

Abstract
Immune exhaustion is an important feature of chronic infections such as HIV and a barrier to effective immunity against cancer. This dysfunction is in part controlled by inhibitory immune checkpoints. Blockade of the PD-1 or IL-10 pathways can reinvigorate HIV-specific CD4 T cell function in vitro, as measured by cytokine secretion and proliferative responses upon antigen stimulation. However, whether this restoration of HIV-specific CD4 T cells can improve help to other cell subsets impaired in HIV infection remains to be determined. Here, we examine a cohort of chronically infected subjects prior to initiation of antiretroviral therapy (ART) and individuals with suppressed viral load on ART. We show that IFN-γ induction in NK cells upon PBMC stimulation by HIV antigen varies inversely with viremia and depends on HIV-specific CD4 T cell help. We demonstrate in both untreated and ART-suppressed individuals that dual PD-1 and IL-10 blockade enhances cytokine secretion of NK cells via restored HIV-specific CD4 T cell function, that soluble factors contribute to these immunotherapeutic effects and that they depend on IL-2 and IL-12 signaling. Importantly, we show that inhibition of the PD-1 and IL-10 pathways also increases NK degranulation and killing of target cells. This study demonstrates a previously under-appreciated relationship between CD4 T-cell impairment and NK cell exhaustion in HIV infection, provides a proof-of-principle that reversal of adaptive immunity exhaustion can improve the innate immune response, and suggest that immune checkpoint modulation that improves CD4-NK cell cooperation can be used as adjuvant therapy in HIV infection.
INTRODUCTION
The progressive loss of immune functions occurring during chronic antigen exposure, termed exhaustion, is an important feature of chronic infections such as HIV (1) and is also a barrier to effective immunity in cancer, along with tolerance (2). A key advance in the field has been the demonstration that T cell exhaustion is in part under the active control of inhibitory immune checkpoints, such as PD-1, whose selective blockade can reinvigorate antigen-specific CD8 T cell function, including in HIV infection (3–5). The relevance of this reversibility is illustrated by the recent dramatic progresses made in clinical oncology by targeting co-inhibitory receptors such as PD-1 (6, 7). These strategies hold promise for treating HIV infection as well, where modulation of immune checkpoints may be relevant to enhance immune function, as HIV-specific immunity is not restored by antiretroviral therapy alone (8, 9), and to reactivate latent reservoirs for targeting (10, 11). Importantly, exhaustion mechanisms are not restricted to CD8 T cells, but also affect other cell subsets that are critical for control of chronic infection such as CD4 T cells, B cells and NK cells. Virus-specific CD4 T cell dysregulation results from the combination of an exhaustion program and skewing in T helper lineage differentiation which impacts function (12–14), presenting both similarities and differences with their CD8 counterparts. While PD-1 and IL-10 mediate HIV-specific CD4 T cell exhaustion (15–17), consistent with their impact on CD8 T cell responses, some co-inhibitory receptors are differentially expressed between subsets, with a preferential upregulation of CTLA-4 on CD4 T cells (18, 19). Combined blockade of PD-1 and IL-10 has an additive effect on HIV-specific CD4 T cell function compared to blockade of either pathway alone (20). A positive feedback loop between IFN-γ produced by HIV-specific CD4 T cells and IL-12 produced by antigen presenting cells (APCs) contribute to this additive effect. As Thelper act mostly by modulating function of other immune cell types, it is important to define whether reversion of HIV-specific CD4 T cell exhaustion can improve the efficacy of effector arms of the immune system.
Natural Killer (NK) cells are another important component of the immune response to HIV (21) and other viral infections, providing a link between the adaptive and innate immune system. NK cells can directly kill infected cells (22, 23), provide pressure on viral evolution (24), and can mediate antibody-dependent cellular cytotoxicity (ADCC) against infected cells (25). Studies have shown that NK cells can develop an exhausted phenotype akin to T-cell exhaustion in cancer and chronic viral diseases, including in HIV and SIV infections (26). Exhausted NK cells can present a range of defects (27), including lower proliferative capacity, decreased expression of activating receptors(28–31), increased expression of co-inhibitory receptors (30), as well as loose ability to degranulate, secrete cytokines, and promote ADCC (32).
Several studies demonstrate an important crosstalk between CD4 T cells and NK cells. Murine models have shown that CD4 T cell help contributes to optimal NK cell function (33). In SIV infection of non-human primates, IL-2-secreting virus-specific CD4 T cells activate NK cells, and this cooperation is impaired in progressive infection (34). In humans, CD4 T cell help enhances responsiveness of NK cells to influenza and to CMV-infected cells in vitro (35, 36), and results in P. falciparum malaria suggest a similar link in vivo (37). Notably, restoration of virus-specific CD4 T cell help by therapeutic vaccine candidates improved NK cell responses in both HIV-uninfected human donors (38) and SIV-infected rhesus macaques (39).
However, whether immune checkpoint blockade in HIV infection can lead to NK cell functional restoration via reinvigorated CD4 T cell help remains to be determined. Here we show that IFN-γ expression by NK cells upon PBMC stimulation by HIV antigen varies inversely with viral load and depends on HIV-specific CD4 T cell help. We demonstrate both in untreated and ART-suppressed individuals that PD-1 and IL-10 blockade enhances cytokine secretion, degranulation and killing capacity of NK cells via restored HIV-specific CD4 T cell function, and that soluble factors contribute to these immunotherapeutic effects, which depend on IL-2 and IL-12 signaling. This study demonstrates a previously under-appreciated relationship between CD4 T-cell impairment and NK cell exhaustion in HIV infection, provides a proof-of-principle in vitro that reversal of adaptive immunity exhaustion can improve an important arm of the innate immune response, and suggests that immune checkpoint modulation that improves CD4-NK cell cooperation can be used as adjuvant therapy in HIV infection.
MATERIALS AND METHODS
Clinical samples
Peripheral blood was obtained from HIV-infected individuals at the Massachusetts General Hospital (MGH) in Boston, and at the Centre Hospitalier de l’Université de Montréal (CHUM) and the McGill University Health Centre (MUHC) in Montreal. The study was approved by the respective Institutional Review Boards and written informed consent was obtained from all study participants prior to enrollment in the study. All participants were adults (18 years old or older). All clinical investigations were conducted according to the Declaration of Helsinki principles. PBMCs from chronically HIV-infected individuals with a broad range of viral loads prior to initiation of antiretroviral therapy (ART) and individuals treated for 0.6–28 years with undetectable levels of viral RNA (˂50 copies/ml) were isolated from blood samples by Ficoll density centrifugation. Freshly isolated PBMCs were cultured in RPMI-1640 containing 10% heat-inactivated Fetal Bovine Serum (FBS; Sigma) supplemented with 50 IU Penicillin, 50 μg/ml Streptomycin, 2 mM L-glutamine, and 10mM HEPES (Mediatech) (R10 medium).
