

SUMMARY
Legionella pneumophila elicits caspase-11-driven macrophage pyroptosis through guanylate-binding proteins (GBPs) encoded on chromosome 3. It has been proposed that microbe-driven IFN upregulates GBPs to facilitate pathogen vacuole rupture and bacteriolysis preceding caspase-11 activation. We show here that macrophage death occurred independently of microbial-induced IFN signaling and that GBPs are dispensable for pathogen vacuole rupture. Instead, the host-intrinsic IFN status sustained sufficient GBP expression levels to drive caspase-1 and caspase-11 activation in response to cytosol-exposed bacteria. In addition,endogenous GBP levels were sufficient for the release of DNA from cytosol-exposed bacteria, preceding the cyclic GMP-AMP synthase/stimulator of interferon genes (cGAS/ STING) pathway for Ifnb induction. Mice deficient for chromosome 3 GBPs were unable to mount a rapid IL-1/chemokine (C-X-C motif) ligand 1 (CXCL1) response during Legionella-induced pneumonia, with defective bacterial clearance. Our results show that rapid GBP activity is controlled by host-intrinsic cytokine signaling and that GBP activities precede immune amplification responses, including IFN induction, inflammasome activation, and cell death.
Graphical Abstract

In Brief
Guanylate-binding proteins act upstream of many cytosolic pathogen sensors. It is assumed that infection-associated IFN signaling precedes GBP induction. Liu et al. find that host-intrinsic IFN signaling maintains GBPs in naive macrophages to mediate the disruption of cytosol-accessible bacteria. The findings elucidate a crucial role of tonic cytokines in maintaining immune readiness.
INTRODUCTION
Cytosolic presence of bacterial lipopolysaccharide (LPS) activates caspase-11, resulting in a lytic form of macrophage death known as pyroptosis (Kayagaki et al., 2011; Meunier and Broz, 2015; Yang et al., 2015). Vacuolar pathogens such as S. typhimurium and L. pneumophila predominantly bypass cas-pase-11 activation by maintaining pathogen replication vacuoles that protect the bacterium from cytosolic sensing (Isberg et al., 2009; LaRock et al., 2015). However, interferon (IFN)-activated macrophages can mount a caspase-11 response to vacuole-resident bacteria (Broz et al., 2012; Case et al., 2013) in a process dependent on guanylate-binding proteins (GBPs), a family of IFN-dependent immune guanosine triphosphatases (GTPases) (Meunier et al., 2014; Pilla et al., 2014). The mechanistic steps in which GBPs function remain controversial.
GBP proteins are undetectable by standard immunoblot probing procedures at a cellular resting state but are highly upregulated by IFNɣ and, to a lesser extent, IFNα/β (Kim et al., 2011; Yamamoto et al., 2012). In IFN-activated macrophages, several GBPs localize to pathogen-containing vacuoles (Kim et al., 2011; Meunier et al., 2014; Yamamoto et al., 2012) and have been functionally associated with pathogen vacuole rupture, bacterial killing, or both (Meunier et al., 2014, 2015; Man et al., 2016). In addition, induction of an IFN response during infection (Broz et al., 2012; Meunier et al., 2014; Man et al., 2015) has resulted in a model postulating that infection-driven IFN signaling is required for GBP synthesis and function (Meunier and Broz, 2015).
During L. pneumophila challenge, bacterial DNA/RNA trigger cytosolic sensors to induce Ifnb transcription within 4–6 hr of infection (Monroe et al., 2009; Lippmann et al., 2011), although secreted IFNβ protein is not detected until 20 hr post-infection (Coers et al., 2007). The disconnect between Ifnb gene transcription and protein accumulation is presumably due to inhibition of host cell protein translation during Legionella infection (Fontana et al., 2011; Ivanov and Roy, 2013; Asrat et al., 2014). In line with these findings, caspase-11 activation in response to vacuolar-resident L. pneumophila requires IFN priming. The need for IFN priming can be bypassed by a L. pneumophila mutant lacking the SdhA protein (⊿sdhA) (Monroe et al., 2009; Aachoui et al., 2013). This mutant strain forms an unstable vacuole that exposes the bacterium to the host cytosol (Laguna et al., 2006; Creasey and Isberg, 2012). Despite the cytosolic exposure of the bacterium, caspase-11 activation remains dependent on GBPs encoded on chromosome 3 (Pilla et al., 2014). The differential requirement of IFN priming for pyroptosis when challenged with vacuole-stable or cytosol-accessible bacteria calls for a re-evaluation of the function of GBPs upstream of caspase-11.
In this work, we investigated the initiation of antimicrobial responses toward a cytosolic bacterium. We found that GBPs are not involved in pathogen vacuole disruption and that cytosol-exposed L. pneumophila drove macrophage pyroptosis in the absence of microbial-induced IFN signaling. Constitutive IFN signaling maintains GBP expression at steady-state levels that are low but sufficient for function upstream of caspase-11 and caspase-1. We also found that chromosome 3 GBPs are required for the release of bacterial DNA, preceding the cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) induction of IFN. During Legionella-induced pneumonia, chromosome 3 (chr3) GBPs are required for the interleukin (IL)-1/ chemokine (C-X-C motif) ligand 1 (CXCL1) response for timely bacterial clearance.
RESULTS
Infection-Driven IFN Signaling Is Dispensable for Pyroptosis
All L. pneumophila strains used in the present study are deficient for flagellin (⊿flaA) to bypass the Naip5/NLRC4/caspase-1 cell death pathway that occurs on the C57BL/6 genetic background (Ren et al., 2006). L. pneumophila deficient for the SdhA protein (⊿sdhA) becomes cytosolic exposed (Laguna et al., 2006; Creasey and Isberg, 2012) and drives macrophage pyroptosis (Aachoui et al., 2013). Maximal caspase-11-mediated macrophage pyroptosis toward the L. pneumophila ⊿sdhA strain was observed by 6 hr post-infection (Figure 1A). Using L. pneumophila-GFP+ to distinguish infected and bystander cells, we confirmed that cell death occurred in the subpopulation that harbored bacteria (Figures S1A and S1B). To rule out the contribution of necroptosis, another form of necrotic cell death (Wallach et al., 2016), we used Necrostatin-1 (Nec1) to inhibit RIP1/RIP3 activity, which did not affect cell death (Figure S1C).
Figure 1. Infection-Driven IFN Signaling Is Dispensable for Pyroptosis.
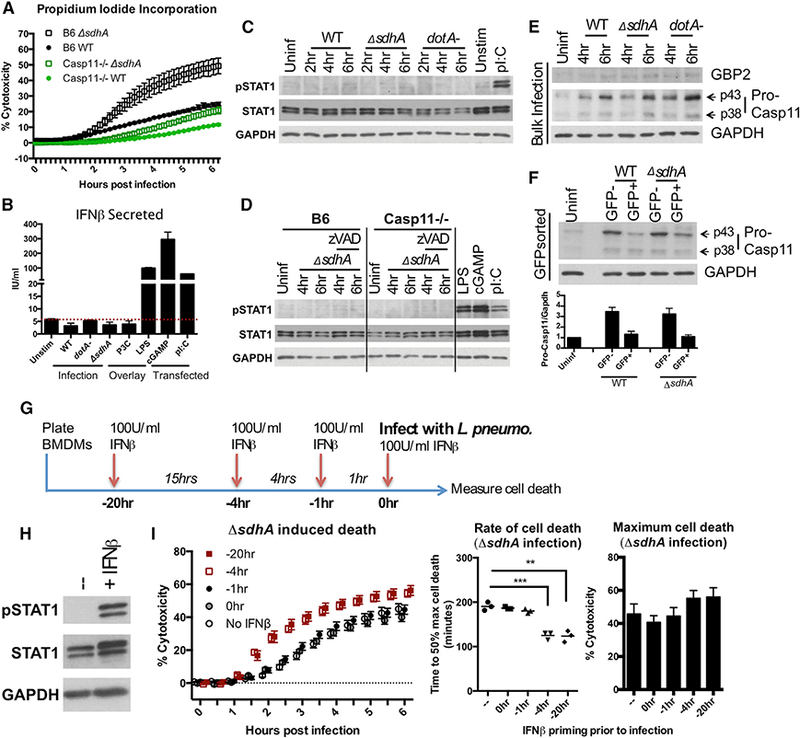
(A) Macrophages were infected with L. pneumophila WT and ⊿sdhA strains. Propidium iodide (PI) incorporation was used to monitor death.
(B) B6 macrophages were infected with WT, dotA ¯, or ⊿sdhA strains or stimulated by agonists: P3C (Pam3Cysk4), LPS, cGAMP (transfected), or polyI:C (transfected). At 8 hr, supernatants were collected and measured by ELISA for IFNβ protein.
(C) B6 macrophages were challenged with WT, dotA ®, or ⊿sdhA strains. Whole-cell lysates were used for immunoblot. 2-hr challenge with polyI:C (overlay) as control.
(D)Macrophages were challenged with the ⊿sdhA mutant in the absence or presence of zVAD (pan-caspase inhibitor). Whole-cell lysates were used for immunoblot. 6-hr transfection with polyI:C and cGAMP as controls.
B6 macrophages were challenged with WT, dotA ®, or ⊿sdhA strains. Whole-cell lysates were used for immunoblot.
Macrophages challenged for6hrwithWT or ⊿sdhA strains were fluorescence-activated cell sorted intoGFP+ (infected) and GFP_ (bystander) populations and lysed for immunoblotting. The blot shows one representative experiment. The bar graph is quantified from n = 3 experiments, normalized to pro-caspase-11 levels in unstimulated macrophages.
(F) Timeline that applies to (H) and (I) and Figure S1 F. B6 macrophages were pre-treated with 100 IU/mL of IFNβ before or during challenge by L. pneumophila ⊿sdhA.
(F) Immunoblot of whole-cell lysates from BMDMs that were unstimulated IFNβ or 1 hr of IFNβ stimulation.
(I) PI incorporation asafunction of time after challeng of B6 macrophages by L. pneumophila ⊿sdhA treated with IFNβ atvarioustimes (left). The rate ofcell death is displayed as time to 50% of maximal cell death (middle). Maximum cell death was measured at 6 hr post infection (hpi) (right).
