

Abstract
Acid sphingomyelinase deficiency (ASMD) is a rare lysosomal storage disorder with heterogeneous clinical manifestations, including hepatosplenomegaly and infiltrative pulmonary disease, and is associated with significant morbidity and mortality. Olipudase alfa (recombinant human acid sphingomyelinase) is an enzyme replacement therapy under development for the non‐neurological manifestations of ASMD. We present a quantitative systems pharmacology (QSP) model supporting the clinical development of olipudase alfa. The model is multiscale and mechanistic, linking the enzymatic deficiency driving the disease to molecular‐level, cellular‐level, and organ‐level effects. Model development was informed by natural history, and preclinical and clinical studies. By considering patient‐specific pharmacokinetic (PK) profiles and indicators of disease severity, the model describes pharmacodynamic (PD) and clinical end points for individual patients. The ASMD QSP model provides a platform for quantitatively assessing systemic pharmacological effects in adult and pediatric patients, and explaining variability within and across these patient populations, thereby supporting the extrapolation of treatment response from adults to pediatrics.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ QSP is an emerging field and models have been developed across several therapeutic areas. However, rare diseases like ASMD remain a new frontier for QSP and present a unique challenge due to the clinical heterogeneity of these diseases, incomplete biological knowledge, and the small, variable patient populations.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study examined how QSP modeling can be applied to support drug development for a rare disease with heterogeneous clinical manifestations, and how the model could be used to provide insight into clinically relevant scenarios.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study presents the first QSP model for ASMD and its response to ERT. This study demonstrates that the QSP approach can successfully describe patient‐specific responses across multiple clinical end points and PD markers, and can provide mechanistic insight into clinically observed patient variability.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study demonstrates that QSP modeling is a versatile and powerful tool to address specific challenges associated with clinical development in the rare disease therapeutic area.
Acid sphingomyelinase deficiency (ASMD), historically known as Niemann‐Pick disease types A and B, is a rare lysosomal storage disorder caused by mutations in the gene SMPD1, which encodes the enzyme acid sphingomyelinase (ASM). ASM catalyzes the conversion of sphingomyelin, a major constituent of cell membranes, to ceramide. The reduced functional ASM leads to accumulation of sphingomyelin in multiple cell types, including cells of the monocyte‐macrophage lineage and hepatocytes.1, 2 The consequent cell and tissue damage affects multiple organ systems, and causes heterogeneous disease manifestations, including, but not limited to, hepatosplenomegaly, infiltrative lung disease, hematological abnormalities, and dyslipidemia.1
ASMD is a serious and potentially fatal disease. It is associated with a spectrum of disease subtypes, ranging from the severe infantile neurovisceral phenotype (Niemann‐Pick type A) to the chronic visceral form (Niemann‐Pick type B), with an intermediate chronic neurovisceral phenotype (Niemann‐Pick type A/B, Niemann‐Pick B variant).3, 4, 5 The infantile neurovisceral form is typically fatal by the age of 3 years. Patients with the chronic visceral phenotype may have a normal lifespan, or may experience early mortality due to disease complications, including hepatic and pulmonary disease.6 The prevalence of ASMD is estimated at 0.4–0.6 per 100,000 births.3 There is currently no approved treatment for the disease, and only supportive care for management of symptoms is possible.
Olipudase alfa (recombinant human acid sphingomyelinase) is an enzyme replacement therapy (ERT) under development for the treatment of the non‐neurological manifestations of ASMD. By supplementing the deficient functional enzyme, ERTs for lysosomal storage diseases can clear the accumulated substrate and alleviate disease symptoms.7 A recent phase Ib trial of olipudase alfa included five adults and demonstrated improvements in multiple clinically relevant end points, including spleen volume, infiltrative lung disease, and in levels of stored sphingomyelin over 26 weeks of treatment.2, 8 The ongoing, long‐term trial demonstrated that these patients continue to show clinical improvements in hepatosplenomegaly, infiltrative lung disease, and lipid profiles through 30 months of treatment.9 A phase II/III adult trial (http://ClinicalTrials.gov Identifier: NCT02004691; clinicaltrialsregister.eu EudraCT number: 2015‐000371‐26) and phase I/II pediatric trial (http://ClinicalTrials.gov Identifier: NCT02292654; clinicaltrialsregister.eu EudraCT Number: 2014–003198‐40) for olipudase alfa are currently ongoing. The primary objective of this phase II/III study is efficacy on spleen volume and hemoglobin (Hb)‐adjusted % predicted diffusion capacity of the lungs for carbon monoxide (Hb‐adjusted % predicted DLco).
To support the clinical development of olipudase alfa, we developed a quantitative systems pharmacology (QSP) model describing ASMD and the pharmacological effects of olipudase alfa. QSP is a mechanistic modeling approach to describe and simulate the system‐wide and multiscale pathophysiology of a disease and response to pharmacological intervention.10 The QSP model incorporates the drivers of disease at the molecular level (e.g., deficient enzymatic activity and substrate accumulation) and the consequent effects at the cellular level (e.g., aberrant macrophage function) and at the organ level (e.g., organomegaly and impaired organ function). Hence, the model applications include providing a mechanistic basis and support for the dose, for pharmacological effects on biomarkers and the translation of these effects at an organ level (linking biomarker effects to clinical end points), as well as for evaluating different dosing regimens and simulating the effects of treatment on the overall disease burden through the relative pharmacological impact on multiple organs. It also provides a framework for understanding and evaluating the variability within and across different patient populations, including adult and pediatric patients.
