

Abstract
Carbonic anhydrase-8 (Car8; murine gene symbol) is an allosteric inhibitor of inositol trisphosphate receptor-1 (ITPR1), which regulates neuronal intracellular calcium release. We previously reported that wildtype Car8 overexpression corrects the baseline allodynia and hyperalgesia associated with calcium dysregulation in the waddle (wdl) mouse due to a 19 bp deletion in exon 8 of the Car8 gene. In this report, we provide preliminary evidence that overexpression of the human wildtype ortholog of Car8 (CA8WT), but not the reported CA8 S100P loss-of-function mutation (CA8MT); inhibits nerve growth factor (NGF)-induced phosphorylation of ITPR1, TrkA (NGF high affinity receptor); and ITPR1-mediated cytoplasmic free calcium release in vitro. Additionally, we show that gene-transfer using AAV8-V5-CA8WT viral particles via sciatic nerve injection demonstrates retrograde transport to dorsal root ganglia (DRG) producing prolonged V5-CA8WT expression, pITPR1 and pTrkA inhibition, and profound analgesia and anti-hyperalgesia in male C57BL/6J mice. AAV8-V5-CA8WT mediated overexpression prevented and treated allodynia and hyperalgesia associated with chronic neuropathic pain produced by the spinal nerve ligation (SNL) model. These AAV8-V5-CA8 data provide a proof-of-concept for precision medicine through targeted gene therapy of NGF-responsive somatosensory neurons as a long-acting local analgesic able to prevent and treat chronic neuropathic pain through regulating TrkA signaling, ITPR1 activation, and intracellular free calcium release by ITPR1.
Keywords: Carbonic Anhydrase-8; hypersensitivity; Allodynia; Inflammatory Pain; Neuropathic Pain; Persistent Pain; Cytosolic Free Calcium; Inositol Trisphosphate; ITPR1; Nerve Growth Factor (NGF); Neurotrophic Tyrosine Kinase Receptor A (TRKA, NTRK1); Adeno Associated Virus (AAV8); Gene Therapy
INTRODUCTION
According to the IOM Report (2011) on Chronic Pain in America, chronic pain affects more than 100 million U.S. adults.1 Our understanding of the natural history of persistent pain and the specific environmental and shared genetic factors thought to impact susceptibility to these pain disorders is lacking.2 Moreover, effective mechanism-based treatments for persistent pain represent a major unmet need in medicine today. Systemic analgesics, including opioids, can be limited by side effects and are inadequate in relieving pain.3 Regional anesthesia can be considered a potential therapy for these patients. Unfortunately, a major limitation in treating acute, subacute, and certain forms of chronic pain with regional techniques is the lack of available local anesthetics exceeding 12 hours in duration.4–6 Moreover, local anesthetics block motor and desirable sensory functions. Agents with target selectivity and the potential for localized somatosensory selective prolonged effects (e.g., lasting weeks to months, or longer) represent a critical goal for ongoing translational research in regional anesthesia and pain medicine today.4
Many critical cellular functions are regulated by intracellular free calcium concentrations including neuronal excitability, neurotransmitter release, neurite outgrowth, apoptosis, and neuroplasticity associated with long-term adaptive responses.7–13 Increased cytosolic free calcium contributes to chronic pain by activating CaMK IIα14–16 and PKC; sensitizing and depolarizing afferent neurons.17–19 In addition, nuclear free calcium integrates communications between the synapse and the nucleus, thereby regulating ‘spinal genomic responses’ required for persistent pain.20, 21 Any imbalance in these regulatory inputs associated with increased intracellular free calcium is likely to affect pain processing in the central or peripheral nervous system leading to altered pain thresholds.22–24 Activation of metabotropic receptors produces IP3 ligand triggering entry into the cell of calcium through receptor-gated and voltage-gated calcium channels (such as N-methyl-D-aspartate receptors); all downstream of Inositol 1,4,5-trisphosphate (IP3) receptors (ITPRs).25–28 IP3, ATP and calcium are significant co-regulators of ITPRs and thereby intracellular free calcium.29, 30 Mouse ITPR1 contains the IP3 ligand-binding core, the ‘modulatory’ domain responding to intracellular regulators such as ATP, calcium and carbonic anhydrase-8 (Car8)31, 32 and undergoes phosphorylation to enhance ATP-dependent calcium release by several protein kinases.33–37 Not surprisingly, these molecular actions are known to play an vital role in persistent pain behaviors.20 Calcium dependent mechanisms that lead to chronic pain may be persistent, difficult to reverse and resistant to current therapies. So far, no available treatments exist that would effectively reverse these mechanisms and produce specifically long lasting analgesia. However, agents that have the potential to shift the balance of cytoplasmic free calcium and thereby decrease neuronal and glial transduction/transmission for extended times, could potentially produce prolonged analgesia and greatly advance this field. In this context, gene therapies may be very promising as emerging novel therapeutic approaches.
Therapeutic gene transfer into somatosensory neurons, including sciatic nerve, dorsal root ganglia (DRG) and the spinal cord, represents a potentially transformational approach to treat chronic pain. Adeno-associated viral vectors (AAV) seldom induce immune responses or produce cytotoxicity, facilitating their targeted delivery of therapeutic genes for overexpression to somatosensory tissues.38 AAV8 can be effectively delivered to the somatosensory pathway by different routes.39–43 In our previous investigation, we demonstrated that Car8 is involved in persistent pain regulation via the ITPR1-intracellular free calcium-signaling pathway.24 We showed that overexpression of murine Car8 wildtype protein downregulated pITPR1, inhibited ATP-stimulated intracellular free calcium release, and abolished mechanical and thermal hypersensitivity. Additionally, we showed that Car8 overexpression in the somatosensory system alleviated chronic inflammatory pain in mice. Herein, we provide the first preclinical proof-of-concept for gene therapy using AAV subtype-8 (AAV8)-mediated CA8 overexpression targeting somatosensory neurons. We show for the first time that CA8 overexpression inhibited NGF-stimulated ITPR1 and TrkA phosphorylation, and ITPR1-mediated calcium release in vitro. We also show AAV8 gene therapy delivered via sciatic nerve injection overexpressed V5-CA8WT and inhibited DRG TrkA phosphorylation; produced prolonged mechanical and thermal analgesia and anti-hyperalgesia; and treated chronic neuropathic pain.
RESULTS
In vitro CA8 expression models
As a prelude to testing the functions of wildtype human CA8WT in the murine somatosensory system, we utilized two AAV vectors to assess whether CA8WT would express well in murine and human cell lines. A V5 sequence was fused to the C-terminal end of the CA8WT cDNA so that exogenously introduced CA8 gene product could be distinguished from the endogenously expressed mouse ortholog with an anti-V5 antibody. These constructs included AAV-V5-CA8WT and AAV-V5-CA8MT vectors. Human derived HEK293 and NBL cell lines were chosen for vector transfection on the basis of higher ITPR1 and of lower endogenous pITPR1 and Car8 levels. Immunostaining data in Fig. 1 shows that there is almost no detectable endogenous Car8 in NBL cells (Fig. 1A and E). In contrast, we observed high levels of ITPR1 in NBL cells (Fig. 1C and E). We also observed almost no detectable pITPR1 at baseline in NBL cells (Fig. 1D-E). In contrast, we observed high levels of TrkA expression at baseline in NBL (Fig. 1B and E). DAPI is used to stain all nuclei for normalizing immune-positive cells. Endogenous expression of CA8, ITPR1, pITPR1 and TrkA in HEK293 cells are similar as that in NBL cells (data not shown). Western blot analyses in Fig. S1 also demonstrate expression of CA8, TrkA and ITPR1 in HEK293 cells.
Figure 1. Endogenous expression of CA8, TrkA, ITPR1 and pITPR1 in cell lines.
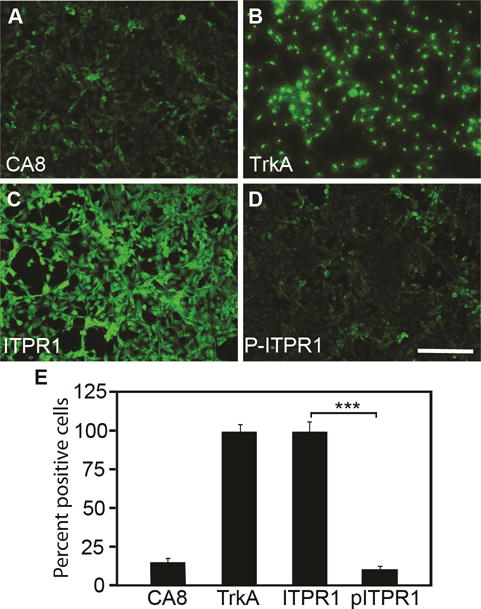
Baseline expression of CA8, TrkA, ITPR1, and pITPR1 are shown for NBL cells using immunohistochemistry (A-D). Quantitation (E) was relative to DAPI staining, which was used to normalize positive staining cells. We observed low levels of CA8 and pITPR1 relative to IPTR1. N=4 from 2 independent cultures in duplicate. Scale bar: 100 μM. (Error bars are SEM; ***P<0.001; Student’s t-test).
