

Abstract
Combination antiretroviral therapy effectively controls human immunodeficiency virus (HIV) viral replication, delaying the progression to acquired immune deficiency syndrome and improving and extending quality of life of patients. However, the inability of antiretroviral therapeutics to target latent virus and their poor penetration of viral reserve tissues result in the need for continued treatment for the life of the patient. Side effects from long‐term antiretroviral use and the development of drug resistance due to patient noncompliance are also continuing problems. Nanostructured systems of antiretroviral therapeutics have the potential to improve targeted delivery to viral reservoirs, reduce drug toxicity, and increase dosing intervals, thereby improving treatment outcomes and enhancing patient adherence. Despite these advantages, very few nanostructured antiretroviral delivery systems have made it to clinical trials due to challenges in preclinical and clinical development. In this context, we review the current challenges in HIV disease management, and the recent progress in leveraging the unique performance of nanostructured systems in therapeutic delivery for improved treatment and prevention of this incurable human disease.
Keywords: nanoformulation, HIV treatment, HIV prevention, long‐acting
1. INTRODUCTION
Acquired immune deficiency syndrome (AIDS) was first described in 1981.1, 2, 3, 4 Human immunodeficiency virus (HIV) was isolated and identified in 1983 before being definitively linked to AIDS in 1984.1, 2, 3, 4 HIV targets CD4+ T lymphocytes, leading to a decline in their numbers that ultimately causes the immune dysfunction known as AIDS.3, 5 As of 2016, 36.7 million people are infected with HIV, with 1.0 million people dying and 1.8 million new infections occurring each year.6 This represents a 48% decline in deaths from a peak of 1.9 million in 2006, believed to be a result of increased efficacy of and access to antiretroviral (ARV) therapeutics.6
While treatment of HIV infected individuals with three drug combinations of food and drug administration (FDA) approved ARV therapeutics has been extremely effective at controlling viral suppression and preventing the infection from progressing to AIDS, as demonstrated by the global decline in HIV related deaths, it does not clear the virus from the infected individual and must be continued for the lifetime of the patient.5, 6, 7, 8, 9, 10, 11 This lack of a cure is attributed to the inability of current therapeutics to target inactive viruses and possibly to limited accumulation in high enough concentrations in viral reservoir tissues.5, 9, 10, 11, 12, 13 Further drawbacks associated with long‐term use of current HIV therapeutics are the need for frequent dosing (once or twice daily), development of resistance due to patient noncompliance, significant adverse effects, and related toxicities.7, 8, 9, 10, 11, 14, 15 Consequently, there is an unmet need for a targetable, long‐acting therapeutic delivery system that can increase the time between dosages, reduce fluctuation of drug levels, and increase delivery of the therapeutic agent to viral reservoirs. Nanostructured delivery systems of current ARV drugs have shown promise at addressing these issues but few have progressed to clinical trials due to challenges in preclinical and clinical development.11, 14
HIV is a lentivirus known for delayed onset and chronic infection with a double‐stranded RNA genome of 9,300 base pairs.5, 16 The HIV RNA genome contains three major open reading frames: gag, pol, and env and six small genes that encode regulatory proteins.5, 16 The gag encoded protein is split to form the structural components of the virus.5, 16 The pol encoded polyprotein contains the viral enzymes reverse transcriptase, integrase, and protease, all of which are critical for successful viral infection of host cells.5, 16 The env gene produces the transmembrane protein responsible for viral binding and entry to human immune cells.5, 16 Each HIV particle consists of a nucleocapsid core that encapsulates two copies of the viral genome and the critical viral enzymes, and that is itself surrounded by a lipid envelope formed from the plasma membrane of the host cell.5, 17 There are two distinct species of HIV, designated as HIV‐1 and HIV‐2.5, 14, 16 HIV‐1 is the more prevalent virus globally whereas HIV‐2 is more commonly found in West Africa and is believed to progress to AIDS more slowly than HIV‐1.5, 14, 16
The endocytic pathways of HIV invasion, replication and spread are now well understood. The envelope protein gp160 (env) of HIV is responsible for HIV's tropism for immune cells.5 Env is split into two domains, gp120 and gp41.18 Gp120 is a surface protein responsible for binding to the CD4 receptor and either the CCR5 or CXCR4 co‐receptors found on the surface of helper T‐Cells and other lymphocytes and macrophages, which is necessary for cellular entry.5, 16, 17 Gp41 regulates fusion of the viral envelope with the host cell membrane by undergoing a structural change once the viral particle is bound to the host cell, thereby allowing entry.5, 10 Once the HIV RNA genome is inside the cell, reverse transcriptase utilizes the RNA strands as templates to create an RNA‐DNA duplex of original viral genome and its DNA replica.5, 10, 17 HIV reverse transcriptase is inaccurate and lacks a proof reading function, leading to an average of three incorrect base pairs for each full length viral RNA copied.5 The RNA templates are then degraded and the DNA replicas are used to make double‐stranded DNA copies of the viral genome.5, 10, 17 Following transportation into the nucleus, this double‐stranded cDNA replica is inserted into the host genome by viral integrase.5, 10, 17 The integrated viral cDNA is transcribed to create both the complete viral RNA genome and mRNAs that code for the necessary structural, enzymatic, and regulatory proteins of HIV.5, 10, 17 The mRNAs are translated and processed into the viral protein precursors that assemble into immature HIV particles at the cell membrane: the structural protein precursors form around the viral RNA genome, reverse transcriptase, integrase, and protease.5, 10, 17 These protein precursors are cleaved by viral protease as the immature virus is escaping from the cell via budding, completing the formation of a mature viral particle.5, 10, 17, 19 The understanding and elucidation of the HIV life cycle has led to the development of six classes of ARV agents with distinct action mechanisms that block HIV activities at different stages (Figure 1).
Figure 1.
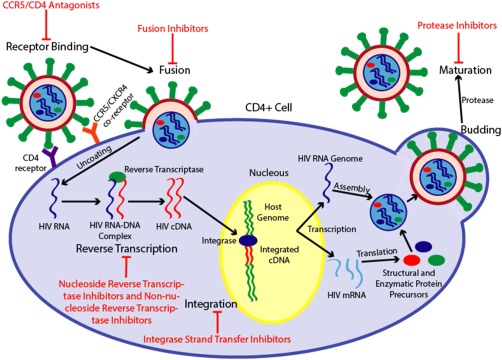
Depiction of the HIV life cycle and the mechanisms of action of the six classes of antiretroviral therapeutics. Mature HIV particles bind with CD4 receptors and either a CCR5 or CXCR4 co‐receptor, triggering a conformational change in the gp41 portion of the HIV env protein that enables fusion of the viral and cellular membranes. CCR5 and CD4 antagonists competitively bind with the host cell receptors, preventing viral binding and subsequent entry. Fusion inhibitors bind with gp41, preventing the conformational change needed for cellular entry. Once the viral membrane has fused with the cellular membrane, viral RNA and enzymes are released into the CD4+ cell, where viral reverse transcriptase transcribes the viral RNA into double‐stranded HIV cDNA. Nucleoside reverse transcriptase inhibitors competitively bind with reverse transcriptase and are incorporated into the DNA strand in place of native nucleotides, resulting in chain termination. Non‐nucleoside reverse transcriptase inhibitors non‐competitively bind with a distal hydrophobic pocket in HIV‐1 reverse transcriptase, inducing a conformation change that drastically reduces enzyme activity. The fully synthesized cDNA is incorporated into the host genome by viral integrase, a process that integrase strand transfer inhibitors impede by preventing the formation of covalent bonds between host and viral DNA. Once incorporated into the genome, normal cellular pathways transcribe the cDNA into a full length viral RNA genome and HIV mRNA strands that are translated into precursors of structural and enzymatic viral proteins. The precursor proteins and HIV RNA genome assemble into a viral capsid that buds from the cell, triggering viral protease to cleave the precursor peptides, resulting in full maturation of the viral particle. Protease inhibitors prevent this proteolytic cleavage, which is necessary for the complete development of the viral particle into a mature infectious agent.
2. CURRENT HIV ARV TREATMENT
Combination antiretroviral therapy (CART), also known as high activity antiretroviral therapy, is currently the standard treatment for HIV‐1 infected individuals.5, 8, 9, 10, 11, 14, 20 A minimum of three ARV drugs are used in combination with one another in order to suppress the viral load of a patient to virtually undetectable levels (defined as <50 copies of virus/mL of blood).5, 8, 9, 10, 11, 14, 20, 21 There are currently 27 FDA approved ARV therapeutics and 1 FDA approved pharmacokinetic enhancer meant to be used to improve the efficacy of ARVs (Tables 1, 2, 3).22 ARV therapeutics are divided into six classes: nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non‐nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors (FIs), CCR5 and CD4 antagonists, and integrase strand transfer inhibitors (INSTIs).5, 10, 20, 22
Table 1.
FDA approved nucleoside reverse transcriptase inhibitors for the treatment of HIV
| Generic name (abbreviation) | Brand name | Manufacturer | IC‐50 (nM) | Bioavailability (%) | Elimination half‐life (hr) | Plasma protein binding (%) | CSF‐plasma ratio (%) | Dosage form | Adult dosage | Approval year |
|---|---|---|---|---|---|---|---|---|---|---|
| Abacavir (ABC) | Ziagen | ViiV Healthcare | 70–5,800 | 83 | 1.54 ± 0.63 | ∼50 | 30 | Tablet, oral solution | 300 mg twice daily or 600 mg once daily | 1998 |
| Didanosine (ddl or enteric‐coated didanosine ddl EC) | Videx | Bristol‐Myers Squibb | 10–10,000 | 42 | 1.5 ± 0.4 | <5 | 21 | Powder (oral solution) | 125–200 mg twice daily or 250–400 mg once daily | 1991 |
| Videx EC | Bristol‐Myers Squibb | 10–10,000 | 42 | 1.19 ± 0.21 | <5 | 21 | capsule | 200–400 mg once daily | 2000 | |
| Emtricitabine (FTC) | Emtriva | Gilead Sciences, Inc | 2–30 | 93 (capsule) 75 (solution) | ∼10 | <4 | 26 | Capsule, oral solution | 200 mg once daily (capsules) 240 mg once daily (solution) | 2003 |
| Lamivudine (3TC) | Epivir | ViiV Healthcare | 2–670 | 86 (tablet) 87 (solution) | 5–7 | <36 | 15 | Tablet, oral solution | 150 mg twice daily or 300 mg once daily | 1995 |
| Stavudine (d4T) | Zerit | Bristol‐Myers Squibb | 9–4,000 | 86.4 | 1.6 ± 0.23 | <5 | 40 | Capsule, powder | 30–40 mg twice daily | 1994 |
| Tenofovir disoproxil fumarate (TDF) | Viread | Gilead Sciences, Inc | 2–7 | 25 | ∼17 | 7.2 | 5 | Powder, tablet | 300 mg once daily | 2001 |
| Tenofovir alafenamide (TAF) | Vemlidy | Gilead Sciences, Inc | 5 (EC50) | 80 |
0.51 (plasma) 150–180 (cellular) |
80 | ND | Tablet | 25 mg once daily | 2016 (for combo therapy) |
| Zidovudine (AZT or ZDV) | Retrovir | ViiV Healthcare | 10–48 | 64 | 0.5–3 | <38 | 60 | Capsule, sirup, injection | 300 mg twice daily (oral) or 1 mg per kg every 4 hr (infusion) | 1987 |
Table 2.
FDA approved protease inhibitors and associated pharmacokinetic enhancers for the treatment of HIV
| Generic name (abbreviation) | Brand name | Manufacturer | IC‐50 (nM) | Bioavailability (%) | Elimination half‐life (hr) | Plasma protein binding (%) | CSF‐plasma ratio (%) | Dosage form | Adult dosage | Approval year |
|---|---|---|---|---|---|---|---|---|---|---|
| Atazanavir (ATV) | Reyataz | Bristol‐Myers Squibb | 2–15 | ND | ∼7 | 86 | <3 | Capsule, oral powder | 400 mg once daily or 300 mg with 100 mg ritonavir once daily | 2003 |
| Darunavir (DRV) | Prezista | Janssen Pharmaceuticals, Inc. | 1–5 | 37 (alone) 82 (with ritonavir) | ∼15 | 95 | 0.3‐1.8 | Oral suspension, tablet | 800 mg with 100 mg ritonavir once daily (treatment naïve) or 600 mg with 100 mg ritonavir twice daily (treatment experienced) | 2006 |
| Fosamprenavir (FOS‐APV or FPV) | Lexiva | ViiV Healthcare | 12–410 | ND | 7.7 | 90 | 12 | Oral suspension, tablet | 700 mg with 100 mg ritonavir twice daily | 2003 |
| Indinavir (IDV) | Crixivan | Merck and Co., Inc. | 25–100 (IC95) | 60–65 | 1.8 ± 0.4 | 60 | 14.7 | Capsule | 800 mg three times daily | 1996 |
| Nelfinavir (NFV) | Viracept | Agouron | 7–196 (EC95) | 20–80 | 3.5–5 | >98 | UD | Oral powder, tablet | 1250 mg twice daily or 750 mg three times daily | 1997 |
| Ritonavir (RTV) | Norvir | AbbVie Inc. | 4–150 | Absolute: ND >60 based on animal studies | 3–5 | 98–99 | <0.5 | Capsule, tablet, oral solution, oral powder | 600 mg twice daily | 1996 |
| Saquinavir (SQV) | Invirase | Hoffman‐La Roche | 3.5–10 | 4 | 1–2 | 98 | <0.5 | Capsule, tablet | 1000 mg with 100 mg ritonavir twice daily | 1995 |
| Tipranavir (TPV) | Aptivus | Boehringer Ingelheim | 30–70 | ND | 6 | >99.9 | ND | Capsule, oral solution | 500 mg with 200 mg of ritonavir twice daily | 2005 |
| Cobicistat (COBI)a | Tybost | Gilead Sciences, Inc | ND | ND | 3–4 | 97–98 | ND | Tablet | 150 mg once daily | 2014 |
Table 3.