Phenotypic analysis of cytokine secretion
To investigate the impact of combined blockade on cytokine secretion, CD8 T cell-depleted PBMCs (RosetteSep CD8 depletion reagent; StemCell) were incubated at 37°C in 5% CO2 for 48 h with an HIV-1 Gag peptide pool (66 overlapping peptides spanning the Clade B consensus sequence; 14–18 amino acids long and overlapping by 11 aa; 1 μg/ml/peptide) or left unstimulated in the presence of blocking antibodies against PD-L1 (clone 29E.2A3 [10 μg/ml]) and IL-10Rα (clone 37607/MAB274; R&D [10 μg/ml])) or the corresponding isotype control antibodies (IgG2b [10 μg/ml] plus IgG1 [10 μg/ml]). For selected control experiments, total T cells were depleted (RosetteSep CD3 depletion reagents; StemCell, or with Dynabeads CD8 positive isolation kit: Invitrogen ). For all samples, brefeldin-A (5ug/ml; Sigma), golgi stop (containing monensin) (0.3uL/mL BD) (and anti-CD107α (clone H4A3, PE-Cy5, BD, or BV786, BD) were added for the last 12 hours of stimulation. After 48 h, cells were stained with viability dye (LIVE/DEAD fixable dead cell dye; Invitrogen/ThermoFisher) for 20 min at room temperature and subsequently stained for fluorescent antibodies against CD3 (clone SK7,APC-Cy7, BD or PerCP-eFluor710, eBioscience), CD4 (clone RPA-T4, V450, BD or BV605, BD), CD8 (clone 3B5, Qdot 605, Invitrogen/ThermoFisher, or clone RPA-T8, V500, BD), CD19 (clone HIB19, V500, BD or APCeFluor780, eBioscience), CD14 (clone M5E2, V500 BD or BUV737, BD), and CD56 (clone NCAM16.2, APC, BD or BV421 BD). Intracellular cytokine staining (ICS) for IFN-γ (clone B27, PE-Cy7, BD), TNF-α (clone MAb11, Alexa 700, BD, or APC, BD), and IL-2 (clone 5344.111, FITC, BD, or clone MQ1–17H12 AF488, BD) was performed using BD Cytofix/Cytoperm Fixation/Permeabilization solution according to the manufacturer’s instructions. Cells were acquired on an LSR Fortessa (BD Biosciences, La Jolla, CA).
To evaluate IL-10 and PD-L1 expression, CD8-depleted PBMCs were stimulated with an HIV Gag peptide pool or left unstimulated. For all samples, brefeldin-A (5ug/ml; BD) was added for the last 12 hours of stimulation. After 18 h, cells were stained with viability dye (LIVE/DEAD fixable dead cell dye; Invitrogen/ThermoFisher) for 20 min at room temperature and subsequently stained for fluorescent antibodies against CD3 (clone UCHT1 APC, BD), CD4 (clone RPA-T4 BV605, BD),CD8 (clone RPA-T8 BUV395, BD), CD19 (clone H1B19 APCeFluor780, eBioscience), CD14 (clone 61D3 PerCPCy5.5, BD), CD56 (clone SK1 BUV737, BD). ICS for IL-10 (clone JES3–19F1 PE, BD) was performed using BD Cytofix/Cytoperm Fixation/Permeabilization solution according to the manufacturer’s instructions. Cells were acquired on an LSR Fortessa (BD Biosciences, La Jolla, CA).
Analysis of NK cell function after HIV-peptide stimulation
CD8-depleted PBMCs were stimulated with an HIV-1 Gag peptide pool or left unstimulated in the presence of isotype control or PD-L1/IL-10Rα blocking antibodies as described above. To investigate mechanisms of action of combined blockade on NK cells, we also used anti-IL-12 (Clone # 24910, MAB219; R&D) or anti-IL-2 (Clone: 22722, MAB223; R&D) neutralizing antibodies or their respective isotype controls. For transwell experiments, we used 24-well plates with 0,4 μmM polycarbonate membrane inserts (EK scientific). 2.5 million CD8-depleted PBMCs were stimulated for 48h in the bottom well in the presence of isotype control or PD-L1/IL-10Rα blocking antibodies. 0.5 million purified NK cells (StemCell kit) were placed in the top chamber, corresponding to a 1:5 cell number ratio between the two compartments of the transwells. Brefeldin-A, monensin and anti-CD107α/LAMP1 (clone H4A3 BV786, BD) were added for the last 12 hours of incubation. Subsequent staining was performed using viability dye (LIVE/DEAD fixable dead cell dye; Invitrogen/ThermoFisher) for 20 min at room temperature and fluorescent antibodies against CD3 (clone SK7 PerCP-eFluor710, eBioscience), CD4 (clone RPA-T4 BV605, BD), CD8 (clone RPA-T8 V500, BD), CD19 (clone HIB19 APC-eFluor780, eBioscience), CD14 (clone M5E2 BUV737, BD), and CD56 (clone NCAM16.2 BV421, BD). (ICS for IFN-γ (clone B27 PE-Cy7,BD), TNF-α (clone MAb11 APC, BD), and IL-2 (clone MQ1–17H12 AF488, BD) was performed using BD Cytofix/Cytoperm Fixation/Permeabilization solution according to the manufacturer’s instructions. Cells were acquired on an LSR Fortessa (BD Biosciences, La Jolla, CA).
NK cell killing assay
CD8-depleted PBMCs were stimulated overnight (16 h) with an HIV-1 Gag peptide pool (1μg/mL/peptide), (SEB (1μg/mL) or left unstimulated in the presence of isotype control or combined anti-PD-L1/anti-IL-10Rα antibodies as described above. After 16 h, equal numbers of carboxyfluorescein succinimidyl ester (CFSE)-stained RAJI cells (ATCC) and CellTrace™ Violet (CTV; ThermoFisher)-stained K562 target cells (ATCC) were added to the CD8-depleted PBMCs at a total effector-to-target cell (E:T) ratio of 10:1 and incubated for an additional 16 h. Cells were collected and stained with LIVE/DEAD viability dye (ThermoFisher) and fluorescent antibodies against CD3, CD4, CD8, CD56, CD14 and CD19 (BD/Pharmingen) before fixation and acquisition by flow cytometry as described above. Lysis of K562 cells was quantified by the frequency of LIVE/DEAD+ cells among CTV+ cells. NK-resistant RAJI cells were included as controls to determine the frequency of background, spontaneous cell death. Specific lysis was calculated as (percent sample lysis − percent background lysis)/(100 − percent background lysis).
Statistical analyses
Flow cytometry data were analyzed with FlowJo version 10 (TreeStar). Statistical analyses were performed using Prism 6.0 (GraphPad). Pairwise comparisons for cytokine secretion were performed using the Wilcoxon matched-pair test. We used a Friedman test with Dunn posttest for comparison of more than three groups with paired data. Correlation coefficients were calculated using the Spearman test.
RESULTS
Stimulation of PBMCs by HIV Gag induces a CD4 T cell-dependent production of cytokines by NK cells.