To explore whether AAV-V5-CA8 are expressed in both human-derived and murine-derived neuronal and non-neuronal cells, we transfected human HEK293, NBL and murine N2A cells with AAV-V5-CA8WT, AAV-V5-CA8MT and control vectors. As expected, using quantitative real-time PCR we show that the relative transcript expression for the V5-CA8WT and V5-CA8MT constructs did not differ in NBL cells (Fig. 2A), HEK293 and N2A cells (data not shown); but the steady-state CA8MT protein expression on western blots was significantly lower than CA8WT protein due to rapid turnover of the CA8MT form in NBL (Fig. 2B), HEK293 and N2A cell lines (Fig. S2A-B). Similar results are demonstrated with immunocytochemistry to that observed with western blotting data. CA8 expression is easily observed after cells are transfected with AAV-V5-CA8WT vector. In contrast, CA8 is difficult to detect after cells are transfected with AAV-V5-CA8MT vector (Fig. 2C-F, S2C-I). Vector controls show no V5 signal in cultures (Fig. 2D, S2D and S2G). Both human cell lines and the N2A murine cell line exhibited high transfection efficiency (Fig. 2E, Fig. S2E and H, and Fig. S3). DAPI was used to stain all nuclei for normalizing V5-positive cells (Fig. S3). Our prior work demonstrates that overexpression of V5-Car8 can inhibit ITPR1 phosphorylation (pITPR1) in murine-derived N2A cells.24 To further investigate if V5-CA8 overexpression in N2A cells also inhibits murine pITPR1, we used forskolin to stimulate N2A cells two days after CA8 transfection. Western blots analyses show forskolin induces pIPTR1 in a dose-dependent manner (Fig. S4A). Overexpression of V5-CA8 inhibited forskolin-induced murine pITPR1 (Fig. S4B). We conclude that V5-CA8WT protein expression is significantly higher in both human and murine cell lines compared to the V5-CA8MT form. Our data implicate that the human protein (i.e., V5-CA8WT) functions similar to the V5-Car8WT protein on murine ITPR1 and would be a suitable reagent for preclinical studies in rodent pain models facilitating further preclinical testing in preparation for therapeutic development.
Figure 2. Expression of V5-CA8WT and V5-CA8MT in NBL cell culture.
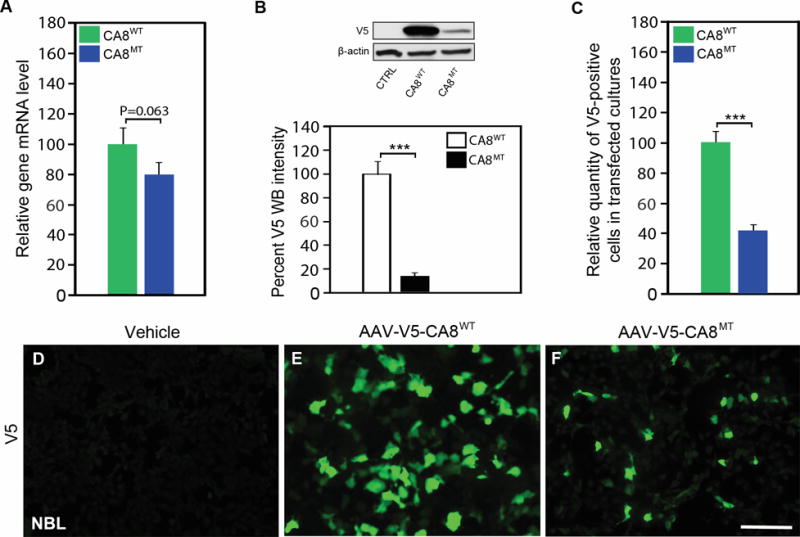
NBL cultures were untransfected, or transfected with AAV-V5-CA8WT or AAV-V5-CA8MT encoding vectors expressing CA8 fused to V5 tag as wildtype or the S100P point mutation. There was no difference in expression of CA8WT as compared to CA8MT as measured by qPCR (A). In contrast, there was little detectable CA8MT protein detected in comparison to CA8WT by western blotting (B). Immunofluorescence data (C, D-F) corroborates our western data showing more positive V5 staining NBL cells after transfection with CA8WT (E) as compared with the CA8MT (F) or untransfected NBL cells (D). N=4 from 2 independent cultures in duplicate. Scale bar: 100 μM. (Error bars are SEM; ***P<0.001; Student’s t-test).
CA8 overexpression down regulates ITPR1 phosphorylation and inhibits NGF-induced free calcium release in vitro
ITPR1 translates IP3 signaling into Ca2+ signaling and thereby plays a key role in many cellular functions.25, 44 Forskolin increases cyclic adenosine monophosphate (cAMP) to phosphorylate ITPR1 via protein kinase A (PKA). Car8 regulates affinity of the ITPR1 intracellular calcium release channel for its IP3 ligand. This allosteric regulator of ITPR1 reduces calcium release from the ER and thereby modulates excitatory calcium signaling.31, 32 To explore whether the interaction of CA8 with ITPR1 will affect its phosphorylation and thereby affect intracellular free Ca2+, we tested whether V5-CA8WT overexpression inhibited the phosphorylation of ITPR1 (pITPR1) by NGF in human HEK293 and NBL cells. NGF-induced increases of ITPR1 phosphorylation (pITPR1) in NBL (Fig. 3A) in a dose-dependent manner and overexpression of V5-CA8WT inhibited the increase of NGF-induced pITPR1 in murine NBL cells, but the V5-CA8MT expressing and empty vectors did not impact pITPR1 (Fig. 3B). CA8WT inhibition of pITPR1 in NBL cells suggests that it may alter ITPR1 mediated intracellular free Ca++ release. Therefore, we next tested the dose-response of NGF stimulated calcium release in NBL and HEK293 cells (Fig. 4A-I and S5A-I). Increasing NGF (1, 10 and 100 ng/mL) produced increasing calcium steady state intracellular calcium, compared to vehicle. The overexpression of CA8WT apparently inhibited the effects of even the highest NGF dose tested (10 ng/mL) but V5-CA8MT could not inhibit NGF-induced calcium release (Fig. 4J and S5J). These findings suggest that NGF signals through ITPR1 to induce calcium release and V5-CA8 inhibits NGF-stimulated ITPR1 activity and thereby reduces steady-state intracellular free calcium levels.
Figure 3. CA8WT and CA8MT Overexpression in cell cultures inhibit NGF-induced ITPR1 phosphorylation (pITPR1).
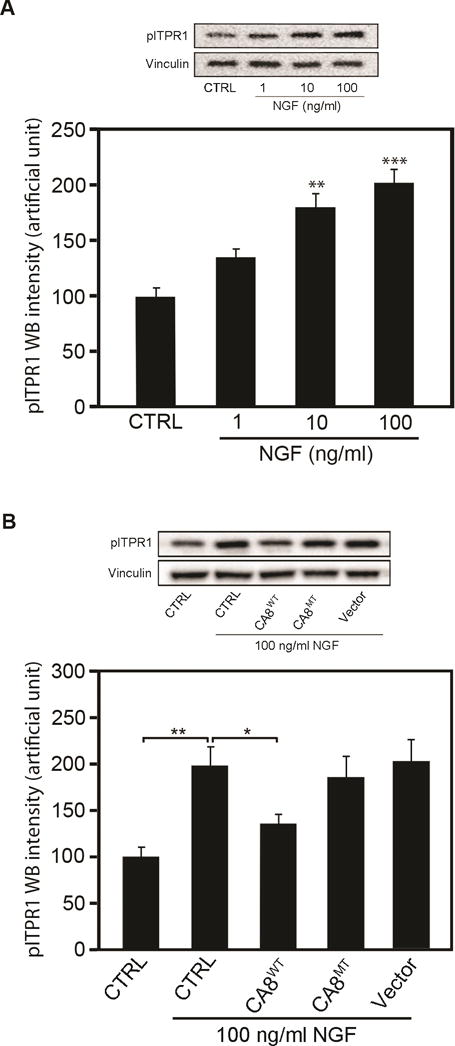
NBL cells were transfected with AAV8-V5-CA8WT or AAV8-V5-CA8MT encoding viral particles. The cultures were collected 48 h after transfection for western blots. Western blotting analyses of pITPR1 demonstrate dose-dependent NGF increase in pITPR1 levels in NBL cultures (A). Overexpression of V5-CA8WT protein significantly reduced NGF-induced pITPR1 increases in NBL cultures (B) but transfection with vectors overexpressing V5-CA8MT failed to effect pITPR1 levels (B). N=6 from 2 independent cultures in triplicate. (Error bars are SEM; *P<0.05; **P<0.01; ***P<0.001; Student’s t-test or one-way ANOVA.)
Figure 4. NGF induces release of cytoplasmic free calcium in a dose-dependent manner and inhibition by CA8WT overexpression in vitro.