QSP modeling has previously been applied in a number of therapeutic areas, including cardiovascular,11, 12 respiratory,13 and neurological14 diseases, as well as diabetes15 and cancer.16 However, QSP modeling largely remains a new frontier for the lysosomal storage diseases. Although prior modeling studies have assessed the evolution of storage diseases based on residual enzyme activity levels17 and the dynamics of pathways related to ASMD, such as sphingolipid metabolism,18, 19, 20 these models have focused on behavior at the molecular level. The QSP approach links representations of molecular‐level dynamics to other submodels describing different biological scales, enabling assessment of therapeutic effects and of different sources of variability on the entire system.
Here, we describe the development, calibration, and qualification of a QSP model for ASMD, with a focus on application to pediatric extrapolation and drug development. We show that the model can replicate the time course of disease development observed in the natural history of ASMD, and the response to treatment with olipudase alfa observed in clinical trials. We also demonstrate the capabilities of the QSP model in helping to understand variability among patients and in predicting the outcomes of novel clinically relevant scenarios.
METHODS
Data sources
Due to the mechanistic and multiscale nature of the ASMD QSP model, a diverse set of data sources was used to develop and calibrate the model (Table 1). These include natural history studies,6, 21 in vitro cell line data, preclinical studies in the ASM knockout (ASMKO) mouse, and data from completed and ongoing clinical trials.2, 8, 9, 22
Table 1.
Overview of data sources used to develop and calibrate the QSP model
| Data source | Model level | Description |
|---|---|---|
|
Preclinical studies (ASMKO mouse)22 |
PBPK | • Estimation of PK and biodistribution parameters from ASMKO PBPK model, which were then scaled to human PBPK model |
|
Natural history study (59 adult and pediatric patients, from 1 year to up to 11 years of assessment)20 |
Organ | • Estimation of disease progression rates in lungs and spleen |
|
Phase Ia (11 adult patients)21 |
PBPK | • Dose response and calibration of human PK model |
|
Phase Ib (5 adult patients)8 |
PBPK, molecular, organ | • Calibration of end points in molecular‐level and organ‐level models |
| Literature | Molecular, cellular, organ |
• Rates of enzyme kinetics and uptake and clearance processes • Sphingomyelin accumulation amounts and rates • Cellular and intracellular compartment volumes |
ASMKO, acid sphingomyelinase knockout mice; PBPK, physiologically based pharmacokinetic; PK, pharmacokinetic; QSP, quantitative systems pharmacology.
Description of model structure
The ASMD QSP model consists of four submodels: a pharmacokinetic (PK) submodel, a molecular‐level submodel, a cellular‐level submodel, and an organ‐level submodel (Figure 1). The model describes four key outputs related to the clinical assessment of ASMD severity and the response to treatment with olipudase alfa: plasma ceramide and plasma lysosphingomyelin from the molecular level, and spleen volume and Hb‐adjusted % predicted DLco from the organ level. Ceramide is central to the mechanism of action of olipudase alfa and is a safety biomarker.8, 23 Lysosphingomyelin is substantially elevated in ASMD and is a disease biomarker.24, 25 Spleen volume and Hb‐adjusted % predicted DLco are key clinical end points because splenomegaly and interstitial lung disease are prominent clinical features of the disease.
Figure 1.
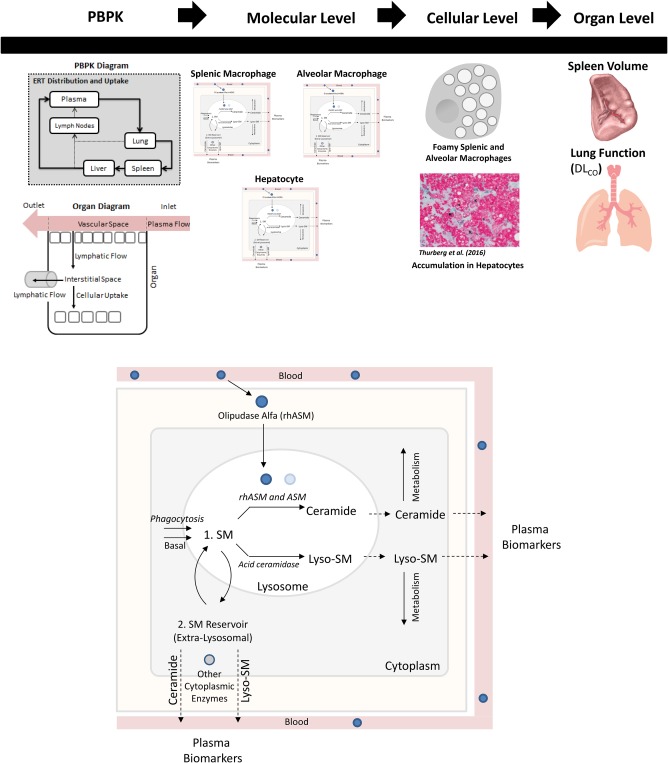
(upper panel) Overview of the structure of the quantitative systems pharmacology (QSP) model for acid sphingomyelinase deficiency (ASMD) and the response to olipudase alfa. (lower panel) Enhanced view of molecular level of model, describing the production of sphingomyelin (SM), its conversion to ceramide by endogenous ASM and olipudase alfa (rhASM), and to lysosphingomyelin (lyso‐SM) by deacylases such as acid ceramidase, the intracellular metabolism of both ceramide and lysosphingomyelin, and the export of these biomarker species into the plasma. All reactions are described in all three cell types, except SM uptake via phagocytosis is limited to the splenic and alveolar macrophages. DLco, diffusing capacity of the lung for carbon monoxide; PBPK, physiologically‐based pharmacokinetics.
Overall, the model is a system of 52 ordinary differential equations using generalized mass action expressions, including Michaelis‐Menten and Hill kinetics (Supplementary Material).
The PK submodel is a reduced physiologically based pharmacokinetic (PBPK) model, based on the full model published in ref. 26. The reduced model focuses on the three organ compartments associated with prominent manifestations of ASMD: the spleen, lung, and liver. The reduced model also includes the plasma and lymph node compartments, as well as organs upon which the liver is dependent. The biodistribution of olipudase alfa across compartments predicted by this submodel serves as input to the molecular level of the model.