To define the impact of CD4 T cell help on NK cell function in HIV-infected subjects, we first measured the modulation of NK cells by virus-specific CD4 T cells stimulated by an HIV Gag peptide pool (Figure 1) in PBMCs of 21 chronically infected individuals prior to initiation of ART (see Supplemental Table 1 for characteristics of study participants). We reasoned that a delayed intracellular staining (ICS) assay, in which cell subsets would be allowed to freely interact and secrete cytokines in the extracellular medium for several hours before addition of protein transport inhibitors, would be suitable to measure a secondary activation of NK cells by autologous CD4 T cells. We thus performed kinetic experiments in which PBMCs were stimulated for 12 or 36 hours before addition of brefeldin for 12 hours prior to ICS. We depleted CD8 T cells prior to the assays to avoid a confounding impact of this subset. We observed that while IFN-γ production by HIV-specific CD4 T cells was clearly detectable at all time points, IFN-γ expression by NK cells was only apparent at the later, 48-h time point (Figure 1A), which we thus selected for subsequent experiments. Stimulation of CD8-depleted PBMCs with HIV Gag peptides resulted in a significant increase in the fraction of IFN-γ producing NK cells compared to the unstimulated condition (Figure 1B, median-fold increase: 2.0; p<0.0001). As previous studies have shown that progressive HIV disease is associated with NK dysfunction (22), we next examined the association of the inducible IFN-γ secretion by NK cells with markers of disease progression. We observed a strong inverse correlation between the frequency of IFN-γ+ NK cells and viral load (Figure 1C; r=−0.59, p=0.005), whereas no significant correlation was found with CD4 T cell count (data not shown). Notably, we found a direct correlation between frequency of IFN-γ+ Gag-specific CD4 T cells and net increase in the IFN-γ+ NK cells in the stimulated CD8-depleted PBMCs (Figure 1D; r=0.54, p=0.01), suggesting a functional link between these responses. To confirm this hypothesis, we compared a subgroup of 7 subjects for IFN-γ secretion by NK cells in PBMC depleted of CD8 T cells alone, versus PBMC depleted of both CD4 and CD8 T cells by CD3+ cell removal (Figure 1EF). CD4 T cell depletion in addition to CD8 T cell removal resulted in abrogation of the NK response measured after HIV antigen addition, contrasting with the enhancement observed after CD8 T cell depletion alone. Thus, these data show that HIV antigens can stimulate NK cells via HIV-specific CD4 T cell-dependent mechanisms, and that this response decreases with higher viral loads.
Figure 1. Stimulation of PBMCs by HIV Gag induces a CD4 T cell-dependent production of cytokines by NK cells.
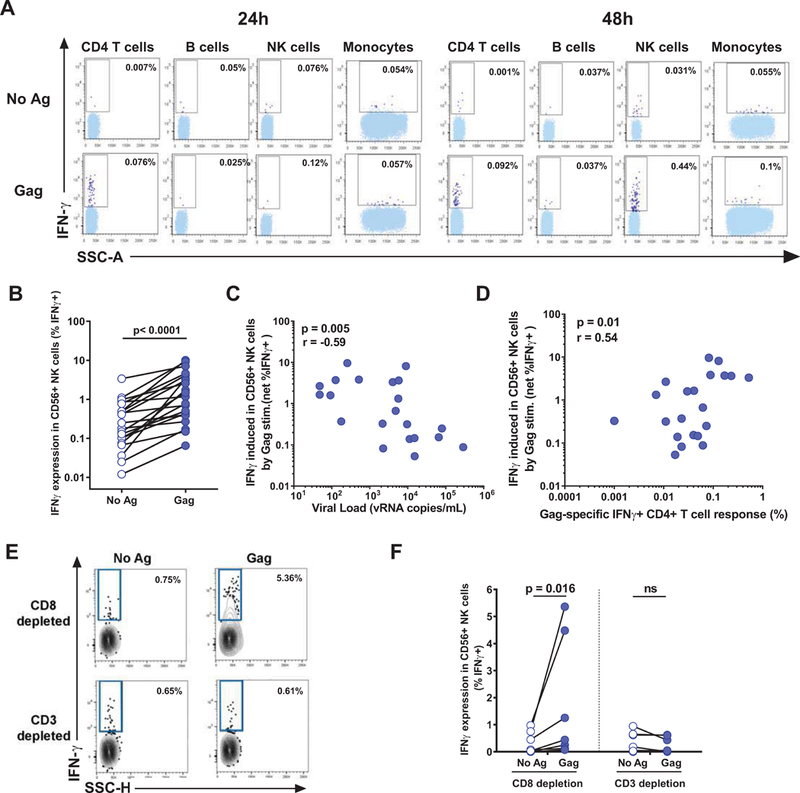
PBMCs were stimulated in vitro with an HIV Gag peptide pool or left unstimulated (negative control) and assessed by a delayed ICS assay, in which cells were incubated with Ag for 12–36 before addition of brefeldin and monensin for 12h and collection after 24–48h. The flow cytometry plots show IFNγ production by CD4 T cells (CD3+CD4+), B cells (CD19+), NK cells (CD56+) and monocytes (CD14+) after doublet exclusion on FSC-A/FSC-H and dead cell exclusion. (A) Example plots for one representative donor showing IFN-γ production by CD4 T cells (CD3+CD4+), B cells (CD3-CD19+), NK cells (CD3-CD56+) and CD14+ monocytes, after doublet exclusion on FSC-A/FSC-H and dead cell exclusion after 24 or 48h of stimulation. (B) ICS frequencies of IFN-γ-producing NK cells 48h after Gag stimulation of PBMCs (n=21 untreated subjects) correlated (C) with HIV viral loads; and (D) with HIV Gag-specific CD4 T cell responses. (E) Representative example and (F) summary data on 7 subjects of the impact of CD3+ or CD8+ cell depletion on IFN-γ production by NK cells. Statistical tests: Wilcoxon paired test (BF) and Spearman correlation (CD).
Combined blockade of the PD-1 and IL-10 pathways leads to a CD4 T cell-dependent enhancement of NK cell function.
We previously showed that blockade of either the PD-1 or the IL-10 pathways reinvigorated HIV-specific CD4 T cell function in vitro and that combined blockade of these immune checkpoints resulted in additive effects compared to inhibition of a single pathway (20). We thus investigated whether combined blockade with anti-PD-L1 and anti-IL10Rα antibodies could improve NK cell function via augmented CD4 T cell help. We first examined PD-L1 and IL-10 expression by four major PBMC subsets: monocytes, CD4 T cells, NK cells and B cells (Figure 2ABC). In line with previous findings (16, 17), monocytes expressed the highest levels of PD-L1 (Figure 2A) and/or IL-10 (Figure 2B), whether the cells were stimulated or not with the Gag peptide pool. IL-10 was frequently co-expressed with PD-L1 by monocytes (Figure 2C). We verified that the delayed ICS assay described above gave results consistent with our previous data using Luminex beads arrays on cell culture supernatants (40), confirming the additive effect of dual blockade on IFN-γ secretion by Gag-specific CD4 T cells (Figure 2D). We next compared the respective impact of blockade on NK and CD4 T cell function. Compared to isotype control antibody conditions, we observed a significant increase of several NK functions upon simultaneous addition of PD-L1/IL-10Rα antibodies and Gag antigen, including IFN-γ production (Figure 2E and 2G; p˂0.0001), degranulation as measured by cell-surface CD107a co-expression with intracellular IFN-γ (Figure 2H; p=0.013) and TNF-α expression (Figure 2I; p=0002). IFN-γ expression by Gag-specific CD4 T cells increased concurrently (Figure 2F and 2J; p<0.0001). However, at this later 48h time point after antigen stimulation, production of other cytokines such as IL-2 was not measurable by ICS in most subjects investigated (data not shown). We noted a direct significant correlation between the gain in CD4 and NK function upon combined PD-L1 and IL-10 blockade, as measured by the fold-increase in immune function observed by combined blockade compared to isotypic control antibodies (Figure 2K; r=47; p=0.03). Thus, blockade of inhibitory immune checkpoints that restore HIV-specific CD4 T cell function also improves NK cell activity in chronically infected individuals in vitro.
Figure 2. CD4 T cell-dependent enhancement of NK cell function after combined blockade of the PD-1 and IL-10 pathways.
