

Abstract
The execution of shock following high dose E. coli lipopolysaccharide (LPS) or bacterial sepsis in mice required pro-apoptotic caspase-8 in addition to pro-pyroptotic caspase-11 and gasdermin d. Hematopoietic cells produced MyD88- and TRIF-dependent inflammatory cytokines sufficient to initiate shock without any contribution from caspase-8 or caspase-11. Both proteases had to be present to support TNF- and interferon β-dependent tissue injury first observed in the small intestine and later in spleen and thymus. Caspase-11 enhanced the activation of caspase-8 and extrinsic cell death machinery within the lower small intestine. Neither caspase-8 nor caspase-11 was individually sufficient for shock. Both caspases collaborated to amplify inflammatory signals associated with tissue damage. Therefore, combined pyroptotic and apoptotic signaling mediated endotoxemia independently of RIPK1 kinase activity and RIPK3 function. These observations bring to light the relevance of tissue compartmentalization to disease processes in vivo where cytokines act in parallel to execute diverse cell death pathways.
eTOC
Endotoxic shock requires inflammatory cytokines and cell death; however, initiation and execution signaling pathways remain unresolved. Mandal et al. describe a collaboration between pro-apoptotic caspase-8 and pro-pyroptotic caspase-11, independent of pro-necroptotic RIPK1 kinase activity or RIPK3, to execute TNF- and type I interferon-mediated inflammatory tissue damage underlying endotoxic shock.
INTRODUCTION
Lipopolysaccharide (LPS) contributes to inflammation underlying Gram-negative (GN) bacterial sepsis in clinical settings (Hurley et al., 2015). Sepsis remains a prominent cause of mortality (Singer et al., 2016). In addition to the acknowledged role of the pathogen sensor toll-like receptor (TLR)4, high dose (≥50 mg/kg body weight; ≥5X LD100) LPS challenge in mice requires inflammatory cytokine signaling via TNF and type I interferon (IFN) (Beutler and Rietschel, 2003; Karaghiosoff et al., 2003; Sheehan et al., 1989). Endotoxemia has been shown to depend on the IFN-induced cysteine protease, caspase (Casp)11 (a homolog of human Casp4 and Casp5) as well as its substrate gasdermin d (Gsdmd) (Kayagaki et al., 2015; Kayagaki et al., 2011). Both are also necessary for pyroptosis, a leakage form of programmed cell death implicated in LPS shock. Nevertheless, apoptotic markers accumulate in association with septic shock (Singer et al., 2016). The interplay of inflammatory signaling and cell death pathways during shock remains unresolved.
LPS signals via membrane-associated TLR4 as well as the noncanonical inflammasome where Casp11 senses endotoxin directly in the cytosol (Hagar et al., 2013; Kayagaki et al., 2013 Shi et al., 2014). Canonical inflammasome components, pathogen sensor NOD-like receptor and pyrin domain-containing (NLRP)3, adaptor ASC and cysteine protease Casp1, as well as cytokines IL-1β and IL-18, are all dispensable for high dose LPS shock (Kayagaki et al., 2011; Man et al., 2017; Vanden Berghe et al., 2014). These shock conditions override impacts of passenger mutations that influence the response to lower doses of LPS (Vanden Berghe et al., 2015). TLR4-dependent signaling induces inflammatory cytokine production via adaptors MyD88 and TRIF that together drive activation of transcription factors NF-κB and IFN regulatory factor (IRF)3 (Beutler and Rietschel, 2003; Sakaguchi et al., 2003; Yamamoto et al., 2003). This signaling also contributes to E. coli sepsis in mice (Roger et al., 2009).
IFN-induced Casp11 cleaves Gsdmd to mediate pyroptosis in cultured macrophages and endothelial cells (Kayagaki et al., 2015). Casp11 has also been implicated in LPS-dependent activation of pro-apoptotic Casp3 and Casp7 in vivo (Kang et al., 2002), activation long known to depend on TNF acting via TNFR1 in mice (Haimovitz-Friedman et al., 1997; Williams et al., 2013). Apoptotic markers correlate with severity of bacterial sepsis in mice and humans (Hotchkiss et al., 2000a; Hotchkiss et al., 2000b). Despite these observations, the interplay between cytokines during endotoxic shock remains unresolved. It is known that TNF and type I IFN signaling combine to mediate parallel apoptosis and necroptosis that drives perinatal death of receptor interacting protein (RIP) kinase (RIPK)1-deficient mice (Kaiser et al., 2014). We set out to dissect the relationship of TNF and type I IFN with cell death pathways during the execution of endotoxemia.
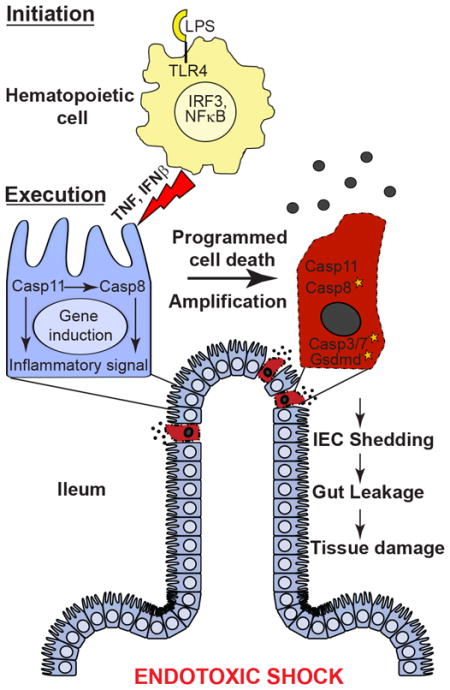
Casp8 deficiency results in embryonic lethality due to unleashed death mediated by pro-necroptotic kinase, RIPK3, that is reversed in Casp8−/−Ripk3−/− mice (Kaiser et al., 2011; Oberst et al., 2011). Comparisons of wild type (WT) mice to Ripk3−/−, Casp8−/−Ripk3−/−, littermate Casp8+/−Ripk3−/− and RIPK3 kinase-inactive derivatives (Mandal et al., 2014; Newton et al., 2014; Newton et al., 2016) provide insights into the contributions of Casp8-dependent (extrinsic) apoptosis and RIPK3-dependent necroptosis during acute hepatic injury (Kaiser et al., 2011), kidney insult (Linkermann, 2016), colitis (Moriwaki et al., 2014), systemic inflammation (Newton et al., 2016) and death-independent inflammatory signaling (Chan et al., 2015; Gurung and Kanneganti, 2015; Lawlor et al., 2015). Here, we established a requirement for Casp8 in endotoxic shock as well as E. coli sepsis. RIPK3 function was dispensable for LPS shock but influences sepsis. The production of critical cytokines by hematopoietic initiator cells (Beutler and Rietschel, 2003) occurred independently of Casp8 or Casp11. TNF and type I IFN mediated crosstalk with target tissues, starting within the small intestine followed by spleen and thymus, during execution of shock. Casp8 collaborated with Casp11 to execute damage and inflammation within target tissues, driving leakage of gut bacteria, splenic congestion and associated acute temperature drop to execute endotoxemia in the intact animal.
RESULTS
Casp8 is required for endotoxic shock
In mice, TLR4-dependent and -independent models of LPS shock are monitored by body temperature loss (Kayagaki et al., 2011; Vanden Berghe et al., 2014). WT C57BL/6 mice exhibited a sharp drop in temperature between 9 and 15 h post challenge (hpc) with high dose (54 mg/kg) LPS (Fig. 1A). C57BL/6-backcrossed Casp8−/−Ripk3−/− mice (Kaiser et al., 2011) resisted (Fig. 1A) even though littermate control Casp8+/−Ripk3−/− and C57BL/6-backcrossed Ripk3−/− mice succumbed to endotoxemia. Independently derived Casp8−/−Ripk3−/− mice (Oberst et al., 2011) also resisted while littermate Casp8+/−Ripk3−/− controls succumbed (Fig. S1A). Given that Ripk1−/−Casp8−/−Ripk3−/− and Ripk1−/−Casp8−/−Ripk3+/− mice (Kaiser et al., 2014) both resisted LPS (Fig. S1B), Casp8 function appeared to be independent of RIPK3. Casp8-deficient mice on a kinase-inactive RIPK3 (Casp8−/−Ripk3K51A/K51A) background (Mandal et al., 2014) also resisted LPS shock (Fig. S1C), confirming the involvement of this apoptotic protease in endotoxemia. In the presence of Casp8 function, male Ripk3K51A/K51A mice succumbed and females resisted, a pattern that acknowledged gender as a factor (Marriott et al., 2006) separate from the contribution of Casp8 to inflammatory insult. With the exception of Casp11−/− mice, the mutant strains used here all carried the C57BL/6 Casp11 allele and showed comparable LPS-dependent Casp11 induction in bone marrow derived macrophages (BMDM) (Figs. S1D and S1E; data not shown). Trif−/−, Myd88−/−, Ifnar1−/− and Irf3−/− mice all resisted temperature drop and shock (Figs. 1B and S1F), consistent with the contribution of type I IFN to Casp11 induction (Rathinam et al., 2012). Ifngr−/− mice succumbed to high dose LPS, as did Asc−/−, Nlrp3−/− and Il1b−/− mice (Figs. S1G–S1I) (Kayagaki et al., 2011; Vanden Berghe et al., 2014), showing that neither canonical inflammasome nor type II IFN signaling contributed. These data reveal a crucial requirement for Casp8 in TLR4-dependent endotoxemia beyond the recognized role of the type I IFN-Casp11-Gsdmd axis (Kayagaki et al., 2015; Kayagaki et al., 2011).
Figure 1. Casp8 drives endotoxic shock.