FDA approved non‐nucleoside reverse transcriptase inhibitors, including Efavirenz, Etravirine, Nevirapine, and Rilpivirine
| Generic name (abbreviation) | Brand name | Manufacturer | IC‐50 (nM) | Bioavailability (%) | Elimination half‐life (hr) | Plasma protein binding (%) | CSF‐plasma ratio (%) | Dosage form | Adult dosage | Approval year |
|---|---|---|---|---|---|---|---|---|---|---|
| Efavirenz (EFV) | Sustiva | Bristol‐Myers Squibb | 3–9 | 50 | 52–76 | 99.5 | 0.26–1.19 | Capsule, tablet | 600 mg once daily | 1998 |
| Etravirine (ETR) | Intelence | Janssen Pharmaceuticals, Inc. | 1–5 | ND | ∼41 | 99.9 | ND | Tablet | 200 mg twice daily | 2008 |
| Nevirapine (NVP and extended‐release nevirapine NVP XR) | Viramune | Boehringer Ingelheim | 10–100 | 93 (tablet) 91 (solution) | 25 ‐ 30 | ∼60 | 45 | Tablet, oral suspension | 200 mg twice daily | 1996 |
| Viramune XR | Boehringer Ingelheim | 10–100 | 80–94 | 25–30 | ∼60 | 45 | Tablet | 400 mg once daily | 2011 | |
| Rilpivirine (RPV) | Edurant | Janssen Pharmaceuticals, Inc. | 42 | ND | ∼50 | 99.7 | ND | Tablet | 25 mg once daily | 2011 |
| Enfuvirtide (T‐20) | Fuzeon | Hoffman‐La Roche; Genentech | 0.1–1,700 | 84 (subcutaneous injection) | 3.8 ± 0.6 | 92 | UD | Subcutaneous injection | 90 mg twice daily | 2003 |
| Ibalizumab | Trogarzo | TaiMed Biologics USA Corp. | 53 | NA | 2.7–64 (dose dependent) | ND | ND | Intravenous injection | 800 mg every two weeks | 2018 |
| Maraviroc (MVC) | Selzentry | ViiV Healthcare | 0.1–4.5 | 23–33 | 14–18 | 76 | 2.8 | Tablet, oral solution | 150, 300, or 600 mg twice daily (depending on combination therapeutics) | 2007 |
| Bictegravir (BIC) | Biktarvy | Gilead Sciences, Inc | 7.5 | ND | 14.9–20.9 | >99 | ND | Tablet | 50 mg once daily (in combination with 200 mg FTC and 25 mg TAF) | 2018 (for combo therapy) |
| Elvitegravir (EVG) | Vitekta | Gilead Sciences, Inc | 44 | ND | 8.7 | 98–99 | ND | Tablet | 85 or 150 mg once daily (depending on combination therapeutics) | 2014 |
| Dolutegravir (DTG) | Tivicay | ViiV Healthcare | 2.7–12.6 | ND | 14 | 98.9 | 0.11–0.66 | Tablet | 50 mg once daily or 50 mg twice daily (depending on past and combinedtherapeutics) | 2013 |
| Raltegravir (RAL) | Isentress | Merck and Co., Inc. | 6–50 (IC95) | ND | 9 | 83 | 5.8 | Tablet, oral suspension | 400 mg twice daily or 1200 mg once daily | 2007 |
| Isentress HD | Merck and Co., Inc. | 6–50 (IC95) | ND | 9 | 83 | 5.8 | Tablet | 400 mg twice daily or 1200 mg once daily | 2017 |
2.1. ARVs mechanisms of action
NRTIs competitively bind with reverse transcriptase when the viral DNA strand is being synthesized, resulting in the addition of an NRTI rather than a native nucleotide.5, 10 This results in chain termination of the cDNA because NRTIs lack the 3′ OH group necessary for the covalent attachment of another nucleotide to the strand.5, 10 NNRTIs non‐competitively bind to a distal hydrophobic pocket in HIV‐1 reverse transcriptase, inducing a conformational change that disrupts the catalytic site of the enzyme and drastically reduces reverse transcriptase activity.5, 10 PIs prevent the cleavage of gag and pol precursor polypeptides at the phenylamine‐proline target peptide sequence.5, 10, 36 If the precursor polypeptides are not cleaved then the virus particles are incapable of developing into mature viral particles.5, 10, 36 FIs are a type of entry inhibitor that disrupt the fusion of the viral and cell membranes by binding to the gp41 domain of env, preventing a necessary structural change that enables fusion.5, 10 CCR5 and CD4 antagonists are unusual in their mechanism of action because they interfere with host cell receptors rather than viral components.5 They competitively bind to the host cell CCR5 co‐receptor or CD4 receptor, preventing binding of HIV gp120 and subsequent fusion and entry (Figure 2).5, 10 CCR5 tropic HIV utilizes CCR5 as its binding co‐receptor with CD4 rather than CXCR4.5 CCR5 tropic HIV is responsible for new infections, with all HIV isolated in the early stages of infection identified as CCR5 tropic.37 INSTIs block the integration of viral DNA into host DNA by binding to HIV integrase and preventing the formation of covalent bonds between host and viral DNA, thereby blocking strand transfer into the host genome.5, 10
Figure 2.
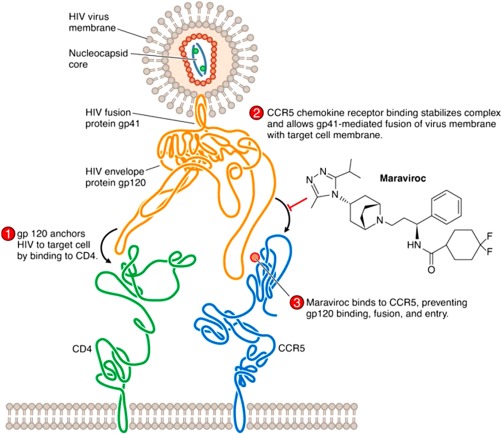
Mechanism of action for the CCR5 antagonist maraviroc. The drug competitively binds to the host cell's CCR5 receptors, preventing the gp120 portion of the HIV env protein from binding to the co‐receptor. By disrupting the interaction between gp120 and CCR5 necessary for fusion of the viral and cellular membranes, CCR5 antagonists prevent cellular entry. Used with permission from Ref. 5.
2.2. FDA therapeutic standards
As per the FDA guidelines on HIV treatment, CART consists of a combination of at least three ARVs from at least two different classes.8, 10, 20 The recommended HIV CART regimen utilizes two NRTIs in combination with an INSTI, NNSTI, or pharmacokinetically enhanced PI (Table 4).20 The goal of ARV therapy is to suppress viral replication to the greatest extent possible and for the longest time possible.5, 20 When used as recommended, CART is extremely effective at reducing viral load; it suppresses viral replication in more than 80% of those beginning treatment.5, 8, 9, 10, 11, 14, 20 Plasma viral load (the number of HIV RNA copies detected per ml of a patient's blood) is the best stand‐alone, prognostic marker for progression to AIDS.38 Furthermore, it has been definitively linked to the efficiency of viral transmission from infected individuals to healthy individuals, with infected individuals with viral loads below 500 copies/mL being extremely unlikely to transmit the virus to others.10, 39
Table 4.
FDA recommended initial regimens of CART20
| Preferred | INSTI + 2 NRTIs |
| regimens | Dolutegravir, Abacavir, Lamivudine |
| Dolutegravir, Tenofovir, Emtricitabine | |
| Elvitegravir, Cobicistat, Tenofovir, Emtricitabine | |
| Raltegravir, Tenofovir, Emtricitabine | |
| Recommended in certain situations | Boosted PI (either Cobicistat or Ritonavir) + 2 NRTIs |
| Darunavir or Atazanavir, Tenofovir, Emtricitabine | |
| Darunavir or Atazanavir, Abacavir, Lamivudine | |
| NNRTI + 2 NRTIs | |
| Efavirenz or Rilpivirine, Tenofovir, Emtricitabine | |
| INSTI + 2 NRTIs | |
| Raltegravir, Abacavir, Lamivudine | |
| Regimens if Abacavir and Tenofovir cannot be used | |
| Darunavir, Raltegravir | |
| Lopinavir, Lamivudine |
2.3. Shortcomings of CART
While CART has demonstrated success at suppressing the viral load of new patients, thereby delaying the progression to AIDS and decreasing the chance of viral transmission, CART is unable to clear HIV from infected individuals.5, 8, 9, 10, 11, 14 Consequently, there is no cure for HIV and CART must be continued for the life of the patient.5, 8, 9, 10, 11, 14 Discontinuation of the ARV therapy leads to a rapid spike in viral load and the development of drug resistance (Figure 3).5, 8, 9, 10, 11, 14, 15 The continued presence of HIV in infected individuals despite CART causes this viral rebound and is believed to be a result of latent viral DNA found in quiescent cells, and possibly active HIV infected cells in physiological compartments that ARVs do not easily penetrate.5, 9, 10, 11, 12, 13 Currently approved ARVs are incapable of targeting the latent viruses because their mechanisms of action are dependent on viral replication.5 However, the presence of residual viral reserves in tissues that CART does not efficiently enter (such as the lymphoid tissues, the central nervous system (CNS) and the testis) could be addressed by targeted delivery of approved ARVs to these reserves. The concentrations of ARV drugs in lymph node mononuclear cells (LNMCs) have been shown to be 66–99% lower than their respective concentrations in peripheral blood mononuclear cells (PBMCs).12 Residual viruses isolated from these reserve tissues are still known to be responsive to the CART regimen of the patient.9, 40, 41 This indicates that the continued existence of these active viruses may be due to insufficient concentrations of ARV therapeutics in the tissue rather than acquired resistance to the drugs, especially since sequencing of the recovered viral genome revealed no known drug resistance sequences.42 Therefore, in order to better control HIV infections and potentially eliminate HIV from the body, treatments that target these viral reservoirs could be explored.
Figure 3.
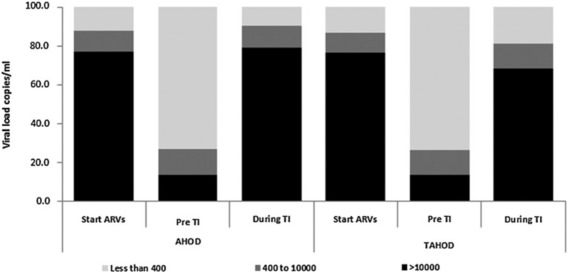
The percent of individuals with viral loads <400 copies/mL, 400–10,000 copies/mL, and >10,000 copies/mL in the Australian HIV Observational Database (AHOD) and Treat Asia HIV Observation Database (TAHOD) at the start of therapy, prior to a treatment interruption (TI) consisting of the discontinuation of ARV therapeutics for >30 days, and during the TI. Used with permission from Ref. 44.
Furthermore, ARVs are known to have significant adverse side effects, toxicity due to off‐target binding or accumulation, and common and often complicated interactions with other drugs.5, 8, 9, 10, 11, 14, 20, 22 CART requires once or twice daily doses for the lifetime of the patient, thereby increasing the likelihood that patients will develop serious adverse effects due to long‐term use and making patient compliance to the regimen difficult.9, 11, 15 Up to 20% of the HIV infected population in the United States has failed to attain full HIV viral load suppression (<200 copies/mL) under sustained CART,43 a problem that is attributed to patient noncompliance to the treatment regimen.6, 15 Patients with difficulty swallowing such as children, others with neurologic, gastrointestinal, or psychiatric disease, and intravenous drug users are at particular risk of not adhering to this self‐administered, oral regimen.9, 15 Fluctuating levels of ARVs in the body can lead to a rebound in viral load and the development of drug resistance.11, 14, 15 It is the rapid mutation of HIV due to the inaccuracy of reverse transcriptase that causes this sudden drug resistance and that makes the development of a broadly functional HIV vaccine difficult.5, 8 Viral replication must be suppressed to prevent the formation of mutated viruses that may contain novel resistance mutations to the administered therapy.11 While inconsistent ARV concentrations are often due to patient failure to take medications as prescribed,11, 15 it is also possible that currently available formulations of ARVs cannot sustain drug concentrations at the necessary levels for the desired dosing interval.14
A better, long‐acting, targetable delivery system is needed to increase the intervals between doses, limit fluctuation of medication levels in between doses, reduce the side effects and toxicity associated with off‐site delivery, and increase the concentration of ARV drugs delivered to HIV viral reserve tissues. HIV infected patients have shown interest in less frequent dosage administration: 73% of HIV infected patients surveyed had a favorable response to less frequent intravenous doses, a number that rises to 84% if the intravenous doses can be administered monthly.45 In order to address this desire for longer dosing intervals, there has been an increase in the investigation and development of drugs with longer terminal half‐lives that could be orally administered once a week.5, 46 However, such new ARVs tend to be expensive and increased access to effective therapies can better be obtained through a delivery system designed to extend the circulation and reduce the toxicity of older, generically available drugs.14 Furthermore, such long‐acting oral formulations do not address the issues of targeting to the viral reserve tissues and only a few drugs exhibit long enough half‐lives, effectively limiting the combination therapies possible.15 Nanostructured delivery systems for traditional ARVs are a potential strategy to address the issues of toxicity, resistance, and insufficient accumulation in HIV viral reserve tissues affiliated with CART.
3. NANOSTRUCTURED SYSTEMS FOR THE TREATMENT OF HIV
Nanostructured ARV drug delivery systems have been explored for both systemic treatment to control HIV replication in infected individuals and for local delivery to prevent the acquisition of the infection. When therapeutics are delivered via nanoparticles (NPs), the nanosystem for the most part governs the pharmacokinetic profile rather than the drug itself.47, 48 Nanoscale delivery systems of various types are advantageous in that they can be created with a variety of physicochemical properties to control drug release, protect the drug from metabolic break down and clearance from the body, enable targeting of specific types of cells or tissues, limit adverse side effects and toxicity, and maintain constant therapeutic concentrations.10, 47 Furthermore, NPs are known for their ability to deliver therapeutics to regions of the body that are not otherwise attainable.49 Nanotherapeutic platforms typically used in HIV ARV delivery include polymer, inorganic, and solid lipid NPs, polymer micelles, liposomes, nanosuspensions, and dendrimers (Figure 4). Initially, nanostructured delivery systems were designed to increase the plasma half‐lives of ARVs in order to create a long‐acting therapy.7, 10, 11 While longer dosing intervals would be well received by patients and could lead to improved adherence,15, 45 these systems do not necessarily result in improved targeting of viral reservoirs.9
Figure 4.
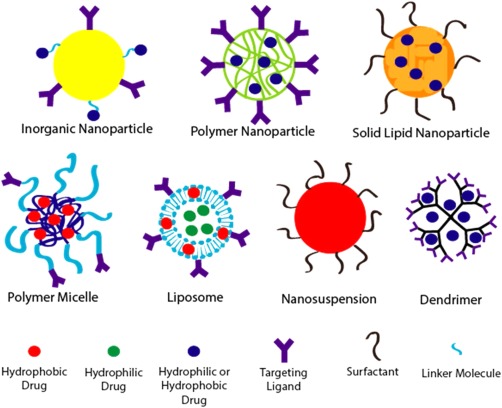
Commonly used nanostructured systems for the investigational delivery of HIV antiretroviral therapeutics. Inorganic NPs have gained popularity due to the inherent antiviral activity of some materials such as silver. Liposomes and solid lipid NPs have also experienced increased use as a result of their ability to permeate the BBB.