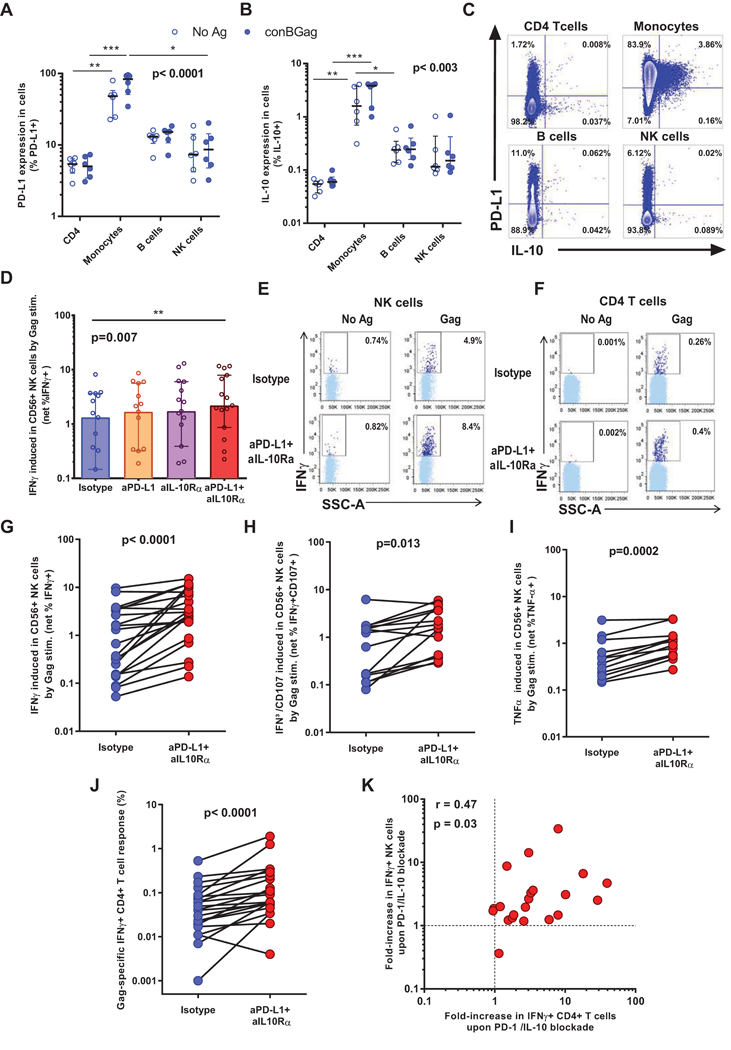
(ABC) PBMCs from untreated donors were stimulated for 48h with an HIV Gag peptide pool or left unstimulated and subsequently stained for (A) PD-L1 and (B) IL-10. (C) shows representative dot plots illustrating PD-L1 and IL-10 co-expression. (D-K) PBMCs were stimulated in vitro with an HIV Gag peptide pool or left unstimulated, this in the presence of blocking antibodies against PD-L1 and IL-10Rα or corresponding isotype controls. A delayed ICS assays performed after 48h. (D) Net IFNγ induction after Gag stimulation combined with single or dual immune checkpoint blockade are shown. (EF) Example plots for one representative donor showing IFN-γ production by NK (E) cells and CD4 (F) T cells after doublet exclusion on FSC-A/FSC-H and dead cell exclusion in the presence of combined blockade with anti-PD-L1 and anti-IL-10Rα or isotype controls. (GHI) Summary data for the frequencies of (G) IFN-γ-producing; (H) IFN-γ/CD107a co-expressing and (I) TNF-α-producing NK cells observed in the presence of combined blockade with anti-PD-L1 and anti-IL-10Rα compared with isotype controls. (J) Summary data for the frequencies of IFN-γ-producing CD4 T cells observed in the presence of combined blockade with anti-PD-L1 and anti-IL-10Rα compared with isotype controls. (K) Correlation between the HIV Gag-specific IFN-γ+ CD4 T cell response to dual PD-1/IL-10 blockade and the increase in the fraction of IFN-γ+ NK cells with this intervention. Number of subjects: (D) n=15; (GJK) n=21; (HI) n = 14. Statistical tests: Non-parametric Friedman test for multiple paired comparisons with Dunn’s post tests (p˂0.01) (A,D). Wilcoxon paired test (G-J) and Spearman correlation (K).
Having demonstrated the effect of combined PD-L1/IL-10R blockade in individuals prior to initiation of therapy, we next sought to define whether NK cell activity could also be restored by this in vitro intervention in subjects with suppressed viremia on ART. Compared to isotype control antibody conditions, IFNγ (Figure 3A), IFNγ/CD107 (Figure 3B) and TNFα (Figure 3C) production was increased in NK cells upon blockade of the PD-1 and IL-10 pathways. These results paralleled the increased function observed in autologous CD4 T cells upon dual blockade (Figure 3D). The fold-increase in the expression of these cytokines upon anti-PD-L1 and anti-IL10Rα blockade was similar between viremic and ART-suppressed individuals (Figure 3E). Therefore, blockade of inhibitory immune checkpoints can also reinvigorate NK function via improved CD4 T cell help in aviremic, ART-treated individuals.
Figure 3. NK cells from ART-suppressed individuals respond to combined blockade of the PD-1 and IL-10 pathways.
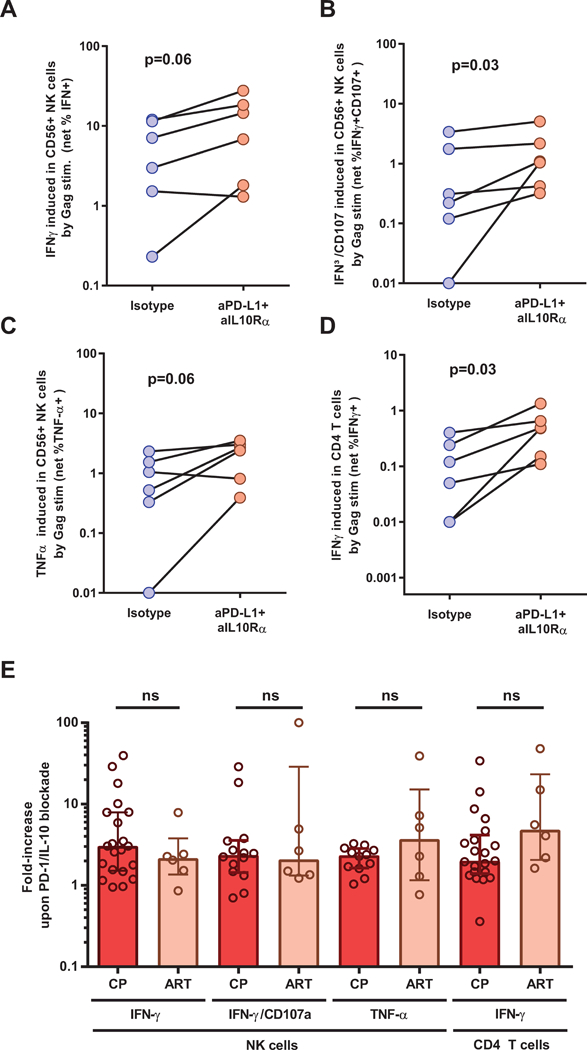
(A-E) PBMCs from ART donors were stimulated in vitro with an HIV Gag peptide pool or left unstimulated, this in the presence of blocking antibodies against PD-L1 and IL-10Rα or corresponding isotype controls. A delayed ICS assays performed after 48h. (A-D) Summary data of the frequencies of (A) IFN-γ-producing NK cells, (B) IFN-γ/CD107a co-expressing NK cells and (C) TNF-α-producing NK cells and (D) IFN-γ-CD4 T cells. (E) Fold increase in net cytokine expression upon PD-L1/IL-10 dual blockade in ART donors compared to untreated individuals presented in Figure 2. Number of ART subjects: (A-E) n=6. Wilcoxon paired test (a A-D). Non-parametric Mann-Whitney U test (p˂0.05) (E).
