Cnp qPCR Forward: CTACTTTGGCAAGAGACCTCC | This paper | N/A |
| Rat Cnp qPCR Reverse: AGAGATGGACAGTTTGAAGGC | This paper | N/A |
| Rat Plp1 qPCR Forward: TCTTTGGCGACTACAAGACCACCA | This paper | N/A |
| Rat Plp1 qPCR Reverse: CAAACAATGACACACCCGCTCCAA | This paper | N/A |
| Rat Hes5 qPCR Forward: ACCAGCCCAACTCCAAAC | This paper | N/A |
| Rat Hes5 qPCR Reverse: AGTAACCCTCGCTGTAGTCC | This paper | N/A |
| Rat Myrf qPCR Forward: ACTGCCAACAACATGCGGAAGAAG | This paper | N/A |
| Rat Myrf qPCR Reverse: TGGGTTAGAGGCCCGAACAATGAT | This paper | N/A |
| Rat Mag qPCR Forward: ACAGCGTCCTGGACATCATCAACA | This paper | N/A |
| Rat Mag qPCR Reverse: ATGCAGCTGACCTCTACTTCCGTT | This paper | N/A |
| Rat Ash2L qPCR Forward: GCTCTGTGGATGAGGAGAATG | This paper | N/A |
| Rat Ash2L qPCR Reverse: GACGTGTCTATTCCAAAGGTGTC | This paper | N/A |
| Rat Wdr5 qPCR Forward: GGCCACAAGAATGAGAAGTAC | This paper | N/A |
| Rat Wdr5 qPCR Reverse: AGGTTATCTTCAGAGCCAGAC | This paper | N/A |
| Rat Brg1 qPCR Forward: TGAGAAGCTGGAGAAGCAGCAGAA | This paper | N/A |
| Rat Brg1 qPCR Reverse: ACAGCCTTGGTGAGTTTCTGGAGT | This paper | N/A |
| Rat Baf45 qPCR Forward: AACACACAGCCAAGAAAGCACCAG | This paper | N/A |
| Rat Baf45 qPCR Reverse: ACCGTGAACTGTAAACACGAGGGA | This paper | N/A |
| Rat Baf45d qPCR Forward: AAAGAAGACTCCCAACCACCCACT | This paper | N/A |
| Rat Baf45d qPCR Reverse: ACAGTAGTTGTTAGGCAGGGCCAA | This paper | N/A |
| Rat Baf60a qPCR Forward: ATGATTGGCTTCAATCCCAGTGCC | This paper | N/A |
| Rat Baf60a qPCR Reverse: TATGTGTTTCGGATTCCCAGGGCT | This paper | N/A |
| Rat SMARCA2 qPCR Forward: TAATGGCACCCTGAAGCACTACCA | This paper | N/A |
| Rat SMARCA2 qPCR Reverse: TGAGTGCAATGGTCTGGATGGTCT | This paper | N/A |
| Rat Baf250a qPCR Forward: AGATCGTGCAGAAGAATGACCCGT | This paper | N/A |
| Rat Baf250a qPCR Reverse: AAGGCAGCAGCTCTATCTTGCTCT | This paper | N/A |
| Rat Olig2 ChIP qPCR Forward: AGTAGCGTTTGGAATGTGAACTC | This paper | N/A |
| Rat Olig2 ChIP qPCR Reverse: TCAAACGCATCCGCCTTATC | This paper | N/A |
| Rat Pdgfra ChIP qPCR Forward: GTACCTTTGGGATGGATGTTCT | This paper | N/A |
| Rat Pdgfra ChIP qPCR Reverse: TGAAGAAACGAGGACCAGATAAC | This paper | N/A |
| Rat Sox10 ChIP qPCR Forward: TGTGAACAAGTCAAGGGTGAG | This paper | N/A |
| Rat Sox10 ChIP qPCR Reverse: GCTTTGCTATCTCTCCATTCTG | This paper | N/A |
| Rat Zfp365 ChIP qPCR Forward: CCTGGCTCAGCCTGATTTAC | This paper | N/A |
| Rat Zfp365 ChIP qPCR Reverse: TCCAGCCGAGACCACTTA | This paper | N/A |
| Rat ChIP qPCR Negative Control Forward: CATGCGAATCCTATTGGGAACC | This paper | N/A |
| Rat ChIP qPCR Negative Control Reverse: GTTCCCAATGGGACATGCTTG | This paper | N/A |
| Chd8 siRNA: CCAAGUACUUCCAUGGUUU[dT][dT] | Sigma-Aldrich | SASI_Rn02_00263627 |
| Ash2L siRNA: UCCAAGGAUAAGGAUAUUA[dT][dT] | Sigma-Aldrich | SASI_Rn02_00215522 |
| WDR5 siRNA: GCUCUGAAGAUAACCUGGU[dT][dT] | Sigma-Aldrich | SASI_Rn02_00204788 |
| Recombinant DNA | ||
| p3XFLAG-CMV-14-WDR5 | Addgene | #59974 |
| hCHD8 | (Durak et al. 2016) | N/A |
| Lentiviral vector pSin4-EF2-Ash2L-IRES-Puro | Laboratory of Jiang Wu, UT Southwestern | N/A |