Figure 1 shows the molecular‐level submodel. The processes described occur in each cell type (i.e., hepatocytes and splenic and alveolar macrophages), and are represented separately for each cell type. Differentiating these cell types allows us to appropriately represent local tissue biology in the model, such as differences in ceramide production between hepatocytes and splenic macrophages, and facilitates linkage to the organ‐level submodel. Molecular‐level processes are separated into those occurring within lysosomal or extralysosomal compartments. Within the lysosome, the model describes the production of ceramide from sphingomyelin, catalyzed both by endogenous residual ASM and exogenously administered olipudase alfa. The input to this level of the model is the drug concentration within each organ compartment, which drives the latter reaction. The production of lysosphingomyelin, another product of sphingomyelin, is also represented within the lysosome. This reaction is thought to be catalyzed by one or more of the ceramidases (e.g., acid ceramidase). In addition, the production of both ceramide and lysosphingomyelin outside the lysosome, catalyzed by other endogenous enzymes (e.g., neutral sphingomyelinases), is represented. This submodel also describes the metabolism of both ceramide and lysosphingomyelin to untracked sink products in the extralysosomal space, and the export of both species into the plasma.
The cellular level of the model is described as algebraic empirical Hill function expressions representing the normality of splenic and alveolar macrophage behaviors. The rationale for this approach is that disease status in both the spleen and the lung in ASMD are linked to the presence of “foamy” macrophages (i.e., those with substantial sphingomyelin accumulation).6, 27, 28, 29 The input to this level of the model is the amount of accumulated sphingomyelin in each macrophage type, which is an output of the molecular‐level submodel. As these amounts increase in untreated ASMD, splenic and alveolar macrophage functions decline. Treatment with olipudase alfa clears accumulated sphingomyelin, leading to improvement in macrophage function. The degree of functionality of each type of macrophage is the output of the cellular‐level submodel, and an input to the organ‐level submodel.
The organ‐level submodel is separated into models of spleen volume and Hb‐adjusted % predicted DLco. Because the biology regulating the processes of splenomegaly and decreased lung function is complex and not yet fully understood, these processes are described empirically. Natural history data were used to assess the long‐term behavior of these clinical end points in untreated patients with ASMD and to help develop the model equations.21 The rates of disease development in both organs are based on the splenic and alveolar macrophage functionalities, respectively, as defined in the cellular level of the model. Both submodels also include terms describing the restoration of the organ towards normal volume (spleen) or function (lung), which are reliant on the macrophage functionalities and the degrees of intracellular sphingomyelin accumulation.
Parameterization and calibration
In the ASMD QSP model, many parameters were obtained from the literature or from nonclinical data. Examples of these fixed parameters include enzyme kinetics rates, average cell volumes, and most parameters related to the PBPK model.26 Some of these fixed parameters (e.g., volumes for the PBPK model), are assigned separately for adults and for different pediatric age cohorts due to scaling considerations. Other parameters were estimated based on literature values, preclinical data, or clinical data, but were not varied among subjects. Examples of this second category include the reflection coefficients in the PBPK model, which were initially calibrated to nonclinical biodistribution data from ASMKO mice and then translated to a human model, and rate constants for unmeasured or lumped processes, such as the intracellular metabolism of ceramide to its products.
The third category consists of parameters that were allowed to vary among subjects, and that were used to calibrate the model to individual‐level clinical data. Examples include the residual endogenous ASM activity and the rate of ASMD‐associated spleen enlargement. Parameters in this category were selected based on prior clinical knowledge (e.g., residual endogenous ASM activity is known to vary both among patients and among cell types from the same patient) as well as analytical techniques for model assessment. These include local sensitivity analysis with respect to the molecular‐level and organ‐level outputs and the system structure‐based Linear‐in‐Flux‐Expressions methodology.30 This category of parameters was further subdivided into those related to the PK model, the molecular‐level submodel, and the organ‐level spleen and lung submodels.
The model was calibrated to adult clinical trial data,8, 9, 22 and parameters were optimized separately for each submodel. Both global (e.g., covariance matrix adaptation evolution strategy (CMA‐ES) and local (e.g., Levenberg‐Marquardt with a multiple initialization strategy) optimization approaches were tested. Estimated parameters were evaluated in terms of biological plausibility, coefficient of variation, and 95% confidence intervals. Alternative optimization solutions were also evaluated using clustering to gain insight into individual‐level and population‐level variability.
Qualification scenarios
Model qualification was approached through comparisons of model‐predicted trends against those observed in natural history21 and preclinical23 data. As an additional qualification step, the model was used to simulate two scenarios assessing (i) differential response to alternative dosing regimens for patients with varying disease severity, and (ii) steady‐state exposure‐responses in the spleen and the lung in a virtual ASMD population. These predictions were also consistent with trends observed in clinical and preclinical data. These scenarios illustrate the capabilities of the model in predicting the outcomes to novel questions of interest that are not readily addressed in the clinic or experimentally.
The virtual population for scenario (ii) was generated through variation of the subject‐specific parameters spanning the PK, molecular, and organ levels. One thousand virtual patients were generated by sampling from a multivariate log‐normal distribution. Mean values for each parameter and the covariance matrix were obtained from the set of all individual calibrations to data from patients in the phase Ib study and its long‐term extension.