NK cell activation after HIV antigen stimulation is dependent on IL-2 and IL-12 secretion.
Having shown that NK cell activation can be improved by CD4 T cell help by interruption of inhibitory pathways, we next sought to determine immune mediators that were involved in this cross talk. We hypothesized that cytokines secreted earlier upon addition of Gag antigen to the CD8-depleted PBMC led to the secondary activation of NK cells. While the data from Figures 1 and 2 show correlations between IFN-γ HIV-specific CD4 T cell responses and NK cell activity, we reasoned that this cytokine was likely here a correlate of robust T helper responses and that the cytokines IL-2 and IL-12 were strong candidates for the improvement seen in NK stimulation. We previously demonstrated the expression of IL-2 and IL-12 in our system by CD4 T cells (16) and monocytes (20), respectively. We thus determined the impact of IL-2 or IL-12 blockade on the augmented NK responses elicited by combined anti-PD-L1 and anti- IL10Rα antibodies, again using a delayed ICS assay (Figure 4). Compared to the isotype control condition, addition of either anti-IL-12 or anti-IL2 lead to significant decrease in the percentage of NK cells producing IFN-γ (Figures 4A and 4B; p<0.001 and p<0.05, respectively) and co-expressing IFN-γ and CD107a (Figures 4A and 4C, p<0.05 and p<0.01 respectively) while the inhibitory effect was also observed but somewhat less pronounced for TNF-α (Figures 4D, p>0.05 and p<0.05, respectively). As in Figure 2, we could not reliably quantitate the induction of single CD107a+ cells because of the high level of pre-expression of this molecule by NK cells. These results show that blockade of either the IL-2 or IL-12 pathways, which are important positive regulators of NK activity, can partly inhibit the robust NK responses induced by the HIV Gag-specific CD4 T cells reinvigorated by combined PD-1/IL-10 blockade.
Figure 4. NK cell activation after HIV antigen stimulation is mainly dependent on soluble IL-2 and IL-12 secretion.
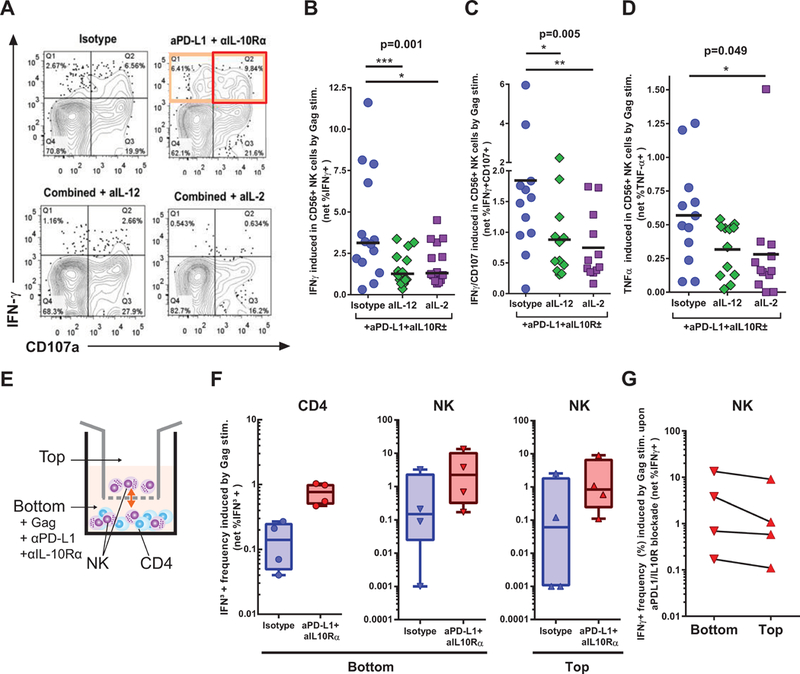
(A-D) CD8-depleted PBMCs were stimulated for 48h with an HIV Gag peptide pool or left unstimulated, this in the presence of combinations of blocking antibodies against PD-L1, IL10Rα, IL-2 and/or IL-12. The last 12 hours were performed in the presence of Brefeldin A and monensin and anti-CD107a antibody before staining and flow cytometric acquisition. (A) Representative example of IFN-γ and CD107a expression by NK cells in the different Gag stimulated conditions. (BCD) Summary plots of the impact of IL-12 or IL-2 blockade on NK cell functions observed in the presence of combined blockade of the PD-1 and IL-10 pathways (n=14): (B) IFN-γ expression; (C) Degranulation as measured by CD107a and IFN-γ co-expression; baseline expression of CD107a precludes reliable analysis of the single positive CD107a+ cells; (D) TNF-α expression. Statistical tests: Non-parametric Friedman test for multiple paired comparisons with Dunn’s post tests. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001. (E) Schematic representation of the transwell experiment. (FG) CD8-depleted PBMCs and purified NK cells were placed in the bottom and top compartment, respectively. PBMCs were then stimulated for 48h with an HIV Gag peptide pool or left unstimulated, this in the presence of a combination of blocking antibodies against PD-L1 and IL10Rα. BFA was added 12h before harvesting the cells for staining. (F) Summary plots of the impact of blockade on IFNγ+ CD4 and NK cells are shown. (G) Net frequencies upon blockade for matched top and bottom NK cell populations are also shown to illustrate the effect of the transwell on crosstalk.
To determine if soluble mediators are the principal contributors of this NK-CD4 T cell cross-talk, we conducted a transwell experiment with samples from 2 untreated, chronically infected individuals and 2 ART-suppressed donors who were good responders to PD-L1 and IL-10 blockade as defined in Figures 2 and 3. CD8-depleted PBMCs were placed in the bottom chamber and purified NK cells in the top chamber of the transwell (Figure 4E). Upon Gag stimulation and PD-L1/IL-10 blockade, we observed a clear increase in IFN-γ expression both in the bottom (CD4 T and NK cells) and top (NK cells) compartments (Figure 4F). These results show that secreted factors able to diffuse through the small-pore membrane efficiently stimulated the purified NK cells. Importantly, the expression of IFNγ by the NK cells of the top chamber was close to that observed in the NK cells from the CD4 T cell-containing bottom chamber (Figure 4G). We therefore conclude that NK-CD4 T cells crosstalk upon PD-L1 and IL-10 blockade depends mainly on soluble mediators.
Combined blockade of the PD-1 and IL-10 pathways restores NK cell killing.
A critical goal of immune checkpoint blockade in infectious diseases and cancer is to improve target cell killing. Therefore, we investigated whether the NK cells stimulated by HIV-specific CD4 T cell help restored by dual PD-L1/IL-10R blockade had superior killing capacity compared to NK cells stimulated by the unmanipulated exhausted CD4 T cells (Figure 5). Killing assays with primary clinical samples are technically challenging and tend to show greater variability than the ICS used in the experiments described above. To overcome this issue, we adapted to our purpose a recently described method of NK killing measurement, which uses internal controls to minimize inter-assay variability (32) (See experimental design, Figure 5A). CD8-depleted PBMC, which contain the NK effector cells and the CD4 T helper cells, are first stimulated with the HIV Gag peptide pool or controls, in the presence or absence or PD-1 and IL-10 blockade. After incubation, the primary cells are mixed with fluorescently labeled NK-killing sensitive (K562, devoid of MHC expression) and NK killing-resistant (RAJI) cell lines. Differential lysis rate of these two lines as measured by a live/dead cell dye assay (Figure 5B) allows calculation of specific NK lysis under the different conditions of stimulation by the formula: (percent sample lysis[K562 cells] − percent background lysis[RAJI cells])/(100 − percent background lysis[RAJI cells]). We observed that compared to the unstimulated conditions, addition of HIV Gag peptides elicited specific killing, while the positive control SEB confirmed the robustness of the assay (Figure 5C). Importantly, combined PD-L1/IL-10Rα augmented specific killing by NK cells in the HIV Gag-stimulated conditions (Figure 5D). Therefore, consistent with the impact observed on IFN-γ and degranulation (CD107a), inhibition of the immune checkpoints PD-1 and IL-10 improves the cytotoxic activity of NK cells stimulated by HIV-specific CD4 T cells.