See Figure S1.
L. pneumophila infection of macrophages results in robust Ifnb transcription via cytosolic DNA- and RNA-sensing pathways STING and RIG-I/MDA5, respectively (Monroe et al., 2009; Lippmann et al., 2011). Because Legionella translocates effectors that selectively block host cell protein translation, it is unclear whether type I IFNs bypass the L. pneumophila-elicited translational block (Fontana et al., 2011; Ivanov and Roy, 2013; Asrat et al., 2014). Transcriptional induction of Ifnb in response to the L. pneumophila ⊿sdhA mutant was similar in magnitude to that of LPS (Figure S1D). However, we did not detect secretion of IFNβ protein from Legionella-infected cells, in contrast to cells stimulated with LPS, cytosolic cyclic [G(2’,5’)pA(3’,5’)p] (cGAMP), and cytosolic polyinosine-polycytidylic acid (polyI:C) (Figure 1B; Figure S1D). To determine whether infected macrophage populations were experiencing IFN signaling, we probed for STAT1 phosphorylation downstream of IFN-α/β receptor (IFNAR). We were unable to detect increased STAT1 phosphorylation within 6 hr of L. pneumophila infection, in contrast to that observed in the presence of LPS, cGAMP, and polyI:C (Figures 1C and 1D). To dismiss the possibility of missed STAT1 phosphorylation in dying cells, STAT1 phosphorylation was measured in Casp11−/− cells or B6 cells treated with pan-caspase inhibitor zVAD (Figure 1D). No increase in STAT1 phosphorylation were detected under these conditions (Figure 1D).
IFN feedback is proposed to drive de novo synthesis of GBPs and pro-caspase-11, so we investigated whether these proteins were produced before cell death. We were unable to detect accumulation of GBP2 by 4 and 6 hr post-infection (Figure 1E), although we did see clear accumulation of GBP2 protein in response to the avirulent L. pneumophila dotA− mutant by 10 hr post-infection, consistent with its inability to block protein synthesis (Figure S1E) (Ivanov and Roy, 2013). We observed an increase of pro-caspase-11 within 6 hr of infection (Figure 1E). To determine whether pro-caspase-11 was synthesized by infected or bystander cell populations, macrophages challenged with L. pneumophila-GFP strains were sorted to separate infected GFP+ cells from bystander GFP ¯ cells. Pro-caspase-11 accumulated in the GFP ® bystander population, but not in the GFP+-infected population (Figure 1F). The lack of GBP2 and pro-caspase-11 accumulation in infected cells suggest that low protein abundance present in resting macrophages is sufficient for pyroptosis.
To determine whether IFN signaling concurrent with infection stimulates pyroptotic death, cells were incubated with 100 IU/mL of recombinant IFNβ at various times leading up to bacterial challenge and cell death was monitored (Figures 1G-1I). We found that despite robust IFNAR signaling in response to exogenous IFNβ (Figure 1H), IFNβ stimulation simultaneous with, or 1 hr before, infection had no effect on cell death induced by either the ⊿sdhA mutant (Figure 1I, left) or wild-type (WT) (Figure S1F). IFNβ pre-treatment at least 4 hr before infection was needed to significantly increase the rate of cell death, as demonstrated by a reduction in the time to 50% of maximal cell death (Figure 1I, middle). Extensive IFN treatment did not significantly increase the maximal cell death in response to the ⊿sdhA mutant (Figure 1I, right). In contrast, cell death toward the WT strain required 4 hr of IFN pre-treatment (Figure S1F). In both cases, at least 4 hr of pre-activation with IFNβ was necessary to enhance cell death, indicating that cell death depends on pre-established IFN status.
Constitutive IFNAR Signaling Controls Macrophage Cell Death Rate
Sub-threshold amounts of IFNα/β are thought to maintain expression of a set of IFN-stimulated genes (ISGs) required for cellular responses against infection (Gough et al., 2012). To determine the impact of constitutive IFN signaling on caspase-11 activation, we treated macrophages with an IFN receptor (IFNAR)-blocking antibody (clone MAR1–5A3) for various lengths of time (Figures 2A–2D). Blocking IFNAR 1 hr before infection did not affect cell death kinetics toward the ⊿sdhA mutant (Figures 2C and 2D), despite efficient block of STAT1 phosphorylationin response to exogenous IFNβ (Figure 2B). However, 20-hr blockade of IFNAR before infection reduced cell death in response to the ⊿sdhA mutant (Figures 2C and 2D). Macrophages from mice lacking type I IFN receptor (Ifnar−/−) also exhibited defective cell death, characterized by delayed onset with a significant increase in time to 50% maximal cell death (Figures 2E and 2F). Prolonged inhibition of IFNAR signaling did not alter cell death in Casp11−/− macrophages, indicating that cell death in the absence of IFN signaling remained caspase-11 dependent (Figure S2A).
Figure 2. Constitutive IFNAR Signaling Controls Macrophage Cell Death Rate.

(A-D) IFN receptor blocking antibody (αIFNAR) or immunoglobulin G (IgG) control were added before or during bacterial challenge.
(A) Antibody block and infection setup.
(B) Immunoblot of macrophages in response to 1 hr of treatment with 100 IU/mL of exogenous IFN, in the presence of antibodies for 1 or 20 hr before IFNIβ addition.
(C) L. pneumophila ⊿sdhA infection-driven PI incorporation of macrophages that were treated with control IgG or aIFNAR antibodyfor 1 or20 hr before infection.
(D) PI uptake by 6 hpi; n = 3.
(E) Cell death of macrophages at 6 hpi with ⊿sdhA mutant, as measured by PI incorporation. aIFNAR: 20 hr pre-treatment. Triplicate averages from n = 5 shown with each marker representing one experiment.
(F) Left: kinetics of macrophage death with ⊿sdhA infection. Right: average time to 50% maximal death from cells from 5 animals.
(G) Total STAT1 protein in the presence of aIFNAR blocking antibody or in untreated Ifnar−/− macrophages were assessed by immunoblotting.
(H) Immunoblot (left) and mRNA (right) of caspase-11 expression in the presence or absence of IFN signaling. Left: the arrow marks the p43 subunit of pro-caspase-11. Caspase-11 antibody also shows a non-specific band at 37 kDa (close to the published p38 subunit). Right: the dotted red line marks background amplification by qPCR from Casp11−/− macrophages.
(I) Steady-state protein levels determined by immunoblotting of macrophages from various mouse strains. Lysate in lane 1 is from B6 macrophages treated with 100 IU/mL of IFNβ for 8 hr. Other samples were unstimulated macrophages.
(J) qRT-PCR of indicated genes. The dotted red line marks background amplification by qPCR from Gbpchr3−/− macrophages.
See Figure S2.
We next determined whether loss of constitutive IFN signaling perturbed basal levels of proteins in the caspase-11 pathway. STAT1 is an ISG whose protein abundance is sustained via constitutive IFNAR signaling (Gough et al., 2010, 2012). 20-hr IFNAR block reduced total STAT1 protein levels (Figure 2G), as well as mRNA expression for several ISGs, such as Irf7, Isg15, and Mx1 (Figure S2B). We did not observe a reduction in baseline expression of pro-caspase-11 protein or Casp11 mRNA under conditions in which constitutive IFN signaling is lost (Figures 2H and 2I). Looking upstream at GBP expression, consistent with published literature, we were unable to detect GBP2 protein in B6 macrophages in the absence of IFN priming (Figure 2I). Of the 5 Gbps encoded on the chr3 locus, only Gbp2, Gbp3, and Gbp7 were expressed in macrophages in the absence of priming (Figure S2C), with expression of Gbp2 and Gbp3 reduced in Ifnar−/− and Ifnb−/− macrophages (Figure 2J; Figure S2C). Expression of Gbp2 and Gbp3 was also significantly reduced by blocking constitutive IFNAR signaling (Figure 2J). Not all GBPs required constitutive IFNAR signaling for basal expression, such as Gbp7 on chr3, as well as Gbps encoded on the chromosome 5 locus (Figure 2J; Figure S2D).
Endogenous GBP Expression and Rate of Cell Death Are Controlled by a Narrow Window of Low-Dose IFN Signaling
Pyroptosis of Ifnb−/− macrophages exhibited delayed onset after challenge with L. pneumophila ⊿sdhA compared to B6, consistent with reduced Gbp2 and Gbp3 expression (Figure 3A). To determine the amount of IFN needed to rescue this defect, Ifnb−/− bone marrow derived macrophages (BMDMs) were treated with increasing amounts of IFNβ 20 hr before challenge with bacteria (Figure 3B). ⊿sdhA mutant-induced cell death was restored with an IFNβ dose of 0.5 IU/mL (Figure 3B). 0.5 or 1 IU/mL of IFNβ restored expression of STAT1 (Figure 3C), Gbp2, and Gbp3 (Figure 3D), as well as several other ISGs (Isg15, Mx1, and Irf7) (Figure S3) to levels observed in B6 macrophages at steady state.
Figure 3. Endogenous GBP Expression and Rate of Cell Death Are Controlled by a Narrow Window of Low-Dose IFN Signaling.
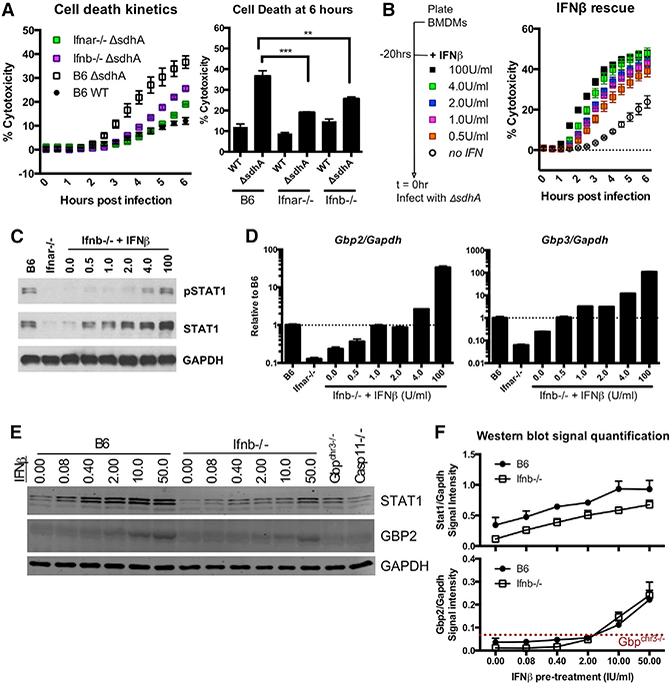
(A) Macrophages were challenged with L. pneumophila ⊿sdhA, and macrophage death was measured by PI incorporation. Left: representative kinetics of cell death. Right: average cell death at 6 hpi; N = 3.