Implementation
The model was implemented in MATLAB (The MathWorks, Natick, MA) and in C through the Multiple Interfaces Solver Toolkit.31
RESULTS
Model calibration – molecular level
The molecular‐level submodel was calibrated to clinical measurements of plasma ceramide and plasma lysosphingomyelin. The model captures individual‐specific responses for both biomarkers, reproducing both different pretreatment levels and dynamics over the course of treatment. The model represents both short‐term (i.e., transient, following each infusion) and longer‐term (i.e., over multiple infusions) responses in plasma ceramide to olipudase alfa across the five adult patients (Figure 2). The short‐term response is mechanistically explained by the clearance of sphingomyelin accumulated in the lysosome by each infusion of olipudase alfa. This lysosomal sphingomyelin is rapidly converted to ceramide, which is then exported from the cell, resulting in a peak in plasma ceramide. The clearance of the lysosomal sphingomyelin enables excess sphingomyelin stored in extralysosomal reservoirs, such as the plasma membrane, to transit into the lysosome for degradation. Repetitions of this cycle over multiple infusions of olipudase alfa leads to the debulking of accumulated cellular sphingomyelin, which is reflected in the long‐term decrease in plasma ceramide trough levels. Export of ceramide to the plasma is described primarily through hepatocytes, based on the predominant distribution of ceramide in very low‐density lipoprotein and low‐density lipoprotein.32 Model predictions of the clearance of liver sphingomyelin over time were also verified against clinical measurements (Supplementary Figure S1).
Figure 2.
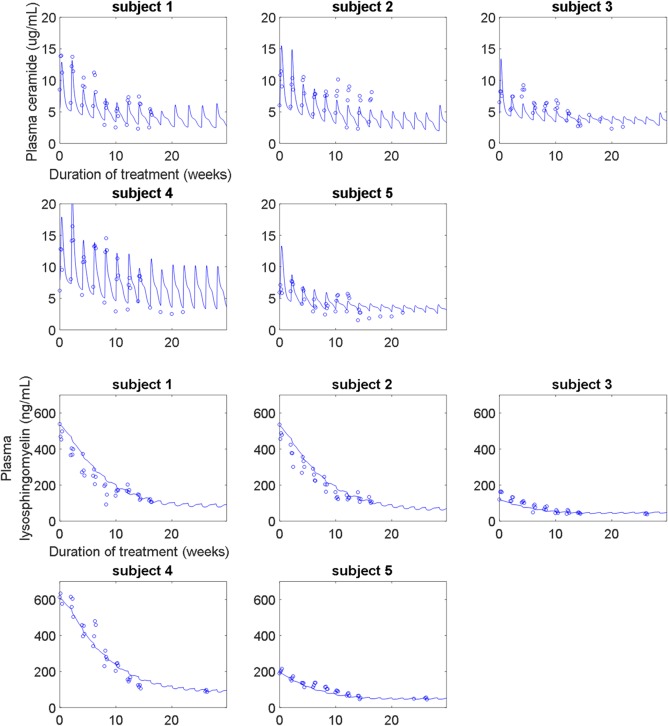
(upper panel) Calibration of the molecular‐level submodel to plasma ceramide clinical data from the phase Ib study. The model describes both the short‐term and long‐term trends in plasma ceramide (normal range: 1.8–6.5 μg/mL) over time and across increasing doses (0.1–3 mg/kg). (lower panel) Calibration of the molecular‐level submodel to plasma lysosphingomyelin clinical data from the phase Ib study. The model describes the long‐term improvement in plasma lysosphingomyelin (normal range: <10 ng/mL) due to olipudase alfa treatment.
The model represents the long‐term responses in plasma lysosphingomyelin for the five adult patients in the phase Ib study (Figure 2). Lysosphingomyelin levels decrease as the accumulated sphingomyelin is cleared from both lysosomal and extralysosomal intracellular compartments through multiple infusions of olipudase alfa. Export of lysosphingomyelin to the plasma is described from all organs, based on its distribution within very low‐density lipoprotein, low‐density lipoprotein, and high‐density lipoprotein.33
Model calibration – organ level
The organ‐level submodel was calibrated to clinical measurements of spleen volume and Hb‐adjusted % predicted DLco (Figure 3). Improvements in both clinical end points over the course of the phase Ib study and its long‐term extension are clinically meaningful.8, 9 The model captures the individual‐specific improvement in spleen volume and Hb‐adjusted % predicted DLco, recapitulating the data well even across varying initial severities in the two clinical manifestations.
Figure 3.
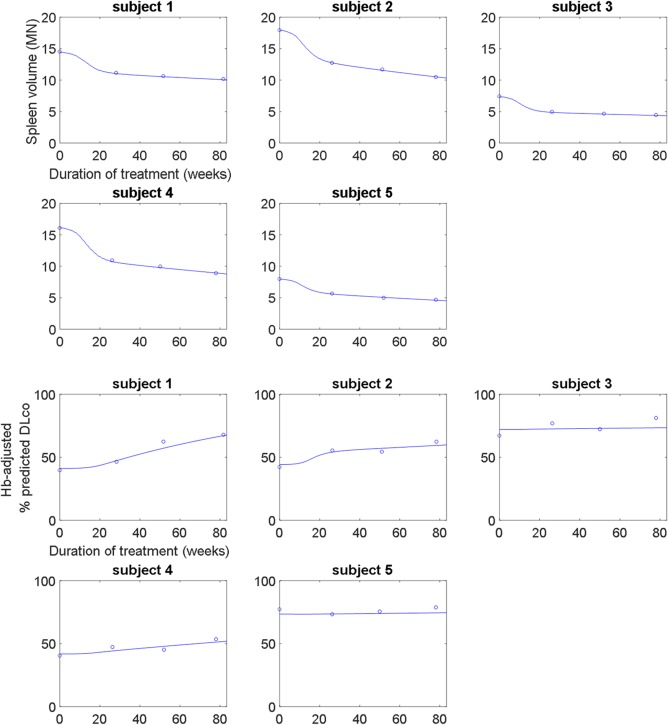
(upper panel) Calibration of the organ‐level submodel to spleen volume (in multiples of normal (MN)) clinical data from the phase 1b study and its long‐term extension. The model describes the long‐term improvement of spleen volume due to olipudase alfa treatment. (lower panel) Calibration of the organ‐level submodel to lung function clinical data in terms of hemoglobin (Hb)‐adjusted % predicted diffusing capacity of the lung for carbon monoxide (DLco) from the phase Ib study and its long‐term extension. The model describes the long‐term improvement of lung function due to olipudase alfa treatment.