Figure 5. Combined blockade of the PD-1 and IL-10 pathways restores NK cell killing.
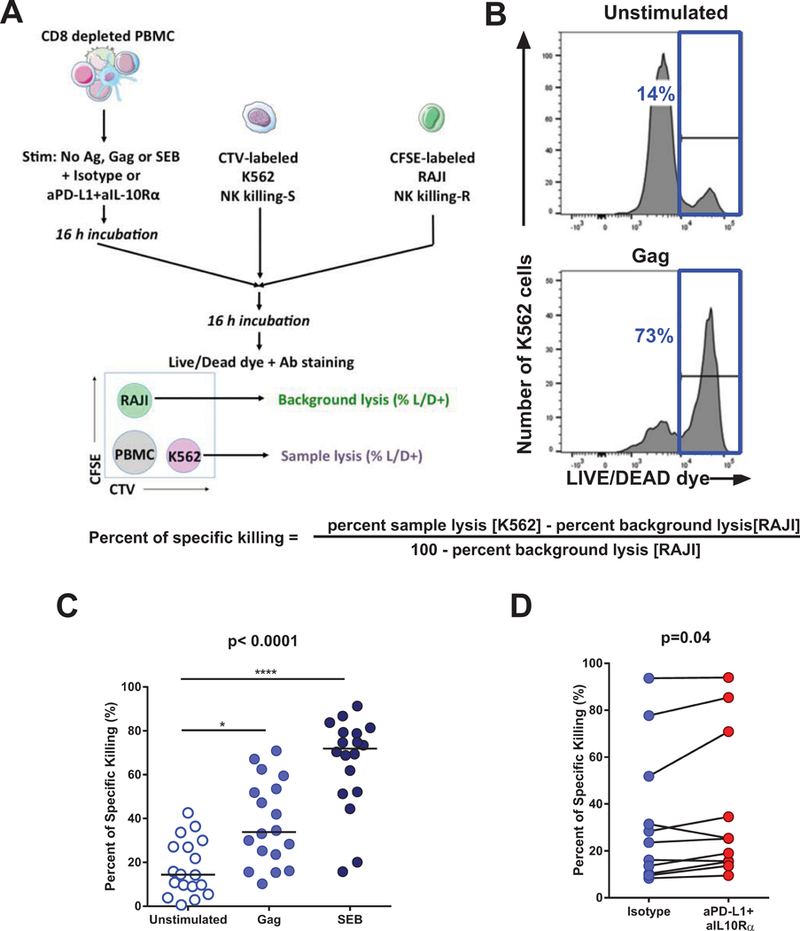
CD8-depleted PBMCS were incubated overnight with an HIV Gag peptide pool, SEB or left unstimulated in the presence of isotype control antibodies or blocking antibodies against PD-L1 or IL-10Rα. On the next day, the PBMCs were mixed with target K562 cells, with effector and target cells identified by cell tracker dyes. Killing was measured by a live/dead dye. (AB) Representative examples of (A) the gating strategy used and (B) results obtained in the killing assay. (C) Summary of the pooled data of the specific killing of K562 cells observed in the Unstimulated, Gag-stimulated and SEB-stimulated conditions. (D) Increase in specific killing elicited by combined PD-1/IL-10 blockade in the Gag-stimulated conditions. Statistical tests: (C) Non-parametric Friedman test for multiple paired comparisons with Dunn’s post tests; (D) Wilcoxon matched-pairs test. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001.
Discussion
Immune checkpoint blockade can restore T cell immunity in chronic infections and cancer, is currently a major therapeutic strategy in clinical oncology, and is considered as a potentially attractive adjuvant therapy for HIV cure strategies. While the benefit of such an approach remains to be evaluated for individuals on ART, reinvigorating the host’s immune system could maximize the effect of therapeutic vaccination and reservoir clearance -for example through “shock and kill” strategies - because both approaches rely on an efficient immune response to eventually succeed. Here, we show that blockade of two major inhibitory pathways contributing to HIV-specific T cell exhaustion, PD-1 and IL-10, increases NK cell function through reinvigorated HIV-specific CD4 T cell help. These results suggest that reversion of CD4 T cell exhaustion in HIV infection can improve co-operation of adaptive immunity and a major effector arm of the innate response, and may thereby facilitate durable viral control.
CD4 T cell responses play a critical role in the development of effective cellular and humoral antiviral immunity (41). While the role of CD4 T cell help for CD8 T cell and B cell maturation has been extensively studied (42, 43), less is known about the role of CD4 T cell help in modulating NK cell function. Cytokines are important mediators of CD4 T cell help to NK cells: IL-2 secreted by virus-specific CD4 T cells can stimulate NK cells directly, and elicit secretion of cytokines such as IL-12 by antigen-presenting cells, which in turn is a potent activator of NK cell function. Indeed, IL-2 is a potent stimulator of IFN-γ production by NK cells (44) and synergize with IL-12 (45). Our results in HIV infection are consistent with such a model of cooperative interaction between CD4 T cells and NK cells and with previous results obtained with SIV-infected macaques. The ability of NK cells to respond to PBMC by HIV antigen inversely correlated with viral load, directly correlated with the magnitude of the HIV Gag-specific CD4 T cells and, importantly, was strongly dependent on the presence of CD4 T cells. While it is likely that some of the NK cells were initially stimulated by direct recognition by Killer Ig-like receptors (KIRs) of HIV peptides bound to HLA molecules (24, 46), a phenomenon that may have been below the sensitivity of our T cell depletion experiments, CD4 T cell help appears clearly to be a limiting factor in the potentiation of NK cell activation.
Restoration of HIV Gag-specific CD4 T cell responses by dual PD-1 and IL-10 blockade resulted in stronger enhancement of IFN-γ NK cell responses compared to interruption of either pathway, consistent with our previous results showing an additive effect of anti-PD-L1 and ani-IL-10Rα on Thelper function (20). Given the multiplicity of conditions, we thus choose the combined intervention in subsequent experiments and demonstrated that immune checkpoint blockade restored both HIV Gag-specific CD4 T cell responses and NK functions, with a direct correlation in the extent of exhaustion reversal by blockade for these two immune subsets. Combined PD-1/IL-10 blockade did not only result in increased IFN-γ expression by NK cells, but also increased NK cell degranulation, suggesting improved killing capacity, and TNF-α secretion. We observed the enhancement of NK responses during a time frame (48h) during which no significant cell proliferation occurred (16). While in vivo data suggest that some immune interventions such as in PD-1 blockade in SIV infection (47) affects magnitude of virus-specific CD4 T cell response less than that of CD8 T cells, our results suggest that improved CD4 T cell help (e.g, improved IL-2 production) may contribute to enhanced NK cell antiviral activity. Of note, dysfunctional NK cells can also upregulate co-inhibitory receptors (27). However, only a small fraction (usually less than 10%), of peripheral blood NK cells express PD-1 in HIV infection (48). Thus, direct impact of PD-L1 blockade on NK cells may only make a minor contribution, if any, to our observations. Our study was mostly focused on viremic subjects before initiation of ART, a group in which we expect to see the highest levels of CD4 T cell and NK cell exhaustion. However, a clinically important group of ART-treated individuals with suppressed viremia responds well to PD-1 and/or IL-10 blockade in vitro. We demonstrated that restored CD4 T cell help through immune checkpoint blockade can also improve NK function in these subjects, which suggest that immune checkpoint modulation that improves CD4-NK cell cooperation could be used as adjuvant therapy in persons receiving potent ART, the current standard of care.