(B) lfnb−/− macrophages were treated for 20 hr with recombinant IFNβ. Cells were then challenged with L. pneumophila ⊿sdhA, and macrophage death was measured by PI incorporation.
(C and D) Immunoblot (C) and qRT-PCR (D) after 20 hr of treatment of Ifnb-/ macrophages with low-dose IFNβ. The dotted black line indicates steady-state expression of genes of interest in B6 macrophages.
(E and F) B6 and Ifnb−/−BMDMs were stimulated with recombinant IFNβ for 8 hr. Immunoblot shown in (E). Quantification of band intensity over GAPDH shown in (F). The dotted red line indicates the threshold of antibody detection on the LI-COR Biosciences. Representative of 3 experiments.
See Figure S3.
We found that 4 IU/mL of IFNβ added to Ifnb−/− macrophages was sufficient to induce a cytotoxic response reminiscent of cells stimulated with 100 IU/mL of IFNβ, a standard dose used for IFN activation (Figure 3B). This parallels the amount necessary to mimic STAT1 phosphorylation levels in B6 macrophages (Figure 3C). Of particular interest is that for detection of GBP2 protein by immunoblotting, at least 10–50 IU/mL of IFNβ stimulation was needed (Figures 3E and 3F).
GBPs Perturb Cytosolic Bacteria after Pathogen Vacuole Disruption
The localization of GBPs around pathogen-containing vacuoles has been associated with bacterial death (Meunier et al., 2014, 2015; Man et al., 2016), although this association has not been demonstrated for restriction of either L. pneumophila or Chlamydia muridarum (Pilla et al., 2014; Finethy et al., 2015). In addition, there is disagreement on the exact step or steps promoted by GBPs, with earlier studies suggesting a role for GBPs in pathogen vacuole disruption and later studies showing GBPs to function downstream of pre-destroyed vacuoles (Meunier et al., 2014, 2015; Man et al., 2015, 2016). We therefore sought to determine the role of GBPs in the disruption of Legionella-containing vacuole (LCV).
The loss of LCV integrity can be assayed by immunofluorescent probing of bacteria in the absence of membrane permeabilization with detergents (Figure 4A) (Creasey and Isberg, 2012). Loss of the ⊿sdhA mutant vacuole integrity was unaltered in Ifnar−/−, Gbpchr3−/−, and Casp11−/− macrophages (Figure 4B). Nevertheless, when challenged with L. pneumophila, ⊿sdhA and Gbpchr3−/− macrophages exhibited delayed and reduced maximal cell death (Figure 4C). Cytosolic L. pneumophila assumes an aberrant morphology after breakdown of the LCV, which can be detected as the normally rod-shaped organism assuming a swollen, frayed, or truncated morphology (Figure 4A) (Laguna et al., 2006; Creasey and Isberg, 2012). Within the cytosol-accessible bacterial population, we observed that cytosolic L. pneumophila retained their rod shape in Gbpchr3−/− macrophages up to 10 hr post-infection, indicating that GBP function was required for inflicting damage on cytosolic bacteria (Figures 4D and 4E). In Ifnar−/− macrophages, cytosolic bacteria retained their rod-shaped morphology at early time points post-vacuole disruption (Figures 4E and 4F) but eventually gained aberrant morphology (Figures 4D, 4E, and 4G).
Figure 4. GBPs Perturb Cytosolic Bacteria after Pathogen Vacuole Disruption.
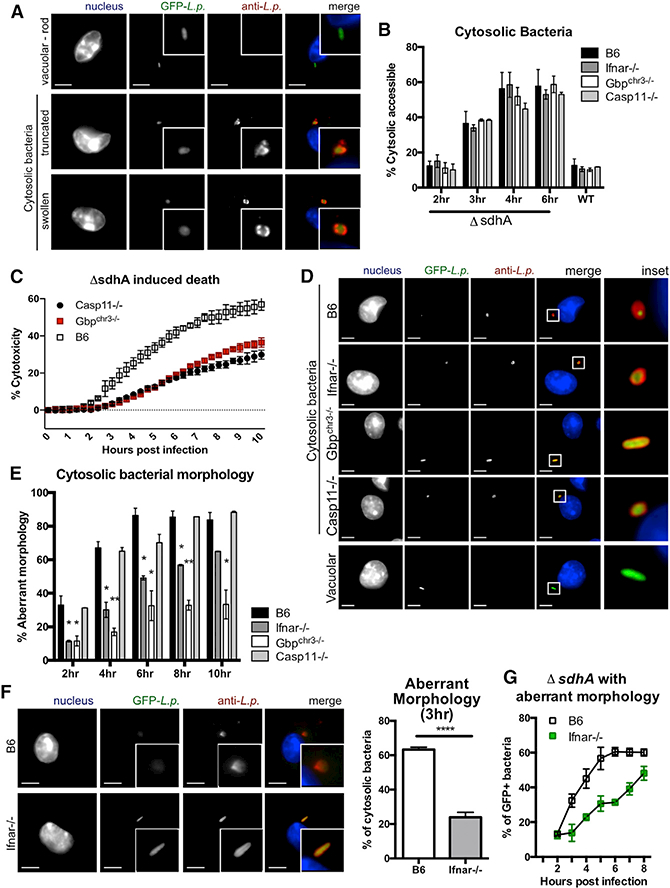
(A)Representative images of vacuolar and cytosolic bacteria in B6 macrophages to demonstrate various morphologies. 100× lens; scale bar, 5 μm.
(B)At the indicated time points, macrophages challenged with L. pneumophila-GFP WT and ⊿sdhA strains were fixed and stained with an antibody against Legionella with no detergent permeabilization. The percentage of antibody-stained bacteria relative to GFP+ bacteria is plotted as cytosolic bacteria.
(C)Macrophages were challenged with L. pneumophila ⊿sdhA, and macrophage death was measured by PI incorporation representative from n = 3.
(D)Representative images of cytosolic bacteria at 8 hpi. 63× lens; scale bar, 5 μm.
(E)Percentage of cytosol-exposed bacteria with aberrant morphology. 100+ bacteria were counted per technical replicate.
(F)Left: representative images of cytosolic ⊿sdhA in macrophages at 3 hpi. Images were taken with 100× lens; scale bar, 5 μm. Right: percentage of bacteria with aberrant morphology quantified at 3 hpi.
(G)Percentage of cytosol-accessible bacteria with aberrant morphology, quantified by microscopy kinetically. 100+ bacteria were counted per technical replicate.
See Figure S4.
WT L. pneumophila triggered pyroptotic cell death only when macrophages were pre-activated with IFNs (Figures S1F, S4A, and S4B) (Case et al., 2013). This process requires the chr3-encoded GBPs (Pilla et al., 2014). 100 IU/mL of IFNα pre-activation rapidly destabilized the vacuoles of WT L. pneumophila in macrophages sufficient or deficient for chr3 GBPs (Figure S4C). Morphologically, IFN activation resulted in the loss of rod-shaped appearance in 80%−90% of cytosol-accessible L. pneumophila within 3 hr of infection in B6 and Casp11−/− macrophages. Almost all bacteria within the cytosol-accessible subpopulation retained their rod-shaped morphology in Gbpchr3−/− macrophages (Figures S4D and S4E). We therefore conclude that IFN pre-activation drives the synthesis of factors other than GBPs that can destabilize an intact bacterial vacuole. While GBPs were dispensable for vacuole disruption under all conditions tested, they were required for the disruption of bacteria exposed to the cytosol, a crucial event upstream of caspase-11 activation and cell death.
Human Macrophages Require JAK/STAT Signaling for Constitutive Antimicrobial Responses
Caspase-4 and caspase-5 are human orthologs of murine cas-pase-11 that drive activation-induced cell death in response to intracellular LPS (Shi et al., 2014; Casson et al., 2015). Similar to our observations with mouse macrophages, both human bronchoalveolar lavage (BAL) cells and human peripheral blood monocyte-derived macrophages (MDM) challenged with L. pneumophila ⊿sdhA exhibited cell death in the absence of an IFN priming event (Figures 5A–5D; Figure S5).
Figure 5. Human Macrophages Require JAK/STAT Signaling for Constitutive Antimicrobial Responses.

(A and B) Human bronchoalveolar lavage (BAL) cells were challenged with L. pneumophila WT or ⊿sdhA strains.
(A) cell death as a function of time assayed by PI uptake, compiled from 5 donors with mean ± SEM plotted.
(B) Magnitude of cell death in individual BAL samples by 4 hpi, represented as one marker per donor.
(C and D) Human peripheral blood monocyte-derived macrophages (MDM) were challenged with L. pneumophila WT or ⊿sdhA strains with kinetics of PI incorporation in (C) and maximal cell death by 4 hr post-infection (representative of 3 experiments) in (D).
(E) Experimental setup for (F)-(L), in which human MDMs were treated with JAK1/2 inhibitor (JAKi) before or during challenge with L. pneumophila ⊿sdhA.
(F) 200 lU/mL of recombinant human IFNβ was used to stimulate human MDMs, incubated for the indicated times with JAKi. 1 hr after IFN stimulation, lysates were collected and probed by immunoblot.
(G) Gene expression in human MDMs with or without JAKi for 40 hr.
(H and I) PI incorporation of human MDMs infected with L. pneumophila ⊿sdhA, with simultaneous or prolonged JAK inhibition. Representative kinetics ofdeath are in (H). Maximum cytotoxicity from 7 individual donors is in (I).
(J) Percentage of cytosol-accessible L. pneumophila ⊿sdhA based on antibody staining without detergent permeabilization. Data are shown from 2 individual donors.