Qualification scenarios
The model was applied to two qualification scenarios to illustrate its capabilities, including investigation of questions not readily amenable to direct clinical or experimental assessment.
The first scenario (Figure 4) examines how two virtual patients with ASMD with varying disease severities at treatment initiation will respond to alternative theoretical maximum doses (3 mg/kg vs. 1 mg/kg). The 1 mg/kg dose is not under clinical consideration, and is simulated here only as an exploratory assessment. The model predictions highlight how the differences in response are both subject‐specific and end‐point‐specific. The right‐most column plots the same temporal predictions as a trajectory representation. For each virtual patient, the sequence of vectors represents the direction and magnitude of change in the spleen volume (vertical axis) and Hb‐adjusted % predicted DLco (horizontal axis) at multiple intermediate time points (e.g., 0.5 year, 1 year, 5 years, etc.) over 15 years of simulated treatment. In terms of spleen volume, for the virtual patient with milder disease at onset (bottom row), there is almost no long‐term effect predicted between the two maximum doses. In comparison, an offset of approximately 4 multiples of normal (MN) between the two dosing regimens is predicted for the more severe patient (top row). This indicates that reducing the dose can limit the amount of improvement that can be achieved, and, depending on patient disease severity, this difference may potentially be clinically significant.
Figure 4.
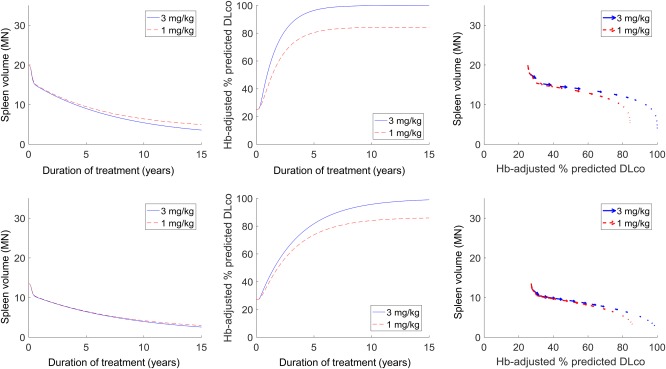
Predicted differential responses in the spleen and lung for alternative olipudase alfa maintenance doses (3 mg/kg in solid blue vs. 1 mg/kg in dashed red) for virtual patients with varying disease severities at onset. The left and center columns show the temporal predictions of treatment response over 15 years of treatment, whereas the right column shows the response trajectory representations.
A similar offset is seen for the Hb‐adjusted % predicted DLco response for the more severe virtual patient. This trend is in agreement with data from the ASMKO mouse showing that clearance of sphingomyelin from the spleen23 and lungs34 is dependent on the administered dose. Interestingly, for the virtual patient with milder disease, an offset is predicted in the Hb‐adjusted % predicted DLco response between the two maintenance doses, even though there was no such difference in the spleen for the same virtual patient. This is supported by ASMKO mice data showing reduced sphingomyelin clearance in the lung compared to the spleen for the same dose ranges,23, 35 indicating that the lung response may be more sensitive to a dose reduction. These patterns emphasize that representing the differential pharmacology across organs is important for capturing interindividual variability in response. The modular structure of the QSP model (i.e., separate molecular‐level submodels evaluated for splenic and alveolar macrophages, which in turn drive separate organ submodels) enables and facilitates such analyses.
Figure 5 describes the second qualification scenario. This builds upon the previous scenario, which examined two virtual patients, by using 1,000 virtual patients with ASMD to investigate long‐term exposure‐responses in terms of spleen volume and Hb‐adjusted % predicted DLco. For each virtual patient, treatment with a maximum dose of 0.6 mg/kg or 3 mg/kg of olipudase alfa was simulated over a period of 15 years. The 0.6‐mg/kg dose is not under clinical consideration, and was simulated only as an exploratory assessment. In the left panel, each dot in the scatterplot represents one virtual patient. The location of a virtual patient on the axis represents their disease severity in terms of spleen volume and Hb‐adjusted % predicted DLco, with those in the upper left quadrant having the most severe disease in both end points. For this assessment, virtual patients with spleen volume >15 MN and Hb‐adjusted % predicted DLco ≤50% at steady‐state before treatment were considered. Black dots represent the virtual patients prior to treatment. Blue and red dots represent the same virtual patients after receiving maximum doses of 0.6 mg/kg (blue) or 3 mg/kg (red) over a 15‐year period. At the 0.6‐mg/kg dose, most virtual patients shift downward, indicating improvement in spleen volume, with little overlap with the pretreatment distribution. They also shift rightward, indicating improvement in Hb‐adjusted % predicted DLco, but not to the same extent; most do not improve beyond 80%. At the 3‐mg/kg dose, more substantial improvement is seen in both organs, with many virtual patients approaching normal spleen volume and 100% Hb‐adjusted % predicted DLco at the lower right corner of the axes.
Figure 5.