One of the advantages of the delayed ICS technique used here is that it allowed us to examine CD4 and NK cell responses in the same sample. This avoided inter-assay variation that might unduly influence potential correlations. One limitation is that we could not reliably quantify IL-2 production by HIV-specific CD4 T cells at 48h, given the faster expression kinetics of this cytokine. To address the role of IL-2 in the restoration of NK function by combined PD-1/IL-10 blockade, we thus used an anti-IL2 antibody in similar functional assays, and compared its impact to the effect of anti-IL12 blockade. Both interventions decreased, without fully abrogating, the boosting of NK cell responses induced by combined PD-1/IL-10 blockade. The partial inhibition observed suggests some redundancy in the CD4/NK crosstalk. These major roles played by the cytokines IL-2 and IL-12 are consistent with our transwell experiments, which show that most of the observed effect of CD4 help on NK function is mediated by soluble factors in this experimental system. While cell-cell contact-dependent factors may contribute to this crosstalk, our data suggest that their contribution is limited. Previous studies showed that other NK stimulatory cytokines besides IL-2 and IL-12, such as IL-15 and IL-18, can reverse NK cell dysfunction (49, 50), and they may also play a role in the improved NK cell activation obtained here.
Our report also shows that the enhanced NK cell activation elicited by combined PD-1/IL-10 blockade improved the killing capacity of these cells against MHC I -negative targets. While this experiment demonstrated improvement of NK-mediated cytotoxicity, further investigation will be needed to determine the relevance of our findings in vivo. A key question that will determine the impact of CD4/NK crosstalk in HIV infection is the level of anatomical co-localization of HIV-specific CD4 T helper cells, NK cells and HIV-infected cells. While previous studies have shown that NK cells are detected in lymphoid tissues at rather low frequencies in healthy uninfected individuals (44), studies in SIV-infected macaques have shown an accumulation of exhausted NK cells in lymph nodes compared to uninfected animals (51), suggesting that reversing dysfunction of these effector cells by immunotherapy could contribute to viral control in situ. Tissue studies and in vivo investigations are warranted to address these important issues.
This study demonstrates a direct relationship between CD4 T-cell impairment and NK cell exhaustion in HIV infection, provides an in vitro proof-of-principle that reversal of CD4 T helper exhaustion by PD-1 and IL-10 blockade can boost an important arm of the innate immune response. Our results suggest that improved CD4-NK cell cooperation may contribute to potential antiviral effects of immune checkpoint blockade as adjuvant therapy in HIV infection.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff at Massachusetts General Hospital and Centre Hospitalier de l’Université de Montréal and all study participants; Dr. Dominique Gauchat, the CRCHUM Flow Cytometry Platform, Dr. Olfa Debbeche and the CRCHUM BSL3 Platform.
Funding: This work was supported by National Institutes of Health (NIH) grants RO1 HL-092565 (D.E.K); NIAID UM1AI100663 (CHAVI-ID) (D.E.K; Dennis Burton, principal investigator), P01AI056299 (G.F); the Canadian Institutes for Health Research (project grant #137694: D.E.K), a Canada Foundation for Innovation Grant (D.E.K) and the FRQS AIDS and Infectious Diseases Network. D.E.K is supported by a FRQS Senior Research Scholar Award. A.F. is supported by a Canada Research Chair. J.R. and M.V. were supported by CIHR fellowships. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
AUTHORSHIP
D.E.K. was responsible for the overall design and conduct and provided supervision; F.P, J.R, A.F, M.D and D.E.K provided intellectual input and contributed to the experimental design; F.P, M.G.H, A.M, H.L.E, A.M, N.B, M.V, M.H and N.L.N did experiments; G.J.F provided novel reagents; and F.P and D.E.K interpreted the data and wrote the paper with all co-authors’ assistance.
Conflict-of-interest disclosure: GJF has patents and receives patent royalties on the PD-1 pathway. The remaining authors declare no competing financial interests.
REFERENCES
- 1.Zuniga EI, Macal M, Lewis GM, and Harker JA. 2015. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu Rev Virol 2: 573–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schietinger A, and Greenberg PD. 2014. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 35: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, and Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443: 350–354. [DOI] [PubMed] [Google Scholar]
- 4.Trautmann L, Janbazian L, Chomont N, Said EA, Wang G, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, and Sekaly RP. 2006. Upregulation of PD-1 expression on HIV-specific CD8 + T cells leads to reversible immune dysfunction. Nat Med. [DOI] [PubMed] [Google Scholar]
- 5.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, and Koup RA. 2006. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med 203: 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, and Sznol M. 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, and Wigginton JM. 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyasere CA, Tilton JC, Johnson AJ, Younes S, Yassine-Diab B, Sekaly RP, Kwok WW, Migueles SA, Laborico AC, Shupert WL, Hallahan CW, Davey DF, Dybul M, Vogel S, Metcalf J, and Connors M. 2003. Diminished proliferation of Human Immunodeficiency Virus-Specific CD4+ T cell Is Associated with Diminished Interleukin-2 (IL-2) Production and IS Recovered by Exogenous IL-2. Journal of Virology 77: 10900–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porichis F, and Kaufmann DE. 2012. Role of PD-1 in HIV pathogenesis and as target for therapy. Curr HIV/AIDS Rep 9: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, Killian M, Epling L, Hoh R, Sinclair E, Hecht FM, Bacchetti P, Deeks SG, Lewin SR, Sekaly RP, and Chomont N. 2016. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog 12: e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufmann DE, and Walker BD. 2009. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol 182: 5891–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morou A, Palmer BE, and Kaufmann DE. 2014. Distinctive features of CD4+ T cell dysfunction in chronic viral infections. Current opinion in HIV and AIDS 9: 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, and Brooks DG. 2011. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. The Journal of experimental medicine 208: 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, and Wherry EJ. 2014. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 40: 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, Porichis F, Le Gall S, Waring MT, Moss K, Jessen H, Pereyra F, Kavanagh DG, Walker BD, and Kaufmann DE. 2009. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 114: 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porichis F, Kwon DS, Zupkosky J, Tighe DP, McMullen A, Brockman MA, Pavlik DF, Rodriguez-Garcia M, Pereyra F, Freeman GJ, Kavanagh DG, and Kaufmann DE. 2011. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood 118: 965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwon DS, Angin M, Hongo T, Law KM, Johnson J, Porichis F, Hart MG, Pavlik DF, Tighe DP, Kavanagh DG, Streeck H, Addo MM, and Kaufmann DE. 2012. CD4+ CD25+ Regulatory T cells Impair HIV-Specific CD4 T Cell Responses by Upregulating IL-10 Production in Monocytes. Journal of virology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khaitan A, and Unutmaz D. 2011. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep 8: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, Palmer S, Brockman M, Rathod A, Piechocka-Trocha A, Baker B, Zhu B, Le Gall S, Waring MT, Ahern R, Moss K, Kelleher AD, Coffin JM, Freeman GJ, Rosenberg ES, and Walker BD. 2007. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nature immunology 8: 1246–1254. [DOI] [PubMed] [Google Scholar]