3.1. Targeted systemic delivery systems
The inability of CART to fully eradicate the virus from the body is in part attributed to the limited concentration of ARVs present in the lymphatic tissues and other viral reserve tissues.7, 12 In order to fully eliminate these viral reservoirs, the ARV concentrations in the lymphatic and central nervous systems must be maintained at effective levels. This requires not just the use of nanoscale systems for ARV delivery, but also the ability of these systems to target the drugs to HIV viral reservoirs, especially the CNS and lymph nodes. These tissues pose unique challenges for drug accumulation due to the exclusivity of the blood brain barrier (BBB), which tightly controls the transport of substances into the CNS,50 and the rapid rate at which small molecules diffuse out of the lymph and back into the blood.9, 51
3.1.1. Inorganic NPs
Inorganic materials such as copper, iron, silver, gold, or silica are used to form inorganic NPs and the therapeutic agents are conjugated to the surface along with any desired peptides or antibodies for functionalization. Gold nanoparticles (AuNPs) have been commonly used in HIV nanotherapy because of their biocompatibility.52 Silver nanoparticles (AgNPs) are of particular interest in the treatment of HIV due to their innate antiviral activity, thereby enabling the NP to act as an HIV therapeutic itself.53 AgNPs between 1 and 10 nm are believed to inhibit HIV infection by binding to the gp120 protein responsible for viral binding to the CD4 receptor and co‐receptors, preventing cellular entry.7, 54 However, AgNPs perforate brain micro‐vascular endothelial cells (BMECs) and induce BBB inflammation,50 making them unsuitable for use in delivery systems that target the HIV reservoir in the brain.
Some nanostructured systems are focused on iron NPs because their use in magnetic resonance imaging allows for rapid assessment of biodistribution. Surface modification of superparamagnetic iron oxide (SPIO) NPs has been used by Gendelman and coworkers to target the delivery systems to monocytes and macrophages.55, 56 Immunoglobulin (IgG) was attached to the surfaces of SPIO NPs.55 The Fc receptor mediated phagocytosis of IgG coated particles led to a 10‐fold increase in uptake of IgG‐SPIO NPs by monocytes and macrophages in comparison to that of uncoated SPIO NPs.55 MRI imaging of the NPs revealed increased accumulation of the IgG‐SPIO NPs in lymphatic tissues such as the spleen.55 Folic acid (FA) surface‐modified SPIO NPs achieved similar results by conjugating FA modified polyethylene glycol (PEG) to the SPIO NPs, with a more than twofold increase in accumulation in the spleen (13 µg/mL for FA‐PEG‐SPIO NPs vs. 6 µg/mL for PEG‐SPIO NPs).56 ARVs can be conjugated to the surfaces of these targeted SPIO NPs to increase the drug accumulation in the viral reservoirs.55, 56 In an attempt to target the brain specifically, the Corsi lab developed amphiphilic polymer coated iron oxide NPs to improve the BBB penetration of enfuvirtide (ENF).57 Such NPs were found to have 170% better permeability through an in vitro double layer of rat BMVECs supported by astrocytes than free ENF, and comparatively higher accumulation in the brains of mice during an in vivo study.57 The mechanism of penetration by the NPs is believed to be some form of non‐endocytotic transcytosis through the BMECs due to their tendency to attach to the plasma membranes of the cells.57
Conjugation to inorganic NPs has been shown by Margolis and coworkers to improve the antiviral activity of ineffective therapeutics. A delivery system with potent anti‐HIV activity can be created from a low‐affinity small molecule that displays no therapeutic activity on its own by conjugating several of the inactive molecules to an AuNP.58 Conjugation of SDC‐1721, a biologically inactive derivative of the ARV maraviroc, to 2.0 nm AuNPs at a ratio of 12:1 SDC‐1721 to NPs resulted in the creation of a multivalent AuNP with ARV activity similar to that of maraviroc.58 Neither SDC‐1721 or AuNPs exhibited independent suppression of viral replication, but the multivalent SDC‐1721 AuNPs inhibited viral replication with an average IC50 value of 10 nM, matching the average IC50 value of maraviroc.58 The uptake of 2–10 nm AuNPs by macrophages, PBMCs, and human‐brain micro‐vascular endothelial cells (HBMECs) and the accumulation of unmodified AuNPs in the spleen and, to a much lesser extent, the brain, was reported.59 By conjugating thiolated raltegravir (RAL), an inactive form of RAL, to AuNPs in a 4:1 ratio, the researchers were able to suppress viral replication to 25.23%.59 While free RAL demonstrates better HIV suppression, the RAL‐AuNPs did exhibit better cellular uptake, BBB penetration, and accumulation in the spleen and brain.59 The accumulation in viral reservoirs was accompanied by increased off‐site accumulation in the liver, kidneys, lungs, and heart.59
Another proposed delivery system of glyceryl monostearate‐modified, nevirapine loaded gold shell NPs displayed similar increased uptake by PBMCs and sustained accumulation in viral reservoirs such as the spleen, lymph nodes, thymus, bone marrow, and brain.60 But once again, the improved viral reserve tissue accumulation was accompanied by increased off‐site accumulation in the liver and ovaries.60 Such off‐site accumulation of nanomaterials is concerning given the potential cytotoxicity of inorganic NPs, making the use of a more biocompatible system necessary. Additionally, since most inorganic NPs are not biodegradable, their size is often limited to several nanometers in order to facilitate renal clearance.
3.1.2. Liposomes
The superior biocompatibility and ease of functionalization of liposomes make them an attractive alternative to inorganic NPs.14 Cationic liposomes are rapidly gaining popularity as delivery vesicles for ARVs due to their ability to penetrate the BBB via adsorption‐mediated transcytosis through BMECs.50 They consist of a phospholipid bilayer that encloses an aqueous core and they can be used to transport either hydrophobic drugs dispersed in the phospholipid tails or hydrophilic drugs encapsulated in the aqueous core. Targeting moieties or short strands of PEG can be attached to the surface to direct accumulation in specific tissues or protect the liposomes from immune clearance.
By attaching sugars to the surface of liposomes, it is possible to increase cellular uptake of the encapsulated ARV via carbohydrate binding receptors found on the surface of lymphocytes, monocytes, and macrophages.61 In a study conducted by the Jain lab, galactosylated liposomes loaded with zidovudine (ZDV) exhibited increased ZDV uptake by alveolar macrophages in vitro.61 The sugar modified liposomes also demonstrated increased plasma concentrations, decreased kidney uptake, and higher distribution to the liver, spleen, lymph nodes and lungs by almost 10‐fold higher than free ZDV following intravenous administration in a rat model.61 While this study sought to demonstrate the ability of modified liposomes to accumulate in viral reserve tissue, another property that make liposomes an interesting delivery vehicle for ARVs is their transdermal capabilities.62 The transdermal delivery of liposomes can be further enhanced by incorporating ethanol into indinavir (IDV) liposomes.62 The incorporation of 45% ethanol in liposomes increased the transdermal flux through cadaver skin from 3.2 µg/cm2/hr for free IDV drug solution and 6.3 µg/cm2/hr for 0% ethanol liposomes to 27.2 µg/cm2/hr.62 This transdermal delivery system of ARVs would likely be an attractive alternative to daily oral therapy or other nanomedicine platforms administered via injection. Its potential to produce improved macrophage uptake and enhanced drug accumulation in viral reserve tissues, as demonstrated by the earlier mentioned liposomes produced by the same lab, make such a delivery system worthy of further investigation.
An alternative approach to improve BBB penetration of liposomes utilizes the application of an external magnetic field to magnetic liposomes.63, 64 Arginylglycylaspartic acid (RGD) modified, magnetic liposomes exhibited a 9.1‐fold increase in brain concentration of the encapsulated drug in comparison to free drug, leading to the design of magnetic liposomes that could both directly penetrate the BBB and induce monocyte‐mediated trans endothelial migration across the BBB.63, 64 The RGD sequence enhanced the uptake of the liposomes by monocytes and neutrophils and the magnetic nature of the liposomes induced the liposome loaded cells to cross the BBB via monocyte/neutrophil‐mediated penetration following the application of an 8.0 kg magnetic field near the brains of rats.63 By encapsulating zidovudine triphosphate (ZDVTP), the active form of ZDV produced in vivo, and magnetite particles within 150 nm liposomes and applying an external magnetic field, BBB transmigration across a single layer of HBMECs supported by human astrocytes (HAs) was enhanced by almost 300% in comparison to free ZDVTP.64 These magnetic liposomes were also capable of uptake by PBMCs, and PBMCs that contained the magnetic liposomes exhibited more than twofold increased penetration of the in vitro BBB model upon application of a magnetic field compared to PBMCs that did not contain magnetic liposomes.64 The magnetic liposomes did not display cytotoxicity of PBMCs, and the encapsulation of the ZDVTP did not negatively affect the anti‐HIV‐1 efficacy of the drug.64 Building on this success, magneto‐plasmonic NPs that consist of gold coated, iron chloride particles encapsulated in tenofovir disoproxil fumarate (TDF) loaded liposomes were developed.65 The combination of iron and gold imparts on the hybrid NPs the unique functionalities of both magnetic NPs and AuNPs.65, 66 These hybrid magneto‐plasmonic liposomes can be easily imaged by different techniques, including MRI and X‐ray computed tomography, and display improved BBB penetration via magnetic targeting with an external magnetic field.65 Magneto‐plasmonic liposomes displayed a 1.8‐fold increase in transmigration across an in vitro BBB model of HBMECs supported by HAs and pericytes than liposomes that did not contain the magneto‐plasmonic particles.65 They also demonstrated an improved therapeutic effect with increased suppression of viral replication in HIV infected microglial cells in comparison to free TDF.65
Although magnetic targeting of magnetic liposomes to the viral reserve tissues is possible, it is not necessary to improve accumulation of ARVs at the sites of action, and it complicates the production of the liposomes and the execution of the therapy.9, 67 The Ho lab was able to demonstrate improved lymph node accumulation of PEGylated liposomes encapsulating three ARV drugs.9, 67 Hydrophilic tenofovir (TFV) was trapped inside the aqueous core and attached to the hydrophilic heads of the phospholipids, and hydrophobic ritonavir (RTV) and lopinavir (LPV) were stored within the hydrophobic core of the phospholipid bilayer.9, 67 24 hr after subcutaneous delivery of the three free ARVs in a primate animal model, virtually no detectable concentration of RTV or LPV was found in LNMCs and 224.2–287.2 ng/mL of TFV was found in LNMCs.67 Subcutaneous injection of liposomes encapsulating the ARVs resulted in accumulation of more than 1,200 ng/mL of LPV and 1,600 ng/mL of RTV in the LNMCs.67 Liposomal encapsulation did not improve the LNMC concentration of TFV, but it did improve the plasma concentration of the ARV.67 While increased lymph node accumulation was demonstrated by attaching CD4 targeting peptides to the surface of the liposomes, such additions add complexity to system and make scale up difficult.9 Given the accumulation of unmodified liposomes in the lymph nodes, it is desirable to first pursue the development of the simpler delivery system.9 While many nanomedicine platforms have been explored for the delivery of one ARV, very few have encapsulated three ARVs. Since monotherapy is not an option for treatment, and all recommended initial HIV regimens consist of a minimum of three anti‐HIV therapeutics, it is extremely important for clinical development that combination nanotherapeutics be investigated. While the combination of RTV, LPV, and TFV is not currently recommended as a therapy by the FDA,20 the delivery system proposed by the lab could be adjusted to incorporate other ARV combinations. However, the poor storage stability and limited loading capacity of liposomes must be addressed if they are to have true clinical potential.14
3.1.3. Solid lipid NPs
Solid lipid nanoparticles (SLNs) are a type of colloidal NP composed of physiologically solid lipids dispersed in an aqueous surfactant solution often containing phospholipids (Figure 4).68, 69 SLNs exhibit similar biocompatibility to liposomes with better entrapment efficiency of hydrophobic drugs, lower cost, and improved scalablity.14, 69 They can be used to encapsulate hydrophilic or hydrophobic drugs and the surface can be modified with ligands for tissue targeting.68, 70 The properties and therapeutic release of the SLNs can be controlled by changing the lipid components and surface modifications.68 Encapsulation efficiency of hydrophilic substances in SLNs is generally low unless double emulsion formulation is used, but trace solvent residues from this technique can cause toxicity.70, 71 SLNs made with cationic lipids show improved cellular uptake believed to be a result of charge‐facilitated interactions with cellular receptors.72 This increased uptake by cells can contribute to increased penetration of the BBB and accumulation of SLNs in the brain.68, 73
One of the early applications of SLNs to HIV treatment was carried out in 2006. The BBB permeability of three different ARVs, stavudine, delavirdine, and saquinavir (SQV), were compared between two polymeric NPs made of polybutylcyanoacrylate (PBCA) and methylmethacrylate‐sulfoproprylmethacrylate and microemulsion‐prepared, slightly anionic SLNs74 All three delivery systems showed improved BBB penetration for all three ARVs, with 3–16‐fold increases in uptake by a monolayer of HBMECs in comparison to uptake of the free drugs.74 The positively charged PBCA NPs displayed the best BBB permeability of the three delivery systems for all ARVs encapsulated due to their potential for electrostatic interactions with the negatively charged HBMECs.74 In order to improve the permeability of SLNs, novel cationic SLNs were developed.75 By incorporating cationic stearylamine and dioctadecyldimethyl ammonium bromide in the lipid components, SQV encapsulating cationic SLNs were created via the microemulsion technique.75 The nonionic lipids passively accumulate in the center of the SNL and the cationic lipids are found on the peripheral edges of the lipid core, the ratios of the lipid components can be adjusted to attain the lipid composition with the best SQV entrapment and release profiles.75, 76 These cationic SLNs were further modified by altering the composition of the nonionic surfactant solution to better bring out their cationic nature.77 The surfactant that resulted in the more positively charged SLN surface produced a stronger attraction between the cationic SLNs and HBMECs based on the electrical interaction energy between the NPs and the cells.77 HBMEC permeability was improved from 4.3 × 10−6 cm/s to > 5.6 × 10−6 cm/s by altering the lipid composition of the SLNs to have a higher weight percentage of cholesterol and cationic esterquat 1.78 The use of cholesterol as one of the nonionic lipids in the cationic SLN is believed to improve BBB penetration via cholesterol receptor‐mediated transcytosis across the HBMECs.78 All of this suggests the highly tunable nature of SLN biodistribution via surfactant and composition alteration.