(A–B) Kaplan-Meier survival plot (left) and body temperature plot (right) over time (hpc) in LPS-challenged WT (n=19), Ripk3−/− (n=15), Casp8+/−Ripk3−/− (n=8), Casp8−/−Ripk3−/− (n=26) and Casp11−/− (n=11) mice (A) or WT (n=6), Ifnar1−/− (n=8) and Irf3−/− (n=5) mice (B). (C) Kaplan-Meier survival plot for WT (n=6), littermate Casp8+/−Ripk3−/− (n=6) and Casp8−/−Ripk3−/− (n=7) as well as Casp11−/− mice (n=6) given a poly(I:C) prime (4 mg/kg) followed 6 h later with low dose LPS challenge (5 mg/kg). (D) Kaplan-Meier survival plot for WT (n=19), littermate Casp8+/−Ripk3−/− (n=20) and Casp8−/−Ripk3−/− (n=15) mice over time, h post infection (hpi) with E. coli (ATCC 25922). Groups here included balanced numbers of male and female animals. Statistical comparisons (to WT) was by Log-Rank (Mantel-Cox) test (* p<0.05; ** p<0.01). Please also see Figure S1.
We next investigated the role of Casp8 in TLR4-independent endotoxemia, where the Casp11 noncanonical inflammasome senses LPS directly (Kayagaki et al., 2013). WT and Casp8+/−Ripk3−/− control mice, primed with the TLR3 agonist poly(I:C) (4 mg/kg), succumbed to subsequent low dose (5 mg/kg) LPS challenge (Figs. 1C and S1J). Both groups of Casp8−/−Ripk3−/− mice (Kaiser et al., 2011; Oberst et al., 2011) resisted. These data demonstrate the contribution of Casp8 to endotoxemia in all settings where Casp11 has been implicated.
Casp8 contributes to E. coli. sepsis
We next assessed the role of Casp8 and RIPK3 during E. coli sepsis, an infection model requiring TLR4 and MyD88 (Roger et al., 2009). C57BL/6-backcrossed Casp8−/−Ripk3−/− mice all survived an E. coli (strain ATCC 25922) infection sufficient to kill 40 percent of WT mice (Fig. 1D). Littermate Casp8+/−Ripk3−/− mice exhibited an intermediate pattern of susceptibility. Thus, both Casp8 and RIPK3 contribute to E. coli sepsis. The exclusive dependence of endotoxemia on Casp8 and Casp11 prompted focus on high dose LPS for further mechanistic assessment in C57BL/6-backcrossed mice.
LPS shock is triggered by a distinct hematopoietic initiator compartment
Even though endotoxic shock is known to depend on inflammatory cytokines produced by hematopoietic cells (Beutler and Rietschel, 2003), the requirements for initiation and execution of tissue damage (Fig. 2A, left) are not yet clear. A transient cell transfer approach (Fig. 2A, right) was employed to investigate these signaling requirements. Due to a lack of type I IFN production (Sakaguchi et al., 2003), >75% of Irf3−/− mice resisted LPS shock as assessed by body temperature drop (Fig. 1B). Transfer of 20 million WT BM cells reconstituted IFNβ production in these mice (Fig. 2B). At 3.5 h post transfer, WT BM cells from donor CD45.1+ mice comprised 0.1 to 0.5% of total leukocytes (70 to 90% viable) from blood or lungs (Figs. 2C and 2D). Transfer of Irf3−/− BM cells failed to confer susceptibility (Fig. 2E), confirming the role of this factor downstream of TLR4 (Beutler and Rietschel, 2003) during initiation of shock. BM from Myd88−/− mice conferred partial susceptibility (Fig. 2F). This strategy identified a distinct initiator compartment where IRF3 induced type I IFN production to mediate cytokine-dependent crosstalk with target tissues. BM from Casp8−/−Ripk3−/−, Ifnar1−/−, Casp11−/− or Gsdmd−/− mice all conferred full susceptibility upon Irf3−/− recipients (Figs. 2E, 2G and 2H), showing that Casp8, RIPK3, IFNAR1, Casp11 and Gsdmd are dispensable for initiation.
Figure 2. Casp8 and Casp11 are dispensable for initiation of shock.

(A) Crosstalk model of myeloid cell-induced inflammatory signal transduction during shock, with design of in vivo complementation assay using BM cell (BMC) transfer shown to the right. (B) Quantification of serum IFNβ in PBS- or LPS-challenged (2 hpc) WT mice (n=3), Irf3−/− mice (n=3) and Irf3−/− recipients (n=2) of WT BM transfer. (C) Percentage of WT CD45.1+ donor and Irf3−/− CD45.2+ recipient leukocytes in the lung (left) and in peripheral blood mononuclear cells (PBMC, right) from recipient mice. (D) Viability of CD45.1+ donor cells (annexin V negative or a combination of annexin V and 7AAD negative) in lungs (left) and PBMC (right) at 3.5 h post transfer. (E–J) Kaplan-Meier survival plots of LPS-challenged Irf3−/− (n=4), WT (n=3) and Casp8−/−Ripk3−/− (n=3) mice (left), and Irf3−/− recipients of BMC from WT (n=7), Irf3−/− (n=4) or Casp8−/−Ripk3−/− (n=4) mice (right) (E), Irf3−/− recipients of BMC from Myd88−/− (n=4) mice (F), Casp11−/− (n=3) mice or Irf3−/−recipients of BMC from Casp11−/− (n=4) or Ifnar1−/− (n=6) mice (G), Irf3−/− mice (n=3) or recipients (n=3) of BMC from Gsdmd−/− mice (H). Statistical comparisons as in Fig. 1 (* p<0.05; ** p<0.01, *** p<.001). Please also see Figure S2.
To examine the contribution of Casp8 and Casp11 to myeloid cell-derived cytokine production, we applied conditioned medium from cultured, LPS-treated (1 μg/ml) BMDM or BM-derived dendritic cells (BMDC) to nonmyeloid SVEC4-10 cells, a TLR4-deficient endothelial cell line. These cells succumbed to death triggered by purified TNF or TNF plus IFNβ, medium from WT BMDM or BMDC, as well as medium from Casp8−/−Ripk3−/−, Casp11−/− or Ifnar1−/− BMDM; but not from Myd88−/− or Tnfa−/− BMDM, or from SVEC4-10 cells (Figs. S2A–S2F). Thus, myeloid cells produced sufficient cytokines to drive death independently of Casp8 or Casp11. SVEC4-10 cells treated with WT BMDM-conditioned medium exhibited cleavage of Casp8 and Casp3 as well as induction of Casp11 (Fig. S2G). In line with the expectation that Casp8 and Casp11 function after this initiation step (Fig. 2A), WT BM cells failed to transfer susceptibility to either Casp8−/−Ripk3−/− or Casp11−/− mice (Figs. S2H and S2I).
Casp8-dependent small intestinal and splenic damage underlie endotoxemia
To identify critical target organs affected by LPS, we assessed histopathologic evidence of tissue damage in WT, Ripk3−/− and Casp8−/−Ripk3−/− mice at 9 hpc when susceptible mice reach euthanasia criteria. Damage was evident only in spleen and small intestine, not in liver, heart, stomach, pancreas, thymus or brain (Figs. 3A–3D and S3A–S3D; data not shown). Small intestinal tissue sections from susceptible mice exhibited atrophy marked by sloughed and shed cells with condensed nuclei. Splenic sections showed extramedullary hematopoiesis and congestion resulting from vascular disruption. Challenged Casp8−/−Ripk3−/− mice resisted small intestinal damage and splenic congestion in a manner similar to PBS-treated WT mice. Despite these stark differences, intestinal fluid (edema) as well as leukocyte infiltration in intestine and lung appeared comparable in all genotypes of mice (Figs. 3C and S3D). Thus, small intestine and spleen emerge as two key execution compartments affected by Casp8-dependent damage.
Figure 3. Casp8 drives small intestinal and splenic damage.

(A–B) Representative (3 mice of each genotype in two independent experiments) images of hematoxylin and eosin (H&E) stained sections of small intestine (A) and spleen (B), from WT, Ripk3−/− and Casp8−/−Ripk3−/− mice at 9 hpc with PBS or LPS, as indicated (bar=100 μm). Arrows indicate damaged and sloughed cells with condensed nuclei. (C–D) Histological score of leukocyte infiltrate (inflammatory cells), condensed nuclei (apoptosis), atrophy (tissue damage) and edema (fluid accumulation) in small intestine (C), or extramedullary hematopoiesis (EMH) as well as congestion in spleen (D), in tissue sections from WT, Ripk3−/− and Casp8−/−Ripk3−/− mice at 9 hpc with PBS or LPS (n=3 to 5 for each group). N.D., not detected. Mean and range in scores are shown. Please also see Figure S3.
Casp8 mediates appearance of early cell death markers in the small intestine
When intestines from WT and Ripk3−/− mice were assessed early, within 1 to 1.5 hpc with LPS, apoptotic cell death markers, including cleaved Casp8 (p43 and p18), cleaved Casp3 (p22, p19 and p17) and cleaved p89 form of caspase substrate poly (ADP-ribose) polymerase (PARP) were evident in duodenal mucosa (Figs. 4A–4C and S4A–S4C). At this time, these markers were detected by immunoblot of duodenal scrapings or by immunohistochemistry in sloughing villus tip intestinal epithelial cells. LPS-resistant Casp8−/−Ripk3−/− mice lacked cell death markers (Fig. 4D).
Figure 4. Casp8 and Casp11 collaborate for lower small intestinal injury.

(A–C) Immunoblot for cleaved (Cl)-Casp (C)8; 43 and 18 kDa), Cl-C3 (22, 19 and 17 kDa) or Cl-PARP (89 kDa) as well as total C11 (43, 38.5 and 25 kDa) and uncleaved C8 (55 kDa) or C3 (31 kDa), showing mucosa from duodenum (Du) in challenged WT and Casp8−/−Ripk3−/− mice (only uncleaved 43 and 38.5 kDa forms of C11 detected) (A), PBMC and Du from WT mice (B), mucosa from Du, as well as stomach (S), proximal jejunum (Jp), distal jejunum (Jd), ileum (Ile), cecum (Ce) and colon (Co) from challenged WT mice (only 19 and 17 kDa forms of Cl-C3 detected) (C). Graphical depiction of intestinal tract segments is above C. Molecular weight markers shown to the right. (D) Immunohistochemistry (IHC), showing Cl-C3 in situ with hematoxylin counterstain, or histology following H&E stain of the indicated intestinal segments from challenged WT and Casp8−/−Ripk3−/− mice (bar=100 μm) representative of 8 mice from 4 independent experiments. Insets show magnified images of the sections (bar=40 μm). Arrows indicate Cl-C3 positive cells in IHC sections and condensed nuclei in H&E stained sections. (E) Immunoblot of mucosa from Du, Jp, Jd and Ile from WT, Casp8−/−Ripk3−/− and Casp11−/− mice at 1.5 hpc with LPS. (F) IHC of Cl-C3 in ileum from two WT and two Casp11−/− mice at 1.5 hpc with PBS or LPS representative of 5 mice from 3 independent experiments. Arrows indicate Cl-C3 positive cells. (G) Immunoblot of ileal mucosa from WT and Irf3−/− mice at 1.5 hpc with PBS or LPS, with two LPS-challenged Irf3−/− mice shown. Please also see Figure S4.