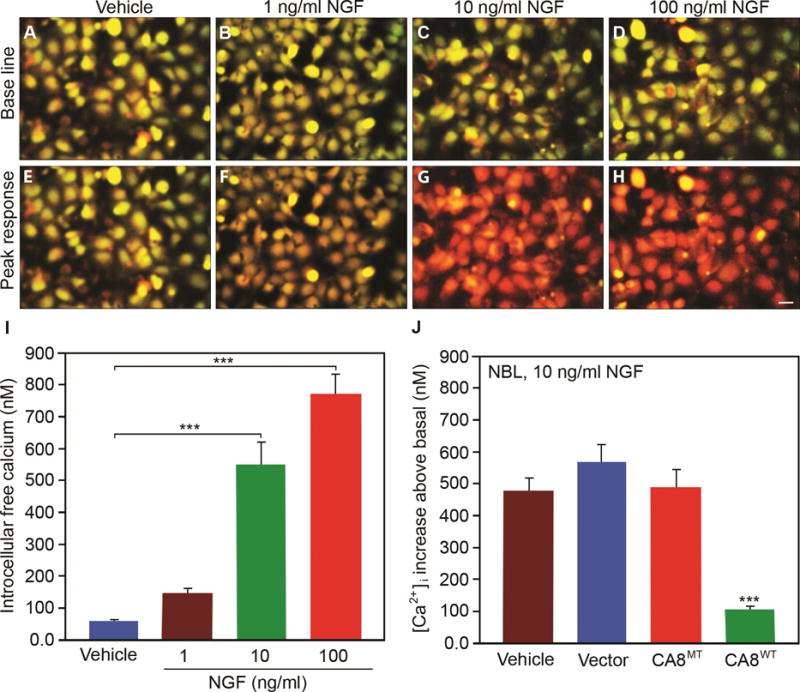
NGF induced intracellular free calcium release as shown by fura-2 labeling in representative NBL cell cultures in a dose-dependent manner from 1ng/mL to 100 ng/mL as compared with vehicle control (A-I). Panels E-H show the peak response for each condition. Panel J shows NBL cells transfected with V5-CA8WT encoding vector inhibited NGF-induced (10 ng/mL) cytoplasmic free calcium release (green bar), but V5-CA8MT encoding vector, empty vector and vehicle could not. N=9 from 3 independent cultures in triplicate. Scale bar: 50 μM. (Error bars are SEM; ***P<0.001; One-way ANOVA.)
NGF calcium release is ITPR1-dependent
We next tested whether NGF-induced calcium release in vitro is ITPR1 dependent. ITPR-specific antagonist 2-aminoethoxydiphenyl borate (2-APB) inhibited the response to 10 ng/mL of NGF in both NBL and HEK293 cultures (Fig. 5 and Fig. S6) in a dose-dependent manner. These findings suggest that NGF signaling is predominantly through ITPR1 and not ryanodine receptor.
Figure 5. NGF induced calcium release is ITPR1-dependent in NBL cells.
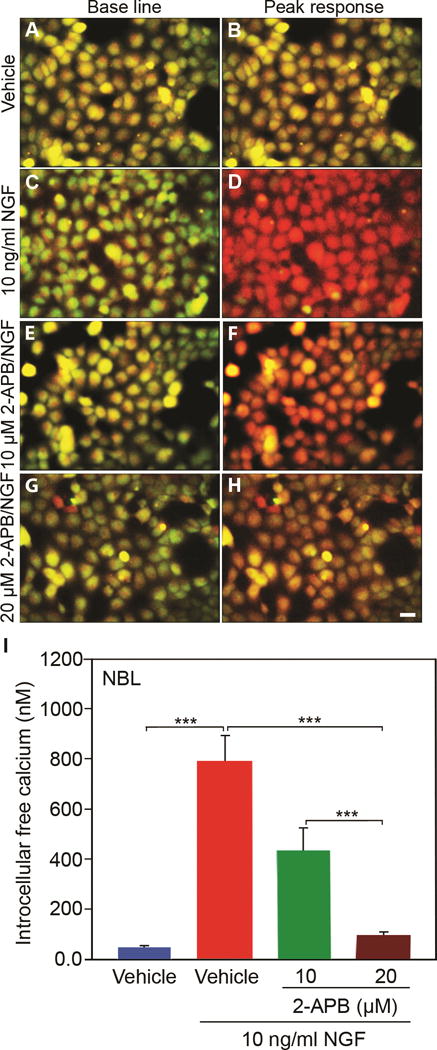
NGF (10ng/mL) stimulated intracellular calcium release was inhibited in a dose-dependent manner by ITPR1 selective inhibitor 2-APB in NBL cells. N=9 from 3 independent cultures in triplicate. Scale bar: 50 μM. (Error bars are SEM; ***P<0.001; One-way ANOVA.)
Overexpression of CA8 in DRG reduces SNL-induced phosphorylation levels of TrkA and ITPR1
To explore if exogenous CA8 can block phosphorylation of TrkA and ITPR1 following lumbar level 5 (L5) spinal nerve ligation (SNL), we analyzed DRG immunofluorescence of V5, pTrkA and pITPR1 Day 21 after SNL. AAV8-V5-CA8 viral particles were injected into sciatic nerve (SN) 3 days before or after SNL (pre-SNL or post-SNL). Using immunohistochemistry, the percentage of V5-positive cells in pre-SNL AAV8-V5-CA8WT group is clearly higher than that in post-SNL AAV8-V5-CA8WT group in L5 DRG (Fig. 6). L4 DRG in Pre-SNL AAV8-V5-CA8MT group showed rare V5-positive cells, however the percentage of V5-positive cells was similar between pre-SNL CA8WT group and post-SNL AAV8-V5-CA8WT group and there were no V5-positive structures in contralateral DRG (data not shown). Overexpression of V5-CA8WT inhibits SNL-induced phosphorylation of TrkA and ITPR1 in L5 DRG in both pre-SNL and post-SNL groups, in comparison with post-SNL V5-CA8MT (Fig. 6 and S7).
Figure 6. AAV8-mediated V5-CA8 gene transfer via sciatic nerve injection transduces L5 DRG and inhibits spinal nerve ligation (SNL)-induced TrkA phosphorylation.
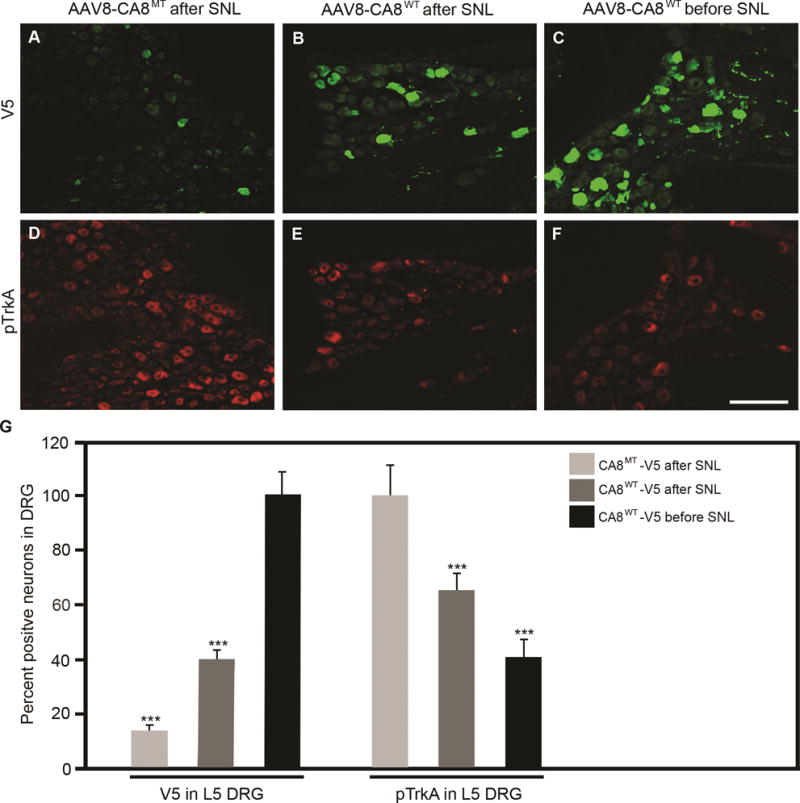
AAV8-V5-CA8WT and AAV8-V5-CA8MT viral particles (1.5μl, 2.65E+14 genome copies/ml) were injected into the sciatic nerves 3 days either before, or after SNL. L5 DRG were harvested on D21 after SNL and immunohistochemistry (IHC) was used to measure V5-CA8WT, V5-CA8MT (A-C) and pTrkA (D-F). SNL L5 DRG V5 IHC was increased significantly at the end of the experiment with SN injection before and after SNL with AAV8-V5-CA8WT, but not after SN injection with AAV8-V5-CA8MT viral particles. There was a significant reduction in pTrkA IHC detected with SN injection before and after SNL with AAV8-V5-CA8WT, but not after SN injection with AAV8-V5-CA8MT. Quantitation was relative to DAPI staining which was used to normalize positive staining cells. N=4. Scale bar: 100 μM. (Error bars are SEM; ***P<0.001; One-way ANOVA.)