Tissue distribution studies revealed the potential of SLNs for lymphatic tissue targeting in addition to CNS penetration.79 Even nonionic SLNs displayed improved uptake by reticuloendothelial system tissues in comparison to polylactic‐co‐glycolic acid (PLGA)‐poloxamer 188 NPs, with an increased concentration of the loaded ARVs found in the spleen and liver when delivered via SLNs.79 SLNs are naturally capable of accumulating in intestinal lymphatic tissues because they induce the formation of chylomicrons by enterocytes, which promotes the absorption of their lipid matrix by the intestinal lymphatic system.80, 81 The concentration of LPV when administered directly into the duodenum via glycerol behenate SLNs was 4.91 times greater in the intestinal lymph than LPV administered in a methyl cellulose suspension.80 Similarly, efavirenz (EFV) encapsulated in multi‐lipid SLNs was found in reduced concentrations in the liver (44.70% lower) and increased concentrations in the spleen in comparison to free EFV.81 This indicates that EFV SLNs bypassed the liver by entering the lymph following oral administration in rats.81 Even ignoring the potential for BBB penetration and lymphatic targeting, encapsulation of ARVs within SLNs can drastically improve the pharmacokinetics of the drugs.82 Loading of EFV in glycerol monostearate SLNs increased the peak plasma concentration of the drug by more than five times that of free EFV and the AUC (area under the curve of the plasma drug concentration versus time) by more than 10‐fold.82 Despite these advantages, the limited batch to batch reproducibility, sterilization difficulties, and poor stability of SLNs pose challenges for their widespread use.14
3.1.4. Polymeric NPs
Improved stability and reproducibility can be obtained by using polymeric NPs as the delivery system. Polymeric NPs can consist of a polymer membrane that encapsulates a drug reservoir, a NP of polymer matrix with drug evenly distributed throughout, or a solid polymer NP with drugs and targeting moieties conjugated to the surface. The NPs tend to be made of biodegradable polymers such as polylactic acid (PLA), polyglycolic acid (PGA), or a copolymer of the two (PLGA) or to be made of biocompatible polymers such as PEG. They are capable of encapsulating both hydrophilic and hydrophobic drugs and the release profile of the incorporated therapeutics can be tuned by altering polymer structure, particle size, pore size, degradation rate, and conjugation chemistry. The shape and size of the NPs are dependent on production technique rather than polymer structure.
The Kuo lab has demonstrated the improved BBB penetration of ARVs encapsulated within the matrix of surface‐modified PLGA NPs.83, 84, 85 Initially, SQV was entrapped in the PLGA matrix to form SQV‐PLGA NPs.83, 84 BBB penetration was enhanced by conjugating poly‐γ‐glutamic acid (γ‐PGA), a biosynthesized hydrophilic polymer that has been shown to improve cellular uptake, to the surface of the SQV‐PLGA NPs.83 85.2% grafting efficiency of 6 kDa γ‐PGA increased penetration of the NPs through a monolayer of HBMECs supported by HAs by six times that of free SQV.83 Conjugation of polyethyleneimine (PEI) to the ends of the γ‐PGA strands already conjugated to the SQV‐PLGA NPs was shown to decrease the release rate of SQV from the NPs, thereby improving control of the drug release profile, limiting the initial burst release, and prolonging the therapeutic lifetime of the NPs.84 The addition of PEI also has the potential to improve BBB penetration due to its cationic nature.84 BMECs have negatively charged surfaces, and electrostatic interactions draw positively charged materials to these surfaces, resulting in increased endocytic transport of the materials through the BMECs into the CNS.84 Administering these PEI/γ‐PGA/SQV‐PLGA NPs and then applying an electromagnetic field increased endocytosis of the NPs into an HA supported monolayer of HBMECs by 2.38 times the uptake of the NPs administered without the electromagnetic field.86 Improved BMEC uptake in comparison to free drug resulted from decorating the surface of nevirapine PLGA NPs with transferrin, a transport vector that has been shown to increase BBB penetration.85
Other systems have attempted to penetrate viral reservoirs by targeting macrophages.87, 88 Macrophages can enter the lymphoid tissue and CNS, thus cellular uptake of ARV NPs could result in improved drug biodistribution by increasing accumulation of ARVs in the reservoir tissues.87 Coating materials with FA has demonstrably improved macrophage cellular uptake via highly expressed folate receptors.87 Encapsulating dolutegravir and europium‐doped cobalt ferrite in a lipid coated, FA surface‐modified, polycaprolactone matrix led to improved macrophage uptake in comparison to uncoated NPs and sustained ARV activity.88 The cobalt ferrite distributed throughout the polymer matrix also enabled the rapid determination of drug biodistribution using MRI. 88 Despite these successes, polymer NPs are hampered by their potential polymer toxicity and their limited scalability, making the investigation of other systems necessary.14
3.1.5. Polymer micelles
Above their critical micellar concentration, amphiphilic block copolymers can self‐assemble into micellar aggregates. It is this capability to self‐assemble that differentiates polymer micelles from polymer NPs. Unlike polymer NPs, the molecular design plays an important role in defining the architecture and size of the micelles. Because the self‐assembly is a result of energy minimization, the micelles demonstrate high thermodynamic stability .14 Hydrophobic therapeutics can either be encapsulated in the hydrophobic core of the micelle during self‐assembly or they can be directly conjugated to the polymer chain to form a polymer prodrug. They demonstrate high targetability since the hydrophilic ends of the polymers can be highly functionalized with specific ligands.
Encapsulation of EFV in different polymeric micelles formed from both linear triblock polyethylene oxide‐polypropylene oxide‐polyethylene oxide (PEO‐PPO‐PEO) polymers known as poloxamers and four‐arm‐branched PPO chains with PEO blocks at the end of each arm known as poloxamines resulted in improved oral bioavailability of EFV.89, 90 Poloxamine micelles were found to be less stable than poloxamer micelles, but both micelles led to a more than 5,000 fold increase in EFV solubility from 4 µg/mL to more than 20 mg/mL.89 Polymer micelle encapsulation resulted in an 87.6% increase in maximum plasma concentrations of EFV and a 56.5% increase in AUC values in comparison to a suspension of free EFV orally administered in a rat model.89 Furthermore, polymer micelles led to a decrease in the variability between individual's pharmacokinetic EFV profiles.89 To balance the oral bioavailability with the micelle stability and release kinetics of the different block copolymers, a multi‐component polymer micelle composed of both poloxamers to confer stability and poloxamines to improve EFV encapsulation capacity and oral bioavailability was created.90, 91 Intranasal administration of these EFV loaded, multi‐component polymer micelles resulted in a CNS concentration of EFV four times higher than intravenous administration of the delivery platform.91 Despite their potential for intranasal delivery of targeted drug delivery systems, such polymer micelles are not suitable for use in a sustained release delivery system.92 The micelles do not exhibit long‐term release and disassemble upon binding to mucosa, making it necessary to develop an alternative delivery system if sustained release via mucosal administration is a desired function.92 It was found that mucosal adhesion could be improved by using a chitosan‐g‐oligo(N‐isopropylacrylamide) copolymer to form polymer micelles with the mucoadhesive capabilities of chitosan.93 The polymer micelles were stabilized by ionotropically crosslinking the chitosan blocks between different micelles to create a supramolecular nanogel.93 This improves the cytocompatibility of the polymer micelles while still enabling the amine groups of chitosan to adhere to the mucosal layer and creates a polymer micelle system potentially capable of sustained drug release.93 However, further research is needed into the release profile of the delivery platform and such polymer micelles still exhibit polymer dependent biocompatibility.
3.1.6. Nanosuspensions/drug NPs
Arguably the most successful nanostructured delivery system for ARV delivery in terms of clinical development, with two long‐acting, injectable nanosuspensions of ARV therapeutics in phase III clinical trials,94, 95 nanosuspensions are gaining attention for the treatment of HIV. They consist of crystallized drugs milled into NPs, stabilized with surfactants, and suspended in an aqueous phase. They can then be injected either subcutaneously or intramuscularly to form a gel‐like drug depot. Because the NPs are pure drug, nanosuspensions have highly efficient drug loading capabilities.96 Nanosuspensions are also simple to manufacture in comparison to other nanoscale delivery systems and thus are easy to scale up for mass production.97 However, drugs must display the necessary hydrophobicity to be suitable for nanoformulation.96, 97
Tibotec Pharmaceuticals, in partnership with Johnson & Johnson, developed NPs of varying size of crystalline, free base rilpivirine (RPV) stabilized with different surfactants.98 Subcutaneous injection of the 200 nm particles at a dose of 5 mg/kg resulted in constant plasma concentrations of 25 ng/mL for 20 days in dogs with a slow decline to 1–3 ng/mL 3 months after injection.98 Further study of the 200 nm RPV nanosuspension revealed that intramuscular injection at a dose of 5 mg/kg led to a 100‐fold higher concentration of RPV in the lymph nodes surrounding the injection site than the plasma one month after injection and a concentration 3–6 fold higher after 3 months.99 Phase I clinical trials showed plasma concentrations of 16.2 ng/mL in healthy volunteers 84 days after an intramuscular injection of 600 mg of the long‐acting RPV.100, 101, 102 Following formulation refinement, the nanosuspension currently being explored in phase III clinical trials is a 300 mg/mL formulation of 200 nm RPV free base particles stabilized by the surfactantPoloxamer 338, administered intramuscularly at a dose of 600 mg every 4 weeks or 900 mg every 8 weeks.103 ViiV Healthcare, with the support of GlaxoSmithKline, created a nanosuspension of 200 nm NPs of crystalline, free acid cabotegravir stabilized with a surfactant and mixed with an aqueous polymer solution.96 Cabotegravir (CAB) is an analog of the FDA approved INSTI dolutegravir currently in phase III clinical trials for long‐acting intramuscular injections.96, 104 Phase I clinical trials of 100–800 mg doses of the long‐acting CAB suspension injected either subcutaneously or intramuscularly produced plasma concentrations above 0.166 µg/mL (the PA‐IC90 value of CAB) for at least 24 weeks for all doses of at least 200 mg and detectable plasma concentrations for all doses at 48 weeks.96 Current phase III clinical trials are investigating the efficacy of a 200 mg/mL concentration nanosuspension of 200 nm crystalline, free acid CAB NPs stabilized with polysorbate 20 (the surfactant) and combined in an aqueous solution of PEG 3350 and mannitol, administered intramuscularly at a dose of 400 mg every 4 weeks.102
Despite the ability of long‐acting RPV and CAB used in combination to decrease the viral load in mouse animal models and their progression to phase III clinical trials, viral replication was reported in macrophages in vitro.106 One solution to this lack of penetration is to improve viral reserve accumulation by modifying the nanoformulations to enhance uptake by macrophages.105 The Gendelman lab demonstrated improved lymph node and spleen accumulation and increased off‐site accumulation in the liver and lungs of an IDV NP delivered via bone marrow‐derived macrophages in humanized mice.107 However, intravenous delivery of foreign macrophages loaded in vitro with NPs is not practical for scale up. A more promising approach is the formulation of a myristoylated CAB prodrug that can still crystallize and that can be formed into NPs (NMCAB) via high pressure homogenization.105 NMCAB was shown to be more effectively taken up by human monocyte derived macrophage cells in vitro than the long‐acting CAB nanosuspension, with NMCAB obtaining an intracellular concentration 60‐fold higher (Figure 5).105 Intramuscular injection of NMCAB in mice resulted in CAB concentrations above the PA‐IC90 in the lymph nodes, spleen, gut, kidney, and lungs 28 days after the injection whereas all tissue concentrations were below the PA‐IC90 value for mice treated with the CAB nanosuspension.105 Another challenge is the lack of available long‐acting ARVs for use in combination therapy.15 Since RPV and CAB are the only long‐acting formulations currently in clinical development, they are being explored in phase III clinical trials as a combination therapy.101 Phase IIb clinical trials of the long‐acting, injectable RPV/CAB therapy demonstrated comparable anti‐HIV activity to an oral regimen consisting of CAB, abacavir, and lamivudine.104
Figure 5.
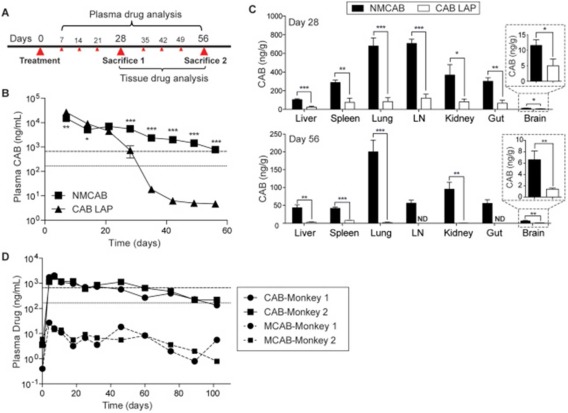
(a) Experimental timeline. (b) Plasma drug concentrations analyzed by UPLC/MS/MS. (c) Tissue drug concentrations analyzed by UPLC/MS/MS. (d) Plasma drug concentrations in the non‐human primate animal models. Used with permission from Ref. 105.
It is also possible to create nanosuspensions of RPV and CAB because of their poor water solubility.15, 96 Most other FDA approved ARVs, including NRTIs, which are a component of almost all FDA approved combination ARV therapeutics, do not have the necessary chemical properties to enable the development of a nanosuspension. If more effective long‐acting combination therapies are to be developed, an alternative approach must be used to create long‐acting delivery systems of NRTIs including TFV, abacavir, emtricitabine, and lamivudine, which are recommended for use in combination with RPV and dolutegravir.