Long-standing evidence implicates the second messenger ceramide C2 triggered by TNF signaling in intestinal injury (Haimovitz-Friedman et al., 1997). C2 was detected at 1 hpc in duodenal mucosa from both WT and Casp8−/−Ripk3−/− groups (Fig. S4D), although the mutant mice resisted insult-associated duodenal fluid accumulation at this time (Fig. S4E). Casp11 was detected in duodenal mucosa from Casp8−/−Ripk3−/− mice (Figs. 4A and 4E), indicating that the expression of this pyroptotic protease occurred independently of Casp8 or RIPK3. However, Casp11 expression was not sufficient to mediate tissue injury in the absence of Casp8. Taken together, the presence of C2 in duodenum of Casp8−/−Ripk3−/− mice showed that this ceremide was not sufficient to drive tissue injury and demonstrated ability of these mice to respond to endotoxin.
In WT mice, cleaved forms of Casp8, Casp3, Casp7 and PARP appeared throughout the small intestine, in duodenum, jejunum and ileum by 1.5 hpc, but were absent in stomach, cecum, colon and peripheral blood mononuclear cells (Figs. 4B–4D and S4F). Furthermore, spleen, thymus, lung, liver, kidney, heart and lymph node all lacked these markers at early times. Tissue injury was absent from the small intestine of Casp8−/−Ripk3−/− mice; whereas, damage was comparable in WT and Ripk3−/− mice (Figs. 4D, S4F and S4G). Thus, Casp8 supports the appearance of cell death markers in the small intestine.
Casp8 and Casp11 collaborate for injury to lower small intestine
IRF3-dependent induction of Casp11 was necessary for the appearance of cell death markers, including activation of Casp8 as well as Casp3, in the lower small intestine (distal jejunum and ileum; Figs. 4E–4G and S4H; data not shown). In upper small intestine, this activation proceeded independently of Casp11 (Figs. 4E and S4I). Casp11 was detected in some control mice, possibly representing tonic IRF3-dependent expression at this bacteria-rich site (Mowat and Agace, 2014). Given that Casp8-dependent cell death proceeded comparably in WT and Casp11−/− BMDM or fibroblasts (Fig. S4J), cultured cells did not reflect the collaboration observed in vivo. Thus, in ileum and adjacent lower segments of the small intestine, Casp11 enhanced extrinsic cell death markers. The ileum emerged as a critical site where Casp8 and Casp11 function during endotoxemia.
Casp8−/−Ripk3−/− mice support expression of apoptotic and pyroptotic machinery in ileum
To better understand the correlates of injury, we assessed LPS-dependent signaling events in susceptible WT and resistant Casp8−/−Ripk3−/− ileum through 180 min post challenge (mpc) (Fig. 5A). Apoptotic markers such as cleaved Casp8 (Figs. 4A and S4A) became elevated between 60 and 180 mpc in susceptible mice, times when there was comparable phospho-IκBα induction in all mice (Fig. 5A). Thus, NF-κB induction in ileum occurred independently of Casp8 or RIPK3. In contrast, LPS-dependent phosphorylation of mitogen-activated protein kinases (phospho-SAPK and phospho-JNK) were reduced in Casp8−/−Ripk3−/− ileum. Critical apoptotic signaling components, cFLIP (long and short forms), RIPK1 and Casp3 were all detected even though cFLIPL and RIPK1 were reduced in Casp8−/−Ripk3−/− tissues by 180 mpc. These data demonstrate the overall ability of critical tissue to respond and set-up apoptotic machinery following LPS challenge independent of Casp8, without progressing to tissue injury.
Figure 5. Casp8 regulates inflammatory signaling in ileum.

(A) Immunoblot of ileal mucosa from unmanipulated (time=0) or from WT and Casp8−/−Ripk3−/− mice at the indicated mpc with LPS, showing expression of phosphorylated forms (p) of IκBα (40 kDa), SAPK and JNK (54 and 46 kDa, respectively), as well as Cl-C8, C8, cFLIPL (50 kDa) and cFLIPs (25 kDa), RIPK1 (75 kDa), IgH (50 kDa), C3, C11, C1 (43 kDa) and Gsdmd (55 kDa, with an * indicating the ~30 kDa cleaved form). (B) Principal component (PC) analysis (PCA) of ileal mucosa from WT, Casp8−/−Ripk3−/− and Casp11−/− mice (n=5 for each group) including 27,578 genes with detectable base mean expression where the contribution of each PC is indicated as a percentage. (C) Heat map comparing expression patterns of genes in ileal mucosa from WT, Casp8−/−Ripk3−/− and Casp11−/− mice (n=5 for each group) at 1.5 hpc, where mean log2 expression of each gene was compared to normalized expression of all genes. Blue, white and red indicate downregulated, unchanged and upregulated expression of genes, respectively. Please also see Figure S5.
Casp11 and Gsdmd, as well as Casp1, were detected in WT and Casp8−/−Ripk3−/− tissues (Fig. 5A); however, Casp11 and Gsdmd were not sufficient to execute shock in the absence of Casp8. Gsdmd cleavage (p30) was evident in WT ileum by 10 mpc and was modestly reduced in Casp8−/−Ripk3−/− tissues. Therefore, it appears that (i) both Gsdmd processing and Casp8 activation were associated with tissue injury, (ii) Gsdmd cleavage preceded Casp8 autoprocessing, (iii) Casp8 was necessary for Casp3 and Casp7 cleavage, and, (iv) Casp8−/−Ripk3−/− tissues expressed pyroptotic signaling components without sustaining injury. These data highlight the need for both pro-pyroptotic and pro-apoptotic machinery to execute inflammatory tissue damage in situ.
Casp11-dependent macrophage pyroptosis proceeds independently of Casp8
We next evaluated the contribution of Casp8 to Casp11-dependent pyroptosis in cultured macrophages, a recognized correlate of endotoxemia (Kayagaki et al., 2013). BMDM and splenic macrophages from WT, Ripk3−/−, Casp8−/−Ripk3−/− and Casp11−/− mice were primed with poly(I:C) and subsequently transfected with LPS (Figs. S5A and S5B). Cell leakage and death showed the expected dependence on Casp11 but not on Casp8 or RIPK3. Compared to WT, Casp8−/−Ripk3−/− and Casp11−/− BMDM produced less IL-1β (Fig. S5C), as expected (Gurung and Kanneganti, 2015; Kayagaki et al., 2013; Philip et al., 2016). Thus, pyroptosis in primary macrophages does not require Casp8.
Casp8 and Casp11 dictate ileal inflammation
To further explore the signaling requirements for tissue injury, we compared gene expression patterns at a time (1.5 hpc) when cell death markers were evident in WT but absent in Casp8−/−Ripk3−/− and Casp11−/− mucosal scrapings from ileum. Unfiltered and unsupervised principle component analysis (PCA) with 27,578 common transcripts revealed tightly clustered transcriptional profiles defining each group (Fig. 5B). Casp8−/−Ripk3−/− and Casp11−/− tissues exhibited non-overlapping gene expression patterns suggesting distinct contributions by Casp8 and Casp11 to tissue injury. When transcripts associated with cell death (including apoptosis and pyroptosis), inflammation and intestinal damage were compared to pooled reference (Fig. 5C), Casp11−/− tissues exhibited reduced expression of Tnfrsf1 (TNFR1), Casp9, Casp3, Casp2, Tnfsf12 (Tweak), Tnfaip1, Lta and Fasl. These observations implicated Casp11 in transcriptional activation of extrinsic cell death genes beyond any contribution to pyroptosis (Figs. 4 and S4). Ileal mucosa from Casp11−/− mice also showed reduced Il12rb and Il17rc as well as elevated Il10, Il22, Vegfa, Vegfc, and Egfr in a recognized anti-inflammatory pattern attributed to tissue repair (Ouyang et al., 2011). When compared to WT tissues, Casp11−/− ileal mucosa exhibited significantly fewer Casp8 transcripts (Fig. S5D). Collectively, these data implicate Casp11 as a promoter of extrinsic cell death machinery as well as inflammation in situ.
The key mediators of apoptotic and pyroptotic signal transduction, including Tnfrsf1 (TNFR1), Fadd, Cflar (cFLIP), Casp3, Casp7 and Gsdmd were all transcribed in WT and Casp8−/−Ripk3−/− tissues, consistent with immunoblot analyses (Figs. 4E and 5A). However, WT and Casp8−/−Ripk3−/− tissues showed distinct inflammatory gene profiles, particularly related to TNF and IFN signaling. Notable amplifiers of TNF signaling, a disintegrin and metalloproteinase (Adam) gene family (Jones et al., 2016), Tnfaip2 and Tnfaip6 were downregulated in Casp8−/−Ripk3−/− tissues. Type I IFN receptor subunits (Ifnar1 and Ifnar2), type II IFN receptor (Ifngr1), TLRs (Tlr1, Tlr4, Tlr5 and Tlr8), adaptors (Trif, denoted as Ticam1, and Ticam2), Ripk1, Gsdm family genes (Gsdma, Gsdmc2, Gsdmc4 and Gsdmc12) as well as inflammatory transcripts (Il1rl1, Il4ra, Il6, Il6st, Il12rb1, Il17ra, Il17rb, Il34, Il1ra1, Ccl2, Ccl11, Ccl24 and CxCl1) were all downregulated in Casp8−/−Ripk3−/− tissues. Even though Casp8 was dispensable for initiation (Fig. 2), this protease contributed to the inflammatory gene signature associated with tissue injury analogous to studies of LPS-treated BMDM (Philip et al., 2016). Fasl, inflammatory Il18 as well as type I IFN-associated transcripts (Isg15, Isg20, Irf7 and Casp11, denoted as Casp4) were all upregulated in Casp8−/−Ripk3−/− tissues. Despite distinct gene profiles, Casp8−/−Ripk3−/− and Casp11−/− ileum exhibited higher Cox gene transcripts than WT tissues. Thus, each pro-death protease exerts a distinct and dramatic impact on cell death and inflammation in the intestinal mucosa.