Overexpression of V5-CA8WT in DRG produces prolonged analgesia, preventing and treating neuropathic pain
We next tested whether if V5-CA8 could prevent or reverse neuropathic pain using the SNL model in mice. Surprisingly, we found that when AAV8-V5-CA8WT producing viral particles (Fig 7 A and B, blue bars) were injected into SN 3 days after SNL, mechanical thresholds returned to baseline as early as Day 10. Moreover, AAV8-V5-CA8WT produced analgesia (starting from Day 24 through Day 38) by significantly raising mechanical thresholds above baseline. However, when AAV8-V5-CA8MT producing viral particles (Fig 7 A, red bars) were injected 3 days after SNL, this failed to produce any effect on mechanical responses through Day 24, with a slow return to baseline on Days 31-35 (Days 28-35 post-SNL). When AAV8-V5-CA8WT encoding viral particles (Fig S8 A, green bars) were injected into SN 3 days before SNL, mechanical thresholds returned to baseline as early as Day 10, once again producing significant analgesia on Days 31-38. Similar findings were observed for thermal responses (S8 B) after sciatic nerve injections of AAV8-V5-CA8WT and AAV8-V5-CA8MT. Before viral SN injections, the average baseline withdrawal threshold was about 2 gms (2.16 ± 0.06 gms) and thermal latency in naïve mice was about 8 secs (8.03 + 0.12 secs). Differences in withdrawal responses were found between groups {F (2, 21) = 29.5, P=7.8E-7 mechanical threshold; F (2, 21) = 28.6, P=1E-5 thermal latency} and across the 8 time points {F (8,168) = 38.8, P=1.9E-34 mechanical threshold; F(8, 168)=18.7, P=1E-20 thermal latency}. There were also significant interactions between time and group {F (16, 168) = 3.9, P=1E-6 mechanical threshold; F (16,168) = 2.7, P=6.5E-4 thermal latency}. Bonferroni’s pairwise comparisons of time*group interaction indicated that there were no differences between all groups at baseline, Day 1 and D3. Thermal withdrawal latencies in the AAV8-V5-CA8WT group were greater than the AAV8-V5-CA8MT group at all time points after Day 10 and greater than Baseline for the AAV8-V5-CA8WT group on Day 31 post-SNL. Mechanical thresholds in the AAV8-V5-CA8WT group were greater than the AAV8-V5-CA8MT group at all time points after Day 17 and beyond. Both thermal withdrawal latencies and mechanical thresholds in the pre-SNL AAV8-V5-CA8WT treated group show significantly anti-hyperalgesia and/or analgesia from Day 7 or beyond (Fig S8).
Figure 7. AAV8-mediated V5-CA8 gene transfer via sciatic nerve injection treats chronic neuropathic pain associated with the SNL model.
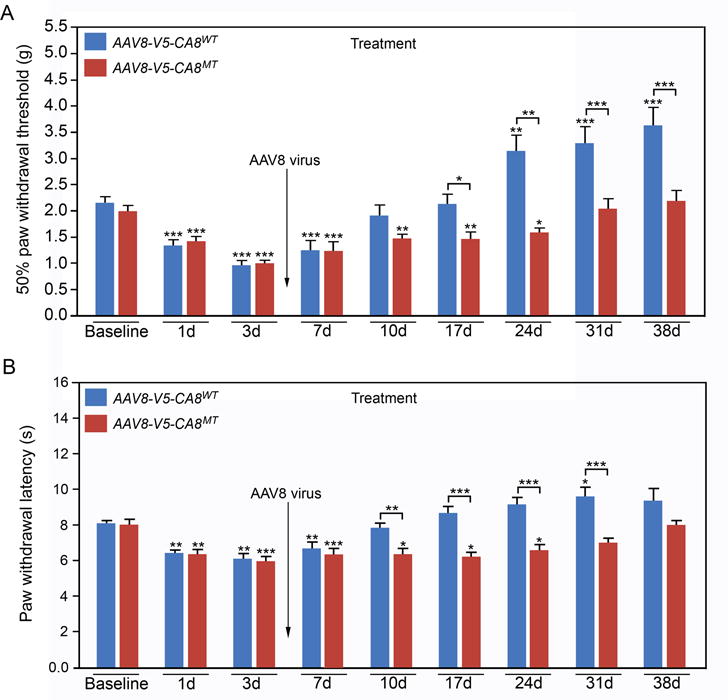
AAV8-V5-CA8WT and AAV8-V5-CA8MT encoding viral particles (1.5μl, 2.65E+14 genome copies/mL) were injected into the sciatic nerves 3 days after SNL. Mechanical withdrawal thresholds (A) were tested by von Frey fibers and thermal paw withdrawal latencies (B) were tested using the Hargreaves method. AAV8-V5-CA8WT inhibited SNL-induced hyperalgesia starting at Day 10 and beyond after virus injection for both mechanical (A) and thermal behaviors (B); but AAV8-V5-CA8MT did not. N = 8. (Error bars are SEM; *P<0.05; **P<0.01; ***P<0.001 by two-way repeated-measures ANOVA in SPSS)
DISCUSSION
In this report, we demonstrate several novel and important findings related to the role of CA8 in regulation of the ITPR1 pathway in chronic pain. First, we show in vitro that overexpression of the human ortholog of murine Car8 (CA8WT) inhibits NGF-stimulated phosphorylation of Serine-1755 of murine ITPR1. Second, we demonstrate that NGF-induced calcium release in NBL and HEK293 cells is inhibited by transfection with AAV vectors encoding V5-CA8WT but not V5-CA8MT. Third, we show in NBL and HEK293 cells that NGF-induced calcium release is almost entirely suppressed by ITPR1-specific inhibitor 2-aminoethoxydiphenyl borate (2APB) suggesting NGF signals almost exclusively through ITPR1 in these cells. Fourth, we demonstrate that SN injection of AAV8-V5-CA8WT viral particles can undergo retrograde transport and transduce DRG somatosensory neurons producing long-lasting expression. Fifth, we show overexpression of CA8 in DRG by sciatic injection inhibits phosphorylation of TrkA and ITPR1. Sixth, we show that sciatic nerve injection of AAV8-V5-CA8WT, but not AAV8-V5-CA8MT viral particles produce significant long-acting analgesia and anti-hyperalgesia in C57BL/6J male mice. Finally, we show AAV8-V5-CA8WT but not AAV8-V5-CA8MT viral particles can prevent or treat SNL induced neuropathic pain. Collectively, these findings are consistent with the actions of CA8 regulating the neuronal ITPR1-cytosolic free calcium pathway downstream of NGF signaling associated with chronic pain.
CA8 inhibits ITPR1-mediated calcium release by regulating TrkA and ITPR1 phosphorylation downstream of NGF
Nerve growth factor45 with activation of its TrkA receptor are sufficient to cause persistent pain.46, 47 We also show that NBL cells express TrkA (NGF high affinity receptor) and when transfected with AAV-V5-CA8WT NGF-induced pITPR1, pTrkA and calcium release were inhibited, but not after transfection with AAV-V5-CA8MT. Similarly, we show that SN injection 3 days after SNL with AAV-V5-CA8WT but not AAV-V5-CA8MT inhibits L5 DRG pTrkA. We believe that this is the first demonstration of CA8 downstream of NGF/TrkA and CA8 inhibition of NGF signaling in vitro and in vivo. We also confirmed that the observed NGF signaling is mediated by ITPR1 using ITPR-specific antagonist 2APB, which inhibited calcium release by NGF in a dose-dependent manner.48 These findings confirm TrkA-dependent NGF signaling requires TrkA phosphorylation (pTrkA) and is associated with ITPR1 activation (pITPR1) and cytoplasmic Ca++ release, each inhibited by CA8 overexpression. After peripheral nerve injury, a positive feedback loop may lead to overproduction of NGF and inflammatory mediators, promoting the upregulation of alpha1A-adrenoceptors expression increasing the production of pro-inflammatory mediators with noradrenaline release. Consequently, nociceptive afferents become primed to respond to adrenergic mediators during continuing inflammation.49 If these mechanisms are relevant to sympathetically maintained pain (e.g., such as cutaneous neuromas, post-herpetic neuralgia, Causalgia and amputation stump pain),50 they may be amenable to CA8 gene therapy. This is a promising approach similar in concept to the anti-NGF therapies in late-stage clinical trials for cancer pain, severe osteoarthritis, and low back pain.51–54 However, anti-NGF therapeutics are currently systemic reagents and significant drug-related adverse events have been described for the class.55, 56 Using precision medicine with targeted overexpression of CA8 via gene therapy could potentially avoid systemic anti-NGF side effects while providing long-lasting analgesia and anti-hyperalgesia.
Many critical cellular functions are regulated by cytoplasmic free calcium concentrations including synaptic plasticity, neurotransmitter release, neuronal excitability, and neuroplasticity.7, 10 Many neuronal biological functions, including memory and pain are dependent to synaptic plasticity.57 It is well known that intracellular signals, including intracellular calcium regulate the activation of pain receptors.58, 59 Pain transmission and perception are affected by physiological as well as pathophysiological modulators of cytosolic free calcium. Generally, alterations of intracellular free calcium in nociceptive neurons will result in a modulation of pain signals. Intracellular free calcium is increased by entry from the extracellular space and/or by consequent Ca2+ release from the intracellular stores. Increased intracellular calcium activates adenylate cyclase and subsequently increases intracellular cAMP, which activates the PKA.60, 61 Similarly, a forskolin-induced rise of intracellular cAMP also subsequently activates (phosphorylates) PKA.60, 61 Activated PKA increases the sensitivity of ITPR1 for IP3 and induces calcium release from endoplasmic reticulum stores. Increased [Ca2+]i also activates PKC and CaM-kinase.62, 63 Interestingly, prior publications implicate PKC in persistent pain associated with the depolarization of afferent neurons and enhanced currents activated by noxious thermal stimuli.18, 57, 64 CaMKIIα also plays an essential role in the initiation of persistent pain.14, 15 CaMKII-dependent ATP sensitive potassium channel (KATP) activation following elevated intracellular free calcium may limit excitotoxicity and cell injury.65 KATP channel opening results in decreased neuronal excitability, reduced neurotransmission, and possibly analgesia and anti-hyperalgesia. Previous data shows that KATP regulation is altered by nerve injury.66 Specifically, activation of KATP channels via intracellular free calcium and the CaM/CaMKII pathway are suppressed by nerve injury, resulting in neuronal hyperexcitability. Therefore, neuronal hyperexcitability represents a molecular lynchpin of pathologic neuroplasticity associated with chronic pain. Based on the significant baseline analgesia and hyperalgesia produced in our CA8 preclinical gene therapy model of neuropathic pain, it will be critical to understand in the future how CA8 impacts the Calcium/CaM/CaMKII pathway and KATP activation in this model.