(K and L) Representative images (K) and quantification (L) of cytosolic L. pneumophila ⊿sdhA in human MDMs in the presence or absence of 40 hr of JAKi pretreatment. Images taken with 63× lens; scale bar, 5 mm.
See also Figure S5.
We used JAK/STAT inhibitors to block human IFNAR signaling (Zurney et al., 2007; Jackson et al., 2016; Mostafavi et al., 2016). Human MDMs were treated with JAK1/2 inhibitor ruxolitinib (JAKi) for either 40 hr before infection (pre-infection block) or exclusively during infection (co-infection block) (Figure 5E). Treatment of human MDMs with JAKi abolished all IFNα-stimulated STAT1 phosphorylation, independent of treatment protocol (Figure 5F). Treatment with JAKi for 40 hr also led to a drastic reduction of Stat1 protein levels (Figure 5F) and hISG15 and hMX1 mRNA expression (Figure 5G), indicating that there was loss of ISG signatures. The long-term block with JAKi before infection significantly reduced L. pneumophila DsdhA-induced cell death in human MDMs (Figures 5H and 5I), mimicking the results we observed with antibody blockade of IFNAR in mu-rin macrophages (Figure 2)
Examining steps upstream of cell death, we observed loss of vacuole integrity with the L. pneumophila ⊿sdhA mutant (Figure 5J). Long-term Jak1/2 inhibition before infection did not reduce cytosol permeability (Figure 5J). In contrast, within the cytosol-exposed ⊿sdhA population, long-term Jak1/2 inhibition before infection significantly decreased the percentage of bacteria with aberrant morphology (Figures 5K and 5L). These results point to the global importance of this host-derived constitutive signaling pathway in protecting against cytosolic Gram-negative pathogen.
GBPs Mediate the Release of DNA to Activate Cytosolic DNA-Sensing Pathways
To examine whether the observed bacterial morphological changes resulted in release of bacterial content into the macrophage cytosol, we probed for the presence of bacterial DNA in the cytosol of the host cell. Using L. pneumophila carrying the non-transferable pJB908 plasmid, the presence of pJB908 in the macrophage cytosol provides a measure of DNA released from disrupted bacteria (Ge et al., 2012). We found 2.5–3 times more plasmid in cytosolic extract of B6 macrophages challenged with L. pneumophila ⊿sdhA(pJB908) than with WT infection (Figure 6A), consistent with previous reports (Geetal., 2012). In contrast, cytosolic extracts from Ifnar−/− macrophages and Gbpchr3−/− macrophages infected with the ⊿sdhA mutantharbored significantly lower amounts of plasmid than did similarly infected B6 macrophages (Figure 6A). During WT L. pneumophila challenge, the release of pJB908 plasmid was enhanced by IFN pre-activation in B6 and Casp11−/− macrophages (Figure 6B). This enrichment was lost in Gbpchr3−/− macrophages (Figure 6B).
Figure 6. GBPs Mediate the Release of DN to Activate Cytosolic DNA-Sensing Pathways.

(A) pJB908 plasmid extracted from cytosolic fractions of macrophages challenged with L. pneumophila (pJB908) WT or ⊿sdhA strains at 6 hpi.
(B) pJB908 plasmid extracted from cytosolic fractions of macrophages challengedwith WT L. pneumophila (pJB908) at3hpiinthe presence or absence of IFNβ 20 hr pre-treatment.
(C) Ifnb transcriptional induction from WT and L. pneumophila ⊿sdhA-infected macrophages of the indicated strains measured by qRT-PCR at 4 hpi.
(D) Cell death of B6, Casp11−/− and Casp1−/−Casp11−/− macrophages when challenged with L. pneumophila ⊿sdhA.
(E) Representative images of ⊿sdhA L. pneumophila and ASC immunofluorescence by antibody staining at 7 hpi. Images were taken with 20× lens; scale bar, 10 μm.
(F) Left: quantification of ASC-positive staining gated within regions of interest (ROIs), defined as regions of positive anti-L. pneumophila signal. Right: quantification of ASC-positive cellsfrom total numberofcells enumerated via Hoechst staining. Each symbol is a technical replicate. One representative experiment out of 3 is shown.
(G)Whole-cell lysate and precipitated supernatant from L. pneumophila-infected cells were probed by western blot for a p17 cleavage product of IL-1β Representative western blot on the left; quantification from n = 3 on the right.
Cytosolic DNA drives cGAS/STING activation and Ifnb induction. We assayed Ifnb mRNA upregulation and found significantly reduced Ifnb induction in Gbpchr3−/− macrophages in response to L. pneumophila ⊿sdhA (Figure 6C). We found significantly heightened Ifnb induction in Casp11−/− macrophages, consistent with reports that caspase-1/11 activation curtails cGAS/STING activation (Corrales et al., 2016; Wang et al., 2017).
Casp1−/−Casp11−/− macrophages had a further reduction in cell death compared to Casp11−/− macrophages, implicating a role for the canonical caspase-1 inflammasome (Figure 6D). Next, we assayed for apoptotic speck protein (ASC) oligomerization microscopically (specks) and IL-1β cleavage during L. pneumophila ⊿sdhA challenge (Figures 6E–6G). We observed enrichment of ASC specks occurs near bacteria, suggestive of DNA-mediated AIM2 activation, and is enhanced in the absence of caspase-1/ 11 activity (Figure 6F, left; Figure S6). Total numbers of macrophages harboring ASC specks also showed significant enrichment in the absence of caspase-1 and caspase-11 (Figure 6F, right), consistent with pyroptosis curtailing ASC oligomerization. We observed no enrichment of ASC specks in Gbpchr3−/− macrophages in response to L. pneumophila ⊿sdhA challenge compared to WT bacteria (Figures 6E and 6F). Lastly, L. pneumophila ⊿sdhA infection resulted in the cleavage of 31-kDa pro-IL-1β into the 17-kDa mature IL-1β fragment in B6 macrophages. This cleavage event was significantly reduced in Casp11−/− macrophages and further reduced in Gbpchr3−/− macrophages (Figure 6G).
Chr3 GBPs Are Involved in Restriction of Legionella Bacterium during Pneumonia
To examine the role of GBPs during Legionella-induced pneumonia, B6 and Gbpchr3−/− mice were infected with L. pneumophila ⊿flaA or L. pneumophila ⊿flaA ⊿sdhA bacteria. We observed an acute drop in body temperature of B6 mice within 7 hr of ⊿sdhA L. pneumophila infection (Figure 7A). Mice infected with the ⊿sdhA mutant recovered their temperature by 20 hr post-infection. In contrast to B6 mice infected with the ⊿sdhA mutant, Gbpchr3−/− mice did not exhibit a ⊿sdhA L. pneumophila-specific temperature drop at an early time point post-infection. Temperature recovery 1 and 2 days post-infection was also delayed in Gbpchr3−/− mice when infected with either WT Legionella or ⊿sdhA mutant (Figure 7A).
Figure 7. Chromosome 3 GBPs Are Involved in Restriction of Legionella Bacterium during Pneumonia.
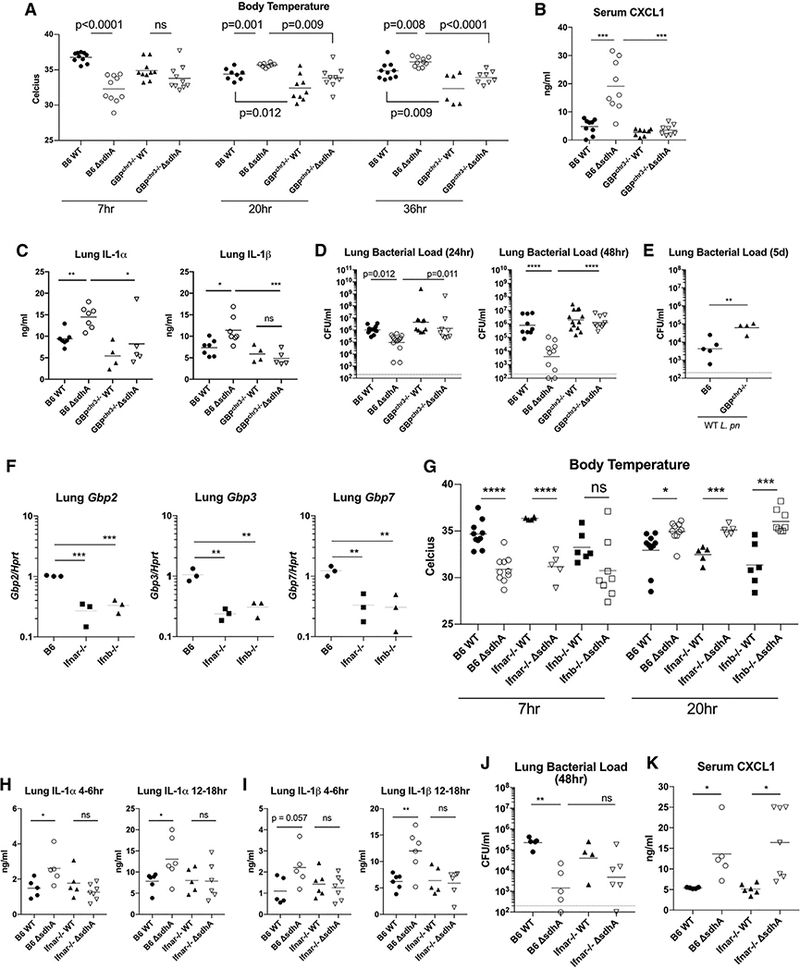
(A) Mice were infected with 1 × 107 L. pneumophila bacteria oropharyngeally. Rectal temperature of mice at various time points post-infection. Each dot represents an animal; results are pooled from 2 experiments.
(B) Serum CXCL1 was measured by ELISA. Each dot represents an animal; results are pooled from 2 experiments in which tissues were harvested between 4 and 6 hr post-infection.
(C) Lung IL-1a and IL-1β were measured by ELISA. Each dot represents an animal; results are pooled from 2 experiments in which tissues were harvested between 12 and 18 hr post-infection.
(D and E) Legionella colony-forming units from whole-lung tissue; the geometric mean is shown. Each dot represents an animal; results are pooled from 2 experiments. Animals were infected with (D) WT or ⊿sdhA bacterium and (E) WT bacterium.