Casp8 and Casp11 contribute to LPS-driven systemic inflammation
We compared serum cytokine patterns in LPS-resistant Casp8−/−Ripk3−/− and Casp11−/− mice to susceptible WT and Ripk3−/− mice in order to determine whether tissue injury correlated with systemic inflammation. At 1 to 2 hpc, Ripk3−/−, Casp8−/−Ripk3−/− and Casp11−/− mice all exhibited lower TNF, IFNβ, IFNα4 and other inflammatory mediators (Figs. S6A–S6D; data not shown). These data acknowledge known contributions of Casp8, Casp11 and RIPK3 to LPS-dependent cytokine production (Chan et al., 2015; Gurung and Kanneganti, 2015; Kayagaki et al., 2013; Saleh and Degterev, 2017) even though BM cells from these mutant groups were nevertheless sufficient to initiate shock in Irf3−/− recipients (Fig. 2). In line with this ability, TNF and IFNβ were readily detectable in WT, Ripk3−/− and Casp8−/−Ripk3−/− mice between 3 and 6 hpc (Figs. S6A–S6D; data not shown) as body temperature dropped (Fig. 1). Casp11−/− mice failed to produce TNF and other inflammatory mediators (Mip1α, Mip1β, Mip2, SDF1, BLC, C5/C5A) observed in WT or Casp8−/−Ripk3−/− mice by 6 hpc (Figs. S6D and S6E), revealing a contribution of Casp11 to systemic inflammation that has not been previously appreciated.
TNF and type I IFN signaling drives Casp8 activation, ileal injury and endotoxemia
WT mice treated with TNF neutralizing antibody, resisted the appearance of apoptotic markers (cleaved forms of Casp8 and Casp3) across the small intestine (Figs. 6A, 6B, S6F and S6G). Casp11 induction was independent of TNF signal transduction in the ileum (Figs. S6G). Ifnar1−/− tissues exhibited Casp11 with reduced Casp8 activation (Fig. S6H), suggesting IRF3 controlled Casp11 induction (Fig. 4G) while IFNAR1-dependent signaling contributed to additional steps. Therefore, TNF, type I IFN, Casp8 and Casp11 all contributed in a non-redundant fashion to ileal injury associated with endotoxemia (Karaghiosoff et al., 2003; Kayagaki et al., 2011; Sheehan et al., 1989).
Figure 6. TNF signaling drives small intestinal injury and endotoxemia.

(A) IHC showing Cl-C3 in situ with hematoxylin counterstain in small intestinal segments of WT mice at 1.5 hpc with LPS (bar=100 μm) treated with either isotype control or anti-TNF antibody for 24 h prior to challenge. Arrows indicate Cl-C3 positive cells. (B) DEVDase (Cl-C3 and Cl-C7) activity assessed as fold increase over PBS-challenged control animals at 1.5 hpc (n=3 for each) in mucosa from intestinal segments following LPS challenge. Mean and range shown. (C–E) Kaplan-Meier survival plots for LPS-challenged WT mice treated as described in A (n=7 for each group) (C), and for Casp8DA/DARipk3+/− males (n=5, left) and females (n=6, right), littermate Casp8+/DARipk3+/− males (n=3, left) and females (n=3, right), as well as female Casp8DA/DARipk3−/− (n=4, right) mice (E). Statistical comparisons were as described in Fig. 1. Please also see Figure S6.
TNF-mediated Casp8 autoprocessing contributes to shock
Cells from Casp8 autoprocessing mutant (Casp8DA/DA) mice (Philip et al., 2016) resist extrinsic apoptosis, but retain scaffold function supporting cytokine production (Kang et al., 2008; Philip et al., 2016). Like Casp8+/−Ripk3−/− mice (Fig. 1), Casp8+/DARipk3+/− and male Casp8DA/DARipk3+/− mice succumbed (Fig. 6D; left); whereas, female Casp8DA/DARipk3+/− and Casp8DA/DARipk3−/− mice resisted LPS shock (Fig. 6D; right). This pattern revealed the contribution of Casp8 autoprocessing as well as scaffolding function to endotoxemia, along with an additional influence of gender. To further evaluate TNF-dependent signaling, we tested RIPK1 kinase-inactive mice known to resist TLR4-dependent cytokine production (Saleh and Degterev, 2017) as well as TNF-dependent inflammation (Berger et al., 2014; Filliol et al., 2017). Both Ripk1K45A/K45A mice (Kaiser et al., 2014) and WT mice treated with RIPK1 kinase inhibitor (Berger et al., 2015) remained susceptible to LPS-mediated tissue injury and shock (Fig. S6I and data not shown). Taken together, these data reveal the contribution of the TNF-Casp8 signaling axis independent of RIPK1 activity for LPS shock.
Casp8 and Casp11 dictate small intestinal and splenic damage underlying shock
In WT mice, Casp8 and Casp11 together contributed to LPS-dependent ileal damage (blunted villus epithelium, debris and sloughed epithelial cells with condensed nuclei and cleaved Casp3) at 6 hpc (Fig. 7A), times when body temperature dropped (Fig. 1). IIeal sections from WT mice or Irf3−/− recipients of WT BM all exhibited damage between 6 and 9 hpc at necropsy (Fig. 7B). In contrast, resistant Casp8−/−Ripk3−/−, Casp11−/− and Irf3−/− mice all lacked evidence of damage at these times. Paralleling susceptibility, LPS-challenged WT and Ripk3−/− mice exhibited increased orally administered FITC-conjugated Dextran-4 in serum; whereas, Casp8−/−Ripk3−/−, Casp11−/− and Ifnar1−/− mice did not exhibit any increase (Fig. 7C). Casp8−/−Ripk3−/− and Casp11−/− mice also lacked appearance of bacterial DNA that was readily detectable in mesenteric lymph node and bloodstream of susceptible WT mice (Fig. 7D). These observations reveal that the collaboration of Casp8 and Casp11 extends to intestinal barrier compromise.
Figure 7. Casp8 and Casp11 mediate gut leakage.

(A) IHC and H&E of ileum from WT, Casp8−/−Ripk3−/− and Casp11−/− mice at 6 hpc with LPS (bar=100 μm). Arrows indicate Cl-C3 positive cells. (B) IHC of ileum from Irf3−/− mice or Irf3−/− recipients of BMC from WT (n=3 each) mice at 6 to 9 hpc with LPS. (C) Detection of FD-4 in sera from WT (n=10), Ripk3−/− (n=10), Casp8−/−Ripk3−/− (n=12), Ifnar1−/− (n=6) and Casp11−/− (n=6) mice at 6 hpc. (Statistical comparisons used Wilcoxan matched pairs analysis (* p<0.05). (D) Fold increase in total eubacterial DNA, measured by quantitative PCR, in mesenteric lymph node (MLN) and blood from WT (n=3), Casp8−/−Ripk3−/− (n=3) or Casp11−/− (n=2) mice at 6 hpc with LPS compared to PBS control. (E) Model for partitioned initiation and execution steps during LPS shock. Please also see Figure S7.
Apoptotic markers (cleaved forms of Casp8, Casp3 and PARP) and Casp11 were detected in spleens of WT mice by 3 hpc (Figs. S7A–S7C). When spleens were fractionated (Schneider et al., 2008), death markers associated with the splenic stromal cell fraction rather than the hematopoietic cell fraction, consistent with reported Casp11-dependent damage to lung stromal endothelial cells (Cheng et al., 2017). In Casp8−/−Ripk3−/− and Casp11−/− mice, cleaved Casp8 was absent from stromal fractions (Fig. S7B) and cleaved Casp3 was absent from stromal cell-rich white pulp (Fig. S7C). At this time, Casp8−/−Ripk3−/− and Casp11−/− mice also lacked Casp3 activation in thymus (Fig. S7C). These observations were consistent with the reported contribution of Casp11 to LPS-triggered splenic injury and thymocyte apoptosis (Kang et al., 2002). Therefore, Casp8- and Casp11-dependent pathways collaborated to mediate injury in two lymphoid tissues as well as lower small intestine of susceptible mice. Histological evidence of apoptosis (condensed nuclei) was detected at 9 hpc in lower jejunum of susceptible WT mice as well as Irf3−/− recipients of WT or Casp8−/−Ripk3−/− BM, but was absent in resistant settings (Fig. S7D). LPS-dependent leukocyte infiltrates appeared in all settings. Casp8- and Casp11-dependent insult to the gut occurred earlier and was more severe than splenic congestion, suggesting that gut damage was the primary determinant of progression to shock. In summary, pro-apoptotic Casp8 and pro-pyroptotic Casp11 collaborate to execute inflammatory tissue injury underlying endotoxemia (Fig. 7E).
DISCUSSION
Here, pro-apoptotic Casp8 is shown to execute endotoxemia together with the Casp11-Gsdmd-noncanonical inflammasome axis. Pyroptosis of cultured cells (Aglietti et al., 2016) does not capture the important contribution of Casp8 to tissue injury and temperature drop defining shock. Likewise, extrinsic apoptosis of cultured cells fails to recapitulate the importance of Casp11. This in situ collaboration between Casp8 and Casp11 depends on parallel TNF and type I IFN signal transduction as well as Casp8 autoprocessing, acting independently of RIPK1 kinase activity and RIPK3 function. Both Casp8 and Casp11 are dispensable in the hematopoietic compartment that produces threshold quantities of cytokines necessary to initiate shock. Our observations showing Casp11 is dispensable in this compartment confirm earlier work (Cheng et al., 2017). Consistent with its role in LPS shock, Casp8 also contributes to E. coli sepsis (Roger et al., 2009). Our observations are in accord with Casp11 acting as a sensor of cytosolic LPS in shock (Kayagaki et al., 2015). Extrinsic apoptosis emerges as a contributor to inflammatory tissue injury as previously reported for experimental death receptor-mediated acute hepatitis and shock (Kaiser et al., 2011; Kang et al., 2008). Our observations are consistent with the role of extrinsic apoptosis during bacterial settings (Peterson et al., 2017), although they contrast the long-held view that apoptotic signaling is strictly anti-inflammatory (Wallach et al., 2016).