Increased calcium can also have far reaching effects on cellular functions through effects on nuclear functions. Recent reports show that calcium transients in the nuclei of spinal dorsal horn neurons are associated with a reprogramming of the transcriptome including many pain-related genes.21 The impact of CA8 on the transcriptome and long-term potentiation is another relevant area for future investigation.
AAV8 demonstrates adequate retrograde transport and DRG transduction after sciatic nerve injection to treat neuropathic chronic pain
We choose AAV8 viral particles using the SN route of injection and observed very high expression in small to medium sized primary sensory neurons as well as glial cells, suggesting AAV8 maybe a desirable serotype for delivering this gene construct for treating chronic pain via this route.24 Foust et al., (2008) previously showed intravenous or intraperitoneal injections of neonatal mice with AAV8-GFP produced infrequent labeling of lower motor neurons across all time points and injections.67 In contrast, they showed widespread labeling of dorsal horn fibers and posterior columns, consistent with dorsal root ganglion transduction. These findings suggested AAV8 might be useful for targeting somatosensory pathways important in chronic pain.67 AAV8 delivered by lumbar puncture showed robust transgene expression in DRG neuronal nuclei and axonal projections into the dorsal horn. Immunohistochemical studies show AAV8 transduction of nociceptive neuronal cells staining for peptidergic marker vanilloid receptor subtype 1; small peptidergic neuron markers calcitonin gene-related peptide and substance P; and the nonpeptidergic neuron marker isolectin-B4 (IB4).68 Despite known limitations in retrograde transport for AAV, our findings extend the above studies by showing adequate retrograde transport and transduction of DRG somatosensory neurons with the AAV8 viral particles using the SN injection route. While prolonged expression was demonstrated in our experiments, it is unclear if repeat injections would be necessary for chronic indications. It is well known that repeat AAV injections may lead to inactivity through immune surveillance.69 Host immune response and more efficient DRG transduction may be limitations to the current AAV approach.69
The role of primary afferents in maintaining persistent pain: Implications of CA8 gene therapy for neuropathic pain
It is now well accepted that pain processing and transmission in primary afferent fibers depend on ion channel function.70, 71 Ion channels determine neuronal excitability and transmitter release by regulating resting membrane potential. Neuronal hyperexcitability (peripheral sensitization) associated with both inflammatory and neuropathic pain can result from alterations in ion channel expression. Intense ongoing efforts are focused on developing analgesic molecules that modulate ion channels in primary afferents.71 Primary afferents are compose of mostly unmyelinated (C fibers) that can be further characterized as peptidergic (TrkA) and non-peptidergic fibers. One mechanism of primary afferent sensitization may depend on TrkA phosphorylation medicated by NGF, which was shown to upregulate expression of peripheral nerve sodium channel Nav1.7 (SCN9A) through release of intracellular calcium stores and activation of phospholipase C-gamma.72 Prototypical nociceptors that respond to a wide variety of noxious stimuli, including thermal, mechanical and chemical insults are polymodal C-fibers. A-fibers can also contribute to persistent pain. Shortland et al. (1997) identified sprouting and aberrant functional synapses in lamina II after SN injury (CCI or SNL L5 lesion), but sprouting was not observed in uninjured A-fibers or C-fibers.73 Nonetheless, deafferentation of A-beta and A-delta primary afferents associated with CCI nerve injury led to spontaneous discharge in adjacent uninjured afferents, likely critical in maintaining neuropathic pain.74 Subtypes of primary afferents differ in their profile as potential therapeutic targets and maybe selectively addressed with precision medicine to produce optimal analgesia for selected indications by using promoter specific expression systems.75 Thus, primary afferents represent attractive targets for regional long acting local analgesic delivered by gene therapy.
Summary
In the present study, we demonstrate that the human carbonic anhydrase-8 (CA8) overexpression inhibited NGF-induced pITPR1 and [Ca2+]i increases in NBL and HEK cells. We also show, once again that sciatic nerve injection of AAV8-V5-CA8WT produced profound analgesia in mice3 and effectively prevented and treated mechanical and thermal hyperalgesia after SNL in mice. Collectively, our data demonstrate that CA8 functions in mouse-derived cell lines and the mouse somatosensory nervous system, to inhibit murine ITPR1-mediated calcium release and pain processing. This work provides the first preclinical therapeutic validation for using CA8 as an allosteric inhibitor of ITPR1 to produce prolonged analgesia; prevent and treat neuropathic pain; all suggesting broad potential applications of this long acting local analgesic approach.
MATERIALS AND METHODS
Animal preparations and care
All procedures related to animal use and care were pre-approved by the University of Miami Institutional Animal Use and Care Committee. All C57BL/6J inbred male mice used in our experiments were 2-3 months of age, weighing 20–35 grams. All animals were housed in a virus/antigen-free facility at a controlled temperature and humidity. A 12 hour light/dark cycle was provided along with water and food ad libitum. These animals randomly assigned to groups using random number tables. All samples were de-identified so the investigator performing an experiment was masked to the group and condition.
Mouse models of neuropathic pain
AAV8-V5-CA8MT injections were used as negative controls, which was shown to produce an unstable protein with rapid proteasome degradation of CA8 leading to an effective null mutant.76 To produce a spinal nerve ligation (SNL) model of neuropathic pain, the L5 transverse lamina was removed to exposure L4 and L5 spinal nerves after male C57BL/6J mice anesthetized as described. The L5 spinal nerve was then isolated and tightly ligated with 6-0 silk thread as previously described.77
Neurobehavioral testing
A quiet room maintained at a temperature of 23–25 °C was used to conduct all behavioral tests. Before collecting data, these mice were trained for 5 consecutive days to adapt to the experimental environment. Prior to each set of measurements, all mice were allowed to acclimate to the testing apparatus for a minimum of 60 min. A set of repeat measurements were used to familiarized all mice with each testing paradigm before collecting baseline measurements. An investigator blind to animal grouping performed behavioral tests essentially as described elsewhere.24
Generation of AAV8 viruses expressing wildtype or mutant CA8 with V5 tag using AAV-MCS vectors
CA8 wildtype (WT) gene cDNA was purchased from Origene (Cat. No. RC210228). An Eppendorf Recycler gradient (Model 5331) was used to amplify the WT gene which was cloned into pcDNA3.1/V5-His A between the BamHI and XhoI (NEB) restriction sites (Invitrogen Life Technologies, Carlsbad, CA) using (TTTGGATCCGCCACC ATGGCGGACCTGAGCTTC) as the forward primer and (TTTCTCGAGCTGAAATGCAGCTCTAATGACTC) as the reverse primer. The V5-CA8 construct was then cloned between the BamHI and BglII restriction sites of the pAAV-MCS vector, one component of AAV Helper-Free System (Agilent Technologies, Santa Clara, CA), after amplification from pcDNA3.1/V5-His A using (CTCGGATCCGCCACCATGGCGGAC) as forward primer and (TTTGTCGACTCACGTAGAATCGAGACCGAG) as reverse primer. The S100P mutation was introduced into the CA8 cDNA (MT) by using the GENEART Site-Directed Mutagenesis System (Invitrogen Life Technologies, Carlsbad, CA) with the forward primer: AGTCAAAATCAGTTCTTCCGGGAGGACCATTGC and the reverse primer: GCAATGGTCCTCCCGGAAGAACTGATTTTGACT. The S100P mutation is associated with proteasome-mediated degradation that represents a null mutation comparable to the causal Car8 deletion mutation of the waddles mouse.82
The Miami Project Viral Vector Core, University of Miami Miller School of Medicine produced the recombinant AAV8-V5-CA8 viral particles, as described elsewhere.24 The purified AAV particles were titrated for genome contents using real-time qPCR method. Titers were in the range 1-3E14 GC (Genome Copy) per mL.
Sciatic nerve injections of AAV8-V5-CA8 viral particles
Male mice were anesthetized by intraperitoneal injection of ketamine, xylazine and acepromazine (VEDCO, Saint Joseph, Mo). After sciatic nerve exposure, 1.5 μl 2.65E+14 genome copies/ml viral particles of AAV8-V5-CA8WT or AAV8-V5-CA8MT were injected into the sciatic nerve through a 35-gauge needle using a NanoFil syringe (World Precision Instruments, Sarasota, FL). The injection site was approximately 45 mm from the tip of the third toe. The needle remained at the injection site for 1 additional min before it was slowly removed.