| pX458 vector | Addgene | #48138 |
| Software and Algorithms | ||
| GSEA | Broad Institute | http://www.broadinstitute.org/gsea/index.jsp |
| ToppCluster | Cincinnati Children’s Hospital | https://toppcluster.cchmc.org |
| GraphPad Prism 6.00 R language | GraphPad | www.graphpad.com |
| R Core Team (2016) The R Project for Statistical Computing | http://www.r-project.org | |
| MEME | University of Queensland | http://meme-suite.org |
| HOMER | University of California San Diego | http://homer.ucsd.edu/homer/motif/ |
| Bowtie2 | University of John Hopkins | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Online CRISPR design tool | Broad Institute | http://www.genome-engineering.org/ |
| Samtools | Wellcome Trust Sanger Institute | http://samtools.sourceforge.net/ |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Q. Richard Lu (Richard.lu@cchmc.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals/Human Subjects
Mice homozygous for floxed alleles of Chd8 (Chd8f/f) were crossed with Olig1-Cre+/− mice to generate Chd8 cKO (Chd8f/f;Olig1-Cre+/−) and heterozygous control (Chd8f/+;Olig1-Cre+/−) mice. PDGFRa-CreERT mice and ccGFP reporter mice (Ai6, Jax laboratory) were crossed with Chd8f/f mice to generate the OPC-specific Chd8 OPC-iKO. Both male and female animals were used in the developmental study. All mice used in experiments were maintained on a mixed C57/Bl6;129Sv background and housed in a pathogen-free vivarium with a 12-hour light/dark cycle with free access to normal chow food and water. Mouse embryos harvested at different ages with E0.5 considered the time of vaginal plug. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Cincinnati Children’s Hospital Medical Center.
All human patient images and tissues were obtained with informed consent as outlined by the institutional review board at the Children’s Hospital of Fudan University, China, the Institut du Cerveau et de la Moelle Épinière - ICM, France, and the Cincinnati Children’s Hospital Medical Center, USA.
Generation of Chd8-floxed mice with CRISPR-Cas9 system
To create genome-edited Chd8 floxed mice, we designed single guide RNA (sgRNA) sequences using the online CRISPR design tool (http://www.genome-engineering.org/). Two sgRNAs that targeted Chd8 introns 3 and 4 were selected based on predictions that their probabilities of off-target sites of action were very low. The annealed complementary oligonucleotides of selected sgRNA sequences were then cloned into a modified pX458 vector (addgene #48138) by flipping an A-T base pair and extending the Cas9-binding hairpin structure. Editing activity was validated via T7E1 assay in mK4 cells, and validated sgRNAs and Cas9 were transcribed in vitro. Both the Cas9 mRNA and the sgRNAs were purified using MEGAclear kit (Life Technologies), and then microinjected using standard methods into the cytoplasm of B6D2F2 fertilized eggs. Injected eggs were transferred into the oviductal ampulla of pseudopregnant CD-1 females. Pups were born and genotyped by PCR screening.