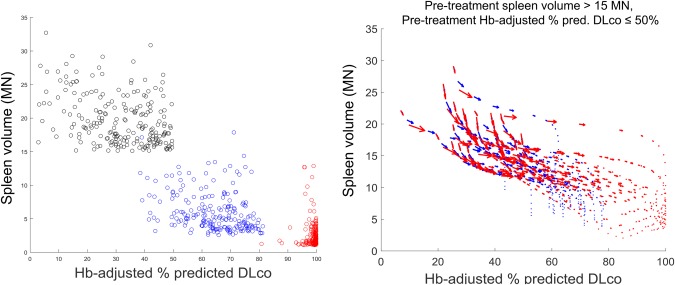
Predicted treatment response in spleen volume and Hb‐adjusted % predicted diffusing capacity of the lung for carbon monoxide (DLco) in a virtual acid sphingomyelinase deficiency (ASMD) patient population with pretreatment spleen volume >15 multiples of normal (MN) and pretreatment hemoglobin (Hb)‐adjusted % predicted DLco ≤50%. The left panel shows a scatterplot comparing virtual patients before treatment (black) with the same virtual patients after receiving 15 years of treatment at a maximum dose of 0.6 mg/kg (blue) or 3 mg/kg (red). The right panel shows the individual response trajectories of a subset of these virtual patients.
The right panel represents treatment responses in terms of the individual trajectories of 25 randomly selected virtual patients administered maximum doses of 0.6 mg/kg (blue) or 3 mg/kg (red). For each individual virtual patient, the response trajectory is represented by a sequence of vectors describing the change in spleen volume (vertical axis) and Hb‐adjusted % predicted DLco (horizontal axis) at multiple intermediate time points (e.g., 0.5 year, 1 year, 5 years, etc.) over 15 years of simulated treatment. For both doses, the virtual patients' trajectories indicate large improvements first, particularly in the spleen – the initial vectors in the trajectories, toward the top left of the figure, have large magnitudes. This is followed by more gradual and long‐term improvements, particularly in the lungs. For the 3‐mg/kg dose, larger improvements (i.e., longer vectors) persist for more time during the simulated treatment period. Overall, these trends are in agreement with data observed in ASMKO mice that ERT response is dose dependent23 and higher systemic ERT doses are necessary to observe clearance from the lungs compared to the spleen.35 This scenario illustrates how the semimechanistic structure of the QSP model enables understanding of the relationship among different end points and how treatment affects different aspects of the disease burden. It also provides an example of how interindividual mechanistic differences can be propagated among levels of the model to help understand population‐level variability.
DISCUSSION
We have presented the first QSP model for ASMD and its response to the ERT olipudase alfa. The model provides a semimechanistic and system‐level description of the disease process and therapeutic response. It successfully replicates both treatment‐naïve disease development patterns and clinical response trends on two pharmacodynamic (PD) biomarkers and two clinical end points. Model performance has been demonstrated on data from five adult patients from the phase Ib clinical trial of olipudase alfa and its ongoing long‐term extension. As illustrated by the qualification scenarios, the model can quantitatively address specific questions about differential patient responses and the propagation of variability across multiple biological levels.
Over the past several years, QSP models have been developed for numerous therapeutic areas. However, rare diseases, including lysosomal storage diseases like ASMD, are a new direction for QSP modeling. QSP provides valuable insight into the specific challenges associated with this therapeutic area, including consideration of pharmacological responses across multiple heterogeneous end points and, critically, mechanism‐based assessment of variability within small patient populations.
In particular, an ongoing application of the ASMD QSP model is to support the extrapolation of olipudase alfa to the pediatric population. Modeling and simulation is becoming an increasingly important tool in pediatric drug development, and has been encouraged by regulatory agencies.36, 37, 38, 39, 40 In particular, the key role of mechanistic modeling approaches (e.g., QSP) as a complement to empirical approaches (e.g., PK/PD modeling) during pediatric extrapolation has been recognized and recommended.41 QSP provides a unique and useful mechanism‐based perspective for evaluating disease and response similarity and the potential factors affecting progression and intervention response in pediatrics compared with adult patients.
To facilitate pediatric extrapolation, the QSP approach leverages the concept that similarity in outputs results from similarity in underlying processes (disease similarity). For example, upon calibration of the model to clinical response data from individual patients in the adult and pediatric cohorts, model‐predicted distributions of clinical and PD end points within and between cohorts can be directly assessed. In addition, the distributions of sensitive parameters governing these outputs can be compared to help understand any observed variability. Moreover, model outputs and parameters can be postprocessed to better understand patterns of variability within the patient population, and to quantify uncertainty. In this regard, the mechanistic framework also provides an opportunity for representing disease progression within the context of normal pediatric development, including scaling of appropriate physiological parameters. Evaluating within‐cohort and between‐cohort distributions at these different levels (e.g., output and parameter) enables the system‐level assessment of response similarity between adult and pediatric patients.
The model will continue to be updated and refined as additional data become available from ongoing adult and pediatric clinical trials, in order to test model performance for new patients based on their baseline disease characteristics and age, improve the assessment of patient variability, and ameliorate uncertainty in the model. However, it is important to acknowledge the inherent data limitations within the rare diseases. In addition, several directions are being explored for expanding the model. One important consideration is integrating knowledge regarding the complex relationship between ASMD genotypes and phenotypes.42, 43 By interpreting genotypes in terms of patterns of pathway‐driven disease development, the model can more directly attribute the diversity in the ASMD population and assist in patient stratification. A second direction is the mechanistic representation of relevant aspects of the inflammatory response. The sphingolipid metabolism pathway contains highly bioactive molecules, including ceramide and sphingosine‐1 phosphate, which are important regulators of the inflammatory response.44, 45 As additional data become available, this will facilitate more mechanistic descriptions of splenomegaly and pulmonary function impairment, including tissue damage1 and feedback between the organ submodels and the PBPK model. Description of other clinical manifestations, including liver disease, is a third key direction. To enable these model development goals, it is necessary to leverage additional diverse data sources, including preclinical and patient registry data, to supplement clinical data.