Tissue and cell culture preparation
Male mice (2-3 months) were anesthetized as described elsewhere.24 Spinal cord and DRG were dissected from the L4 and L5 levels, and put in the same fixative for 2–4 h, then transferred to 20% sucrose overnight or until the tissues sank to the bottom of the vessel. The tissue was embedded with OCT (Andwin Scientific Inc., Schaumburg, IL) on dry ice. Leica 1900 Cryostat (Leica Microsystems Inc., Buffalo Grove, IL) was used to cut 16 μm sections and mounted to slides for immunofluorescence staining. Western blots were run with fresh spinal cord and DRG tissues dissected directly from mice for western blot. The vendor supplied validation of cell lines. N2A (ATCC CCL-131), HEK293 (ATCC CRL-1573) and NBL (ATCC CRL-1622) were plated in 24-well plates on poly-D-lysine-, laminin-coated glass coverslips for immunocytochemistry, in 6-well plates for western blot. Cells were seeded at a density of 2 × 105 cells/ml. Cultures were set up in 6-well plates in 2 mL per well, and in 24-well plates at 0.5 mL. The cultures were incubated in a water-saturated atmosphere containing 5% CO2/95% air at 37°C in Gibco DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and 1X cellgro penicillin-streptomycin (Fisher Scientific, Pittsburg, PA).
Transfections
HEK293, NBL and mouse N2A cells were plated in 6-well plates at 4 × 105 cells and 24-well plates at 1 × 105 cells per well to obtain 90% confluent layer after 24 h and transfected, as described elsewhere.24 Cells were used for measurement of mRNA and protein expression using real-time PCR, immunocytochemistry and western blot after 48 h incubation.
Immunocytochemistry and imaging
Tissue immunostaining was performed, as previously described.84 Tissue sections and cell cultures were fixed by 4% PFA in PBS for 30 min, permeabilized in 0.3% Triton X-100 for 1.5 h at room temperature, and blocked in 4% normal serum (Jackson ImmunoResearch Laboratories, West Grove, PA) for 20 min. Primary antibodies specific for CA8, V5, vinculin, TrkA, pTrkA (Abcam, Cambridge, MA), pITPR1 and ITPR1 (Cell Signaling Technology, Danvers, MA). Antibodies were allowed to incubate with sections and cell cultures overnight at 4°C after dilution in PBS containing 0.1% Triton X-100. Sections and cultures were washed three times for 10 min each in PBS, and incubated with Alexa Fluor 488 and/or Alexa Fluor 594-conjugated second antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at RT. Sections and cultures were mixed with the 2nd antibody to assess the total number of cells, and images were acquired as described elsewhere.24
Western blot assays
HEK293, NBL and N2A cultures were homogenized in RIPA buffer with a mixture of proteinase and phosphatase inhibitors (Sigma). Protein samples were generally separated on 4–15% SDS polyacrylamide gels and transferred to nitrocellulose membrane. For big protein p-ITPR1 western blot, 6% Tris-HCl gel in transfer buffer were used to increase efficiency. Western blots were prepared, handled and analyzed essentially as presented elsewhere24 using anti-CA8 (Santa Crutz, Santa Crutz, CA), anti-V5 (Invitrogen), anti-pITPR1 (Cell Signaling Technology), vinculin, TrkA (Abcam, Cambridge, MA) and anti-β-actin (Sigma). Density analysis was performed using UN-SCAN-IT, standardized to β-actin, and a one-way ANOVA was used for statistical analysis.
RNA extraction, reverse transcription-polymerase chain reaction (RT-PCR) and real time PCR (qPCR)
Total RNA was extracted from cultured N2A, NBL and HEK293 cells essentially as described elsewhere.24 qPCR was used to amplify exogenous CA8-V5 with the forward primer: ACCTTGCAGTGAAGGTGTCA and the reverse primer: CCGAGGAGAGGGTTAGGGAT. GAPDH was used as internal control with the forward primer: GGATTTGGTCGTATTGGGCG and the reverse primer: ATCGCCCCACTTGATTTTGG. qPCR and analysis as described elsewhere24 using CA8WT and CA8MT versions and these results were normalized to the expression of GAPDH. Efficiency of each primer pair was determined via standard curve and used to determine relative quantities of starting mRNA amounts.
Calcium assay methods
Human HEK293 (ATCC CRL-1573), or SH-SY5Y (NBL) neuroblastoma (ATCC CRL-2266) cells were transfected with AAV-V5-CA8WT, AAV-V5-CA8MT vectors or treated as sham 48 h prior to assay, according to the manufacturer’s instructions. Twenty-four hours before assay, 100K to 250K cells/mL in 1 or 0.5 mL 12 or 24-well plates respectively and then split onto 15 mm glass coverslips (Propper, Long Island, NY) coated with poly-lysine overnight and laminin for 2 h at RT. On the day of the assay, cells were rinsed with perfusate buffer (Buffer 1),85 then incubated in Buffer 1 containing 0.1 μM Fura-2/AM (Molecular Probes) and 0.012% pluronic F-127 (Sigma) for 45 min at RT (21-22OC) and washed twice with Buffer 1. After another 10 min incubation in Buffer 1 at RT, coverslips were transferred to a RC-42 LP open bath chamber in the QE-1 platform, loaded onto a Leica DMI 6000 B inverted microscope and perfused with Ca2+ free Buffer 2 (Buffer 2, all concentrations in mM: 130 NaCl; 4.7 KCl; 2.3 MgSO4; 5 Glucose; 20 HEPES; 10 EGTA; 1.2 KH2PO4).86 Cells were allowed to equilibrate for 2-5 min before recording on Leica Application Suite 1.1.0.12420 (Leica, Germany) was initiated. Cells from 2-3 different cultures were visualized every 5s for 7-15 min while perfused with Buffer 2; or Buffer 2 containing NGF or 2-APB/NGF (Sigma), as described elsewhere.85 Six consistent points of peak or base line values of F340/F380 from a special coverslip were collected and averaged as one sample (N=1). Intracellular free calcium concentration [Ca2+]I were calculated, as described elsewhere.85
Quantification and statistics
To quantify immunoreactive staining of CA8, ITPR1, pITPR1, TrkA, pTrkA and V5 expression in the DRG, the percentages of positive neurons in the L5 and L4 DRG from four nonadjacent sections were determined as described previously.72 We have previously shown that SN injection of AAV8 is highly selective for small to medium sized DRG neurons.24 Briefly, the DRGs were serially sectioned at 16 μm. The percentage of positive DRG neurons was estimated by calculating the average total number of V5 positive cells divided by the total number of DAPI positive nuclei from 3-4 sections of each animal. Results represent 3 to 4 mice in each group. Quantitative evaluations were made by an investigator masked to the arrangement of DRG sections analyzed. Groups were compared using Student’s t-test, or ANOVA, followed by Fisher’s PLSD test and data presented as mean ± SEM. The criterion for statistical significance was P < 0.05.
The sample size was N=8 per group for all in vivo experiments. For data presented in Figure 7, a Tukey’s multiple comparison post-hoc test of a two-way repeated-measures ANOVA (treatment X time) was used to analyze these data (IBM SPSS Statistics 24). Time was used as the repeated measure factor to determine main effects of treatment, time, and the interaction. Tests of between-subject effects were performed to show an observed power of 1.000 (computed using alpha = 0.05), which include assumption of sphericity, Greenhouse-Geisser Correction, and Huynh-Feldt Correction. IBM SPSS Statistics 24 was modified to directly calculate the significance between groups at each time point incorporating a Bonferroni correction. The number of mice used per assay group was based on power analyses using the observed variation in prior AAV8 assays; and to ensure sufficient tissues to measure the planned post-mortem endpoints. We estimated that N = 8 animals per group would provide 95% power at P=0.05. No animals were excluded in our analyses.
Supplementary Material
Acknowledgments
A grant from the NIDCR R01DE022903; and funding from the Department of Anesthesiology, Perioperative Medicine, and Pain Management, University of Miami Miller School of Medicine, Miami, Florida were used to perform this work.
Footnotes
Classification: Biological Sciences (Neuroscience)
CONFLICTS OF INTEREST:
Drs. Maixner, Diatchenko, and Smith have financial holdings receive compensation from Algynomics, Inc. The authors declare that their affiliation(s) to Algynomics, Inc., including employment, consultancy, patents, products in development or marketed products, does not alter our adherence to Gene Therapy policies on sharing data and/or materials.
Algynomics Inc. provided support in the form of salaries for authors SBS, WM, LD, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section. The remaining authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS: RCL designed the overall study, setup collaborations, supervised these studies, analyzed data, primarily wrote, edited and submitted this manuscript. GZZ performed the major portion of the experiments; GZZ assisted with experimental design, data collection, analyses and manuscript preparation. XT, YK UU and DME contributed to experimental work included in this manuscript. KDS, UU, ESF, TW, ERM, LD, WM, SBS contributed to the critical review and final editing of the manuscript.