METHOD DETAILS
Genotyping and Sanger sequencing of Chd8 floxed transgenic lines
Mouse tail tip biopsies were collected for genotyping via PCR with the following primer pairs: forward primer 5′-AGTACACTGCGGGATAAAGTGAC-3′ and reverse primer 5′-ACCTAAGATTGAAACAAGAACCTATC-3′ for the 5′ loxP sites. The wild type and floxed alleles gave products of 336 bp and 380 bp, respectively. The 3′ loxP sites were amplified using forward primer 5′-ACACCATGGAAGCTCATGATGC-3′ and reverse primer 5′-CAAGAACTCAACAGGGAAGCAC-3′. Here, the wild type and floxed alleles gave products of 342bp and 387bp, respectively. PCR products were purified using the Qiagen PCR cleanup kit per the manufacturer’s instructions. Purified PCR products were then sent for Sanger sequencing with the responsive forward primers. At least three Chd8 floxed mice were identified and gave rise to the same phenotypes.
Tissue processing and in situ hybridization
Mice at various developmental stages were anesthetized with ketamine/xylazine and perfused with PBS followed by 4% paraformaldehyde (PFA). Spinal cords or brains were dissected, fixed in 4% PFA overnight, dehydrated in 20% sucrose at 4°C, embedded in OCT and cryosectioned at 15 μm. Both male and female animals were used for the study. For in situ hybridization, cryosections of CNS tissues were used with digoxigenin (DIG)-labeled antisense riboprobes specific for Mbp, PDGFRa, Olig2 and Plp1 as previously described (He et al., 2016). Antisense riboprobes were synthesized with T3 or T7 RNA polymerase (Promega, P207B and P2083) and labeled with DIG RNA Labeling Mix (Roche, Cat# 11 277 073 910). An anti-DIG antibody conjugated to alkaline phosphatase (Roche, Cat# 11093274910) was applied to the probe hybridized tissue sections, and stained with 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) chromogenic substrates (Sigma, B5655). The step-by-step in situ hybridization protocol is available upon request.
Transmission Electron microscopy
Tissue processing was performed as described previously (He et al., 2016). In brief, mice were deeply anesthetized with ketamine/xylazine, perfused with 0.1M cacodylate, followed by 2.5% paraformaldehyde/2.5% glutaraldehyde in 0.1 M cacodylate (pH 7.2). Spinal cord and optic nerves were dissected and postfixed in 1% OsO4. Ultrathin sections were stained with lead citrate for electron microscopy imaging.
Cell culture, siRNA and plasmids transfection and transduction
Primary OPCs were obtained from newborn rat brains after preparation of mixed glial cultures and shake-off (Chen et al., 2007). Briefly, mixed glial cells were initially cultured in DMEM-F12 medium supplied with 15% FBS, then switched to B104 conditioned medium for 2 days before isolating OPCs by mechanical detachment in an orbital shaker. Isolated rat OPCs were grown in Sato growth medium supplemented with mitogens 10 ng/ml PDGF-AA and 10 ng/ml bFGF, which maintained OPC in A2B5+/PDGFRα+ state with minimal O4+ differentiating oligodendrocyte. OPCs were differentiated in the oligodendrocyte Differentiation Medium (Sato medium supplemented with 15 nM T3 and 10 ng/ml ciliary neurotrophic factor). Plasmids expressing Chd8, Ash2L, and Wdr5 were transfected into 293 cells using polyJet (SignaGen Laboratories, Catalog #: SL100688) or using AMAXA Nucleofector™ Technology (Lonza). For gene-specific knockdown, primary OPCs were transfected with siRNAs (sigma), and shifted to the differentiation medium one day after transfection and maintained for 72 hours.
Mouse OPCs were isolated from P6-P7 cortices of Chd8 floxed mice by immunopanning with antibodies against Ran-2, GalC and O4 sequentially as previously described (Wang et al., 2017). OPCs were transduced with adenovirus carrying GFP control or Cre-GFP for 4 days in Sato growth medium without mitogens and analyzed by MBP immunofluorescence. Mouse OPCs were isolated from the cortices of control and Chd8 cKO mice by immunopanning and treated with KDM5 inhibitor 15 μM CPI-455 (Selleckchem, cat# S8287) or DMSO vehicle control for 96 hours. For in vivo treatment, control and Chd8 cKO pups from the same litter were treated with 10 μl KDM5 inhibitor CPI-455 (15 mM) or DMSO vehicle by IP injection at P1, P3, P5 and P7. The spinal cord samples were harvested at P14 for analysis.