Overall, the ASMD QSP model provides a mechanistic basis for understanding the variability in the ASMD patient population, both within and between the adult and pediatric populations. The results shown demonstrate that the model recapitulates responses observed in completed and ongoing clinical trials. The qualification scenarios illustrate how the model can help address relevant questions associated with the clinical development of olipudase alfa, and, hence, support decision making. Ongoing model development will further extend these capabilities, with the ultimate goal of helping to improve outcomes for patients with non‐neurological manifestations of ASMD.
Source of Funding
This study was funded by Sanofi.
Conflict of Interest
C.D.K., B.N., R.B., J.L., Z.V.R., M.T., C.O.R., J.S.B., and K.A. are current employees of Sanofi. R.E., S.T., and A.C.P. were employees of Sanofi while this research was conducted and this manuscript was being prepared. B.N., P.J., J.P., and J.T. were contracted by Sanofi while this research was conducted. E.H.S. is a Sanofi consultant. The authors declared no competing interests for this work.
Author Contributions
C.D.K., B.N., A.C.P., E.H.S., J.S.B., and K.A. wrote the manuscript. C.D.K., E.H.S., J.S.B., and K.A. designed the research. C.D.K., B.N., P.J., J.P., J.T., and K.A. performed the research. C.D.K., B.N., R.B., P.J., J.P., J.T., J.L., C.O.R., R.E., S.T., A.C.P., E.H.S., and K.A. analyzed the data. C.D.K., Z.V.R., M.T., and K.A. contributed new reagents/analytical tools.
Supporting information
Additional supporting information may be found in the online version of this article.
Supporting Information 1
Supporting Information 2
References
- 1. McGovern, M.M. , Avetisyan, R. , Sanson, B.J. & Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 12, 41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thurberg, B.L. , Wasserstein, M.P. , Jones, S.A. , Schiano, T.D. , Cox, G.F. & Puga, A.C. Clearance of hepatic sphingomyelin by olipudase alfa is associated with improvement in lipid profiles in acid sphingomyelinase deficiency. Am. J. Surg. Pathol. 40, 1232–1242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McGovern, M.M. et al Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet. Med. 19, 967–974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schuchman, E.H. The pathogenesis and treatment of acid sphigomyelinase‐deficient Niemann‐Pick disease. Int. J. Clin. Pharmacol. Ther. 47(suppl. 1), S48–S57 (2009). [DOI] [PubMed] [Google Scholar]
- 5. Wasserstein, M.P. , Aron, A. , Brodie, S.E. , Simonaro, C. , Desnick, R.J. & McGovern, M.M. Acid sphingomyelinase deficiency: prevalence and characterization of an intermediate phenotype of Niemann‐Pick disease. J. Pediatr. 149, 554–559 (2006). [DOI] [PubMed] [Google Scholar]
- 6. McGovern, M.M. , Lippa, N. , Bagiella, E. , Schuchman, E.H. , Desnick, R.J. & Wasserstein, M.P. Morbidity and mortality in type B Niemann‐Pick disease. Genet. Med. 15, 618–623 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Desnick, R.J. & Schuchman, E.H. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat. Rev. Genet. 3, 954–966 (2002). [DOI] [PubMed] [Google Scholar]
- 8. Wasserstein, M.P. et al Successful within‐patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol. Genet. Metab. 116, 88–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wasserstein, M.P. et al Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): safety and efficacy in adults treated for thirty months. J. Inherit. Metab. Dis.; e‐pub ahead of print 5 January 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danhof, M. Systems pharmacology – towards the modeling of network interactions. Eur. J. Pharm. Sci. 94, 4–14 (2016). [DOI] [PubMed] [Google Scholar]
- 11. Ming, J.E. et al A quantitative systems pharmacology platform to investigate the impact of alirocumab and cholesterol‐lowering therapies on lipid profiles and plaque characteristics. Gene Regul. Syst. Biol. 11, 1177625017710941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gadkar, K. , Lu, J. , Sahasranaman, S. , Davis, J. , Mazer, N.A. & Ramanujan, S. Evaluation of HDL‐modulating interactions for cardiovascular risk reduction using a systems pharmacology approach. J. Lipid Res. 57, 46–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demin, O. et al Systems pharmacology models can be used to understand complex pharmacokinetic‐pharmacodynamic behavior: an example using 5‐lipoxygenase inhibitors. CPT Pharmacometrics Syst. Pharmacol. 2, e74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberts, P. , Spiros, A. & Geerts, H. A humanized clinically calibrated quantitative systems pharmacology model for hypokinetic motor symptoms in Parkinson's disease. Front. Pharmacol. 7, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dalla Man, C. , Rizza, R.A. & Cobelli, C. Meal simulation model of the glucose‐insulin system. IEEE Trans. Biomed. Eng. 54, 1740–1749 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Byrne‐Hoffman, C. & Klinke, D.J. II. A quantitative systems pharmacology perspective on cancer immunology. Processes 3, 235–256 (2015). [Google Scholar]
- 17. Conzelmann, E. & Sandhoff, K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev . Neurosci. 6, 58–71 (1983). [DOI] [PubMed] [Google Scholar]