References Cited
- 1.Institute of Medicine Committee on Advancing Pain Research C, Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. National Academies Press (US) National Academy of Sciences; Washington (DC): 2011. The National Academies Collection: Reports funded by National Institutes of Health. [PubMed] [Google Scholar]
- 2.Levitt AE, Galor A, Chowdhury AR, Felix ER, Sarantopoulos CD, Zhuang GY, et al. [EXPRESS] Evidence that Dry Eye Represents a Chronic Overlapping Pain Condition. Molecular pain. 2017;13 doi: 10.1177/1744806917729306. 1744806917729306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu ES, Erasso DM, Zhuang GZ, Upadhyay U, Ozdemir M, Wiltshire T, et al. Impact of human CA8 on thermal antinociception in relation to morphine equivalence in mice. Neuroreport. 2017 doi: 10.1097/WNR.0000000000000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berde CB, Athiraman U, Yahalom B, Zurakowski D, Corfas G, Bognet C. Tetrodotoxin-bupivacaine-epinephrine combinations for prolonged local anesthesia. Marine drugs. 2011;9(12):2717–28. doi: 10.3390/md9122717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanelli G, Casati A, Beccaria P, Aldegheri G, Berti M, Tarantino F, et al. A double-blind comparison of ropivacaine, bupivacaine, and mepivacaine during sciatic and femoral nerve blockade. Anesthesia and analgesia. 1998;87(3):597–600. doi: 10.1097/00000539-199809000-00019. [DOI] [PubMed] [Google Scholar]
- 6.Klein SM, Greengrass RA, Steele SM, D’Ercole FJ, Speer KP, Gleason DH, et al. A comparison of 0.5% bupivacaine, 0.5% ropivacaine, and 0.75% ropivacaine for interscalene brachial plexus block. Anesthesia and analgesia. 1998;87(6):1316–9. doi: 10.1097/00000539-199812000-00019. [DOI] [PubMed] [Google Scholar]
- 7.Augustine GJ. How does calcium trigger neurotransmitter release? Curr Opin Neurobiol. 2001;11(3):320–326. doi: 10.1016/s0959-4388(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 8.Inoue T, Kato K, Kohda K, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci. 1998;18(14):5366–5373. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyata M, Finch EA, Khiroug L, Hashimoto K, Hayasaka S, Oda SI, et al. Local calcium release in dendritic spines required for long-term synaptic depression. Neuron. 2000;28(1):233–244. doi: 10.1016/s0896-6273(00)00099-4. [DOI] [PubMed] [Google Scholar]
- 10.Rizzuto R. Intracellular Ca(2+) pools in neuronal signalling. Curr Opin Neurobiol. 2001;11(3):306–311. doi: 10.1016/s0959-4388(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 11.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386(6627):855–8. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 12.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294(5541):333–9. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 13.Gunter TE, Gunter KK. Uptake of calcium by mitochondria: transport and possible function. IUBMB life. 2001;52(3–5):197–204. doi: 10.1080/15216540152846000. [DOI] [PubMed] [Google Scholar]
- 14.Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22(10):4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones TL, Lustig AC, Sorkin LS. Secondary hyperalgesia in the postoperative pain model is dependent on spinal calcium/calmodulin-dependent protein kinase II alpha activation. Anesth Analg. 2007;105(6):1650–6. doi: 10.1213/01.ane.0000287644.00420.49. table. [DOI] [PubMed] [Google Scholar]
- 16.Zeitz KP, Giese KP, Silva AJ, Basbaum AI. The contribution of autophosphorylated alpha-calcium-calmodulin kinase II to injury-induced persistent pain. Neuroscience. 2004;128(4):889–898. doi: 10.1016/j.neuroscience.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 17.Schepelmann K, Messlinger K, Schmidt RF. The effects of phorbol ester on slowly conducting afferents of the cat’s knee joint. Exp Brain Res. 1993;92(3):391–398. doi: 10.1007/BF00229027. [DOI] [PubMed] [Google Scholar]
- 18.Dray A, Rang H. The how and why of chronic pain states and the what of new analgesia therapies. Trends Neurosci. 1998;21(8):315–317. doi: 10.1016/s0166-2236(98)01291-0. [DOI] [PubMed] [Google Scholar]
- 19.Rang HP, Ritchie JM. Depolarization of nonmyelinated fibers of the rat vagus nerve produced by activation of protein kinase C. J Neurosci. 1988;8(7):2606–2617. doi: 10.1523/JNEUROSCI.08-07-02606.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth Analg. 2003;97(4):1108–1116. doi: 10.1213/01.ANE.0000081061.12235.55. [DOI] [PubMed] [Google Scholar]
- 21.Simonetti M, Hagenston AM, Vardeh D, Freitag HE, Mauceri D, Lu J, et al. Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron. 2013;77(1):43–57. doi: 10.1016/j.neuron.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain disorders–pathways of vulnerability. Pain. 2006;123(3):226–30. doi: 10.1016/j.pain.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Obata K, Yamanaka H, Fukuoka T, Yi D, Tokunaga A, Hashimoto N, et al. Contribution of injured and uninjured dorsal root ganglion neurons to pain behavior and the changes in gene expression following chronic constriction injury of the sciatic nerve in rats. Pain. 2003;101(1–2):65–77. doi: 10.1016/s0304-3959(02)00296-8. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang GZ, Keeler B, Grant J, Bianchi L, Fu ES, Zhang YP, et al. Carbonic anhydrase-8 regulates inflammatory pain by inhibiting the ITPR1-cytosolic free calcium pathway. PloS one. 2015;10(3):e0118273. doi: 10.1371/journal.pone.0118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 26.Carrasco MA, Jaimovich E, Kemmerling U, Hidalgo C. Signal transduction and gene expression regulated by calcium release from internal stores in excitable cells. Biol Res. 2004;37(4):701–712. doi: 10.4067/s0716-97602004000400028. [DOI] [PubMed] [Google Scholar]
- 27.Verkhratsky A. Endoplasmic reticulum calcium signaling in nerve cells. Biol Res. 2004;37(4):693–699. doi: 10.4067/s0716-97602004000400027. [DOI] [PubMed] [Google Scholar]
- 28.Wu LJ, Zhuo M. Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics. 2009;6(4):693–702. doi: 10.1016/j.nurt.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuichi T, Simon-Chazottes D, Fujino I, Yamada N, Hasegawa M, Miyawaki A, et al. Widespread expression of inositol 1,4,5-trisphosphate receptor type 1 gene (Insp3r1) in the mouse central nervous system. Receptors Channels. 1993;1(1):11–24. [PubMed] [Google Scholar]
- 30.Worley PF, Baraban JM, Colvin JS, Snyder SH. Inositol trisphosphate receptor localization in brain: variable stoichiometry with protein kinase C. Nature. 1987;325(7000):159–161. doi: 10.1038/325159a0. [DOI] [PubMed] [Google Scholar]
- 31.Hirasawa M, Xu X, Trask RB, Maddatu TP, Johnson BA, Naggert JK, et al. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol Cell Neurosci. 2007;35(1):161–170. doi: 10.1016/j.mcn.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirota J, Ando H, Hamada K, Mikoshiba K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J. 2003;372(Pt 2):435–441. doi: 10.1042/BJ20030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH. Inositol trisphosphate receptor: phosphorylation by protein kinase C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc Natl Acad Sci USA. 1991;88(6):2232–2235. doi: 10.1073/pnas.88.6.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haug LS, Jensen V, Hvalby O, Walaas SI, Ostvold AC. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J Biol Chem. 1999;274(11):7467–7473. doi: 10.1074/jbc.274.11.7467. [DOI] [PubMed] [Google Scholar]
- 35.Maes K, Missiaen L, Parys JB, Sienaert I, Bultynck G, Zizi M, et al. Adenine-nucleotide binding sites on the inositol 1,4,5-trisphosphate receptor bind caffeine, but not adenophostin A or cyclic ADP-ribose. Cell Calcium. 1999;25(2):143–152. doi: 10.1054/ceca.1998.0011. [DOI] [PubMed] [Google Scholar]
- 36.Mignery GA, Johnston PA, Sudhof TC. Mechanism of Ca2+ inhibition of inositol 1,4,5-trisphosphate (InsP3) binding to the cerebellar InsP3 receptor. J Biol Chem. 1992;267(11):7450–7455. [PubMed] [Google Scholar]
- 37.Yamada M, Komatsu N, Okada K, Kato T, Miyazaki H, Miura Y. Thrombopoietin induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in a human thrombopoietin-dependent cell line. Biochemical and biophysical research communications. 1995;217(1):230–7. doi: 10.1006/bbrc.1995.2768. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y, Gu Y, Xu GY, Wu P, Li GW, Huang LY. Adeno-associated viral transfer of opioid receptor gene to primary sensory neurons: a strategy to increase opioid antinociception. Proc Natl Acad Sci USA. 2003;100(10):6204–6209. doi: 10.1073/pnas.0930324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Storek B, Harder NM, Banck MS, Wang C, McCarty DM, Janssen WG, et al. Intrathecal long-term gene expression by self-complementary adeno-associated virus type 1 suitable for chronic pain studies in rats. Molecular pain. 2006;2:4. doi: 10.1186/1744-8069-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Towne C, Pertin M, Beggah AT, Aebischer P, Decosterd I. Recombinant adeno-associated virus serotype 6 (rAAV2/6)-mediated gene transfer to nociceptive neurons through different routes of delivery. Molecular pain. 2009;5:52. doi: 10.1186/1744-8069-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vulchanova L, Schuster DJ, Belur LR, Riedl MS, Podetz-Pedersen KM, Kitto KF, et al. Differential adeno-associated virus mediated gene transfer to sensory neurons following intrathecal delivery by direct lumbar puncture. Mol Pain. 2010;6:31. doi: 10.1186/1744-8069-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Gu Y, Wu P, Li GW, Huang LY. Efficiencies of transgene expression in nociceptive neurons through different routes of delivery of adeno-associated viral vectors. Hum Gene Ther. 2003;14(9):897–906. doi: 10.1089/104303403765701187. [DOI] [PubMed] [Google Scholar]