Immunohistochemistry and immunoblotting
Cell culture on coverslip were fixed with 4% PFA for 8min and quenched with 10mM Glycine for 30min and then blocked in blocking buffer (0.02% Triton X-100 and 5% normal donkey serum in PBS) for 20min and incubate with primary antibodies for 40min at room temperature. Cryosections (12-μm thick) or vibratome sections (50-μm thick) were permeabilized and blocked in blocking buffer (0.3% Triton X-100 and 5% normal donkey serum in PBS) for 1 h at room temperature and overlaid with primary antibodies overnight at 4 °C. Antibodies used in the study were: rabbit anti-Olig2 (Millipore, AB9610), rat anti-PDGFRα (BD Bioscience, 558774), mouse anti-APC (CC1, Oncogene Research, OP80), goat anti-MBP (Santa Cruz Biotechnology, sc-13914), rabbit anti-CHD8 (Abcam, ab84527), rabbit anti-GFAP (Invitrogen, #13-0300), rabbit anti-cleaved caspase 3 (Cell Signaling Technology, #9661), rabbit anti-GFP (Molecular Probes, #A-11122), Rabbit Monoclonal anti-Sox10 (Abcam, ab180862), mouse anti-NeuN (Millipore, MAB377). After washing with 0.2% Triton X-100 in PBS, cells or brain sections were incubated with secondary antibodies conjugated to Cy2, Cy3 or Cy5 (Jackson ImmunoResearch Laboratories) for 2 h at room temperature, stained in DAPI for 5 min, washed in PBS and mounted with Fluoromount-G (SouthernBiotech). Cell counting was carried out in a double-blind manner.
Lysolecithin-induced demyelinating injury
Lysolecithin-induced demyelination was carried out in the ventrolateral spinal white matter of approximately 8-week-old male mice as previously described (He et al., 2016). Briefly, after exposing the spinal vertebrae at the level of T9-T12, meningeal tissue in the intervertebral space was cleared, and the dura was pierced with a dental needle. 0.5 μl of 1% lysolecithin (l-a-lysophosphatidylcholine, Sigma L4129) was injected via a Hamilton syringe attached to a glass micropipette into the ventrolateral white matter using a stereotactic apparatus. Injuries were conducted in a genotype-blinded manner. The date of LPC injection was denoted as 0 dpl. Mice were then left for indicated periods and subsequently perfused for immunohistochemical and electron microscopic analysis. Tamoxifen (Sigma, T5648, 90 mg/ml) was dissolved in 100% ethanol and then diluted in autoclaved sunflower oil (Sigma, S5007) to a final concentration of 10 mg/ml. Tamoxifen was i.p injected into mice at 45 mg/kg once per day.
Histochemistry analysis of multiple sclerosis lesions
Autopsy brain tissue samples from patients with confirmed secondary progressive MS were obtained from the United Kingdom MS tissue bank (Richards Reynolds, Imperial College, London). MS tissue block containing active lesions and periplaque white matter were selected for analysis. Cryosections (14 μm thick) of snap frozen MS brain tissue were permeabilized and blocked in blocking buffer (0.05% Triton X-100 and 10% normal goat serum in PBS) for 1h and overlaid with primary antibodies overnight at 4 °C. Antibodies used in the study were: rabbit anti-CHD8 (Bethyl, A301-225A, 1:1000), goat anti-Sox10 (R&D systems, AF2864, 1:400), rabbit anti-Nogo A (Millipore, AB5888, 1:200), chicken anti-MBP (Millipore, AB9348, 1/50). After washing with 0.05% Triton X-100 in PBS, sections were incubated with secondary antibodies conjugated to Alexa488, Alexa594 or Alexa647 (Thermo, 1:1,000) and DAPI for 1h at room temperature, washed in PBS and mounted with Fluoromount-G (SouthernBiotech). Pictures of were taken with Zeiss microscope using apotome. Z-stack was used to average 5–7 planes and pictures were treated and cells were counted using Zen and imageJ software packages.