- 20.Porichis F, Hart MG, Zupkosky J, Barblu L, Kwon DS, McMullen A, Brennan T, Ahmed R, Freeman GJ, Kavanagh DG, and Kaufmann DE. 2014. Differential Impact of PD-1 and/or Interleukin-10 Blockade on HIV-1-Specific CD4 T Cell and Antigen-Presenting Cell Functions. Journal of virology 88: 2508–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scully E, and Alter G. 2016. NK Cells in HIV Disease. Curr HIV/AIDS Rep 13: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, Streeck H, Waring M, Meier A, Brander C, Lifson JD, Allen TM, Carrington M, and Altfeld M. 2007. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med 204: 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lisovsky I, Isitman G, Song R, DaFonseca S, Tremblay-McLean A, Lebouche B, Routy JP, Bruneau J, and Bernard NF. 2015. A Higher Frequency of NKG2A+ than of NKG2A- NK Cells Responds to Autologous HIV-Infected CD4 Cells irrespective of Whether or Not They Coexpress KIR3DL1. J Virol 89: 9909–9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, Oniangue-Ndza C, Martin M, Li B, Khakoo SI, Carrington M, Allen TM, and Altfeld M. 2011. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 476: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gooneratne SL, Richard J, Lee WS, Finzi A, Kent SJ, and Parsons MS. 2015. Slaying the Trojan horse: natural killer cells exhibit robust anti-HIV-1 antibody-dependent activation and cytolysis against allogeneic T cells. J Virol 89: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schafer JL, Muller-Trutwin MC, and Reeves RK. 2015. NK cell exhaustion: bad news for chronic disease? Oncotarget 6: 21797–21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bi J, and Tian Z. 2017. NK Cell Exhaustion. Front Immunol 8: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, O’Shea MA, Kinter A, Kovacs C, Moretta A, and Fauci AS. 2005. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci U S A 102: 2886–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Maria A, Fogli M, Costa P, Murdaca G, Puppo F, Mavilio D, Moretta A, and Moretta L. 2003. The impaired NK cell cytolytic function in viremic HIV-1 infection is associated with a reduced surface expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44). Eur J Immunol 33: 2410–2418. [DOI] [PubMed] [Google Scholar]
- 30.Mavilio D, Benjamin J, Daucher M, Lombardo G, Kottilil S, Planta MA, Marcenaro E, Bottino C, Moretta L, Moretta A, and Fauci AS. 2003. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci U S A 100: 15011–15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmad R, Sindhu ST, Tran P, Toma E, Morisset R, Menezes J, and Ahmad A. 2001. Modulation of expression of the MHC class I-binding natural killer cell receptors, and NK activity in relation to viral load in HIV-infected/AIDS patients. J Med Virol 65: 431–440. [PubMed] [Google Scholar]
- 32.Schafer JL, Li H, Evans TI, Estes JD, and Reeves RK. 2015. Accumulation of Cytotoxic CD16+ NK Cells in Simian Immunodeficiency Virus-Infected Lymph Nodes Associated with In Situ Differentiation and Functional Anergy. J Virol 89: 6887–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly MN, Zheng M, Ruan S, Kolls J, D’Souza A, and Shellito JE. 2013. Memory CD4+ T cells are required for optimal NK cell effector functions against the opportunistic fungal pathogen Pneumocystis murina. J Immunol 190: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vargas-Inchaustegui DA, Xiao P, Tuero I, Patterson LJ, and Robert-Guroff M. 2012. NK and CD4+ T cell cooperative immune responses correlate with control of disease in a macaque simian immunodeficiency virus infection model. J Immunol 189: 1878–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He XS, Draghi M, Mahmood K, Holmes TH, Kemble GW, Dekker CL, Arvin AM, Parham P, and Greenberg HB. 2004. T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J Clin Invest 114: 1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Z, Frascaroli G, Bayer C, Schmal T, and Mertens T. 2015. Interleukin-2 from Adaptive T Cells Enhances Natural Killer Cell Activity against Human Cytomegalovirus-Infected Macrophages. J Virol 89: 6435–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCall MB, Roestenberg M, Ploemen I, Teirlinck A, Hopman J, de Mast Q, Dolo A, Doumbo OK, Luty A, van der Ven AJ, Hermsen CC, and Sauerwein RW. 2010. Memory-like IFN-gamma response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur J Immunol 40: 3472–3477. [DOI] [PubMed] [Google Scholar]
- 38.Jost S, Tomezsko PJ, Rands K, Toth I, Lichterfeld M, Gandhi RT, and Altfeld M. 2014. CD4+ T-cell help enhances NK cell function following therapeutic HIV-1 vaccination. J Virol 88: 8349–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vargas-Inchaustegui DA, Xiao P, Demberg T, Pal R, and Robert-Guroff M. 2015. Therapeutic envelope vaccination in combination with antiretroviral therapy temporarily rescues SIV-specific CD4(+) T-cell-dependent natural killer cell effector responses in chronically infected rhesus macaques. Immunology 145: 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porichis F, Hart MG, Zupkosky J, Barblu L, and Kaufmann DE. 2013. In vitro assay to evaluate the impact of immunoregulatory pathways on HIV-specific CD4 T cell effector function. J Vis Exp: e50821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Virgin HW, Wherry EJ, and Ahmed R. 2009. Redefining chronic viral infection. Cell 138: 30–50. [DOI] [PubMed] [Google Scholar]
- 42.Laidlaw BJ, Craft JE, and Kaech SM. 2016. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crotty S 2014. T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, and Caligiuri MA. 2003. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood 101: 3052–3057. [DOI] [PubMed] [Google Scholar]
- 45.Chan SH, Perussia B, Gupta JW, Kobayashi M, Pospisil M, Young HA, Wolf SF, Young D, Clark SC, and Trinchieri G. 1991. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med 173: 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saunders PM, Pymm P, Pietra G, Hughes VA, Hitchen C, O’Connor GM, Loiacono F, Widjaja J, Price DA, Falco M, Mingari MC, Moretta L, McVicar DW, Rossjohn J, Brooks AG, and Vivian JP. 2016. Killer cell immunoglobulin-like receptor 3DL1 polymorphism defines distinct hierarchies of HLA class I recognition. J Exp Med 213: 791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, and Amara RR. 2008. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taborda NA, Hernandez JC, Lajoie J, Juno JA, Kimani J, Rugeles MT, and Fowke KR. 2015. Short Communication: Low Expression of Activation and Inhibitory Molecules on NK Cells and CD4(+) T Cells Is Associated with Viral Control. AIDS Res Hum Retroviruses 31: 636–640. [DOI] [PubMed] [Google Scholar]
- 49.Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, Deng W, Ring AM, Fischer S, Garcia KC, and Raulet DH. 2014. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest 124: 4781–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horowitz A, Behrens RH, Okell L, Fooks AR, and Riley EM. 2010. NK cells as effectors of acquired immune responses: effector CD4+ T cell-dependent activation of NK cells following vaccination. J Immunol 185: 2808–2818. [DOI] [PubMed] [Google Scholar]
- 51.Reeves RK, Gillis J, Wong FE, Yu Y, Connole M, and Johnson RP. 2010. CD16- natural killer cells: enrichment in mucosal and secondary lymphoid tissues and altered function during chronic SIV infection. Blood 115: 4439–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.