Casp8 drives extrinsic cell death and licenses associated inflammation in target tissues following LPS challenge. The presence of Casp8 also ensures optimal Gsdmd cleavage without influencing homeostatic production of pro-apoptotic and pro-pyroptotic machinery. Casp11 promotes transcriptional activation of Casp8, licenses inflammatory gene expression and enhances extrinsic cell death machinery. This interplay of caspases results in rapid appearance of TNF- and type I IFN-dependent apoptotic markers in the ileum in response to endotoxin. This vulnerability represents a liability unique to mammals. Therapeutic intervention with pan-caspase inhibitors prevents the elaboration of apoptotic markers and improves survival during sepsis (Hotchkiss et al., 2000a) as well as during non-alcoholic steatohepatitis (Barreyro et al., 2015), consistent with the contribution of multiple caspases in these settings. Therefore, we bring to light the complexity of inflammatory cytokine-dependent crosstalk during acute inflammation in mammals. The presence of these inflammatory checkpoints makes it necessary to consider the impact of combined cell death machinery in settings of clinically relevant organ damage and disease.
Our transient cell transfer experiments reinforce (Beutler and Rietschel, 2003) the importance of the hematopoietic initiator compartment for the production of TNF and IFNβ. Initiation proceeds independent of pro-apoptotic, pro-pyroptotic and pro-necroptotic machinery as well as type I IFN receptor. In target tissues, TNF mediates Casp8 autoprocessing while parallel IRF3-dependent signaling induces IFNβ as well as Casp11, which in association with IFNAR1, contributes to extrinsic cell death. The collaboration between Casp8 and Casp11 drives ileal damage that is crucial to shock progression. Our observations set the stage to identify cell types in which Casp8 and Casp11 function within ileum, as well as to explore the underpinnings of gender and microbiota influence on outcome. Casp8, Casp11 and RIPK3 all contribute to amplification of inflammation associated with tissue injury. The full impact of inflammatory cytokines as well as the autocrine and paracrine signal transduction pathways contributing to Casp11 induction, Casp8 activation and subsequent tissue damage requires future investigations employing tissue-specific mouse mutants.
Splenic apoptotic markers and Casp11 induction associate specifically with the stromal compartment, revealing the significance of combined apoptotic and pyroptotic signaling separate from the hematopoietic compartment in this secondary lymphoid organ. The additional influence of RIPK3 during E. coli infection reflects the complexity of infection compared to intoxication. Propagation of live bacteria requires host processes beyond those involved in LPS shock. Our observations implicate Casp8 and Casp11, but dismiss RIPK1 kinase activity and RIPK3 function during endotoxemia. The concepts emerging as a result of our studies enhance the understanding of inflammatory immune responses at mucosal surfaces and in lymphoid tissues.
The collaboration between pro-apoptotic Casp8 and pro-pyroptotic Casp11 in shock driven by E. coli endotoxin or live bacteria comes as a surprise based on the independence of these death pathways when evaluated in cultured cells. Casp8 should now be considered as a critical player along with TLR4, MyD88, TRIF, type I IFN, TNF, Casp11 and Gsdmd in shock (Beutler and Rietschel, 2003; Man et al., 2017). Insights from mammalian models of inflammation are informative and promise to translate to humans, albeit with additional consideration of Casp10 along with Casp8 as well as the presence of two pyroptotic caspases (Casp4 and Casp5). The essential collaboration of apoptotic and pyroptotic signal transduction during shock in mice suggests that therapeutic intervention with existent pan-caspase inhibitors may prove therapeutically beneficial against inflammatory diseases.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-cleaved Casp8 | Cell Signaling Technology | 8592 |
| Rabbit anti-Casp3 | Cell Signaling Technology | 8610 |
| Rabbit anti-cleaved Casp3 | Cell Signaling Technology | 9661 |
| Rabbit anti-cleaved PARP | Cell Signaling Technology | 9544 |
| Rabbit anti-phospho-IκBα | Cell Signaling Technology | 2859 |
| Rabbit anti-phospho-SAPK-phospho-JNK | Cell Signaling Technology | 9251 |
| Rabbit anti-cFLIP | Cell Signaling Technology | 8510 |
| Rat anti-Casp8 | Enzo | ALX-804-447 |
| Mouse anti-RIPK1 | BD Biosciences | 610458 |
| Rabbit anti-Gsdmd | Sigma | G7422 |
| Rat anti-Casp11 | Novus Bio | 17D9 |
| Rat anti-Casp1 | Novus Bio | NBP-45433 |
| Mouse anti-β-actin | Sigma | A2228 |
| Anti-CD45.1 | BD Biosciences | 582452 |
| Anti-CD45.2 | BD Biosciences | 560693 |
| Peroxidase labeled goat anti-rabbit IgG (H+L) | Vector Laboratories | PI-1000 |
| Peroxidase labeled horse anti-mouse IgG (H+L) | Vector Laboratories | PI-2000 |
| Peroxidase conjugated hamster anti-rat IgG (H+L) | Jackson Laboratories | 712-035-150 |
| Biotinylated goat anti-rabbit IgG (H+L) | Vector Laboratories | BA-1000 |
| Streptavidine, Horseradish Peroxidase for IHC | Vector Laboratories | SA-5004 |
| Armenian hamster anti-TNF alpha monoclonal antibody (TN3-19.12) | eBiosciences | 16-7423 |
| Armenian hamster IgG isotype control | eBiosciences | 16-4888-81 |
| Bacterial and Virus Strains | ||
| E. coli | ATCC | ATCC 25922 |
| Biological Samples | ||
| Sera | Mus musculus | Described in current manuscript |
| Peripheral blood mononuclear cells | Mus musculus | Described in current manuscript |
| Protein from tissues | Mus musculus | Described in current manuscript |
| Paraffin-embedded tissue sections | Mus musculus | Described in current manuscript |
| Chemicals, Peptides, and Recombinant Proteins | ||
| E. coli 0111:B4 LPS (for in vivo studies) | Sigma | L4391 |
| Low molecular weight poly(I:C) (for in vivo studies) | InvivoGen | poly(I:C) LMW |
| E. coli 0111:B4 (for cell culture studies) | InvivoGen | LPS-EB, Ultrapure |
| High molecular weight poly(I:C) (for cell culture studies) | InvivoGen | poly(I:C) HMW |
| FITC-Dextran 4 | Sigma | FD4 |
| Collagenase D | Sigma | 11088858001 |
| collagenase III | Worthington Biochemical Corporation | CLS-3 |
| FuGENE HD | Promega | E2311 |
| SMAC mimetic BV6 | Genentech | Not applicable |
| Cycloheximide | Sigma | C7698 |
| Murine IFN | PBL | 12410-1 |
| Murine TNF | PeproTech | AF-315-01A |
| Murine GSF | PeproTech | AF-315-03 |
| Murine IL-4 | PeproTech | AF-214-14 |
| Critical Commercial Assays | ||
| Cell Titer-Glo Luminescent Cell Viability Assay Kit | Promega | G7570 |
| Caspase-Glo 3/7 Activity Assay System | Promega | G8090 |
| Mouse IL-1beta/IL-F2 Duo Set ELISA | R&D | DY401-05 |
| Mouse TNF-alpha Duo Set ELISA | R&D | DY410-05 |
| Mouser interferon beta ELISA kit | Verikine | 42400-1 |
| Cytokine Array Panel A | R&D | ARY006 |
| DNAeasy Blood and Tissue Kit | Qiagen | 69504 |
| Deposited Data | ||
| Transcriptome (RNASq/RefSeq) analysis | GEO metadata | GSE115094 |
| Experimental Models: Cell Lines | ||
| SVEC 4-10 | ATCC | ATCC CRL-2181 |
| Murine Embryonic Fibroblast (MEF) | Prepared in | Described in |
| Mocarski laboratory | current manuscript | |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Jackson Laboratories | JAX 000664 |
| CD45.1 congenic C57BL/6J | Jackson Laboratories | JAX 002014 |
| C57BL/6-backcrossed mice | Emory University Animal Housing | Described in current manuscript |
| 129 X C57BL/6 mixed background mice | Emory University Animal Housing | Described in current manuscript |
| Oligonucleotides | ||
| 5′-AGGCATATCTATAATCCCTTCACTG-3′ 5′-GGAATATATCAAAGAGATGACAAGAGC-3′ |
(Vanden Berghe et al., 2015) | Casp11-specific primers |
| 5′ TGGCTCAGGACGAACGCTGGCGGC 3′ 5′ CCTACTGCTGCCTCCCGTAGGAGT 3′ |
(Gomez-Hurtado et al., 2011) | Bacterial DNA- specific primers |
| Recombinant DNA | ||
| None | ||
| Software and Algorithms | ||
| DESeq software | Bioconductor Open Source Software | Not applicable (N/A) |
| Partek Genomic Suite software | Partek | N/A |
| Graphpad Prism 5 software | Graphpad Prism | N/A |
| Adobe Illustrator CC 2015 | Adobe | N/A |
| EIS Elements BR 3.10 | Nikon Instruments | N/A |
| Microsoft Excel | Microsoft | N/A |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding authors Edward Mocarski (mocarski@emory.edu) and Pratyusha Mandal (pmanda2@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J (JAX 000664) and CD45.1 congenic C57BL/6J (JAX 002014), C57BL/6-backcrossed (97% based on sequence polymorphisms) Casp8−/−Ripk3−/− and littermate Casp8+/−Ripk3−/− (Kaiser et al., 2011) as well as Casp8−/−Ripk3K51A/K51A (Mandal et al., 2014) have been described. Casp8−/−Ripk3−/− and littermate Casp8+/−Ripk3−/− were independently derived (Oberst et al., 2011) on a 129 x C57BL/6 mixed background. Casp11−/− (Kayagaki et al., 2011), Ticam1lps-2/lps-2 (Trif−/−), Ripk1K45A/K45A (Kaiser et al., 2014) and Ripk3K51A/K51A (Mandal et al., 2014) were all C57BL/6-derived strains. Ripk1−/−Casp8−/−Ripk3−/− and littermate Ripk1−/−Casp8−/−Ripk3+/− (Kaiser et al., 2014) were 129 x C57BL/6 mixed background. Irf3−/− (Sato et al., 2000), Ifnar1−/− (Kolumam et al., 2005; Muller et al., 1994), Ifngr−/− (Huang et al., 1993) backcrossed at Emory (O’Flaherty et al., 2015), Pycard−/− (Asc−/−) (Kayagaki et al., 2011), Il1b−/− (Shornick et al., 1996), Nrlp3−/− (Mariathasan et al., 2006), Casp7−/− (Lamkanfi et al., 2009) and Myd88−/− were all C57BL/6-backcrossed. Irf3−/− mice were developed using C57BL/6 embryonic stem (ES) cells (Sato et al., 2000). Ticam1lps-2/lps-2 (Trif−/−) mice originated from Jackson Laboratories (JAX 005037), were developed on a C57BL/6 background, and demonstrated resistance to high dose LPS (Hoebe et al., 2003). Casp8DA/DARipk3+/−, Casp8DA/DARipk3−/−, Casp8+/DARipk3+/− and Casp8+/DARipk3−/− littermates were derived from a cross of Casp8DA/DARipk3+/+ (Philip et al., 2016) with C57BL/6-backcrossed Ripk3−/− mice (Newton et al., 2004). All mice were bred at Emory University except mice used for Figs. 6C and S6I, which were bred at GlaxoSmithKline. Mice were 6 to 12 weeks of age and gender-matched in each experiment. All animal experiments were conducted with approval according to the guidelines of the Emory University IACUC and GSK Animal Care and Welfare Review Committees.