3.1.7. Other nanostructured systems
The nanostructured systems mentioned above are by no means a comprehensive list of all nanoscale systems applied to the treatment of HIV, they are simply the most common systems that have been attempted (with varying levels of success). Carbon nanotubes and quantum dots have also been explored for the treatment of HIV due to their interesting and unique physical and chemical properties. Carbon nanotubes are hollow, cylindrical, single layers of carbon atoms. Spherical fullerene (C60) was one of the first NPs shown to have inherent anti‐HIV activity in the early 1990s.108 Its therapeutic capabilities were thought to arise from its hydrophobic interactions with the predominantly hydrophobic HIV‐1 protease, which effectively inhibits the activity of the viral enzyme.108 However, more recent studies have shown that while fullerene derivatives do inhibit viral maturation, their mechanisms of action are somehow independent of protease inhibition.109 Further exploration into carbon nanotubes has shown that the cylindrical version of fullerene also displays anti‐HIV activity due to its high binding affinity for HIV gag proteins and its ability to bind with and alter the confirmation of viral integrase.110, 111 While the ARV activity of carbon spheres and nanotubes have made them nanomaterials of interest in the treatment of HIV, they have rarely been used in the delivery of ARVs for the treatment of HIV. This lack of use could be due to their insolubility in aqueous environments without further modification that might inhibit their ARV efficacy.112 Quantum dots are semi‐conductor nanocrystals that display narrow, tunable emission spectra and that are excitable by a broad range of wavelengths.113 Quantum dots rapidly lost their initial popularity as drug delivery vehicles, despite their demonstrated ability to cross the BBB, due to their potential toxicity.50 Carbon based quantum dots were recently developed that display enhanced biocompatibility while retaining their BBB permeation capabilities.50 There has been increasing use of quantum dots for HIV diagnosis and the monitoring of HIV progression.114 One carboxylated quantum dot based system has been functionalized with transferrin and amprenavir (APV) and used to enhance accumulation of the ARV in the brain.115 Despite their limited use in the treatment of HIV, quantum dots have the potential to improve diagnosis, progression monitoring, and treatment through their increased accumulation in the brain.
3.2. Local delivery systems for HIV prevention
ARVs have also been encapsulated within gel‐forming nanosystems and explored for use as vaginal or rectal microbicides. The goal of such microbicides is local delivery of ARVs to the site of exposure to prevent the acquisition of HIV. The application of nanostructured systems to microbicide formulation can result in prolonged ARV retention at the site of action for sustained transmission protection, increased cellular uptake, protection of microbicide agents from inactivation by the vaginal microenvironment, and improved antiviral activity of encapsulated ARVs.116, 117, 118 Many microbicides are ARV encapsulating, insertable solids or hydrogels made from electrospun polymer fibers, dendrimers, or other polymer NPs. While hydrogels have traditionally been used for local treatment or prevention, there has been recent success with the systemic release of locally delivered hydrogels.119 This demonstrates that hydrogels can be used for systemic or local treatment and broadens the scope of use of existing local delivery systems.
3.2.1. Dendrimer based gels
Dendrimers are hyper‐branched, three‐dimensional polymers of well‐defined molecular weight and chain architecture.120 They consist of branched polymer arms originating from a multi‐functional core; drugs can be encapsulated within the core, or chemically conjugated onto the terminal ends. Furthermore, it has been shown that cellular uptake mechanism can be controlled based on surface functionalization of the dendrimers, allowing for targeting to specific regions of the cell if desired.121 In addition to serving as carriers for therapeutic and imaging agents, dendrimers have also gained attention for use in anti‐HIV microbicides due to the antiviral activity of certain polyanionic dendrimers.120, 122 Starpharma has developed a naphthalene‐3,6‐disulfonate terminated, polylysine dendrimer named VivaGel with demonstrated HIV preventive abilities in non‐human primate models.123, 124 Phase I clinical trials for the use of the antiviral microbicide in HIV prevention were completed in 2007 and 2009.125, 126 The targetability of dendrimers has also been demonstrated by mannosylated, polypropyleneimine, ARV encapsulating dendrimers for systemic delivery that exhibited improved uptake by macrophage and T‐cells and increased anti‐HIV activity.127, 128 Enhanced BBB penetration and accumulation in microglia (the immune cells of the brain) have been accomplished by loading therapeutics in systemically delivered, hydroxyl‐terminated, polyamidoamine (PAMAM) dendrimers.129, 130 The mechanism of uptake was altered by changing dendrimer surface charge and size, with 4 nm, neutrally charged, 4‐hydroxyl‐terminated PAMAM dendrimers exhibiting uptake by endocytosis.130 The conjugation of other terminal functional groups, such as amines, to PAMAM dendrimers led to hole formation in cellular membranes when dendrimer concentrations were above 10 nM, allowing the dendrimers to diffuse through and enter the cell.131 FA was conjugated to the terminal amine groups to create dendrimers capable of targeting cells that express FA receptors.132 The described system was initially intended to target epithelial cancer,132 but FA receptors are also highly expressed by macrophages and FA targeting has been used in previously described systems to enhance lymph node accumulation of NPs.87 While PAMAM dendrimers have not been applied to HIV treatment, their enhanced uptake by microglial cells, their ability to penetrate the BBB when intravenously administered, and the ease with which they can be functionalized with targeting ligands make them attractive candidates for the delivery of ARVs to the CNS. The antiviral nature of dendrimers combined with their targetability to immune cells make them nanostructured systems of interest for both systemic and local delivery.
3.2.2. Polymeric hydrogels
Prolonged vaginal retention of ARVs is also important for sustained protection from HIV infection.116 The work of the Saltzman lab demonstrated that vaginal retention of elvitegravir can be increased by encapsulating the drug within a PLA matrix coated with surface‐modified, hyper‐branched polyglycerol to form NPs with improved leukocyte and epithelial cell adherence (Figure 6).116 A similar strategy is to attach antihuman anti‐CD4 antibodies to the surface of PLGA NPs loaded with SQV and formulated within a hydroxyethylcellulose gel, doubling the uptake of the NPs by CD4+ immune cells following vaginal administration and increasing the ARV concentration at the site of infection.133 Incorporation of maraviroc in a silicone elastomer gel produced increased and sustained concentrations of the ARV in the vaginal tissue in comparison to maraviroc loaded hydroxyethylcellulose gel.134
Figure 6.
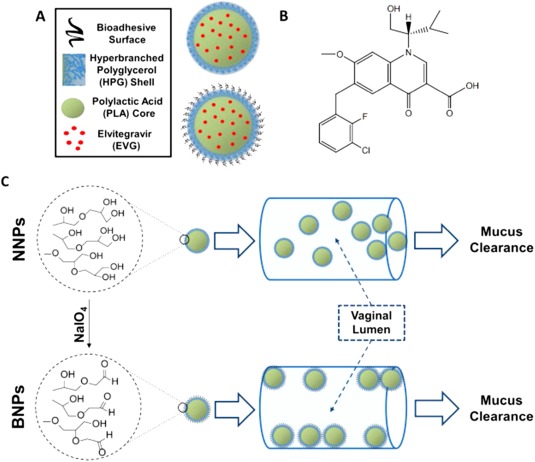
(a) System design of nonadhesive nanoparticles (NNPs) and bioadhesive nanoparticles (BNPs). (b) Chemical structure of elvitegravir. (c) The conversion of NNPs to BNPs using NaIO4 and the hypothesized vaginal adherence of the NPs. Used with permission from Ref. 116
3.2.3. Electrospun fibers
Such vaginal gels can have issues of stability and long‐term storage which can be overcome by using solid, electrospun, ARV‐incorporating fibers instead.118 Electrospinning of hydrophilic and hydrophobic polymer mixtures containing various ARVs (both hydrophilic and lipophilic in nature) resulted in the formation of drug eluting nanofibers or meshes for use as vaginal microbicides.135 It is possible to create electrospun fibers that dissolve rapidly, releasing the loaded ARV in less than 15 min, and fibers that sustain release for more than a week.118, 136 By combining rapid release hydrophilic core polymers with sustained release hydrophobic shell polymers in coaxial electrospun fibers, precise control of the rate of release of maraviroc from the fibers prevents the burst release commonly observed in uniaxial electrospun fibers.137 Altering the core drug loading and the ratio of core to shell thickness of the coaxial spun fibers resulted in drug release that could range from several hours to several days.137 The potential for systemic release from local delivery of vaginal microbicides has been demonstrated by administering chitosan to improve local association with specific immune cells and transcytosis of carboxylate‐modified polystyrene NPs through the vaginal epithelium into the female reproductive tract draining lymph nodes, thereby improving both local function of the microbicide and adapting the vaginally administered NPs for systemic function.138 While the scalability of ARV loaded electrospun fibers has been demonstrated on production‐scale instruments, few in vivo studies have been performed for fiber biocompatibility and there is the potential for residual polymer remnants to remain following use.118, 139 Furthermore, both dendrimer and electrospun fiber biocompatibility are dependent upon polymer biocompatibility and degradation product toxicity.
4. FUTURE PERSPECTIVES
Although nanoscience has shown promise at improving the treatment and prevention of HIV by prolonging the half‐lives of the therapeutics and increasing accumulation in viral reservoir tissues, very few of the proposed nanostructured systems have made it to clinical trials. Many current NP‐based medical approaches face difficulties in part because they do not have the multifunctionality needed to fulfill all the biological and therapeutic requirements.14 The capability of nanosystems to improve targeting to viral reservoirs, limit side effects, prolong plasma half‐life, and increase dosing intervals still make them attractive alternatives to traditional oral treatment. However, issues arising from polymer toxicity, poor scalability, limited storage stability, low loading efficiency, and accumulation of nanomaterials in major organs are barriers to the development of the field.11 Some have even abandoned nanostructured systems altogether in favor of systems with better potential for progression through clinical trials.
The Langer lab recently reported a millimeter scale, multi‐ARV weekly oral drug delivery device with different arms made from different polymer components with varying degradation rates to control and sustain release for the desired duration.140 Another proposed alternative is a sub‐dermal implant of TFV alafenamide capable of releasing 0.92 mg of drug per day with demonstrable zero‐order release kinetics, but that necessitates surgical implantation and removal.141 A simpler approach to improve the pharmacokinetic profile of ARVs is to develop prodrugs of the therapeutics with improved bioavailability and cellular uptake. The ARVs TFV and APV are administered solely in their respective prodrug forms of TDF or tenofovir alafenamide (TAF), and fosamprenavir (FPV) (Figure 7).142, 143 Oral administration of TFV is not possible due to its hydrophilic nature, resulting in poor membrane permeability and subsequently low intestinal absorption.143 TDF hides the negatively charged phosphate group and exhibits improved lipophilicity, thereby increasing oral bioavailability.142 The aryl and l‐alanine isopropyl ester functionalization of TAF serve a similar purpose as the acyloxyalkyl ester functionalization of TDF, but the phosphonamidate bond of TAF results in preferential accumulation of the drug in HIV target cells.143, 144, 145 APV displays an extremely variable oral bioavailability because of its low water solubility.142 FPV has improved water solubility and the prodrug is rapidly converted back to the more membrane permeable APV structure following oral administration.142 While TDF, TAF, and FPV are the only FDA approved ARV prodrugs for HIV treatment, prodrugs of emtricitabine have demonstrated promise in preclinical studies.146
Figure 7.
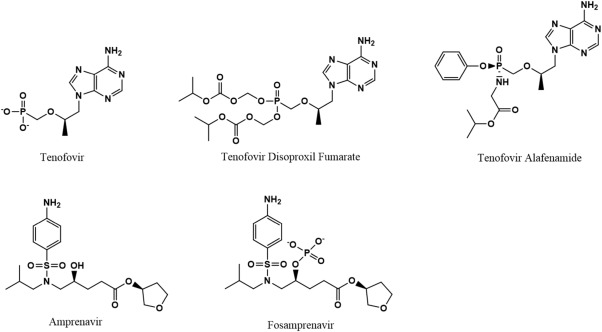
Chemical structures of TFV and APV and their respective prodrugs. The conjugation of acyloxyalkyl esters or aryls to TFV increase its lipophilicity and the addition of l‐alanine isopropyl ester enhances accumulation of the active drug in HIV target cells. The addition of a phosphate group to APV improves its water solubility.
There have been some successes in the use of nanostructured systems to formulate ARVs (RPV and CAB), both of which are currently in Phase III clinical trials and expected to enter the market shortly. If they are to be used as long‐acting therapies, other long‐acting ARVs must be developed to use in combination. The Owen lab, in collaboration with the Rannard lab, developed long‐acting nanosuspensions of the NNRTI EFV and the PI LPV that are currently in Phase I clinical trials.147 Solid drug NPs of LPV and EFV are formed via emulsion‐templated spray or freeze‐drying and solubilized using surfactants (Figure 8).148, 149, 150 The hydrophobic drugs and stabilizing surfactants are dispersed in a chloroform‐in‐water emulsion, with the drug sequestered in the organic phase and the stabilizers in the continuous aqueous phase.150 The emulsion is rapidly frozen, causing the growth of ice crystals and subsequent supersaturation of the surfactants, leading to the phase separation of the drug and the stabilizers.150 A porous solid drug particle, surrounded by a matrix of stabilizers, forms after freeze‐drying the frozen emulsion.150 Dissolving the drug‐surfactant matrix in water results in the formation of surfactant‐stabilized solid drug NPs approximately 300 nm in size that can be administered orally for daily dosing or intramuscularly for extended release.148, 149, 150 The NPs formed have extremely high drug loading capacities (70 wt% relative to the stabilizers) due to the fact that their primary component is the drug itself.150 Oral administration of the solid drug NPs has the potential to reduce the necessary drug dose by up to 50% as a result of increased transport through the gut epithelium and reduced cytotoxicity of the nanoformulation.148, 150 However, the trial assessing the long‐acting intramuscular administration route was suspended in 2016 due to lack of funding and the formulations face many years of clinical trials before they reach the market.147 The development of long‐acting formulations of NRTIs is important, since they are incorporated in all FDA recommended initial regimens,20 but no nanostructured systems involving NRTIs have been brought to clinical trials. Since NRTIs do not have suitable chemical properties for development into nanosuspensions, alternative sustained release systems must be explored.
Figure 8.
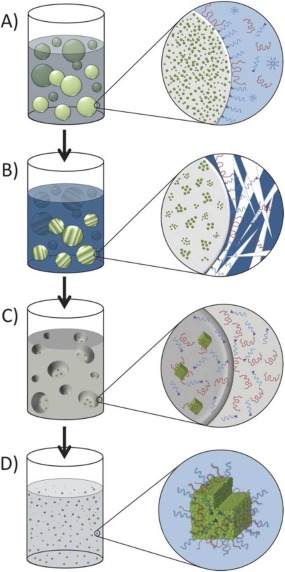
Synthesis of solid drug NPs using emulsion‐templated freeze‐drying. (a) A chloroform‐in‐water emulsion is formed with the hydrophobic drug in the organic phase and the stabilizing surfactants and polymers in the continuous aqueous phase. (b) The growth of ice crystals due to rapid freezing causes supersaturation of the stabilizers and leads to phase separation of the drug and surfactants. (c) Freeze‐drying results in the formation of an emulsion‐templated porous drug monolith surrounded by a matrix of stabilizers. (d) Dissolving the drug monolith and stabilizer matrix in water results in dissolution of the water‐soluble stabilizers and dispersion of the surfactant stabilized, amorphous, solid drug NPs into a nanosuspension. Used with permission from Ref. 150.