Fluorescence activated cell sorting of primary mouse OPC
For OPC sorting, single cell suspension was prepared from cortices with papain dissociation and cells were re-suspended in PBS with 2 mM EDTA and 0.5% BSA and incubated with rat anti- CD140a/PDGFRα (BD Bioscience) and O4+ primary monoclonal antibody. Isotype and fluorescence-minus one controls were used to set appropriate gates. After sorting, cells were directly subjected to ATAC-seq, ChIP-seq, and RNA-seq profiling.
RNA extraction, qRT-PCR and RNA-seq
RNA was extracted from cell culture or optic nerve tissue of mutant mice and their littermate controls. Total RNA was extracted per the Trizol (Life Technologies) protocol. cDNA was generated with iScript™ cDNA Synthesis Kit (Bio-Rad). qRT-PCR was performed using the ABI Prism 7700 Sequence Detector System (Perkin-Elmer Applied Biosystems). qRT-PCR primers are available upon request. RNA-seq libraries from control and mutant OPC cells were prepared using Illumina RNA-Seq Preparation Kit and sequenced using a HiSeq 2500 sequencer.
Assay for transposase-accessible chromatin using sequencing (ATAC-Seq)
ATAC-seq assays were performed as previously described (Buenrostro et al., 2013). Briefly, we isolated nuclei of 30,000 sorted cells from the mouse cortex in a cold lysis buffer. Immediately after the nuclei preparation, we performed the transposase reaction for 30 min at 37 °C. The samples were purified using a Qiagen MinElute kit. Following purification, we amplified library fragments using 1× NEBnext PCR master mix. The libraries were purified using a Qiagen MinElute kit and then run on the Illumina sequencer HS2500.
Chromatin-immunoprecipitation followed by sequencing (ChIP-seq) and ChIP-qPCR
ChIP-seq assays were performed as previously described. In brief, chromatin was cross-linked and sheared with a Covaris S220 sonicator from primary oligodendrocyte cultures of newborn rat or mouse cortex. Immunoprecipitation was overnight at 4°C using antibodies directed against or control immunoglobulins in the presence of protein A/G Sepharose CL-4B beads (Life Technologies) pretreated with BSA. Rabbit anti-CHD8 (Abcam, ab114126), H3K4me3 (Millipore cat# 07-473) and H3K27Ac (active motif cat# 39135) antibodies were used for immunoprecipitation. After reverse crosslink, proteinase K treatment, the precipitated chromatin DNA was purified and prepared library using NEBNext ChIP-seq Library Prep Master Mix Set for Illumina (NEB cat # E6240L), and then to sequence on the Illumina sequencer HS2500.
RNA-Seq, ATAC-seq and ChIP-seq data analysis
All RNA-Seq data were aligned to 10mm using TopHat with default settings. We used Cuff-diff to (1) estimate FPKM values for known transcripts and (2) analyze differentially-expressed transcripts. In all differential expression tests, a difference was considered significant if the q value was less than 0.05 (Cuff-diff default). A heatmap of gene expression was generated using R language (http://www.r-project.org). Reads of ATAC-seq and ChIP-seq data were aligned to mm10 and Rn5 using Bowtie with the following options: -p 8, -m 1 (http://bowtie-bio.sourceforge.net). Peak calling was performed using MACS (Model-based Analysis of ChIP-seq) (http://liulab.dfci.harvard.edu/MACS) with a p value cutoff of 1x e−9. Motif was analyzed using HOMER program (http://homer.salk.edu/homer). GO-analysis of differentially expressed genes was performed using Gene Set Enrichment (GSEA, http://www.broadinstitute.org/gsea/index.jsp). We used ToppCluster (https://toppcluster.cchmc.org/) to construct the network of induced and suppressed genes belonging to over-represented GO-term categories. Enriched motifs of CHD8 binding peaks in oligodendrocytes were identified by MEME (http://meme-suite.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were done using GraphPad Prism 6.00 (San Diego California, www.graphpad.com). All data are shown as mean ± s.e.m. Data distribution was assumed to be normal, but this was not formally tested. Statistical significance was determined using two-tailed Student’s t tests or Wilcoxon rank-sum and signed-rank tests as indicated. One-way ANOVA test was performed by multiple comparisons or pairwise comparisons following Turkey’s ranking tests when comparing multiple groups. Significance was set as * for p < 0.05, ** for p < 0.01, and *** p < 0.001, unless otherwise indicated. In MEME motif analysis, E value shows the significant enrichment of de novo motifs while p value is shown the significant similarity between de novo motif and known motif. Quantifications were performed from at least three independent experiments. No randomization was used to collect all the data, but they were quantified blindly.