Cell culture and reagents
SVEC4-10 cells (ATCC CRL-2181) were maintained at 37C in a humidified 5% CO2 incubator using complete medium (DMEM containing 4.5 g/ml glucose, 10% fetal bovine serum [F2442, Sigma], 2 mM L-glutamine [MT 25005CI, Fisher] with 100 units/ml penicillin and 100 units/ml streptomycin [MT 3002CI, Fisher]) and used within eight passages. To generate BMDM, pooled BM cells from complete medium-flushed tibias and femurs were cultured for 5 to 7 days in macrophage medium (complete medium containing 20% serum and 20% filtered L929-conditioned medium to provide macrophage colony-stimulating factor). To generate BMDC, BM cells were cultured for 12 days in complete medium supplemented with murine granulocyte-macrophage colony stimulating factor (GM-CSF, AF315-03, PeproTech; 20 μg/ml) and murine IL-4 (AF-214-14, PeproTech; 5 ng/ml), with medium replacement every 3 days until harvest on day 12. Suspended cells were collected. For splenic macrophages, each freshly harvested spleen was Dounce homogenized (pestle A), passed through a 20 μm mesh filter to obtain single cell suspension and erythrocytes were lysed as described previously (Mandal et al., 2014). Splenic cells were centrifuged at 500 x g for 5 mins at 4C, suspended in macrophage medium and maintained for 5 to 7 days as described for BMDM. Murine embryonic fibroblasts (MEF) were collected as described previously (Kaiser et al., 2014).
METHOD DETAILS
Casp11 DNA detection and protein expression
DNA was isolated from tail snips using DirectAmp Genomic DNA Amplification Kit (Denville Scientific). Casp11-specific primers, 5′-AGGCATATCTATAATCCCTTCACTG-3′ and 5′-GGAATATATCAAAGAGATGACAAGAGC-3′ employed a PCR protocol (Vanden Berghe et al., 2015) to distinguish C57BL/6 functional and 129 mutant Casp11 alleles. Casp11-specific antibody was used to detect protein in BMDM lysates. BMDM (106) were plated in each well of a 6-well plate and 24 h later cells were treated with E. coli 0111:B4 LPS for the time indicated. Cell lysate preparation and immunoblotting methods are described below. With the exception of Casp11−/−, all mice carried the functional C57BL/6 Casp11 allele, lacked the 5 bp deletion characteristic of 129 allele, and expressed a pattern of intact and cleaved products typical of the C57BL/6 strain.
Endotoxic shock
For TLR4-dependent endotoxic shock, mice were injected intraperitoneally with LPS from E. coli 0111:B4 LPS (L4391, Sigma) at 54 mg/kg (Kayagaki et al., 2011). For TLR4-independent endotoxic shock (Kayagaki et al., 2013), mice were primed by intraperitoneal injection of low molecular weight poly(I:C) (LMW, InvivoGen; 4 mg/kg) followed 6 h later by intraperitoneal challenge with LPS at 5 mg/kg. Injection volume for all experiments was 200 μl made with sterile PBS. Injected mice were monitored by rectal thermometer Type T Thermocouple, Ball Tip Probe, 0.065″, Shaft 0.75″ (Braintree Scientific) eight times a day for five days. Mice exhibiting drop in temperature of ≥8C were euthanized using isoflurane anesthetic. One Casp8+/DA Ripk3+/− littermate control (Fig. 6D) was euthanized at 72 hpc due to ocular pus accumulation, ruffled fur and lack of mobility, even though this mouse did not meet temperature drop criteria. For in vivo complementation assays, single cell suspensions from complete medium-flushed BM were prepared by passage through a 40 μm Becton Dickenson Cell Strainer. Within 30 min of harvest, depending on the number of BM cells recovered, cells were either collected by centrifugation at 500 x g for 5 min at 4C or directly suspended in 200 μl of complete medium at 108 cells/ml. Cells were injected via tail vein into age and gender-matched recipients 1.5 h prior to LPS challenge.
For TNF blockade, WT mice were injected intraperitoneally with 200 μl (100 μg) of anti-TNF (16-7423, eBiosciences) or isotype control antibody (16-4888, eBiosciences) and challenged with LPS 24 h later. To assess intestinal leakage, 11 mg of FITC-Dextran 4 (FD4, Sigma) dissolved in 500 μl of PBS was administered to mice by oral gavage using a 24G × 25 mm needle with a rounded 2 mm tip (Braintree Scientific) (Liang et al., 2014) after 1 hpc with LPS. Mice were euthanized for terminal bleed 5 h later. Serum was prepared from coagulated blood centrifuged at 2000 x g for 90 secs at room temperature. FD-4 absorbance was measured on three-fold PBS-diluted serum using a Nanodrop-4 spectrophotometer.
E. coli sepsis
For E. coli infection, mice were anesthetized with inhaled isoflurane (4% induction and 2% maintenance) and intratracheally inoculated with E. coli (strain ATCC 25922) using the method described (Yoseph et al., 2016). Briefly, 50 μl of bacteria diluted to 3 × 108 CFU/ml in 0.9% NaCl was injected via a 100 mm midline cervical incision using sterile technique. After injection, mice were held head-up for 15 to 30 secs to ensure flow into the lung. The surgical incision was sealed using tissue glue and animals were monitored for survival through 7 dpi. Surviving mice were euthanized by CO2 inhalation.
Flow cytometry
To detect the presence of transferred CD45.1+ cells, single-cell suspensions were prepared from lungs collected from freshly euthanized mice, minced into ~3 mm sections, digested with collagenase D (11088858001, Sigma; 1.5 mg/ml) in PBS, filtered through a metal sieve and subjected to erythrocyte lysis (Daley-Bauer et al., 2012). Viable cells were counted by hemocytometer using trypan blue dye exclusion. Either PBMC (106) or lung cells (2 × 106) were subjected to flow cytometry using the following protocol. Cell surface FcγRII and FcγRIII were blocked prior to incubating with anti-CD45.1 (582452, BD) and anti-CD45.2 (560693, BD) antibodies followed by annexin V and 7AAD (7-aminoactinomycin D) staining using the PE Annexin V Detection Kit (559763, BD) in FACS-staining buffer (Mandal et al., 2014). Data were acquired by flow cytometry (BD LSRII cytometer and FACSDiva Software; BD Biosciences). Cell debris was excluded from measurement using forward and side scatter gating. The single cell population was gated away from doublets and clumped cells using side scatter height versus side scatter width density plot followed by forward scatter height and forward scatter width density plot. Total cells in this single cell gate were analyzed to determine percentage of CD45.1+ and CD45.2+ cells. The viability of CD45.1+ cells was determined by annexin V staining alone or in combination with 7AAD staining. Data were analyzed with FlowJo (TreeStar).
In vitro complementation and cell death assays
BMDM (106), BMDC (106) or SVEC4-10 cells (3 × 105) were plated per well in 6-well tissue culture plates for 18 to 24 h. Cells were treated with control PBS or LPS (1 μg/ml, E. coli 0111:B4, LPS-EB Ultrapure, InvivoGen) for the indicated times when cell-free supernatants were prepared by centrifugation at 1000 × g for 3 min at 4C. To assess the ability of supernatants to induce cytotoxicity of SVEC4-10 cells, cell permeable SYTOX Green dye (S7020, Invitrogen) uptake was used in real time using an IncuCyte instrument (Essen BioScience) as described (Mandal et al., 2014). SVEC4-10 cells, used before passage eight and maintained at no higher than 60% confluency, were plated at 5 × 104 cells per well in 24-well tissue culture plates. Murine TNF (25 ng/ml, AF-315-01A, PeproTech) or IFNβ (1000 U/ml, 12410-1, PBL) were directly added to complete medium. Cycloheximide (CHX) was from Sigma (C7698).