(F) Whole-lung tissue was extracted from B6, ifnar−/−, and Ifnb−/− animals in the absence of bacterial challenge. Each symbol represents one mouse. mRNA transcripts for indicated Gbps were quantified by qRT-PCR.
(G) Body temperature of mice infected with 1 × 107 L. pneumophila bacteria oropharyngeally. Each dot represents an animal; results are pooled from 2 experiments.
(H and I) Lung IL-1 a and IL-1 β measured by ELISA. Each dot represents an animal; results are pooled from 2 experiments in which tissues were harvested between 4 and 6 hpi (H) or 12 and 18 hpi (I).
(J) Legionella colony-forming units from whole-lung tissue of B6 and ifnar−/− animals.(K) Serum CXCL1 was measured by ELISA. Each dot represents an animal; results are pooled from 2 experiments in which tissues were harvested between 4 and 6 hpi.
Pneumonia models using flagellated Legionella had shown that the NLRC4/NAIP5 inflammasome elicits a robust IL-1 response in the lungs. IL-1 receptor signaling was necessary for the induction of CXCL1 to recruit neutrophils, leading to clearance of bacteria (Tateda et al., 2001; Barry et al., 2013). Pyroptotic macrophage corpses have been shown to retain bacteria at the site of infection. CXCL1-driven influx of neutrophils engulfed the macrophage carcasses, along with the trapped bacteria, to facilitate clearance (Jorgensen et al., 2016). CXCL1 thus appears to be crucial for clearance of bacteria that illicit inflammasome activation. Systemically, B6 animals infected with the ⊿sdhA mutant displayed a robust, acute increase in serum CXCL1 that coincides with the dip in body temperature. This induction of serum CXCL1 was absent from Gbpchr3−/− mice (Figure 7B). Locally, we observed a ⊿sdhA-specific increase in IL-1 a and IL-1β in the lungs of B6 animals 4–6 hr post-infection (Figure S7A). This IL-1 a and IL-1β production was further amplified during the next 10 hr in B6 but was absent from the lungs of Gbpchr3−/− mice (Figure 7C).
To determine whether the perturbations in cytokine production affected the rate of bacterial clearance, we measured bacterial colony-forming units (CFUs) in the lungs of B6 and Gbpchr3−/− mice. It is well established that Legionella growth is restricted on a C57BL/6 genetic background. We found the restriction was more severe with ⊿sdhA mutant in B6 animals. In contrast, bacterial load of the ⊿sdhA mutant is significantly higher 1 and 2 days post-infection in Gbpchr3−/− mice (Figure 7D). WT Legionella burden was also higher in Gbpchr3−/− mice by day 5 (Figure 7E).
Finally, to determine whether tonic IFN signaling plays a role in the expression of GBPs in vivo, we performed global RNA sequencing (GEO: GSE110678) from lungs of uninfected B6 and Ifnar−/− animals (Figure S7B). In whole lungs of resting B6 animals, Gbps as a gene family are skewed toward higher expression in B6 as opposed to Ifnar−/− animals, with Gbp1 and Gbp11 being undetectable from Ifnar−/− lungs (Figure S7B). Gbp2, Gbp3, and Gbp7 showed small differences in the two strains by global RNA sequencing (GEO: GSE110678), but on further interrogation by RT-PCR, they were found to be significantly reduced in the lungs of both Ifnar−/− and Ifnb−/− mice (Figure 7F). We similarly observed reduced expression of Irf7, Isg15, and Mx1 in lungs of Ifnar−/− animals, although not all ISGs appearto depend on IFNβ, indicating differential roles for various IFNs in tonic signaling (Figure S7C).
When subjected to Legionella lung infection, we surprisingly found temperature fluctuation of IFN-deficient animals—Ifnar−/− and Ifnb−/− mice—to be similar to that of IFN-sufficient B6 animals (Figure 7G). We found Ifnb−/− mice exhibit more variability in their response; thus, further experiments are conducted using Ifnar−/− animals to bypass possible confounding effects is the various IFNas that may play a role in vivo. In Ifnar−/− animals, local production of IL-1 a and IL-1 β in the lungs was significantly reduced during ⊿sdhA mutant challenge (Figures 7H and 7I), reminiscent of Gbpchr3−/− mice (Figure 7C; Figure S7A). However, Ifnar−/− animals remained restrictive to both WT and mutant Legionella, because we did not detect a difference in bacterial CFUs between Ifnar−/− and B6 animals toward the various Legionella strains at 48 hr post-infection (Figure 7J). Lastly, we found that Ifnar−/− animals retained the ability to induce a robust serum CXCL1 response when infected with the ⊿sdhA mutant (Figure 7K). The discrepancy between IL-1 and CXCL1 levels in IFN-deficient animals suggest the surfacing of a compensatory pathway when IFNAR signaling is lost. Work by Schliehe et al. (2015) elucidated one such mechanism, in which reduced expression of Setdb2 in Ifnar−/− mice, a repressor of CXCL1 expression, is specifically induced by type I IFN signaling. Loss of IFNAR heightened CXCL1 secretion in response to Streptococcus pneumonia infection (Schliehe et al., 2015). To determine whether breakthrough of CXCL1 may be a contributing factor, we measured the local production of CXCL1 in the lungs and found a trend toward higher lung CXCL1 in Ifnar−/− mice when infected with the ⊿sdhA mutant, a response that is quiescent in B6 animals. We did not observe this enrichment in lung CXCL1 production in Gbpchr3−/− animals infected with the ⊿sdhA mutant (Figure S7D). This indicates a possibility that systemic CXCL1 response from Ifnar−/− mice is GBP independent but stems from a CXCL1 self-amplification loop via other cytosolic-sensing mechanisms.
DISCUSSION
Macrophage upregulation of an IFN response facilitates antimicrobial activities, including autophagy, pathogen recognition and attack, and host cell death (Randow et al., 2013; Schneider et al., 2014). Many studies have uncovered the mechanism of hyper-IFN-mediated protection (MacMicking, 2012; Pilla-Moffett et al., 2016). However, in the absence of disease and infection, an immunocompetent host is quiescent for cytokine responses, with intermittent spikes of IFN occurring only during infection or immune dysregulation (Bocci, 1985; Taniguchi and Takaoka, 2001; Gough et al., 2012). Little is known about the initial host-pathogen encounter at the onset of infection, before the infection-driven IFN spike. In this work, we set out to investigate the mechanisms by which naive macrophages respond to vacuolar and cytosolic bacteria while simulating a physiologically relevant host-pathogen initial encounter.
Using the cytosol-accessible bacteria L. pneumophila ⊿sdhA, we found that caspase-11 action occurred independently of infection-driven IFN feedback. Instead, constitutive IFN signaling in the absence of infection was sufficient to maintain GBP expression and enable GBP activity against cytosol-accessible bacteria. We propose that upon challenge with vacuole-resident bacteria, a small percentage of bacteria that are unable to maintain a stable replication vacuole engage the host cytosolic surveillance pathways as a result of GBP-mediated release of bacterial components. For Gram-negative bacteria, LPS activates caspase-11 (caspase-4/5 in humans), resulting in gasdermin D-dependent pyroptotic cell death (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). Simultaneously, GBPs play a role in leakage of bacterial nucleic acids into the cytosol, activating nucleic acid-sensing pathways that can drive transcriptional activity, as well as the canonical caspase-1 inflammasome via AIM2. In the absence of protein translation block, a STING-dependent IFN response would in turn activate neighboring bystander cells, generating an IFN-activated phenotype in uninfected macrophages. Upon further infection, these IFN-activated macrophages would lyse pathogen vacuoles in an event that does not require GBPs encoded on chr3.
The canonical caspase-1 pathway can also contribute to pyroptosis in a GBP-dependent manner to contribute to the local IL-1 response. On a systemic level, IL-1 receptor signaling on endothelial cells amplifies the CXCL1 response to recruit neutrophils to the site of infection. Jorgensen et al. (2016) showed that pyroptotic macrophage corpses can trap cytosolic bacteria, increasing the efficiency by neutrophil internalization and degradation of the invading pathogen. In our Legionella-induced pneumonia model, we found that chr3-encoded GBPs are necessary for heightened IL-1 a and IL-1 β produced locally in the lungs, systemic CXCL1 response, and timely bacterial clearance.