We have extensively explored the use of peptide drug hydrogelators for long‐acting delivery of cancer therapeutics. The formation of amphiphilic peptide prodrugs that self‐assemble into nanofibers and entangle into hydrogels in aqueous environments have been shown to produce controlled release and increased cellular uptake of doxorubicin, paclitaxel, and camptothecin.151, 152, 153 Although ARV hydrogels have been traditionally used for the local delivery of HIV microbicides, the potential for systemic release from locally delivered nanostructured systems has been demonstrated by the injectable ARV nanosuspensions and the codelivery of chitosan with a PS NP microbicide. Based on the successes of TFV, emtricitabine, and APV prodrugs, we believe it is possible to expand these prodrug strategies to other ARVs. Such conversion of hydrophilic NRTIs into amphiphilic peptide prodrugs will promote hydrogel formation in vivo. Ideally, hydrogels could be injected intramuscularly or subcutaneously to form a drug depot capable of sustaining systemic release for months. These peptide drug conjugates are simple to synthesize and to scale up, with precisely‐controlled drug loading capacities,154 demonstrating the feasibility of creating peptide‐based ARV prodrugs.
5. CONCLUSIONS
CART is highly effective at suppressing viral replication in a majority of HIV infected individuals. CART has improved patient survival but the continued presence of the virus in reservoir tissues makes discontinuation of the regimen impossible. Long‐term therapy may be associated with significant adverse side effects and lack of patient compliance to the daily regimen may lead to drug resistance. Consequently, there is a need for an improved delivery system that will prolong dosing intervals, increase accumulation in viral reservoirs, and minimize drug toxicity. Despite numerous examples of increased plasma half‐life and improved viral reservoir penetration of nanostructured systems in vitro, preclinical successes have largely not been taken into clinical trials. FDA approved nanoscale delivery systems of ARV therapeutics have not been forthcoming due predominantly to limited scalability and unknown toxicity. To overcome this issue, research efforts must be focused on easily manufacturable drug delivery systems with extensively demonstrated biocompatibility, treatment efficacy, and improved patient compliance.
ACKNOWLEDGMENTS
We acknowledge financial support from the National Science Foundation (DMR 1255281). CF is supported by the Long Acting/Extended Release Antiretroviral Research Resource Program, R24‐AI118397 from NIAID.
Funding information National Science Foundation, Grant/Award Number: DMR 1255281; Long Acting/Extended Release Antiretroviral Research Resource Program, Grant/Award Number: R24‐AI118397
Contributor Information
Charles Flexner, Email: flex@jhmi.edu.
Honggang Cui, Email: hcui6@jhu.edu.
LITERATURE CITED
- 1. Broder S, Gallo RC. A pathogenic retrovirus (HTLV‐III) linked to AIDS. N Engl J Med. 1984;311(20):1292–1297. doi: 10.1056/NEJM198411153112006 [DOI] [PubMed] [Google Scholar]
- 2. Blattner W, Gallo RC, Temin HM. HIV causes AIDS. Science 1988;241(4865):515–516. doi: 10.1126/science.3399881 [DOI] [PubMed] [Google Scholar]
- 3. Gallo RC, Montagnier L. The discovery of HIV as the cause of AIDS. N Engl J Med. 2003;349(24):2283–2285. doi: 10.1056/NEJMp038194 [DOI] [PubMed] [Google Scholar]
- 4. Gallo RC, Montagnier L. The chronology of AIDS research. Nature 1987;326(6112):435–436. doi: 10.1038/326435a0 [DOI] [PubMed] [Google Scholar]
- 5. Flexner C. Chapter 59: antiretroviral agents and treatment of HIV infection In: Brunton L, Chabner B, Knollmann B, eds. Goodman & Gilman's: The Pharmacological Basis of Therapeutics. 12th ed New York, NY: McGraw‐Hill; 2009:1623–1663. [Google Scholar]
- 6. Programme U‐JUN . UNAIDS Data 2017. Joint United Nations Programme on HIV/AIDS. UNAIDS Data 2017. 2017:1–248. doi:978‐92‐9173‐945‐5.
- 7. Nowacek A, Gendelman HE. NanoART, neuroAIDS and CNS drug delivery. Nanomedicine 2009;4(5):557–574. doi: 10.2217/nnm.09.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mamo T, Moseman EA, Kolishetti N, et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine 2010;5(2):269–285. doi: 10.2217/nnm.10.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shao J, Kraft JC, Li B, et al. Nanodrug formulations to enhance HIV drug exposure in lymphoid tissues and cells: clinical significance and potential impact on treatment and eradication of HIV/AIDS. Nanomedicine 2016;11(5):545–564. doi: 10.2217/nnm.16.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim PS, Read SW. Nanotechnology and HIV: potential applications for treatment and prevention. WIREs Nanomed Nanobiotechnol. 2010;2(6):693–702. doi: 10.1002/wnan.118 [DOI] [PubMed] [Google Scholar]
- 11. Roy U, Rodríguez J, Barber P, das Neves J, Sarmento B, Nair M. The potential of HIV‐1 nanotherapeutics: from in vitro studies to clinical trials. Nanomedicine 2015;10(24):3597–3609. doi: 10.2217/nnm.15.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fletcher CV, Staskus K, Wietgrefe SW, et al. Persistent HIV‐1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci USA. 2014;111(6):2307–2312. doi: 10.1073/pnas.1318249111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Solas C, Lafeuillade A, Halfon P, Chadapaud S, Hittinger G, Lacarelle B. Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus‐infected patients. Antimicrob Agents Chemother. 2003;47(1):238–243. doi: 10.1128/AAC.47.1.238-243.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar L, Verma S, Prasad DN, Bhardwaj A, Vaidya B, Jain AK. Nanotechnology: a magic bullet for HIV AIDS treatment. Artif Cells, Nanomedicine Biotechnol. 2015;43(2):71–86. doi: 10.3109/21691401.2014.883400 [DOI] [PubMed] [Google Scholar]
- 15. Jacobson JM, Flexner CW. Universal antiretroviral regimens: thinking beyond one‐pill‐once‐a‐day. Curr Opin HIV AIDS. 2017;12(4):343–350. doi: 10.1097/COH.0000000000000374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chinen J, Shearer WT. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol. 2010;125(2):S195–S203. doi: 10.1016/j.jaci.2009.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greene WC, Peterlin BM. Charting HIV's remarkable voyage through the cell: Basic science as a passport to future therapy. Nat Med. 2002;8(7):673–680. doi: 10.1038/nm0702-673 [DOI] [PubMed] [Google Scholar]
- 18. McCune JM, Rabin LB, Feinberg MB, et al. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 1988;53(1):55–67. doi: 10.1016/0092-8674(88)90487-4 [DOI] [PubMed] [Google Scholar]
- 19. U.S. Department of Health and Human Services . The HIV Life Cycle | Understanding HIV/AIDS | AIDSinfo. AIDSinfo. https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/19/73/the-hiv-life-cycle. Published 2016.
- 20. Panel on Antiretroviral Guidelines for Adults and Adolescents DHHS . Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. October. doi: 10.3390/v7102887 [DOI]
- 21. Berg KM, Litwin AH, Li X, Heo M, Arnsten JH. Lack of sustained improvement in adherence or viral load following a directly observed antiretroviral therapy intervention. Clin Infect Dis. 2011;53(9):936–943. doi: 10.1093/cid/cir537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. U.S. Department of Health and Human Services . FDA‐Approved HIV Medicines | Understanding HIV/AIDS | AIDSinfo. AIDSinfo. https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/21/58/fda-approved-hiv-medicines. Published 2017. Accessed April 30, 2018.
- 23. Best BM, Letendre SL, Koopmans P, et al. Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. J Acquir Immune Defic Syndr. 2012;59(4):376–381. doi: 10.1097/QAI.0b013e318247ec54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calcagno A, Bonora S, Simiele M, et al. Tenofovir and emtricitabine cerebrospinal fluid‐to‐plasma ratios correlate to the extent of blood‐brainbarrier damage. AIDS 2011;25(11):1437–1439. doi: 10.1097/QAD.0b013e3283489cb1 [DOI] [PubMed] [Google Scholar]
- 25. Chapman H, Kernan M, Rohloff J, Sparacino M, Terhorst T. Purification of PMPA amidate prodrugs by SMB chromatography and x‐ray crystallography of the diastereomerically pure GS‐7340. Nucleosides, Nucleotides and Nucleic Acids. 2001;20(4–7):1085–1090. doi: 10.1081/NCN-100002495 [DOI] [PubMed] [Google Scholar]
- 26. Custodio JM, Fordyce M, Garner W, et al. Pharmacokinetics and safety of tenofovir alafenamide in HIV‐uninfected subjects with severe renal impairment. Antimicrob Agents Chemother. 2016;60(9):5135–5140. doi: 10.1128/AAC.00005-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yilmaz A, Izadkhashti A, Price R, et al. Darunavir concentrations in cerebrospinal fluid and blood in HIV‐1–infected individuals. AIDS Res Hum Retroviruses. 2009;25(4):457–461. doi:doi/abs/10.1089/aid.2008.0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Croteau D, Letendre S, Best BM, et al. Therapeutic amprenavir concentrations in cerebrospinal fluid. Antimicrob Agents Chemother. 2012;56(4):1985–1989. doi: 10.1128/AAC.05098-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haas DW, Stone J, Clough LA, et al. Steady‐state pharmacokinetics of indinavir in cerebrospinal fluid and plasma among adults with human immunodeficiency virus type 1 infection. Clin Pharmacol Ther. 2000;68(4):367–374. doi:S0009-9236(00)09021-4[pii]\r10. 1067/mcp.2000.109391 [DOI] [PubMed] [Google Scholar]
- 30. Lafeuillade A, Solas C, Halfon P, Chadapaud S, Hittinger G, Lacarelle B. Differences in the detection of three HIV‐1 protease inhibitors in non‐blood compartments: clinical correlations. HIV Clin Trials 2002;3(1):27–35. doi: 10.1310/WMWL-6W9Y-PXV2-X148 [DOI] [PubMed] [Google Scholar]
- 31. Kravcik S, Gallicano K, Roth V, et al. Cerebrospinal fluid HIV RNA and drug levels with combination ritonavir and saquinavir. J Acquir Immune Defic Syndr. 1999;21–25. 371–375. http://www.ncbi.nlm.nih.gov/pubmed/10458617. [PubMed] [Google Scholar]
- 32. Azijn H, Tirry I, Vingerhoets J, et al. TMC278, a next‐generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild‐type and NNRTI‐resistant HIV‐1. Antimicrob Agents Chemother. 2010;54(2):718–727. doi: 10.1128/AAC.00986-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Croteau D, Best BM, Letendre S, et al. Lower than expected maraviroc concentrations in cerebrospinal fluid exceed the wild‐type CC chemokine receptor 5‐tropic HIV‐1 50% inhibitory concentration. AIDS 2012;26(7):890–893. doi: 10.1097/QAD.0b013e328351f627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gallant JE, Thompson M, DeJesus E, et al. Antiviral activity, safety, and pharmacokinetics of bictegravir as 10‐day monotherapy in HIV‐1‐infected adults. J Acquir Immune Defic Syndr. 2017;75(1):61–66. doi: 10.1097/QAI.0000000000001306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones GS, Yu F, Zeynalzadegan A, et al. Preclinical evaluation of GS‐9160, a novel inhibitor of human immunodeficiency virus type 1 integrase. Antimicrob Agents Chemother. 2009;53(3):1194–1203. doi: 10.1128/AAC.00984-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flexner C. HIV‐protease inhibitors. N Engl J Med. 1998;338(18):1281–1293. doi: 10.1056/NEJM199804303381808 [DOI] [PubMed] [Google Scholar]
- 37. Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV‐1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17(1):657–700. doi:10.1146/annurev.immunol.17.1. 657 [DOI] [PubMed] [Google Scholar]
- 38. Mellors JW, Muñoz A, Giorgi JV, et al. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV‐1 infection. Ann Intern Med. 1997;126(12):946–954. doi: 10.1059/0003-4819-126-12-199706150-00003 [DOI] [PubMed] [Google Scholar]
- 39. Quinn TC, Wawer MJ, Sewankambo N, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N Engl J Med. 2000;342(13):921–929. doi:10.1056/NEJM200003303 421303 [DOI] [PubMed] [Google Scholar]
- 40. Cavert W, Notermans DW, Staskus K, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV‐1 infection. Science 1997;276(5314):960–964. doi: 10.1126/science.276.5314.960 [DOI] [PubMed] [Google Scholar]
- 41. Freeling JP, Ho RJY. Anti‐HIV drug particles may overcome lymphatic drug insufficiency and associated HIV persistence. Proc Natl Acad Sci USA. 2014;111(25):E2512–E2513. doi: 10.1073/pnas.1406554111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication‐competent HIV despite prolonged suppression of plasma viremia. Science 1997;278(5341):1291–1295. doi: 10.1126/science.278.5341.1291 [DOI] [PubMed] [Google Scholar]