- 18. Reali, F. et al Mechanistic interplay between ceramide and insulin resistance. Sci. Rep. 7, 41231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wronowska, W. , Charzyńska, A. , Nienałtowski, K. & Gambin, A. Computational metabolism of sphingolipid metabolism. BMC Syst. Biol. 9, 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupta, S. , Maurya, M.R. , Merrill, A.H. Jr , Glass, C.K. & Subramaniam, S. Integration of lipidomics and transcriptomics data towards a systems biology model of sphingolipid metabolism. BMC Syst. Biol. 5, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGovern, M.M. et al A prospective, cross‐sectional survey study of the natural history of Niemann‐Pick disease type B. Pediatrics 122, e341–e349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McGovern, M.M. et al Novel first‐dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann‐Pick disease type B (acid sphingomyelinase deficiency). Genet. Med. 18, 34–40 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Murray, J.M. et al Nonclinical safety assessment of recombinant human acid sphingomyelinase (rhASM) for the treatment of acid sphingomyelinase deficiency: the utility of animal models of disease in the toxicological evaluation of potential therapeutics. Mol. Genet. Metab. 114, 217–225 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Chuang, W.L. et al Lyso‐sphingomyelin is elevated in dried blood spots of Niemann‐Pick B patients. Mol. Genet. Metab. 111, 209–211 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Rodriguez‐Lafrasse, C. & Vanier, M.T. Sphingosylphosphorylcholine in Niemann‐Pick disease brain: accumulation in type A but not in type B. Neurochem. Res. 24, 199–205 (1999). [DOI] [PubMed] [Google Scholar]
- 26. Shah, D.K. & Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67–86 (2012). [DOI] [PubMed] [Google Scholar]
- 27. McGovern, M.M. et al Lipid abnormalities in children with types A and B Niemann Pick disease. J. Pediatr. 145, 77–81 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Dhami, R. , He, X. , Gordon, R.E. & Schuchman, E.H. Analysis of the lung pathology and alveolar macrophage function in the acid sphingomyelinase–deficient mouse model of Niemann‐Pick disease. Lab. Invest. 81, 987–999 (2001). [DOI] [PubMed] [Google Scholar]
- 29. Dawson, P.J. & Dawson, G. Adult Niemann‐Pick disease with sea‐blue histiocytes in the spleen. Hum. Pathol. 13, 1115–1120 (1982). [DOI] [PubMed] [Google Scholar]
- 30. McQuade, S.T. , Abrams, R.E. , Barrett, J.S. , Piccoli, B. & Azer, K. Linear‐in‐flux‐expressions methodology: toward a robust mathematical framework for quantitative systems pharmacology simulators. Gene Regul. Syst. Bio. 11, 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Rijn, Z. , Kaddi, C. & Azer, K. Multiple‐interface solver toolkit (MIST): efficient and modular quantitative systems pharmacology modeling framework. 14th Annual Conference on Frontiers in Applied and Computational Mathematics, Newark, New Jersey, June 24–25, 2017.
- 32. Lightle, S. et al Elevation of ceramide in serum lipoproteins during acute phase response in humans and mice: role of serine‐palmitoyl transferase. Arch. Biochem. Biophys. 419, 120–128 (2003). [DOI] [PubMed] [Google Scholar]
- 33. Scherer, M. , Böttcher, A. , Schmitz, G. & Liebisch, G. Sphingolipid profiling of human plasma and FPLC‐separated lipoprotein fractions by hydrophilic interaction chromatography tandem mass spectrometry. Biochim. Biophys. Acta. 1811, 68–75 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Ziegler, R.J. et al Pulmonary delivery of recombinant acid sphingomyelinase improves clearance of lysosomal sphingomyelin from the lungs of a murine model of Niemann‐Pick disease. Mol. Genet. Metab. 97, 35–42 (2009). [DOI] [PubMed] [Google Scholar]
- 35. Miranda, S.R. et al Infusion of recombinant human acid sphingomyelinase into Niemann‐Pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J. 14, 1988–1995 (2000). [DOI] [PubMed] [Google Scholar]
- 36. Ward, R.M. et al Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr. Res. 81, 692–711 (2016). [DOI] [PubMed] [Google Scholar]
- 37. Stockmann, C. , Barrett, J.S. , Roberts, J.K. & Sherwin, C. Use of modeling and simulation in the design and conduct of pediatric clinical trials and the optimization of individualized dosing regimens. CPT Pharmacometrics Syst. Pharmacol. 4, 630–640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun, H. et al Steps toward harmonization for clinical development of medicines in pediatric ulcerative colitis‐a global scientific discussion, part 2: data extrapolation, trial design, and pharmacokinetics. J. Pediatr . Gastoenterol. Nutr. 58, 684–688 (2014). [DOI] [PubMed] [Google Scholar]
- 39.US Food and Drug Administration (FDA), Center for Drug Evaluation and Research. Summary Minutes of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology, March 14, 2012, Gaylord National Resort & Convention Center, National Harbor, Maryland. <https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM306989.pdf≥ (2012).
- 40. Bellanti, F. & Della Pasqua, O. Modelling and simulation as research tools in paediatric drug development. Eur. J. Clin. Pharmacol. 67(suppl. 1), S75–S86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.European Medicines Agency (EMA). Reflection paper on extrapolation of efficacy and safety in paediatric medicine development. Draft EMA/199678/2016. <http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/04/WC500204187.pdf≥ (2016).
- 42. Zhou, Y.F. , Metcalf, M.C. , Garman, S.C. , Edmunds, T. , Qiu, H. & Wei, R.R. Human acid sphingomeylinase structures provide insight to molecular basis of Niemann‐Pick disease. Nat. Commun. 7, 13082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simonaro, C.M. , Desnick, R.J. , McGovern, M.M. , Wasserstein, M.P. & Schuchman, E.H. The demographics and distribution of type B Niemann‐Pick disease: novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 71, 1413–1419 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gomez‐Muñoz, A. , Presna, N. , Gomez‐Larrauri, A. , Rivera, I.G. , Trueba, M. & Ordoñez, M. Control of inflammatory responses by ceramide, sphingosine 1‐phosphate and ceramide 1‐phosphate. Prog. Lipid Res. 61, 51–62 (2016). [DOI] [PubMed] [Google Scholar]
- 45. Maceyka, M. , Harikumar, K.B. , Milstien, S. & Spiegel, S. Sphingosine‐1‐phosphate signaling and its role in disease. Trends Cell Biol. 22, 50–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article.
Supporting Information 1
Supporting Information 2