Macrophage pyroptosis was assessed as described previously (Kayagaki et al., 2013). Briefly, BMDM (106) were plated in each well of 6-well plates or 104 BMDM (or splenic macrophages) were plated in each well of custom 96-well plates (3610, Corning). At 24 h post-plating cells were primed with 5 μg/ml poly(I:C) (HMW, InvivoGen) and then transfected with LPS from E. coli 0111:B4 (2 μg/ml, LPS-EB Ultrapure, InvivoGen) using 0.25% v/v FuGENE HD (E2311, Promega). Cell viability was measured by IncuCyte (for 6-well plates) or change in ATP (for 96-well plates) measured by Cell Titer-Glo Luminescent Cell Viability Assay Kit (G7570, Promega). To assess extrinsic apoptosis, MEF (5 × 103) or BMDM (104) were plated per well in a 96-well plate, and 24 h post-plating, were treated with TNF and CHX in absence or presence of SMAC mimetic BV6 (1 μM, Genentech). ATP was measured 18 h later.
Immunoblot and antibodies
In general, immunoblot analysis on lysates from cultured cells, primary macrophages or PBMCs, collected using a Histopaque-1119/Histopaque-1077 protocol (Sigma), was as described (Mandal et al., 2014). Stomach, cecum, colon and small intestine were excised from freshly euthanized mice and immediately placed in ice-cold PBS. Small intestine was divided into distinct duodenum, jejunum and ileum segments, with an approximate length ratio 1:3:2), from gastric pylorus to cecum. Jejunum was further divided into equal parts, proximal jejunum and distal jejunum. To prepare mucosa from these tissues for immunoblot analysis in Figs. S2, 4 and S4, the inner surface of each segment was exposed under a dissecting microscope, washed with cold PBS and scraped from the muscularis propria layer into fresh, cold PBS using the curved ends of tissue forceps (Haimovitz-Friedman et al., 1997). This mucosal material was Dounce homogenized (pestal A), collected by centrifugation at 1000 x g for 5 min at 4C, and the resulting pellet was weighed and lysed on ice by adding an equal weight-to-volume ratio of ice-cold Triton-X-100 lysis buffer (Mandal et al., 2014). To prepare mucosa for immunoblot analysis in Figs. 5, 6 and S6, a modified protocol was used to increase yield of proteins. Small intestinal segments were excised, inner surface exposed under a dissecting scope and quickly washed in ice cold PBS. Mucosal surface was then scraped from the muscularis propria directly into ice cold Triton-X-100 lysis buffer (1 ml lysis buffer/5 cm of tissue) using curved ends of tissue forceps. The mucosal material was Dounce homogenized (pestal A) and lysed on ice (Mandal et al., 2014). Following either method, lysates were centrifuged at 150,000 rpm at 4C for 20 mins using a Tomy TX-160 high speed refrigerated micro centrifuge and supernatants were subjected to SDS-polyacrylamide gel electrophoresis followed by transfer to PVDF membranes (Immobilon-P, EMD Millipore). β–actin was employed to assure comparable loading on immunoblots. In Fig. 5A, Casp8−/−Ripk3−/− tissues exhibit an excess of IgH due to accumulation of autoantibody (Kaiser et al., 2014).
Antibodies for immunoblot analysis: rabbit anti-cleaved Casp8 (8592), rabbit anti-Casp3 (8610), rabbit anti-cleaved Casp3 (9661), rabbit anti-cleaved PARP (9544), rabbit anti-phospho-IκBα (2859), rabbit anti-phospho-SAPK and phospho-JNK (9251) and rabbit anti-cFLIP (8510) were all from Cell Signaling Technology; rat anti-Casp8 (ALX-804-447, Enzo); mouse anti-RIPK1 (610458, BD Biosciences); rabbit anti-Gsdmd (G7422, Sigma); rat anti-Casp11 (17D9, Novus Bio); rat anti-Casp1 (NBP-45433, Novus Bio) and mouse anti-β-actin (A2228, Sigma).
Splenic stromal and hematopoietic cell separation
Excised spleens from euthanized mice were injected with 1 ml of murine collagenase III (100 u/ml, Worthington Biochemical Corporation) in Hank’s balanced salt solution (HBSS) (H9394, Sigma) supplemented with calcium chloride (140 mg/L) or magnesium sulphate (98 mg/L). Spleens were then incubated in 1 ml of concentrated collagenase III solution (400 u/ml) for 1 h at 37C as described (Schneider et al., 2008). Spleens were forced through a 70 μm Becton Dickenson Cell Strainer using the plunger from a 5 ml syringe, resulting in a filtrate constituting HC. Membranes were washed with cold HBSS and membrane-retained SC were suspended by vigorous pipetting and shaking. SC and HC were washed with cold HBSS and centrifuged at 500 x g for 5 min at 4C. Cells were counted, subjected to lysis with Triton-X-100 lysis buffer as described (Mandal et al., 2014).
Histology and immunohistochemistry
Excised intestine from euthanized mice was fixed in ice-cold 10% normal buffered formalin (5700TS, Fisher) at 4C. 24 to 48 h later, samples were cut longitudinally to reveal the inner surface and either Swiss-rolled or left unrolled (without influencing observations). Paraffin-embedded tissue sections were prepared and scored by the Emory Histology Core at Yerkes National Primate Research Center. For IHC detection, paraffin-embedded sections were prepared and stained as described (Liang et al., 2014) with rabbit anti-cleaved-Casp3 (1:100 dilution) or anti-cleaved-Casp8 (1:100 dilution) at 4C, followed by biotinylated goat anti-rabbit secondary antibody (BA-1000, Vector Laboratories), streptavidine-horseradish peroxidase (HRP, SA-5004, Vector Laboratories) and peroxidase reaction reagents from an ABC kit (PK-4002, Vector Laboratories). Slides were counterstained using hematoxylin (22110639, Fisher) counterstain for 2 to 5 min and washed under tap water and finally with ultrapure water. Images were collected on a Nikon Elements microscope using Imaging Software-EIS Elements BR 3.10 (Nikon Instruments). Original higher magnification images are available upon request.
Transcriptome analysis
For transcriptome (RNASeq or RefSeq) analysis, ileum from each WT, Casp8−/−Ripk3−/− and Casp11−/− mouse (5 male mice per group) harvested at 1.5 hpc was placed in RNAlater (Thermo-Fisher) at 4C. 24 h post-harvest, intestines were cleaned with cold RNAlater and mucus was scraped, homogenized and pelleted as described for immunoblot analysis. RNA isolation and further steps for transcriptome analysis were carried out as described (Sandler et al., 2014) by the NHP Genomics Core, Yerkes National Primate Research Center. Briefly, total RNA was isolated from mucus pellet for each sample using RNeasy Micro and RNA integrity was tested using Agilent Bioanalyzer capillary electrophoresis. cDNA was prepared using Illumina Stranded RNATruSeq kits, barcoded and sequenced on an Illumina HiSeq 3000. Five million 150-base pair mapping reads were obtained per sample, with >90% reads having Q-Scores of >30 (>99.9% estimated accuracy for 150 base reads).
Caspase activity
Effector caspase activity was determined by measuring the DEVDase proteolytic activity in 25 μg of mucosa lysates using a Caspase-Glo 3/7 Activity Assay System (G8090, Promega). Protein quantification employed the DC Protein Assay Kit (500-0116, Bio-Rad).
ELISA and cytokine array
Cytokines were quantified using ELISA kits for murine IL-1beta/IL-F2 (DY401-05, R&D), TNF (DY410-05, R&D) and interferon beta (42400-1, VeriKine). Five serum samples showing similar quantities of TNF as well as IL-1β across the samples were pooled for cytokine array (Cytokine Array Panel A, ARY006, R&D).
Bacterial DNA isolation from MLN and blood
Blood and MLN were collected from PBS- or LPS-challenged mice at 6 hpc. Blood was collected in 10% EDTA solution made in sterile PBS (final pH 8.0). MLN was cut into 0.1 mm pieces and Dounce homogenized (pestal A). DNA was isolated from blood and MLN homogenate using DNAeasy Blood and Tissue Kit (Qiagen) and subjected to quantitative real time PCR analysis (qRT-PCR) in an ABI Real Time PCR 7500 instrument using the following primers, 5′ TGGCTCAGGACGAACGCTGGCGGC 3′ and 5′ CCTACTGCTGCCTCCCGTAGGAGT 3′ (Gomez-Hurtado et al., 2011) corresponding to forward 20 to 43 and reverse 361 to 338, respectively, of E. coli 16S ribosomal RNA gene (Itoi et al., 2009).
Enteropooling assay
To quantify accumulation of fluid in small intestine (enteropooling), food was withdrawn for 7 h after which mice were challenged with LPS or PBS control and euthanized at 1.5 hpc. Both ends of the duodenum were tightly tied with pre-weighed string to prevent fluid escape and then a ratio of weight/length was calculated (Musch et al., 2002).
Mass spectrometry
Baseline standard and position of ceramide C2 peak at m/z 340 (Merrill et al., 2005) was established by adding C2 (C7980, Sigma) at 0.02 mg/ml concentration to lipid samples extracted from duodenal mucosa (Haimovitz-Friedman et al., 1997). Untreated parallel lipid samples from mucosa were then spiked with control lipid backbone N-acetyl-D-sphingosine (A7191, Sigma) to place readings within the instrument’s limit of detection without altering C2 quantity in samples. All samples were run on a TSQ Vantage Triple Quadrupole mass spectrometer (Thermo-Fisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
Biostatistics
For PCA, genes were normalized for library size and adjusted to log scale using DESeq software. All genes (41,128) were included for the unsupervised and unfiltered PCA that was plotted using Partek Genomic Suite software. Of the total genes, 27,578 exhibited base mean expression greater than zero.
For generation of a heat map, expression of 123 genes were baseline normalized to the mean log2 expression of all samples from all groups. Upregulation or downregulation of each gene was based on comparison to the overall base mean expression of all detected genes. Casp11 (annotated as Casp4) exon 5 was reduced in Casp11−/− tissues, as expected (Kayagaki et al., 2011) compared to WT or Casp8−/−Ripk3−/− tissues even though exon 4 was upregulated in Casp11−/− tissues, suggesting abnormal splicing in these animals. Even though overall expression of Casp11 transcript appeared unaffected and comparable in all three groups, in comparison to WT, Casp8−/−Ripk3−/− tissues exhibited upregulation of exon 5.