- 43. Bradley H, Mattson CL, Beer L, Huang P, Shouse RL. Increased antiretroviral therapy prescription and HIV viral suppression among persons receiving clinical care for HIV infection. AIDS 2016;30(13):2117–2124. doi: 10.1097/QAD.0000000000001164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guy R, Wand H, McManus H, et al. Antiretroviral treatment interruption and loss to follow‐up in two HIV cohorts in Australia and Asia: implications for “test and treat” prevention strategy. AIDS Patient Care STDS. 2013;27(12):681–691. doi:10.1089/apc.2012.0 439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Williams J, Sayles HR, Meza JL, et al. Long‐acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV‐infected patients. Nanomedicine 2013;8(11):1807–1813. doi: 10.2217/nnm.12.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stoddart CA, Galkina SA, Joshi P, et al. Oral administration of the nucleoside EFdA (4′‐ethynyl‐2‐fluoro‐2′‐deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob Agents Chemother. 2015;59(7):4190–4198. doi: 10.1128/AAC.05036-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langer R. Drug delivery and targeting. Nature 1998;392:145. doi: 10.1517/14728222.2.1. [DOI] [PubMed] [Google Scholar]
- 48. Macheras P, Iliadis A. Chapter 4 – Drug release In: Pharmacokinetic Characterization of Controlled‐Release Formulations: Homogeneous and Heterogeneous Approaches. New York, NY: Springer; 2006. [Google Scholar]
- 49. Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1(1):10–29. doi: 10.1002/btm2.10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Y, Peng Z, Seven ES, Leblanc RM. Crossing the blood‐brain barrier with nanoparticles. J Control Release. 2018;270:290–303. doi: 10.1016/j.jconrel.2017.12.015 [DOI] [PubMed] [Google Scholar]
- 51. Moghimi SM, Moghimi M. Enhanced lymph node retention of subcutaneously injected IgG1‐PEG2000‐liposomes through pentameric IgM antibody‐mediated vesicular aggregation. Biochim Biophys Acta – Biomembr. 2008;1778(1):51–55. doi: 10.1016/j.bbamem.2007.08.033 [DOI] [PubMed] [Google Scholar]
- 52. Chen YS, Hung YC, Liau I, Huang GS. Assessment of the in vivo toxicity of gold nanoparticles. Nanoscale Res Lett. 2009;4(8):858–864. doi: 10.1007/s11671-009-9334-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang X‐F, Liu Z‐G, Shen W, Gurunathan S. Silver nanoparticles: synthesis, characterization, properties, applications, and therapeutic approaches. IJMS. 2016;17(9):1534. doi: 10.3390/ijms17091534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lara HH, Ayala‐Nuñez NV, Ixtepan‐Turrent L, Rodriguez‐Padilla C. Mode of antiviral action of silver nanoparticles against HIV‐1. J Nanobiotechnol. 2010;8(1):1. doi: 10.1186/1477-3155-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beduneau A, Ma Z, Grotepas CB, et al. Facilitated monocyte‐macrophage uptake and tissue distribution of superparmagnetic iron‐oxide nanoparticles. PLoS One. 2009;4(2):e4343. doi: 10.1371/journal.pone.0004343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li T, Gendelman HE, Zhang G, et al. Magnetic resonance imaging of folic acid‐coated magnetite nanoparticles reflects tissue biodistribution of long‐acting antiretroviral therapy. Int J Nanomed. 2015;10:3779–3790. doi: 10.2147/IJN.S83279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fiandra L, Colombo M, Mazzucchelli S, et al. Nanoformulation of antiretroviral drugs enhances their penetration across the blood brain barrier in mice. Nanomedicine Nanotechnology, Biol Med. 2015;11(6):1387–1397. doi: 10.1016/j.nano.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 58. Bowman MC, Ballard TE, Ackerson CJ, Feldheim DL, Margolis DM, Melander C. Inhibition of HIV fusion with multivalent gold nanoparticles. J Am Chem Soc. 2008;130(22):6896–6897. doi: 10.1021/ja710321g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garrido C, Simpson CA, Dahl NP, et al. Gold nanoparticles to improve HIV drug delivery. Future Med Chem. 2015;7(9):1097–1107. doi: 10.4155/fmc.15.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dalvi BR, Siddiqui EA, Syed AS, et al. Nevirapine loaded core shell gold nanoparticles by double emulsion solvent evaporation: in vitro and in vivo evaluation. Curr Drug Deliv. 13(7):1071–1083. 2016. doi: 10.2174/1567201813666160114093005 [DOI] [PubMed] [Google Scholar]
- 61. Garg M, Jain NK. Reduced hematopoietic toxicity, enhanced cellular uptake and altered pharmacokinetics of azidothymidine loaded galactosylated liposomes. J Drug Target. 2006;14(1):1–11. doi: 10.1080/10611860500525370 [DOI] [PubMed] [Google Scholar]
- 62. Dubey V, Mishra D, Nahar M, Jain V, Jain NK. Enhanced transdermal delivery of an anti‐HIV agent via ethanolic liposomes. Nanomedicine Nanotechnology, Biol Med. 2010;6(4):590–596. doi: 10.1016/j.nano.2010.01.002 [DOI] [PubMed] [Google Scholar]
- 63. Jain S, Mishra V, Singh P, Dubey PK, Saraf DK, Vyas SP. RGD‐anchored magnetic liposomes for monocytes/neutrophils‐mediated brain targeting. Int J Pharm. 2003;261(1–2):43–55. doi: 10.1016/S0378-5173(03)00269-2 [DOI] [PubMed] [Google Scholar]
- 64. Saiyed ZM, Gandhi NH, Nair MPN. Magnetic nanoformulation of azidothymidine 5’‐triphosphate for targeted delivery across the blood‐brain barrier. Int J Nanomedicine. 2010;5:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tomitaka A, Arami H, Huang Z, et al. Hybrid magneto‐plasmonic liposomes for multimodal image‐guided and brain‐targeted HIV treatment. Nanoscale. 2017. doi: 10.1039/C7NR07255D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eustis S, El‐Sayed MA. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem Soc Rev. 2006;35(3):209–217. doi: 10.1039/B514191E [DOI] [PubMed] [Google Scholar]
- 67. Ho RJY, Yu J, Li B, et al. Systems approach to targeted and long‐acting HIV/AIDS therapy. Drug Deliv Transl Res. 2015;5(6):531–539. doi: 10.1007/s13346-015-0254-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Geszke‐Moritz M, Moritz M. Solid lipid nanoparticles as attractive drug vehicles: composition, properties and therapeutic strategies. Mater Sci Eng C. 2016;68:982–994. doi:10.1016/j.msec.2016.05.1 19 [DOI] [PubMed] [Google Scholar]
- 69. Kammari R, Das NG, Das SK. Chapter 6 – nanoparticulate systems for therapeutic and diagnostic applications In: Mitra A, Cholkar K, Mandal A, eds. Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices. Elsevier Inc; 2017:105–144. doi: 10.1016/B978-0-323-42978-8.00006-1 [DOI] [Google Scholar]
- 70. Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev. 2007;59(6):478–490. doi: 10.1016/j.addr.2007.04.007 [DOI] [PubMed] [Google Scholar]
- 71. Becker Peres L, Becker Peres L, de Araújo PHH, Sayer C. Solid lipid nanoparticles for encapsulation of hydrophilic drugs by an organic solvent free double emulsion technique. Colloids Surf B Biointerfaces. 2016;140:317–323. doi: 10.1016/j.colsurfb.2015.12.033 [DOI] [PubMed] [Google Scholar]
- 72. Doktorovová S, Santos DL, Costa I, Andreani T, Souto EB, Silva AM. Cationic solid lipid nanoparticles interfere with the activity of antioxidant enzymes in hepatocellular carcinoma cells. Int J Pharm. 2014;471(1–2):18–27. doi: 10.1016/j.ijpharm.2014.05.011 [DOI] [PubMed] [Google Scholar]
- 73. Hwang T‐L, Aljuffali IA, Hung C‐F, Chen C‐H, Fang J‐Y. The impact of cationic solid lipid nanoparticles on human neutrophil activation and formation of neutrophil extracellular traps (NETs). Chem Biol Interact. 2015;235:106–114. doi: 10.1016/j.cbi.2015.04.011 [DOI] [PubMed] [Google Scholar]
- 74. Kuo YC, Su FL. Transport of stavudine, delavirdine, and saquinavir across the blood‐brain barrier by polybutylcyanoacrylate, methylmethacrylate‐sulfopropylmethacrylate, and solid lipid nanoparticles. Int J Pharm. 2007;340(1–2):143–152. doi: 10.1016/j.ijpharm.2007.03.012 [DOI] [PubMed] [Google Scholar]
- 75. Kuo YC, Chen HH. Entrapment and release of saquinavir using novel cationic solid lipid nanoparticles. Int J Pharm. 2009;365(1–2):206–213. doi: 10.1016/j.ijpharm.2008.08.050 [DOI] [PubMed] [Google Scholar]
- 76. Kuo YC, Wang CC. Cationic solid lipid nanoparticles with primary and quaternary amines for release of saquinavir and biocompatibility with endothelia. Colloids Surf B Biointerfaces. 2013;101:101–105. doi: 10.1016/j.colsurfb.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 77. Kuo Y‐C, Wang C‐C. Electrophoresis of human brain microvascular endothelial cells with uptake of cationic solid lipid nanoparticles: effect of surfactant composition. Colloids Surf B Biointerfaces. 2010;76(1):286–291. doi: 10.1016/j.colsurfb.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 78. Kuo YC, Wang CC. Cationic solid lipid nanoparticles with cholesterol‐mediated surface layer for transporting saquinavir to the brain. Biotechnol Progress. 2014;30(1):198–206. doi: 10.1002/btpr.1834 [DOI] [PubMed] [Google Scholar]
- 79. Sankar V, Nilaykumar Nareshkumar P, Nishit Ajitkumar G, Devi Penmetsa S, Hariharan S. Comparative studies of lamivudine‐zidovudine nanoparticles for the selective uptake by macrophages. Curr Drug Deliv. 2012. 9(5):506–514. doi: 10.2174/156720112802650707 [DOI] [PubMed] [Google Scholar]
- 80. Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42(1–2):11–18. doi: 10.1016/j.ejps.2010.10.002 [DOI] [PubMed] [Google Scholar]
- 81. Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm. 2015;495(1):439–446. doi: 10.1016/j.ijpharm.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 82. Gaur PK, Mishra S, Bajpai M, Mishra A. Enhanced oral bioavailability of Efavirenz by solid lipid nanoparticles: in vitro drug release and pharmacokinetics studies. Biomed Res Int. 2014;2014:1 doi: 10.1155/2014/363404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kuo YC, Yu HW. Transport of saquinavir across human brain‐microvascular endothelial cells by poly(lactide‐co‐glycolide) nanoparticles with surface poly‐(γ‐glutamic acid). Int J Pharm. 2011;416(1):365–375. doi: 10.1016/j.ijpharm.2011.06.037 [DOI] [PubMed] [Google Scholar]
- 84. Kuo YC, Yu HW. Polyethyleneimine/poly‐(γ‐glutamic acid)/poly(lactide‐co‐glycolide) nanoparticles for loading and releasing antiretroviral drug. Colloids Surf B Biointerfaces. 2011;88(1):158–164. doi: 10.1016/j.colsurfb.2011.06.026 [DOI] [PubMed] [Google Scholar]
- 85. Kuo YC, Lin PI, Wang CC. Targeting nevirapine delivery across human brain microvascular endothelial cells using transferrin‐grafted poly(lactide‐co‐glycolide) nanoparticles. Nanomedicine 2011;6(6):1011–1026. doi: 10.2217/nnm.11.25 [DOI] [PubMed] [Google Scholar]
- 86. Kuo YC, Yu HW. Expression of ornithine decarboxylase during the transport of saquinavir across the blood‐brain barrier using composite polymeric nanocarriers under an electromagnetic field. Colloids Surf B Biointerfaces. 2011;88(2):627–634. doi: 10.1016/j.colsurfb.2011.07.053 [DOI] [PubMed] [Google Scholar]
- 87. Puligujja P, McMillan JE, Kendrick L, et al. Macrophage folate receptor‐targeted antiretroviral therapy facilitates drug entry, retention, antiretroviral activities and biodistribution for reduction of human immunodeficiency virus infections. Nanomedicine Nanotechnology, Biol Med. 2013;9(8):1263–1273. doi: 10.1016/j.nano.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kevadiya BD, Woldstad C, Ottemann BM, et al. Multimodal theranostic nanoformulations permit magnetic resonance bioimaging of antiretroviral drug particle tissue‐cell biodistribution. Theranostics 2018;8(1):256–276. doi: 10.7150/thno.22764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chiappetta DA, Hocht C, Taira C, Sosnik A. Efavirenz‐loaded polymeric micelles for pediatric anti‐HIV pharmacotherapy with significantly higher oral bioavailability [corrected]. Nanomedicine 2010;5(1):11–23. doi: 10.2217/nnm.09.90 [DOI] [PubMed] [Google Scholar]
- 90. Chiappetta DA, Hocht C, Taira C, Sosnik A. Oral pharmacokinetics of the anti‐HIV efavirenz encapsulated within polymeric micelles. Biomaterials 2011;32(9):2379–2387. doi:10.1016/j.biomaterials. 2010.11.082 [DOI] [PubMed] [Google Scholar]
- 91. Chiappetta DA, Hocht C, Opezzo JA, Sosnik A. Intranasal administration of antiretroviral‐loaded micelles for anatomical targeting to the brain in HIV. Nanomedicine 2013;8(2):223–237. doi: 10.2217/NNM.12.104 [DOI] [PubMed] [Google Scholar]
- 92. Seremeta KP, Chiappetta DA, Sosnik A. Poly(e‐caprolactone), Eudragit® RS 100 and poly(e‐caprolactone)/Eudragit® RS 100 blend submicron particles for the sustained release of the antiretroviral efavirenz. Colloids Surf B Biointerfaces. 2013;102:441–449. doi: 10.1016/j.colsurfb.2012.06.038 [DOI] [PubMed] [Google Scholar]
- 93. Raskin MM, Schlachet I, Sosnik A. Mucoadhesive nanogels by ionotropic crosslinking of chitosan‐g‐oligo(NiPAam) polymeric micelles as novel drug nanocarriers. Nanomedicine 2016;11(3):217–233. doi: 10.2217/nnm.15.191 [DOI] [PubMed] [Google Scholar]
- 94. US Department of Health and Human Services . Rilpivirine LA (HIV prevention). AIDSinfo. https://aidsinfo.nih.gov/drugs/581/rilpivirine-la-hiv-prevention/0/patient. Accessed February 6, 2018.
- 95. US Department of Health and Human Services . Cabotegravir. AIDSinfo. https://aidsinfo.nih.gov/drugs/513/cabotegravir/0/patient. Accessed February 6, 2018.