Upstream of endogenous GBP expression, there exists a sensitive balance in which constitutive cytokine signaling primes the system for critical immune responses to occur upon infection. This concept was best represented in Ifnb−/− macrophages, in which the loss of ISG expression can be restored with a single low dose of IFNβ. This indicates that even small, intermittent triggering of the IFN receptor can be effective in maintaining ISG steady-state levels. Several studies show support for the model of constitutive IFN production triggered by cell-intrinsic DNA damage via the cytosolic cGAS/STING pathway (Ahn et al., 2012; Rongvaux et al., 2014; Hartlova et al., 2015; West et al., 2015). In necroptotic cell death, cGAS/STING-mediated constitutive IFN signaling is crucial for maintaining the expression of mixed lineage kinase-like (MLKL) (Sarhan et al., 2018). One can speculate that during homeostasis, self-DNA can stem from a multitude of sources, including microbiota components (Abt et al., 2012), tissue turnover (Lienenklaus et al., 2009), and reactivation of endogenous retroelements (Stetson et al., 2008). Low levels of circulating cytokines can thus condition innate immune cells during their maturation process, determining the rate and mechanism of response during infection. The response to low levels of cytokines is conserved in humans, because human macrophages chronically depleted for JAK1/2 signaling are dampened in their cell death response to cytosol-resident L. pneumophila.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Caspase-11 clone 17D9 | Cell Signaling Technology | 14340 |
| Anti-GBP2 | Proteintech | 11854–1-AP |
| Anti-phospho-Stat1 Y701 58D6 | Cell Signaling technology | 9167 |
| Anti-STAT1 | Cell signaling technology | 9172 |
| Rabbit polyclonal serum against L. pneumophila | Isberg Lab | (Laguna et al., 2006; Creasey and Isberg, 2012) |
| Rat polyclonal serum against L. pneumophila | Isberg Lab | (Laguna et al., 2006; Creasey and Isberg, 2012) |
| Anti-ASC | Adipogen | AG-25B-0006 |
| Anti-Gapdh 14C10 | Cell Signaling Technology | 2118 |
| Anti-Gapdh D4C6R | Cell Signaling Technology | 97166 |
| Anti-IFNβ (ELISA capture antibody) | Santa Cruz | sc-57201 |
| Anti-IFNβ (ELISA detection antibody) | R&D Systems | 32400–1 |
| Bacterial and Virus Strains | ||
| L. pneumophila Lp02 ΔflaA | Isberg Lab | (Creasey and Isberg, 2012; Asrat et al., 2014) |
| L. pneumophila Lp02 ΔflaA ΔsdhA | Isberg Lab | (Creasey and Isberg, 2012) |
| L. pneumophila Lp03 (dotA-) ΔflaA | Isberg Lab | (Asrat et al., 2014) |
| L. pneumophila Lp02 ΔflaA Ptac:: PahpC::Gfp | Isberg Lab | (Coers et al., 2007; O’Connor et al., 2012) |
| L. pneumophila Lp02 ΔflaA ΔsdhA Ptac:: PahpC::Gfp | Isberg Lab | (Coers et al., 2007; O’Connor et al., 2012) |
| L. pneumophila Lp02 ΔflaA pJB908 | Isberg Lab | (Laguna et al., 2006; Ge et al., 2012) |
| L. pneumophila Lp02 ΔflaA ΔsdhA pJB908 | Isberg Lab | (Laguna et al., 2006; Ge et al., 2012) |
| Biological Samples | ||
| Human Bronchoalveolar lavage cells | Tufts Medical Center, Alexander Panda, M.D. | De-identified volunteers |
| Human peripheral whole blood | NY Biologics | De-identified whole blood |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Purified NA/LE Mouse anti-mouse IFNARα/β receptor MAR1–5A3 | BD PharMingen | 561183 |
| Purified NA/LE Mouse IgG1 κ Isotype control | BD PharMingen | 553447 |
| Jak inhibitor Ruxolitinib | Cayman Chemical | 11609 |
| Recombinant mouse IFNβ | PBL | 12400–1 |
| TMB substrate | ThermoFisher | N301 |
| Deposited Data | ||
| Mi-Seq GSE110678 | This paper | N/A |
| Experimental Models: Cell Lines | ||
| Murine bone marrow derived macrophages | Mouse long bones | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Jackson Laboratory | 000664 |
| Ifnar−/− (B6.129S2-Ifnar1tm1Agt/Mmjax) | Jackson Laboratory | 32045-JAX |
| Ifnb−/− | Dr. Stephanie Vogel | N/A |
| GBPchr3−/− | Dr. Jorn Coers; Dr. Masahiro Yamamoto | (Yamamoto et al., 2012; Pilla et al., 2014) |
| Casp11−/− | Dr. Vishva Dixit | (Kayagaki et al., 2011,2013) |
| Casp1 −/−Casp11−/− | Jackson Laboratory | (Lietal., 1995) |
| Asc−/− | Dr. Kate Fitzgerald | (Zheng et al., 2011) |
| Oligonucleotides | ||
| Forward and reverse primers for Gbps | Sigma | (Kim et al., 2011) Table S2 |
| Forward and reverse primers for Casp11 | Sigma | (Kayagaki et al., 2011) |
| Murine Irf7: (F) 5’-CTTCAGCACTTTCTTCCGAGA-3’; (R) 5’-TGTAGTGTGGTGACCCTTGC-3’ | Sigma | (Rocha et al., 2015) Table S4 |
| Murine Isg15: (F) 5’-GAGCTAGAGCCTGCAGCAAT-3’; (R) 5’-TTCTGGGCAATCTGCTTCTT-3’ | Sigma | (Pott et al., 2011) |
| Murine Mx1: (F) 5’-TCTGAGGAGAGCCAGACGAT-3’; (R) 5’-ACTCTGGTCCCCAATGACAG-3’ | Sigma | (Pottetal., 2011) |
| human IRF7 (F) 5’-CTTGGCTCCTGAGAGGGCAG-3’; (R) 5’-CGAAGTGCTTCCAGGGCA-3’ | Sigma | (Dill et al., 2014) Table S5 |
| human ISG15 (F) 5’-TCCTGCTGGTGGTGGACAA-3’; (R) 5’ -TTGTTATTCCTCACCAGGATGCT-3’ | Sigma | (Dill et al., 2014) Table S5 |
| human Mx1 (F) 5’-GTGCATTGCAGAAGGTCAGA-3’; (R) 5’-TCAGGAGCCAGCTTAGGTGT-3’ | Sigma | (Dill et al., 2014) Table S5 |
| human GAPDH (F) 5’-GCTCCTCCTGTTCGACA GTCA-3’; (R) 5’-ACCTTCCCCATGGTGTCTGA-3’ | Sigma | (Dill et al., 2014) Table S5 |
| pJB908 plasmid amplification (F) 5’-TCAGGAA GCACAAATGTCAATG-3’; (R) 5’-GGTCTA CACCACCAAAATCACG-3’ | Sigma | (Ge et al., 2012) |
| Primers for pUC18 flanks the M13 cloning site of the vector. (F) 5’-CAGGAAACAGCTATGAC −3’; (R) 5’-GTAAAACGACGGCCAG-3’ |
Sigma | Isberg Lab |
| Recombinant DNA | ||
| pJB908 (pMMB66EH oriRSF1010 ΔoriT tdDi bla+) | Isberg Lab | (Laguna et al., 2006) |
| Ptac:: PahpC::Gfp (pMMB207Dmob267) | Isberg Lab | (O’Connor et al., 2012) |
| Software and Algorithms | ||
| GraphPad Prism7 | GraphPad | N/A |
| Adobe Photoshop | Adobe | N/A |
| Affinity Designer | Affinity | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources and reagents described in manuscript should be directed to Dr. Alexander Poltorak (alexander.poltorak@ tufts.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
L. pneumophila Bacterial Strains
All L. pneumophila strains used in the present study are deficient for flagellin (⊿flaA) to mimic growth in amoeba and human macrophages and bypass the NLRP4/Naip5/Caspase-1 cell death pathway that occurs on the C57BL/6 genetic background. L. pneumophila derivatives used in this study were Lp02⊿flaA (thyA rpsL ⊿flaA; referred to as WT), Lp02⊿flaA⊿sdhA (thyA rpsL ⊿flaA ⊿sdhA; referred to as ⊿sdhA), and Lp03⊿flaA (dotA3 thyA rpsL ⊿flaA; referred to as dotA-). All strains were derived originally from Legionella pneumophila Philadelphia-1(Berger and Isberg, 1993). L. pneumophila was propagated on charcoal-N-(2-acetamido)-2-aminoethanesulfonic acid (ACES)-yeast extract plates with 0.1mg/mL thymidine (CYE/T) and in ACES-yeast extract broth (AYE/T) with 0.2mg/mL thymidine as previously described (Creasey and Isberg, 2012; Asrat et al., 2014).
To generate L. pneumophila-GFP, PCR-amplified gfp gene was fused to PahpC (Coers et al., 2007) and inserted downstream of Ptac in the plasmid pMMB207⊿mob267 {Morales et al. 1991 #1; O’Connor et al. 2012 #2; kind gift of Dr. Kimberly Davis}(Morales et al., 1991; O’Connor et al., 2012). Strains harboring the plasmid were maintained on CYE plates containing 0.1mg/mL thymidine and 5 μg/mL chloramphenicol. Prior to challenge of cultured cells, AYE broth cultures containing 0.2mg/mL thymidine and 2mM IPTG were grown to post-exponential phase, as described {Asrat et al., 2014 #3}. Inoculation into macrophage monolayer cultures was carried out at MOI = 10 in the presence of 2mM IPTG.
The plasmid pJB908 was described previously (pMMB66EH oriRSF1010 ⊿oriT tdAibla+; Laguna et al., 2006). For selection, pJB908-containing strains were grown on CYE plates or in AYE broth in the absence of added thymidine.
Murine Macrophages
8–10-weeks-old male and female mice are used for this study. C57BL/6 and Ifnar−/− mice were obtained from Jackson Laboratory. Ifnb−/− mice and Casp11−/− mice were kind gifts from Dr. Stephanie Vogel and Dr. Vishva Dixit respectively. All animal studies were approved by the Institutional Animal Care and Use Committee of Tufts University. Bone marrow macrophages are from Gbpchr3−/− mice (Pilla et al., 2014; Yamamoto et al., 2012). Bone marrow-derived macrophages were isolated from mice and propagated for 7 days in RPMI containing 20% FBS, 30% L cell supernatant, 2% Penn/Strep on non-tissue culture treated Petri dishes (Asrat etal., 2014).
Murine in vivo infections
Animal infection protocols were approved by the Tufts University Medical School Animal Care and Use Committees. Male and female mice between 8–10-weeks-old are used. C57BL/6, Ifnar−/−, Gbpchr3−/− and Ifnb−/− mice were oropharyngeally inoculated with 1×107 bacteria under isoflurane anesthesia. Temperature was monitored via rectal thermometer. At time points indicated in the text and figures, mice were euthanized via CO2 asphyxiation, and blood and lungs were harvested for downstream cytokine and bacterial load quantification.
Human peripheral blood monocytes derived macrophages (MDM)
De-identified human peripheral blood was obtained from New York Biologics. The use of de-identified human samples followed a protocol approved by the Tufts University School of Medicine Institutional Review Board. No information on age and sex of the donor were provided by NY Biologics. Monocytes were obtained from peripheral blood using the EasySep Direct Monocyte Isolation Kit (STEMCELL technologies). CD14+CD16 ® CD68~ monocytes were extracted and differentiated into CD14+CD16 ® CD68+ macrophages over the course of 7 days in RPMI containing 20% FBS, 200 IU/mL Penicillin and 200 mg/ml Streptomycin. and 100 mg/ml of human monocyte colony stimulating factor (M-CSF; PeproTech) (Davies and Gordon, 2005; Jaguin et al., 2013). Nonpolarized MDMs were cultured for 40 hr further in RPMI containing 10% FBS, 200 IU/ml Penicillin and 200 mg/ml Streptomycin, in the absence of M-CSF prior to infection. Infection was carried out at MOI = 10.