- 43.Zheng H, Qiao C, Wang CH, Li J, Li J, Yuan Z, et al. Efficient retrograde transport of adeno-associated virus type 8 to spinal cord and dorsal root ganglion after vector delivery in muscle. Hum Gene Ther. 2010;21(1):87–97. doi: 10.1089/hum.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mikoshiba K. The InsP3 receptor and intracellular Ca2+ signaling. Curr Opin Neurobiol. 1997;7(3):339–45. doi: 10.1016/s0959-4388(97)80061-x. [DOI] [PubMed] [Google Scholar]
- 45.Langford DJ, Schmidt B, Levine JD, Abrams G, Elboim C, Esserman L, et al. Preoperative Breast Pain Predicts Persistent Breast Pain and Disability Following Breast Cancer Surgery. Journal of pain and symptom management. 2014 doi: 10.1016/j.jpainsymman.2014.11.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang FC, Tan T, Huang T, Christianson J, Samad OA, Liu Y, et al. Genetic control of the segregation of pain-related sensory neurons innervating the cutaneous versus deep tissues. Cell reports. 2013;5(5):1353–64. doi: 10.1016/j.celrep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eskander MA, Ruparel S, Green DP, Chen PB, Por ED, Jeske NA, et al. Persistent Nociception Triggered by Nerve Growth Factor (NGF) Is Mediated by TRPV1 and Oxidative Mechanisms. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35(22):8593–603. doi: 10.1523/JNEUROSCI.3993-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itoh K, Ishima T, Kehler J, Hashimoto K. Potentiation of NGF-induced neurite outgrowth in PC12 cells by papaverine: role played by PLC-gamma, IP3 receptors. Brain research. 2011;1377:32–40. doi: 10.1016/j.brainres.2010.12.075. [DOI] [PubMed] [Google Scholar]
- 49.Drummond ES, Dawson LF, Finch PM, Bennett GJ, Drummond PD. Increased expression of cutaneous alpha1-adrenoceptors after chronic constriction injury in rats. The journal of pain: official journal of the American Pain Society. 2014;15(2):188–96. doi: 10.1016/j.jpain.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Schlereth T, Drummond PD, Birklein F. Inflammation in CRPS: role of the sympathetic supply. Auton Neurosci. 2014;182:102–7. doi: 10.1016/j.autneu.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 51.Niclas Jonsson E, Xie R, Marshall SF, Arends RH. Population pharmacokinetics of tanezumab in phase 3 clinical trials for osteoarthritis pain. British journal of clinical pharmacology. 2015 doi: 10.1111/bcp.12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ekman EF, Gimbel JS, Bello AE, Smith MD, Keller DS, Annis KM, et al. Efficacy and safety of intravenous tanezumab for the symptomatic treatment of osteoarthritis: 2 randomized controlled trials versus naproxen. The Journal of rheumatology. 2014;41(11):2249–59. doi: 10.3899/jrheum.131294. [DOI] [PubMed] [Google Scholar]
- 53.Gimbel JS, Kivitz AJ, Bramson C, Nemeth MA, Keller DS, Brown MT, et al. Long-term safety and effectiveness of tanezumab as treatment for chronic low back pain. Pain. 2014;155(9):1793–801. doi: 10.1016/j.pain.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 54.Spierings EL, Fidelholtz J, Wolfram G, Smith MD, Brown MT, West CR. A phase III placebo- and oxycodone-controlled study of tanezumab in adults with osteoarthritis pain of the hip or knee. Pain. 2013;154(9):1603–12. doi: 10.1016/j.pain.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 55.Hochberg MC. Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2015;23(Suppl 1):S18–21. doi: 10.1016/j.joca.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Hochberg MC, Tive LA, Abramson SB, Vignon E, Verburg KM, West CR, et al. When is osteonecrosis not osteonecrosis? adjudication of reported serious adverse joint events in the tanezumab clinical development program. Arthritis Rheumatol. 2015 doi: 10.1002/art.39492. [DOI] [PubMed] [Google Scholar]
- 57.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26(12):696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 58.Hagenacker T, Ledwig D, Busselberg D. Feedback mechanisms in the regulation of intracellular calcium ([Ca2+]i) in the peripheral nociceptive system: role of TRPV-1 and pain related receptors. Cell Calcium. 2008;43(3):215–227. doi: 10.1016/j.ceca.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 59.Youn DH, Voitenko N, Gerber G, Park YK, Galik J, Randic M. Altered long-term synaptic plasticity and kainate-induced Ca2+ transients in the substantia gelatinosa neurons in GLU(K6)-deficient mice. Brain Res Mol Brain Res. 2005;142(1):9–18. doi: 10.1016/j.molbrainres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 60.Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35(4):721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- 61.Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005;280(14):13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- 62.Piper AS, Yeats JC, Bevan S, Docherty RJ. A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. J Physiol. 1999;518(Pt 3):721–733. doi: 10.1111/j.1469-7793.1999.0721p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123(1):53–62. doi: 10.1085/jgp.200308906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cesare P, McNaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci USA. 1996;93(26):15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamoto S, Tanaka E, Higashi H. Mediation by intracellular calcium-dependent signals of hypoxic hyperpolarization in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;77(1):386–92. doi: 10.1152/jn.1997.77.1.386. [DOI] [PubMed] [Google Scholar]
- 66.Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, et al. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8725–30. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foust KD, Poirier A, Pacak CA, Mandel RJ, Flotte TR. Neonatal intraperitoneal or intravenous injections of recombinant adeno-associated virus type 8 transduce dorsal root ganglia and lower motor neurons. Hum Gene Ther. 2008;19(1):61–70. doi: 10.1089/hum.2007.093. [DOI] [PubMed] [Google Scholar]
- 68.Storek B, Reinhardt M, Wang C, Janssen WG, Harder NM, Banck MS, et al. Sensory neuron targeting by self-complementary AAV8 via lumbar puncture for chronic pain. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(3):1055–60. doi: 10.1073/pnas.0708003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tse LV, Moller-Tank S, Asokan A. Strategies to circumvent humoral immunity to adeno-associated viral vectors. Expert Opin Biol Ther. 2015;15(6):845–55. doi: 10.1517/14712598.2015.1035645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kohane DS, Smith SE, Louis DN, Colombo G, Ghoroghchian P, Hunfeld NG, et al. Prolonged duration local anesthesia from tetrodotoxin-enhanced local anesthetic microspheres. Pain. 2003;104(1–2):415–21. doi: 10.1016/s0304-3959(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 71.Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nature neuroscience. 2014;17(2):153–63. doi: 10.1038/nn.3602. [DOI] [PubMed] [Google Scholar]
- 72.Choi DY, Toledo-Aral JJ, Segal R, Halegoua S. Sustained signaling by phospholipase C-gamma mediates nerve growth factor-triggered gene expression. Mol Cell Biol. 2001;21(8):2695–705. doi: 10.1128/MCB.21.8.2695-2705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shortland P, Kinman E, Molander C. Sprouting of A-fibre primary afferents into lamina II in two rat models of neuropathic pain. European journal of pain. 1997;1(3):215–27. doi: 10.1016/s1090-3801(97)90107-5. [DOI] [PubMed] [Google Scholar]
- 74.Kajander KC, Bennett GJ. Onset of a painful peripheral neuropathy in rat: a partial and differential deafferentation and spontaneous discharge in A beta and A delta primary afferent neurons. Journal of neurophysiology. 1992;68(3):734–44. doi: 10.1152/jn.1992.68.3.734. [DOI] [PubMed] [Google Scholar]
- 75.Beck C, Uramoto H, Borén J, Akyürek LM. Tissue-specific targeting for cardiovascular gene transfer. Potential vectors and future challenges. Curr Gene Ther. 2004;4(4):457–67. doi: 10.2174/1566523043346138. [DOI] [PubMed] [Google Scholar]
- 76.Turkmen S, Guo G, Garshasbi M, Hoffmann K, Alshalah AJ, Mischung C, et al. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS genetics. 2009;5(5):e1000487. doi: 10.1371/journal.pgen.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50(3):355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 78.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 79.Hamamoto DT, Giridharagopalan S, Simone DA. Acute and chronic administration of the cannabinoid receptor agonist CP 55,940 attenuates tumor-evoked hyperalgesia. Eur J Pharmacol. 2007;558(1):73–87. doi: 10.1016/j.ejphar.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 81.Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24(38):8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turkmen S, Guo G, Garshasbi M, Hoffmann K, Alshalah AJ, Mischung C, et al. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 2009;5(5):e1000487. doi: 10.1371/journal.pgen.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28(2):158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 84.Zhuang Z, Yang B, Theus MH, Sick JT, Bethea JR, Sick TJ, et al. EphrinBs regulate D-serine synthesis and release in astrocytes. J Neurosci. 2010;30(47):16015–16024. doi: 10.1523/JNEUROSCI.0481-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levitt RC, Zhuang GY, Kang Y, Erasso DM, Upadhyay U, Ozdemir M, et al. Car8 dorsal root ganglion expression and genetic regulation of analgesic responses are associated with a cis-eQTL in mice. Mammalian genome: official journal of the International Mammalian Genome Society. 2017;28(9–10):407–415. doi: 10.1007/s00335-017-9694-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boehmerle W, Zhang K, Sivula M, Heidrich FM, Lee Y, Jordt SE, et al. Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(26):11103–8. doi: 10.1073/pnas.0701546104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–50. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.