Quantification of bacterial DNA in MLN and blood
Fold increase in bacterial DNA expression in samples from LPS-challenged over PBS-challenged mice of the same genotype was determined using a standard ΔΔCT formula (ABI Biosystems). Briefly, CT value of bacterial DNA in each sample was first normalized to GAPDH DNA from the same sample to obtain ΔCT. ΔCT for each LPS-challenged sample was then normalized with average ΔCT of PBS-challenged mice for the same genotype to obtain ΔΔCT. Fold increase in bacterial DNA was expressed as 2−ΔΔCT.
Quantification of ceramide C2
N-acetyl-D-sphingosine readings at m/z 263 were subtracted from total signal (m/z 263 + m/z 340) to yield C2 quantification for each sample.
Graphing Software
Data were either analyzed on Microsoft excel or GraphPad Prism 5 software. All data were plotted and statistically analyzed on GraphPad. Figures were assembled on Adobe Illustrator CC 2015. Error bars represent standard error of the mean (SEM) or mean and range as specified in figures.
DATA AND SOFTWARE AVAILABLIBITY
Transcriptome analysis data
Transcriptome analysis raw data (accession number GSE115094) is submitted to the Gene Expression Omnibus archive.
Supplementary Material
Highlights.
Pro-apoptotic Casp8 is essential for lethal LPS shock and E. coli sepsis
Initiation of cytokine production proceeds independent of Casp8 and Casp11
TNF and type 1 IFN drive Casp8 and Casp11 collaboration in target tissues
Combined pro-apoptotic and pro-pyroptotic signaling executes LPS shock
Acknowledgments
We dedicate this manuscript to the memory of Stanley Falkow, who, as the father of pathogen-host interactions, enriched the scientific lives of everyone with whom he interacted. We thank Vishva Dixit and Kim Newton (Genentech, LLC) for helpful discussions and for providing Ripk3−/−, Casp11−/− and Asc−/− mice, as well as Alan Aderem (Institute for Systems Biology) for Irf3−/− mice and Michael Diamond (Washington University) for Ifnar1−/− mice. We thank Nobuhiko Kayagaki (Genentech, LLC) for bones from Gsdmd−/− mice. At Emory University, we thank Samuel Speck for Ifnar1−/− and Ifngr−/− mice and Bali Pulendran for Myd88−/− and Trif−/− mice. At the Yerkes National Primate Research Center, we thank Evan Dessasau and AnaPatricia Garcia for histopathology as well as Tershel Batchelor and Nirav Patel for processing RNA from tissues. Supported by NIH (PHS grants R01 GM112547 and R01 AI118853 to E.S.M., RO1 GM072808 to C.M.C., T32 GM095442 and F32 GM117895 to J.D.L., R01 AI065429 and R01 AI107960 to D.M.S. and R01 AI123126 to N.C.D.P.) and by GlaxoSmithKline (S.B.B., M.S., S.H., C.C., P.J.G. and J.B.).
Footnotes
AUTHOR CONTRIBUTIONS
P.M., Y.F., J.D.L., S.B.B., S.O., A.D.L., K.M-S, M.S., S.H., C.C., L.R., C.B.Y., G.K.T. and Z.L. performed experiments and data analysis. P.M. and E.S.M. assembled figure panels and wrote the manuscript. P.J.G., S.B.B., J.B., N.C.D., E.A.O., D.M.S., I.B., C.M.C. and E.S.M. managed the research and edited the text and figures during assembly of the manuscript.
DECLARATION OF INTERESTS
Scott B. Berger, Michelle Schaeffer, Sandra Hoffman, Carol Capriotti, John Bertin and Peter J. Gough are employees of GlaxoSmithKline.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, Carreras MC, Poderoso JJ, Gores GJ. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015;35:953–966. doi: 10.1111/liv.12570. [DOI] [PubMed] [Google Scholar]
- Berger SB, Harris P, Nagilla R, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Ouellette M, et al. Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 2015;1:15001. doi: 10.1038/cddiscovery.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but Is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–4135. doi: 10.1172/JCI94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity. 2012;37:122–133. doi: 10.1016/j.immuni.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol A, Piquet-Pellorce C, Raguenes-Nicol C, Dion S, Farooq M, Lucas-Clerc C, Vandenabeele P, Bertrand MJM, Le Seyec J, Samson M. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J Hepatol. 2017;66:1205–1213. doi: 10.1016/j.jhep.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Gomez-Hurtado I, Santacruz A, Peiro G, Zapater P, Gutierrez A, Perez-Mateo M, Sanz Y, Frances R. Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl4-induced fibrosis. PLoS One. 2011;6:e23037. doi: 10.1371/journal.pone.0023037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P, Kanneganti TD. Novel Roles for Caspase-8 in IL-1beta and Inflammasome Regulation. Am J Pathol. 2015;185:17–25. doi: 10.1016/j.ajpath.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, Garzotto M, McLoughlin M, Gallily R, Edwards CK, 3rd, Schuchman EH, Fuks Z, Kolesnick R. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186:1831–1841. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S, Roy S, Black C, Grimm E, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000a;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Schmieg RE, Jr, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Karl IE, Buchman TG. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. 2000b;28:3207–3217. doi: 10.1097/00003246-200009000-00016. [DOI] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Hurley JC, Nowak P, Ohrmalm L, Gogos C, Armaganidis A, Giamarellos-Bourboulis EJ. Endotoxemia as a diagnostic tool for patients with suspected bacteremia caused by gram-negative organisms: a meta-analysis of 4 decades of studies. J Clin Microbiol. 2015;53:1183–1191. doi: 10.1128/JCM.03531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoi S, Yuasa K, Washio S, Abe T, Ikuno E, Sugita H. Phenotypic variation in Lactococcus lactis subsp. lactis isolates derived from intestinal tracts of marine and freshwater fish. J Appl Microbiol. 2009;107:867–874. doi: 10.1111/j.1365-2672.2009.04266.x. [DOI] [PubMed] [Google Scholar]
- Jones JC, Rustagi S, Dempsey PJ. ADAM proteases and gastrointestinal function. Annu Rev Physiol. 2016;78:243–276. doi: 10.1146/annurev-physiol-021014-071720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111:7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002;9:1115–1125. doi: 10.1038/sj.cdd.4401087. [DOI] [PubMed] [Google Scholar]
- Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, Wallach D. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol. 2008;181:2522–2532. doi: 10.4049/jimmunol.181.4.2522. [DOI] [PubMed] [Google Scholar]
- Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4:471–477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Moreira LO, Makena P, Spierings DC, Boyd K, Murray PJ, Green DR, Kanneganti TD. Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival. Blood. 2009;113:2742–2745. doi: 10.1182/blood-2008-09-178038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Xie Y, Dominguez JA, Breed ER, Yoseph BP, Burd EM, Farris AB, Davidson NO, Coopersmith CM. Intestine-specific deletion of microsomal triglyceride transfer protein increases mortality in aged mice. PLoS One. 2014;9:e101828. doi: 10.1371/journal.pone.0101828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 2016;89:46–57. doi: 10.1016/j.kint.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti TD. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. 2017;7:45126. doi: 10.1038/srep45126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Marriott I, Bost KL, Huet-Hudson YM. Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: a possible mechanism for gender-based differences in endotoxic shock susceptibility. J Reprod Immunol. 2006;71:12–27. doi: 10.1016/j.jri.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Merrill AH, Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–224. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567–578. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–685. doi: 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, Shalowitz D, Mittal N, Efthimiou P, Alnadjim Z, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest. 2002;110:1739–1747. doi: 10.1172/JCI15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, Cristina de-Almagro M, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RAD, Cao TC, van Bruggen, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Diff. 2016;23:1565–1576. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa B signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flaherty BM, Matar CG, Wakeman BS, Garcia A, Wilke CA, Courtney CL, Moore BB, Speck SH. CD8+ T Cell response to gammaherpesvirus infection mediates inflammation and fibrosis in interferon gamma receptor-deficient mice. PLoS One. 2015;10:e0135719. doi: 10.1371/journal.pone.0135719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Peterson LW, Philip NH, DeLaney A, Wynosky-Dolfi MA, Asklof K, Gray F, Choa R, Bjanes E, Buza EL, Hu B, et al. RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J Exp Med. 2017;214:3171–3182. doi: 10.1084/jem.20170347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, et al. Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLoS Pathog. 2016;12:e1005910. doi: 10.1371/journal.ppat.1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, Burns K, Riederer BM, Akira S, Calandra T. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A. 2009;106:2348–2352. doi: 10.1073/pnas.0808146106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- Saleh D, Degterev A. Emerging roles for RIPK1 and RIPK3 in pathogen-induced cell death and host immunity. Curr Top Microbiol Immunol. 2017;403:37–75. doi: 10.1007/82_2015_449. [DOI] [PubMed] [Google Scholar]
- Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schneider K, Loewendorf A, De Trez C, Fulton J, Rhode A, Shumway H, Ha S, Patterson G, Pfeffer K, Nedospasov SA, et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe. 2008;3:67–76. doi: 10.1016/j.chom.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KC, Ruddle NH, Schreiber RD. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J Immunol. 1989;142:3884–3893. [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Ferguson TA, Chaplin DD. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, Vuylsteke M, Roelandt R, Van Wonterghem E, Vandenbroecke J, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282–291. doi: 10.1164/rccm.201308-1535OC. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Hulpiau P, Martens L, Vandenbroucke RE, Van Wonterghem E, Perry SW, Bruggeman I, Divert T, Choi SM, Vuylsteke M, et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity. 2015;43:200–209. doi: 10.1016/j.immuni.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, Sutton R, Hughes KR, Hall LJ, Caamano JH, et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis Model Mech. 2013;6:1388–1399. doi: 10.1242/dmm.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yoseph BP, Klingensmith NJ, Liang Z, Breed ER, Burd EM, Mittal R, Dominguez JA, Petrie B, Ford ML, Coopersmith CM. Mechanisms of intestinal barrier dysfunction in sepsis. Shock. 2016;46:52–59. doi: 10.1097/SHK.0000000000000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.