Human bronchalveolar lavage cells (BAL)
Bronchoalveolar (BAL) lavage of humans was performed at Tufts Medical Center. BAL was obtained with informed consent under a protocol approved by the Tufts University School of Medicine Institutional Review Board. Subjects underwent fiberoptic bronchoscopy and BAL with 180 mL of lavage solution. With the subject under local anesthesia, a 5.5-mm O.D. fiberoptic bronchoscope was advanced through the mouth and into the trachea and was wedged into a segmental or subsegmental bronchus of the right upper lobe. The wedged lung segment was lavaged with three aliquots of 60 mL each of normal sterile saline prewarmed to 37°C; and the fluid was gently aspirated (St-Laurent et al., 2009; Freeman et al., 2015). BAL fluids were provided without information on age and sex of the donors. Cells were strained from BAL fluid using 70 μm cell strainers and allowed to adhere to experimental dishes in RPMI containing 10% FBS and 200 IU/ml Penicillin and 200 mg/ml Streptomycin, at a density of 0.5× 106 cells per cm2. Cells were allowed to attach for 2 hr at 37°C, 5% CO2, at which point non-adherent cells and antibiotics were washed away with RPMI plus 10% FBS (Davies and Gordon, 2005). Bacteria were inoculated at an MOI = 10.
METHOD DETAILS
In vitro L. pneumophila infections
For infection of murine and human macrophages, L. pneumophila was infected at MOI = 10, in the absence of antibiotics.
Kinetic cytotoxicity assay with propidium iodide uptake
Cells were incubated in clear bottom 96 well microplates (Costar 3603) in media containing 10 mg/mL propidium iodide (PI; Life Technologies, P3566). ATECAN Infinite 200 Pro plate reader was used to maintain temperature at 37°C and 5% CO2 during incubations. PI uptake was monitored every 10 min at 535 nm excitation and 617 nm emission, using bottom reading setting. For 100% cytotoxicity control, cells were treated with 0.1% Triton X-100, similar to protocols for measuring Lactate Dehydrogenase (Promega). All propidium iodide incorporation assays were performed in triplicate wells. This protocol was adapted from previous work (Case et al., 2013).
Western blotting
Macrophages were challenged with L. pneumophila as described (Asrat et al., 2014). At the desired time points, cells were lysed directly in 1X Laemmli Buffer with 5% β-mercaptoethanol, boiled for 10 min, and incubated on ice for 10 min prior to loading on SDS-PAGE gels and western blotting (Asrat et al., 2014). Primary antibodies used were anti-Caspase-11 clone 17D9 (Cell Signaling Technology #14340, 1:500), anti-GBP2 (Proteintech 11854–1-AP, 1:1000). Anti-phospho-STAT1(Tyr701) (58D6), anti-total STAT1, and anti-GAPDH were obtained from Cell Signaling Technology. Western blots were imaged using the LI-COR Odyssey CLx or by chemiluminescent detection on film followed by analysis using LI-COR analysis software.
IFNβ ELISA
A sandwich ELISA for IFNβ was used to measure supernantant IFNβ as International Units/ml as calculated based on a bioactive IFNβ recombinant protein standard, as previously described (Roberts et al., 2007; Rathinam et al., 2012). In brief, capture antibody (monoclonal rat anti-mouse IFNβ, Santa Cruz sc-57201) was applied at 4C overnight in 0.1M carbonate buffer. Samples are incubated overnight and further antibody steps are diluted in 10% fetal calf serum in phosphate-buffered saline. Recombinant IFNβ (PBL 12400–1) was used for protein standard. Bound protein was detected using polyclonal rabbit anti-mouse IFNβ (R&D Systems, 32400–1). ELISA was developed using Goat anti-rabbit-HRP (Cell Signaling Technology 7074) in combination with TMB substrate (ThermoFisher N301), reaction stopped with 2N H2SO4.
Quantitative RT-PCR
Cells were lysed with TRIzol (Thermo Fisher Scientific) and mRNA extracted by chloroform/isopropanol in accordance with TRIzol manufacturer’s protocol. Quantitative PCR was performed with SYBR Green (Thermo Fisher Scientific) following cDNA generation by M-MuLV Reverse Transcriptase (NEB). Primers for Gbp genes were previously published (Kim et al., 2011). Primer sequences for Casp11 were same as previously published (Kayagaki et al., 2011). Primers for mouse Irf7·. (F) 5’-CTTCAGCACTTTCTTCC GAGA-3’, (R) 5’-TGTAGTGTGGTGACCCTTGC-3’; Isg15: (F) 5’-GAGCTAGAGCCTGCAGCAAT-3’, (R) 5’-TTCTGGGCAATCTGC TTCTT-3’, Mx1: (F) 5’-TCTGAGGAGAGCCAGACGAT-3’, (R) 5’-ACTCTGGTCCCCAATGACAG-3’; human IRF7 (F) 5’-CTTGGCTC CTGAGAGGGCAG-3’, (R) 5’-CGAAGTGCTTCCAGGGCA-3’; human ISG15 (F) 5’-TCCTGCTGGTGGTGGACAA-3’, (R) 5’-TTGTTAT TCCTCACCAGGATGCT-3´; human Mx1 (F) 5´-GTGCATTGCAGAAGGTCAGA-3´, (R) 5´-TCAGGAGCCAGCTTAGGTGT-3´; human GAPDH (F) 5’-GCTCCTCCTGTTCGACAGTCA-3’, (R) 5’-ACCTTCCCCATGGTGTCTGA-3’.
Immunofluorescence microscopy for bacterial morphology
Both mouse and human cells were seeded at a density of 0.5× 106 cells per cm2 on MatriPlate 0.17mm glass bottom plates (DOT Scientific) in RPMI containing 10% FBS. Cells were challenged with L. pneumophila-GFP at MOI = 10, in the presence of 0.1mg/mL thymidine and 2mM IPTG. Cultures were centrifuged at 1000rpm for 5 min, and at 1 hr post inoculation, extracellular bacteria were washed off with warm medium. At the desired time points, cells were washed, fixed with 2% paraformaldehyde and stained with polyclonal rabbit serum against L. pneumophila (1:5000), and detected with goat a-rabbit IgG-Texas Red (1:500) (Life technologies). Bacteria that were antibody reactive were quantified from the GFP+ pool and counted as having cytosolic-permeable vacuoles. For IFN-pretreated macrophages, a second round of probing with anti-L. pneumophila following 0.1% Triton X-100 permeabilization was performed to expose the total bacterial population, detected with goat a-rabbit IgG-Alexa Fluor 488 (1:500) (Life technologies). Here, the total cytosolic permeable vacuoles was determined as the ratio of bacteria staining prior to permeabilization relative to the number of bacteria visualized after detergent permeabilization (Creasey and Isberg, 2012). Each experiment was a representative of at least 3 independent experiments. 3–4 technical replicates were performed per experiment, with 100+ bacteria viewed per technical replicate.
Cytosolic plasmid extraction and quantification
For pJB908 plasmid extraction, the experimental protocol followed previous protocol (Ge et al., 2012). BMDM were seeded at 12×106 cells in a 10cm non-tissue culture treated dish with RPMI medium containing 10% FBS and 10% L929 cell supernatant to promote adherence. Cells were rested for 16 hr, then challenged with L. pneumophila harboring nontranferrable pJB908 for the amount of time indicated in the text and figure legends. Cells were lifted passively after 10 min incubation with ice-cold PBS, washed twice with ice-cold PBS, and quantified by hemocytometer. Cells were lysed with 1.0×107 cells per mL of hypotonic buffer (20 mM HEPES-KOH [pH 7.5], 10 mM KCl, 1.5 mM MgCl2,1 mM Na EDTA, 1 mM Na EGTA, and the Roche protease inhibitor cocktail) on ice for 15 min. The lysate was then passaged 15 times through a 20G needle attached to a 3ml syringe to complete the mechanical disruption. An unrelated plasmid pUC18 was spiked into the lysate at 100ng/ml after needle disruption to control for plasmid lost during the following extraction. Cellular debris, including the nuclear fraction and membrane vesicles, was removed by serial centrifugation at 5 min at 1500 g and 15 min at 5000 g. The cleared cytosolic lysate was then subjected to 2× phenol/chloroform extraction, 2× chloroform wash, followed by precipitation in 2.5× vol ethanol and chilled to −80°C. DNA was pelleted by centrifugation at 12,000 rpm for 30 min at 4°C. The precipitated DNA pellet was resuspended in 50 μL of H2O then serially diluted for quantitative PCR with primer sets specific to pJB908. Standard curves with known amounts of plasmids were used to back-calculate the amount of plasmid contained in cytosolic fractions. Primer sets for pJB908 was published previously: (F) 5’-TCAGGAAGCACAAATGT CAATG-3’; (R) 5’-GGTCTACACCACCAAAATCACG-3’ (Ge et al., 2012). Primers for pUC18 flanks the M13 cloning site of the vector. (F) 5’-CAGGAAACAGCTATGAC −3’, (R) 5’-GTAAAACGACGGCCAG-3’.
ASC speck quantification
Cells were seeded at a density of 0.5× 106 cells/cm2 on MatriPlate 0.17mm glass bottom plates (DOT Scientific) in RPMI containing 10% FBS. At indicated time point post challenge with L. pneumophila, cells were fixed/permeabilized via ice cold methanol.
Polyclonal rat serum against L. pneumophila (1:5000) and rabbit anti-ASC antibody were used to recognize L. pneumophila and endogenous ASC. Goat anti-rat AF488 and goat anti-rabbit AF594 were used to visualize L. pneumophila and ASC, respectively. 25 fields at 20× magnification were captured and stitched by the Cytation3 automated microscope, generating fields of view with 3000 cells each for image quantification. Signal intensity of L. pneumophila and ASC were analyzed with the Gen5 software, using uninfected wells, and Asc−/− macrophages as negative controls to determine the appropriate signal intensity and puncta size. Puncta sizes less than 3 μm were considered positive events.