DATA AND SOFTWARE AVAILABILITY
Accession codes
All the RNA-seq, ATAC-seq, and ChIP-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE 107919.
Supplementary Material
Highlights.
Autism-associated CHD8 required for oligodendrocyte development and survival
Chromatin regulator CHD8 required for OPC proliferation and remyelination
CHD8-BRG1-CHD7 cascade regulates oligodendroglial lineage progression
CHD8 recruits KMT2 histone methyltransferase to promote oligodendrocyte development
Acknowledgments
The authors thank Drs. Michael Wegner, Ron Waclaw and Ed Hurlock for critical comments. We thank Drs. Lihui Tsai and Jiang Wu for CHD8 and ASH2L-expressing vectors, respectively, Dr. Yuehchiang Hu for generating transgenic lines, and Jinglong Xu for technical support. This study was funded in part by grants from the US National Institutes of Health R01NS072427 and R01NS075243 to QRL, the National Multiple Sclerosis Society (RG1508) to QRL, and the CHARGE syndrome Foundation to QRL and CZ, and National Natural Science Foundation of China 81720108018 to WZ, the National Multiple Sclerosis Society (NMSS RG-1501-02851) to CP and QRL, and the Fondation pour l’Aide à la Recherche sur la Sclérose en Plaques (ARSEP, 2014, 2015, 2017) to CP.
Footnotes
AUTHOR CONTRIBUTIONS
C.Z. and Q.R.L. designed the experiments, analyzed the data and wrote the manuscript with input from all authors. C.Z and C.D. carried out the majority of experiments. Y.D. F.Z., L.X. Z.M., X.D., Y.L., S.K., L.W. assisted in vitro, in vivo, and gene profiling analyses. M.F. C.M., B.N., C.P. examined MS tissues. D.M., M.X., C.P., W.Z. provided resources and inputs. Q.R.L. supervised the project.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Supplemental Information includes seven figures and two movies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banelli B, Daga A, Forlani A, Allemanni G, Marubbi D, Pistillo MP, Profumo A, Romani M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget. 2017;8:34896–34910. doi: 10.18632/oncotarget.16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard RA, Pomaville MB, O’Roak BJ. Mutations and Modeling of the Chromatin Remodeler CHD8 Define an Emerging Autism Etiology. Front Neurosci. 2015;9:477. doi: 10.3389/fnins.2015.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsukh T, Pieper L, Koszucka AM, von Velsen N, Hoyer-Fender S, Elbracht M, Bergman JE, Hoefsloot LH, Pauli S. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet. 2010;19:2858–2866. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- Bercury KK, Macklin WB. Dynamics and Mechanisms of CNS Myelination Developmental cell. 2015;32:447–458. doi: 10.1016/j.devcel.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vultovan Silfhout AT, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof M, Weider M, Kuspert M, Nave KA, Wegner M. Brg1-dependent chromatin remodelling is not essentially required during oligodendroglial differentiation. J Neurosci. 2015;35:21–35. doi: 10.1523/JNEUROSCI.1468-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddaert N, Zilbovicius M, Philipe A, Robel L, Bourgeois M, Barthelemy C, Seidenwurm D, Meresse I, Laurier L, Desguerre I, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e4415. doi: 10.1371/journal.pone.0004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21–29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Rodriguez D, Gjaltema RA, Jilderda LJ, Jellema P, Dokter-Fokkens J, Ruiters MH, Rots MG. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat Commun. 2016;7:12284. doi: 10.1038/ncomms12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF. Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist. 2006;12:435–441. doi: 10.1177/1073858406290375. [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deoni SC, Zinkstok JR, Daly E, Ecker C, Williams SC, Murphy DG Consortium MA. White-matter relaxation time and myelin water fraction differences in young adults with autism. Psychol Med. 2015;45:795–805. doi: 10.1017/S0033291714001858. [DOI] [PubMed] [Google Scholar]