- 96. Spreen WR, Margolis DA, Pottage JC. Long‐acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS. 2013;8(6):565–571. doi: 10.1097/COH.0000000000000002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Malamatri M, Taylor K, Malamatrix S, Douroumis D, Kachrimanis K. Pharmaceutical nanocrystals: production by wet milling and applications. Drug Discov Today. 2018. doi: 10.1016/j.drudis.2018.01.016 [DOI] [PubMed] [Google Scholar]
- 98. Baert L, van ‘t Klooster G, Dries W, et al. Development of a long‐acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur J Pharm Biopharm. 2009;72(3):502–508. doi: 10.1016/j.ejpb.2009.03.006 [DOI] [PubMed] [Google Scholar]
- 99. van 't Klooster G, Hoeben E, Borghys H, et al. Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long‐acting injectable antiretroviral formulation. Antimicrob Agents Chemother. 2010;54(5):2042–2050. doi: 10.1128/AAC.01529-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jackson AGA, Else LJ, Mesquita PMM, et al. A compartmental pharmacokinetic evaluation of long‐acting rilpivirine in HIV‐negative volunteers for pre‐exposure prophylaxis. Clin Pharmacol Ther. 2014;96(3):314–323. doi: 10.1038/clpt.2014.118 [DOI] [PubMed] [Google Scholar]
- 101. U.S. National Library of Medicine . Efficacy, Safety and Tolerability Study of Long‐acting Cabotegravir Plus Long‐acting Rilpivirine (CAB LA + RPV LA) in Human‐immunodeficiency Virus‐1 (HIV‐1) Infected Adultsitle: Clinical trial locator NCT03299049. ClinicalTrials.gov.
- 102. US National Library of Medicine . Study to Evaluate the Efficacy, Safety, and Tolerability of Long‐acting Intramuscular Cabotegravir and Rilpivirine for Maintenance of Virologic Suppression Following Switch From an Integrase Inhibitor in HIV‐1 Infected Therapy Naive Participants: NCT02938. ClinicalTrials.gov.
- 103. Landovitz RJ, Kofron R, McCauley M. The promise and pitfalls of long‐acting injectable agents for HIV prevention. Curr Opin HIV AIDS. 2016;11(1):122–128. doi: 10.1097/COH.0000000000000219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Margolis DA, Gonzalez‐Garcia J, Stellbrink HJ, et al. Long‐acting intramuscular cabotegravir and rilpivirine in adults with HIV‐1 infection (LATTE‐2): 96‐week results of a randomised, open‐label, phase 2b, non‐inferiority trial. Lancet 2017;390(10101):1499–1510. doi: 10.1016/S0140-6736(17)31917-7 [DOI] [PubMed] [Google Scholar]
- 105. Zhou T, Su H, Dash P, et al. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials 2018;151:53–65. doi: 10.1016/j.biomaterials.2017.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Araínga M, Edagwa B, Mosley RL, Poluektova LY, Gorantla S, Gendelman HE. A mature macrophage is a principal HIV‐1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology 2017;14(1):17. doi: 10.1186/s12977-017-0344-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage‐based nanoparticle platform for antiretroviral drug delivery. Blood 2006;108(8):2827–2835. doi: 10.1182/blood-2006-03-012534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Friedman SH, DeCamp DL, Sijbesma RP, Srdanov G, Wudl F, Kenyon GL. Inhibition of the HIV‐1 protease by fullerene derivatives: model building studies and experimental verification. J Am Chem Soc. 1993;115(15):6506–6509. doi: 10.1021/ja00068a005 [DOI] [Google Scholar]
- 109. Martinez ZS, Castro E, Seong CS, Cerón MR, Echegoyen L, Llano M. Fullerene derivatives strongly inhibit HIV‐1 replication by affecting virus maturation without impairing protease activity. Antimicrob Agents Chemother. 2016;60(10):5731–5741. doi: 10.1128/AAC.00341-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Krishnaraj R, Chandran S, Pal P, Berchman S. Investigations on the antiretroviral activity of carbon nanotubes using computational molecular approach. Comb Chem High Throughput Scren 2014; 17(6):531–535. [DOI] [PubMed] [Google Scholar]
- 111. Zhang Z, Wang B, Wan B, Yu L, Huang Q. Molecular dynamics study of carbon nanotube as a potential dual‐functional inhibitor of HIV‐1 integrase. Biochem Biophys Res Commun. 2013;436(4):650–654. doi: 10.1016/j.bbrc.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 112. Bakry R, Vallant RM, Najam‐ul‐Haq M, et al. Medicinal applications of fullerenes. Int J Nanomedicine. 2007;2(4):639–649. doi: 10.1016/S0081-1947(08)60578-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor nanocrystals as fluorescent biological labels. Science. 1998;281(5385):2013–2016. doi: 10.1126/science.281.5385.2013 [DOI] [PubMed] [Google Scholar]
- 114. Mukherjee A, Shim Y, Myong Song J. Quantum dot as probe for disease diagnosis and monitoring. Biotechnol J. 2016;11(1):31–42. doi: 10.1002/biot.201500219 [DOI] [PubMed] [Google Scholar]
- 115. Mahajan SD, Law WC, Aalinkeel R, et al. Nanoparticle‐mediated targeted delivery of antiretrovirals to the brain. Meth Enzymol. 2012;509:41–60. doi: 10.1016/B978-0-12-391858-1.00003-4 [DOI] [PubMed] [Google Scholar]
- 116. Mohideen M, Quijano E, Song E, et al. Degradable bioadhesive nanoparticles for prolonged intravaginal delivery and retention of elvitegravir. Biomaterials 2017;144:144–154. doi: 10.1016/j.biomaterials.2017.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Jiang Y, Cao S, Bright DK, et al. Nanoparticle‐based ARV drug combinations for synergistic inhibition of cell‐free and cell‐cell HIV transmission. Mol Pharmaceutics. 2015;12(12):4363–4374. doi: 10.1021/acs.molpharmaceut.5b00544 [DOI] [PubMed] [Google Scholar]
- 118. Blakney AK, Ball C, Krogstad EA, Woodrow KA. Electrospun fibers for vaginal anti‐HIV drug delivery. Antiviral Res. 2013;100:S9. doi: 10.1016/j.antiviral.2013.09.022 [DOI] [PubMed] [Google Scholar]
- 119. Li Y, Wang F, Cui H. Peptide‐based supramolecular hydrogels for delivery of biologics. Bioeng Transl Med. 2016;1(3):306–322. doi: 10.1002/btm2.10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bourne N, Stanberry LR, Kern ER, Holan G, Matthews B, Bernstein DI. Dendrimers, a new class of candidate topical microbicides with activity against herpes simplex virus infection. Antimicrob Agents Chemother. 2000;44(9):2471–2474. doi: 10.1128/AAC.44.9.2471-2474.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Perumal OP, Inapagolla R, Kannan S, Kannan RM. The effect of surface functionality on cellular trafficking of dendrimers. Biomaterials 2008;29(24–25):3469–3476. doi:10.1016/j.biomaterials.2008.04.0 38 [DOI] [PubMed] [Google Scholar]
- 122. Lüscher MM. Polyanions – a lost chance in the fight against HIV and other virus diseases? Antivir Chem Chemother. 2000;11(4):249–259. doi: 10.1177/095632020001100401 [DOI] [PubMed] [Google Scholar]
- 123. Jiang Y‐H, Emau P, Cairns JS, et al. SPL7013 gel as a topical microbicide for prevention of vaginal transmission of SHIV 89.6P in macaques. AIDS Res Hum Retroviruses. 2005;21(3):207–213. doi: 10.1089/aid.2005.21.207 [DOI] [PubMed] [Google Scholar]
- 124. McCarthy TD, Karellas P, Henderson SA, et al. Dendrimers as drugs: discovery and preclinical and clinical development of dendrimer‐based microbicides for HIV and STI prevention. Mol Pharmaceutics. 2005;2(4):312–318. doi: 10.1021/mp050023q [DOI] [PubMed] [Google Scholar]
- 125. US National Library of Medicine . SPL7013 Gel ‐ Male Tolerance Study: NCT00370357. ClinicalTrials.gov.
- 126. US National Library of Medicine . Retention and Duration of Activity of SPL7013 (VivaGel®) After Vaginal Dosing: NCT00740584. ClinicalTrials.gov.
- 127. Dutta T, Jain NK. Targeting potential and anti‐HIV activity of lamivudine loaded mannosylated poly (propyleneimine) dendrimer. Biochim Biophys Acta – Gen Subj. 2007;1770(4):681–686. doi: 10.1016/j.bbagen.2006.12.007 [DOI] [PubMed] [Google Scholar]
- 128. Gajbhiye V, Ganesh N, Barve J, Jain NK. Synthesis, characterization and targeting potential of zidovudine loaded sialic acid conjugated‐mannosylated poly(propyleneimine) dendrimers. Eur J Pharm Sci. 2013;48(4–5):668–679. doi: 10.1016/j.ejps.2012.12.027 [DOI] [PubMed] [Google Scholar]
- 129. Kannan S, Dai H, Navath RS, et al. Dendrimer‐based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci Transl Med. 2012;4(130):130ra46. doi: 10.1126/scitranslmed.3003162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Nance E, Kambhampati SP, Smith ES, et al. Dendrimer‐mediated delivery of N‐acetyl cysteine to microglia in a mouse model of Rett syndrome. J Neuroinflammation. 2017;14(1):doi:10.1186/s12974‐017‐1004‐5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hong S, Bielinska AU, Mecke A, et al. Interaction of poly(amidoamine) dendrimers with supported lipid bilayers and cells: hole formation and the relation to transport. Bioconjugate Chem. 2004;15(4):774–782. doi: 10.1021/bc049962b [DOI] [PubMed] [Google Scholar]
- 132. Swanson SD, Kukowska‐Latallo JF, Patri AK, et al. Trageted gadolinium‐loaded dendrimer nanoparticles for tumor‐specific magnetic renosance contrast enhancement. Int J Nanomedicine. 2008;3(2):201–210. doi: 10.2147/IJN.S2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yang S, Chen Y, Kaien G, et al. Novel intravaginal nanomedicine for the targeted delivery of saquinavir to CD+ immune cells. Int J Nanomedicine. 2013;8:2847–2858. doi: 10.2147/IJN.S46958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Forbes CJ, Lowry D, Geer L, et al. Non‐aqueous silicone elastomer gels as a vaginal microbicide delivery system for the HIV‐1 entry inhibitor maraviroc. J Control Release. 2011;156(2):161–169. doi: 10.1016/j.jconrel.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ball C, Krogstad E, Chaowanachan T, Woodrow KA. Drug‐eluting fibers for HIV‐1 inhibition and contraception. PLoS One. 2012;7(11):e49792. doi: 10.1371/journal.pone.0049792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ball C, Woodrow KA. Electrospun solid dispersions of maraviroc for rapid intravaginal preexposure prophylaxis of HIV. Antimicrob Agents Chemother. 2014;58(8):4855–4865. doi: 10.1128/AAC.02564-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ball C, Chou SF, Jiang Y, Woodrow KA. Coaxially electrospun fiber‐based microbicides facilitate broadly tunable release of maraviroc. Mater Sci Eng C. 2016;63:117–124. doi: 10.1016/j.msec.2016.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Park J, Ramanathan R, Pham L, Woodrow KA. Chitosan enhances nanoparticle delivery from the reproductive tract to target draining lymphoid organs. Nanomed Nanotechnol Biol Med. 2017;13(6):2015–2025. doi: 10.1016/j.nano.2017.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Krogstad EA, Woodrow KA. Manufacturing scale‐up of electrospun poly(vinyl alcohol) fibers containing tenofovir for vaginal drug delivery. Int J Pharm. 2014;475(1–2):282. doi: 10.1016/j.ijpharm.2014.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kirtane AR, Abouzid O, Minahan D, et al. Development of an oral once‐weekly drug delivery system for HIV antiretroviral therapy. Nat Commun. 2018;9(1):2. doi: 10.1038/s41467-017-02294-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Gunawardana M, Remedios‐Chan M, Miller CS, et al. Pharmacokinetics of long‐acting tenofovir alafenamide (GS‐7340) subdermal implant for HIV prophylaxis. Antimicrob Agents Chemother. 2015;59(7):3913–3919. doi: 10.1128/AAC.00656-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Palombo MS, Singh Y, Sinko PJ. Prodrug and conjugate drug delivery strategies for improving HIV/AIDS therapy. J Drug Deliv Sci Technol. 2009;19(1):3–14. doi: 10.1016/S1773-2247(09)50001-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ray AS, Fordyce MW, Hitchcock MJM. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63–70. doi: 10.1016/j.antiviral.2015.11.009 [DOI] [PubMed] [Google Scholar]
- 144. Hecker S, Erion M. Prodrugs of phosphates and phosphonates. J Med Chem. 2008;51(8):2328–2345. [DOI] [PubMed] [Google Scholar]
- 145. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV‐1 infection: Two randomised, double‐blind, phase 3, non‐inferiority trials. Lancet 2015;385(9987):2606–2615. doi: 10.1016/S0140-6736(15)60616-X [DOI] [PubMed] [Google Scholar]
- 146. Agarwal HK, Chhikara BS, Bhavaraju S, Mandal D, Doncel GF, Parang K. Emtricitabine prodrugs with improved anti‐hiv activity and cellular uptake. Mol Pharmaceutics. 2013;10(2):467–476. doi: 10.1021/mp300361a [DOI] [PubMed] [Google Scholar]
- 147. US National Library of Medicine . PK of Efavirenz & Lopinavir Nano‐formulations in Healthy Volunteers: NCT02632473. ClinicalTrials.gov.
- 148. Liptrott N, Giardiello M, McDonald T, Rannard S, Owen A. Assessment of interactions of efavirenz solid drug nanoparticles with human immunological and haematological systems. J Nanobiotechnology. 2018;16(1):22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Liptrott NJ, Giardiello M, McDonald TO, Rannard SP, Owen A. Lack of interaction of lopinavir solid drug nanoparticles with cells of the immune system. Nanomedicine 2017;12(17):2043–2054. doi: 10.2217/nnm-2017-0095 [DOI] [PubMed] [Google Scholar]
- 150. Mcdonald TO, Giardiello M, Martin P, et al. Antiretroviral solid drug nanoparticles with enhanced oral bioavailability: production, characterization, and in vitro‐in vivo correlation. Adv Healthcare Mater. 2014;3(3):400–411. doi: 10.1002/adhm.201300280 [DOI] [PubMed] [Google Scholar]
- 151. Cheetham AG, Chakroun RW, Ma W, Cui H. Self‐assembling prodrugs. Chem Soc Rev. 2017;46(21):6638–6663. doi: 10.1039/C7CS00521K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Su H, Koo JM, Cui H. One‐component nanomedicine. J Control Release. 2015;219:383–395. doi: 10.1016/j.jconrel.2015.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ma W, Su H, Cheetham AG, et al. Synergistic antitumor activity of a self‐assembling camptothecin and capecitabine hybrid prodrug for improved efficacy. J Control Release. 2017;263:102–111. doi: 10.1016/j.jconrel.2017.01.015 [DOI] [PubMed] [Google Scholar]
- 154. Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2017;110–111:112–126. doi: 10.1016/j.addr.2016.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]