RNA-sequencing
Total RNA was isolated from uninfected mouse lungs using TRIzol and used to make a directional cDNA library using TrueSeq kit. cDNA libraries were sequenced on MiSeq (Illumina) and aligned using TopHat2 and Cufflinks software. Fragments per kilobases mapped was plotted in Prism7. Colors for data visualization is described in the text and figure legends. The data are available at the National Center for Biotechnology Information Gene Expression Omnibus: GSE110678
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses on experiments using murine bone marrow derived macrophages were performed using the Student’s t test (twotailed) using GraphPad Prism7. Experiments involving human macrophages were analyzed using paired analysis, in which each pair consists of cells from the same donor. Bacterial colony forming units from mouse lungs were compared using log-transformed values. (*) p < 0.05; (**) p < 0.01; (***) p < 0.001; (****) p < 0.0001.
ACCESSION NUMBERS
The accession number for the RNA sequencing data reported in this paper is NCBI SRA: GSE110678.
Supplementary Material
Highlights.
Infection-driven IFN is dispensable for pyroptosis against cytosolic Legionella
Constitutive IFN maintains GBP expression in murine and human macrophages
GBPs are needed to release bacterial content from cytosolic Legionella bacteria
GBPs are required for restriction of Legionella bacterial growth in vivo
ACKNOWLEDGMENTS
We thank Dr. Vishva Dixit and Dr. Stephanie Vogel for the donation of Casp11−/− mice and Ifnb−/− mice, respectively. We are grateful to Dr. Katherine Fitzgerald, Dr. Robert O. Watson, and George Papadopulos for insightful discussion and review of the text. This work was supported by NIH grants AI126050 and AI135369 and the Russian Science Fund Project 15–15-00100 to A. Poltorak and by HHMI and NIAID grant R01AI-113211 to R.R.I. B.C.L. was supported by the HHMI MERGE-ID initiative 56006767 and by NIAID training grant T32 AI 007077. V.I. was supported by Government Contract 6.5111.2017/BCH.
Footnotes
AUTHOR CONTRIBUTIONS
B.C.L. designed and conducted experiments. B.C.L., A. Poltorak, and R.R.I. interpreted results and wrote the paper. A. Panda provided human BAL cells. V.I. performed RNA sequencing. A. Poltorak and R.R.I. provided guidance and reagents. J.S. and H.I.M. provided technical aid and discussion. M.Y. and J.C. contributed mice and discussion.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.012.
REFERENCES
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE,Tan MH, Cotter PA, Vance RE, Aderem A, and Miao EA (2013). Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. (2012). Commensal bacteria calibrate theactivation threshold of innate antiviral immunity. Immunity 37, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, and Barber GN (2012). STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 109, 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrat S, Dugan AS, and Isberg RR (2014). The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translationof pro-inflammatory cytokines. PLoS Pathog. 10, e1004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry KC, Fontana MF, Portman JL, Dugan AS, and Vance RE (2013). IL-1 α signaling initiatesthe inflammatory response to virulent Legionella pneumophila in vivo. J. Immunol 190, 6329–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, and Isberg RR (1993). Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol 7, 7–19. [DOI] [PubMed] [Google Scholar]
- Bocci V (1985). The physiological interferon response. Immunol. Today 6,7–9. [DOI] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, and Monack DM (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, and Roy CR (2013). Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. USA 110, 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, and Shin S (2015). Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 112, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J, Vance RE, Fontana MF, and Dietrich WF (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol 9, 2344–2357. [DOI] [PubMed] [Google Scholar]
- Corrales L, Woo S-R, Williams JB, McWhirter SM, Dubensky TW Jr., and Gajewski TF (2016). Antagonism ofthe STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J. Immunol 196, 3191–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasey EA, and Isberg RR (2012). The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. USA 109, 3481–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JQ, and Gordon S (2005). Isolation and culture of human macrophages In Basic Cell Culture Protocols, Helgason CD and Miller CL, eds. (Humana Press; ), pp. 105–116. [DOI] [PubMed] [Google Scholar]
- Dill MT, Makowska Z, Trincucci G, Gruber AJ, Vogt JE, Filipowicz M, Calabrese D, Krol I, Lau DT, Terracciano L, et al. (2014). Pegylated IFN-α regulates hepatic gene expression through transient Jak/STAT activation. J. Clin. Invest 124, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, Yamamoto M, Nagarajan UM, Miao EA, and Coers J (2015). Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect. Immun 83, 4740–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo ZQ, and Vance RE (2011). Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7, e1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman CM, Crugington S, Stolberg VR, Brown JP, Sonstein J, Alexis NE, Doerschuk CM, Basta PV, Carretta EE, Couper DJ, et al. (2015). Design of a multi-center immunophenotyping analysis of peripheral blood, sputum and bronchoalveola rlavage fluid in theSubpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS). J. Transl. Med 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Gong Y-N, Xu Y, and Shao F (2012). Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc. Natl. Acad. Sci. USA 109, 6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson APS, Trapani JA, Levy DE, Hertzog PJ, Clarke CJP, and Johnstone RW (2010). Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 8, e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Clarke CJP, Johnstone RW, and Levy DE (2012). Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlova A, Erttmann SFF, Raffi FAA, Schmalz AMM, Resch U, Anugula S, Lienenklaus S, Nilsson LMM, Kroger A, Nilsson JAA, et al. (2015). DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42, 332–343. [DOI] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1 β secretion. Cell Res. 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, O’Connor TJ, and Heidtman M (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol 7, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SS, and Roy CR (2013). Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat. Immunol 14, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JD, Markert JM, Li L, Carroll SL, and Cassady KA (2016). STAT1 and NF-kB inhibitors diminish basal interferon-stimulated geneexpression and improve the productive infection of oncolytic HSV in MPNST cells. Mol. Cancer Res 14, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaguin M, Houlbert N, Fardel O, and Lecureur V (2013). Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol. 281, 51–61. [DOI] [PubMed] [Google Scholar]
- Jorgensen I, Zhang Y, Krantz BA, and Miao EA (2016). Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med 213, 2113–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, AkashiTakamura S, Miyake K, Zhang J, Lee WP, Muszyriski A, et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kim B-H, Shenoy AR, Kumar P, Das R, Tiwari S, and MacMicking JD (2011). A family of IFN-𝛄-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721. [DOI] [PubMed] [Google Scholar]
- Laguna RK, Creasey EA, Li Z, Valtz N, and Isberg RR (2006).A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. USA 103, 18745–18750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock DL, Chaudhary A, and Miller SI (2015). Salmonellae interactions with host processes. Nat. Rev. Microbiol 13, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. (1995). Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80, 401–411. [DOI] [PubMed] [Google Scholar]
- Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, et al. (2009). Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol 183, 3229–3236. [DOI] [PubMed] [Google Scholar]
- Lippmann J, Müller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, Hellwig K, Kirschning CJ, Taylor GA, Barchet W, et al. (2011). Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell. Microbiol 13, 1668–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol 12, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, Lamkanfi M, and Kanneganti TD (2015). The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol 16, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, et al. (2016). IRGB10 liberates bacterial ligandsforsensing by theAIM2 and caspase-11-NLRP3 inflam-masomes. Cell 167, 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier E, and Broz P (2015). Interferon-induced guanylate-binding proteins promote cytosolic lipopolysaccharide detection by caspase-11. DNA Cell Biol. 34, 1–5. [DOI] [PubMed] [Google Scholar]
- Meunier E, Dick MS, Dreier RF, Schürmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, et al. (2014). Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–370. [DOI] [PubMed] [Google Scholar]
- Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Rühl S, Dussurgey S, Dick MS, Kistner A, Rigard M, et al. (2015). Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol 16, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, and Vance RE (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales VM, Backman A, and Bagdasarian M (1991). A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97, 39–47. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang S-YY, Feng T, et al. ; Immunological Genome Project Consortium (2016). Parsing the interferon transcriptional network and its disease associations. Cell 164, 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor TJ, Boyd D, Dorer MS, and Isberg RR (2012). Aggravating genetic interactions allow a solution to redundancy in a bacterial pathogen. Science 338, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, and Coers J (2014). Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl. Acad. Sci. USA 111, 6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla-Moffett D, Barber MF, Taylor GA, and Coers J (2016). Interferon-inducible GTPases in host resistance, inflammation and disease. J. Mol. Biol. 428, 3495–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, and Hornef MW (2011). IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 108, 7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, MacMicking JD, and James LC (2013). Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science 340, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren T, Zamboni DS, Roy CR, Dietrich WF, and Vance RE (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ZJ, Goutagny N, Perera P-Y, Kato H, Kumar H, Kawai T, Akira S, Savan R, van Echo D, Fitzgerald KA, et al. (2007). The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med 204, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha BC, Marques PE, Leoratti FMS, Junqueira C, Pereira DB, Antonelli LRDV, Menezes GB, Golenbock DT, and Gazzinelli RT (2015).Type I interferon transcriptional signature in neutrophils and low-density granulocytes are associated with tissue damage in malaria. Cell Rep. 13, 2829–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CCD, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan C-YY, et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, et al. (2018). Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. Published online May 21,2018. https://doi.org/ 10.1038/s41418-018-0122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, Bauer L, Kandasamy RK, Griesshammer IM, Kosack L, et al. (2015). The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat. Immunol 16, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- St-Laurent J, Turmel V, Boulet L-PP, and Bissonnette E (2009). Alveolar macrophage subpopulations in bronchoalveolar lavage and induced sputum of asthmatic and control subjects. J. Asthma 46, 1–8. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, and Takaoka A (2001). A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol 2, 378–386. [DOI] [PubMed] [Google Scholar]
- Tateda K, Moore TA, Newstead MW, Tsai WC, Zeng X, Deng JC, Chen G, Reddy R, Yamaguchi K, and Standiford TJ (2001). Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect. Immun 69,2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang T-B, Dillon CP, and Green DR (2016). Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352, aaf2154. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G, et al. (2017). Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404. [DOI] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, et al. (2012). A cluster of interferon-g-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–313. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhao Y, and Shao F (2015). Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol 32, 78–83. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Lilo S, Brodsky IE, Zhang Y, Medzhitov R, Marcu KB, and Bliska JB (2011). A Yersinia effector with enhanced inhibitory activity on the NF-kB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog. 7, e1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurney J, Howard KE, and Sherry B (2007). Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J. Virol 81, 13668–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.