- Ding HF, Fisher DE. Mechanisms of p53-mediated apoptosis. Crit Rev Oncog. 1998;9:83–98. doi: 10.1615/critrevoncog.v9.i1.60. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai LH. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci. 2016;19:1477–1488. doi: 10.1038/nn.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Lu QR. Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb Perspect Biol. 2015;7:a020461. doi: 10.1101/cshperspect.a020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Kawauchi D, Korkel-Qu H, Deng H, Serger E, Sieber L, Lieberman JA, Jimeno-Gonzalez S, Lambo S, Hanna BS, et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat Commun. 2017;8:14758. doi: 10.1038/ncomms14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Franklin RJ. Remyelination of the demyelinated CNS: the case for and against transplantation of central, peripheral and olfactory glia. Brain Res Bull. 2002;57:827–832. doi: 10.1016/s0361-9230(01)00765-1. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Goldman SA. Glia Disease and Repair-Remyelination. Cold Spring Harb Perspect Biol. 2015;7:a020594. doi: 10.1101/cshperspect.a020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, Stradleigh TW, Pride MC, Schaffler MD, Wade AA, Catta-Preta R, et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci. 2017;20:1062–1073. doi: 10.1038/nn.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardan AY, Fung LK, Frazier T, Berquist SW, Minshew NJ, Keshavan MS, Stanley JA. A proton spectroscopy study of white matter in children with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2016;66:48–53. doi: 10.1016/j.pnpbp.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Marie C, Zhao C, Kim B, Wang J, Deng Y, Clavairoly A, Frah M, Wang H, He X, et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat Neurosci. 2016;19:678–689. doi: 10.1038/nn.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Wang J, Lu Y, Deng Y, Zhao C, Xu L, Chen Y, Hu YC, Zhou W, Lu QR. lncRNA Functional Networks in Oligodendrocytes Reveal Stage-Specific Myelination Control by an lncOL1/Suz12 Complex in the CNS. Neuron. 2017;93:362–378. doi: 10.1016/j.neuron.2016.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development. 2016;143:2882–2897. doi: 10.1242/dev.128892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537:675–679. doi: 10.1038/nature19357. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston RE, Tamkun JW. Transcriptional regulation by trithorax-group proteins. Cold Spring Harb Perspect Biol. 2014;6:a019349. doi: 10.1101/cshperspect.a019349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuspert M, Wegner M. SomethiNG 2 talk about-Transcriptional regulation in embryonic and adult oligodendrocyte precursors. Brain Res. 2016;1638:167–182. doi: 10.1016/j.brainres.2015.07.024. [DOI] [PubMed] [Google Scholar]
- Liu L, Lei J, Sanders SJ, Willsey AJ, Kou Y, Cicek AE, Klei L, Lu C, He X, Li M, et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol Autism. 2014;5:22. doi: 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BJ, Yusufzai T. The ATP-dependent chromatin remodeling enzymes CHD6, CHD7, and CHD8 exhibit distinct nucleosome binding and remodeling activities. The Journal of biological chemistry. 2017;292:11927–11936. doi: 10.1074/jbc.M117.779470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet C Semin Med Genet. 2003;117C:57–65. doi: 10.1002/ajmg.c.10020. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Banine F, Feistel K, Foster S, Xing R, Struve J, Sherman LS. Brg1 directly regulates Olig2 transcription and is required for oligodendrocyte progenitor cell specification. Dev Biol. 2016;413:173–187. doi: 10.1016/j.ydbio.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD. Motor skill learning requires active central myelination. Science. 2014;346:318–322. doi: 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Nakatani H, Martin E, Hassani H, Clavairoly A, Maire CL, Viadieu A, Kerninon C, Delmasure A, Frah M, Weber M, et al. Ascl1/Mash1 promotes brain oligodendrogenesis during myelination and remyelination. J Neurosci. 2013;33:9752–9768. doi: 10.1523/JNEUROSCI.0805-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Zhou Y, Slaymaker IM, Shetty AS, Weisbach NR, Kim JA, Sharma J, Desai M, Sood S, Kempton HR, et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017;19:335–350. doi: 10.1016/j.celrep.2017.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy S, D’Mello SR, Narayanan V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics. 2013;10:742–756. doi: 10.1007/s13311-013-0227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AP, Rodgers JM, Goings GE, Miller SD. Characterization of oligodendroglial populations in mouse demyelinating disease using flow cytometry: clues for MS pathogenesis. PLoS One. 2014;9:e107649. doi: 10.1371/journal.pone.0107649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH. Glial specification in the vertebrate neural tube. Nat Rev Neurosci. 2004;5:409–419. doi: 10.1038/nrn1389. [DOI] [PubMed] [Google Scholar]
- Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Steffenburg S, Steffenburg U, Gillberg C. Autism spectrum disorders in children with active epilepsy and learning disability: comorbidity, pre- and perinatal background, and seizure characteristics. Dev Med Child Neurol. 2003;45:724–730. doi: 10.1017/s0012162203001361. [DOI] [PubMed] [Google Scholar]
- Stolerman ES, Smith B, Chaubey A, Jones JR. CHD8 intragenic deletion associated with autism spectrum disorder. Eur J Med Genet. 2016;59:189–194. doi: 10.1016/j.ejmg.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Takahashi YH, Westfield GH, Oleskie AN, Trievel RC, Shilatifard A, Skiniotis G. Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A. 2011;108:20526–20531. doi: 10.1073/pnas.1109360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, Williamson KE, Guler GD, Gangurde P, Manieri W, et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol. 2016;12:531–538. doi: 10.1038/nchembio.2085. [DOI] [PubMed] [Google Scholar]
- Volkmar FR, Nelson DS. Seizure disorders in autism. J Am Acad Child Adolesc Psychiatry. 1990;29:127–129. doi: 10.1097/00004583-199001000-00020. [DOI] [PubMed] [Google Scholar]
- Wakeford S, Hinvest N, Ring H, Brosnan M. Autistic characteristics in adults with epilepsy and perceived seizure activity. Epilepsy Behav. 2015;52:244–250. doi: 10.1016/j.yebeh.2015.08.031. [DOI] [PubMed] [Google Scholar]
- Wang H, Moyano AL, Ma Z, Deng Y, Lin Y, Zhao C, Zhang L, Jiang M, He X, Ma Z, et al. miR-219 Cooperates with miR-338 in Myelination and Promotes Myelin Repair in the CNS. Developmental cell. 2017;40:566–582 e565. doi: 10.1016/j.devcel.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner M, Stolt CC. From stem cells to neurons and glia: a Soxist’s view of neural development. Trends Neurosci. 2005;28:583–588. doi: 10.1016/j.tins.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Weng Q, Chen Y, Wang H, Xu X, Yang B, He Q, Shou W, Higashi Y, van den Berghe V, Seuntjens E, et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron. 2012;73:713–728. doi: 10.1016/j.neuron.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, Lu QR. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci. 2005;25:1354–1365. doi: 10.1523/JNEUROSCI.3034-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–261. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhu X, Gui X, Croteau C, Song L, Xu J, Wang A, Bannerman P, Guo F. Sox2 Is Essential for Oligodendroglial Proliferation and Differentiation during Postnatal Brain Myelination and CNS Remyelination. J Neurosci. 2018;38:1802–1820. doi: 10.1523/JNEUROSCI.1291-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Argaw AT, Gurfein BT, Zameer A, Snyder BJ, Ge C, Lu QR, Rowitch DH, Raine CS, Brosnan CF, et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci U S A. 2009;106:19162–19167. doi: 10.1073/pnas.0902834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng Y, Liu L, Yu K, Zhang L, Wang H, He X, Wang J, Lu C, Wu LN, et al. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat Commun. 2016;7:10883. doi: 10.1038/ncomms10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.