

Abstract
Purpose of Review
Preeclampsia (PE) is a disorder of pregnancy typically characterized by new-onset hypertension and proteinuria after gestational week 20. Although preeclampsia is one of the leading causes of maternal and perinatal morbidity and death worldwide, the mechanisms of the pathogenesis of the disorder remain unclear and treatment options are limited. Placental ischemic events and the release of placental factors appear to play a critical role in the pathophysiology. These factors contribute to a generalized systemic vascular endothelial dysfunction and result in increased systemic vascular resistance and hypertension.
Recent Findings
There is increasing evidence to suggest that endothelin-1 (ET-1) in the maternal vascular endothelium is a critical final common pathway, whereby placental ischemic factors cause cardiovascular and renal dysfunction in the mother. Multiple studies report increased levels of ET-1 in PE. A number of experimental models of PE are also associated with elevated tissue levels of prepro-ET-1 mRNA. Moreover, experimental models of PE (placental ischemia, sFlt-1 excess, TNF-α excess, and AT1-AA infusion) have proven to be responsive to ET type A receptor antagonism. Recent studies also suggest that abnormalities in ET type B receptor signaling may also play a role in PE.
Summary
Although numerous studies highlight the importance of the ET system in the pathogenesis of PE, further work is needed to determine whether ET receptor antagonists could provide an effective therapy for the management of this disease.
Keywords: Preeclampsia, Pregnancy, Hypertension, Endothelin, Endothelium, Placenta, Cardiovascular, Blood pressure, Vascular smooth muscle
Introduction
Preeclampsia (PE) is a pregnancy-specific disorder defined as hypertension > 140/90 mm Hg in pregnancy and proteinuria > 0.3 g/24 h after 20 weeks of gestation. Recently, the American College of Obstetricians and Gynecologists has broadened the definition of PE to blood pressure > 140/90 after 20 weeks of pregnancy and either proteinuria ≥ 300 mg/24 h or protein/creatinine ratio ≥ 0.3 or one of the 35 following: thrombocytopenia, elevated liver transaminases, pulmonary edema, new onset renal insufficiency, or cerebral or visual disturbances [1]. Despite being a leading contributor of maternal and perinatal morbidity and death worldwide, the mechanisms of the pathogenesis of PE remain unclear and treatment options are very limited [2–4]. Current therapy for the management of PE includes antihypertensives, such as methyldopa, labetalol, and nifedipine and magnesium sulfate for prevention of eclamptic seizures [5]; however, these treatments have limited efficacy, and the only “cure” for PE is the delivery of the placenta. Earlyonset PE (< 34 weeks) is more severe, and outcomes are especially serious if disease develops every preterm (<32 weeks). Each week pregnancy is prolonged markedly reduces fetal morbidity and mortality, but only at the expense of an increased risk of maternal morbidity or even death [6]. A therapeutic approach that will prolong pregnancy yet do no harm to the fetus represents a critically important step in identifying agents for treatment of PE.
The Pathogenesis of PE
While the exact causes of PE remain unknown, it is thought that in some cases of the disease, especially early-onset PE, abnormal placentation due to insufficient trophoblast invasion and failure of spiral artery remodeling leads to inadequate blood flow to the placenta and a continuing cycle of repeated ischemia-reperfusion injury [3, 7]. The resulting hypoxic environment within the placenta stimulates oxidative stress and the release of placental factors such as soluble fms-like tyrosine kinase 1 (sFlt-1), soluble endoglin, agonistic autoantibodies to the angiotensin type 1 receptor (AT1-AA), and inflammatory cytokines [8]. These factors, along with the presence of additional maternal risk factors for PE, such as age, obesity, and pre-existing hypertension, contribute to the vascular endothelial dysfunction, vasoconstriction, and hypertension in PE.
It is becoming increasingly evident that an important final common pathway whereby many soluble placental factors elicit cardiovascular and renal dysfunction during PE is by activation the endothelin (ET) system. In order to develop safe therapeutic interventions of this system, it is important to fully understand the role of the ET system in blood pressure control during normal pregnancy and in PE. In this review, we start by discussing biology of the ET system, its role during normal pregnancy, and end with highlighting the critical role of ET in the pathophysiology of PE by discussing the therapeutic potential and limitations for treatment of PE.
The ET System
ET-1 is the most potent vasoconstrictor substance known in the human cardiovascular system [9]. ET-1 is derived from prepro-ET-1 mRNA. The mRNA is then translated to prepro-ET-1, which is first cleaved by furin to produce big- ET-1 and then subsequently acted on by ET converting enzymes (ECEs) or other enzymes to generate bioavailable ET-1 [9]. ET-1 is 21 amino acids in length and is produced and released from endothelial and other cell types. Since approximately 80% of ET-1 is secreted from the basolateral side of endothelial cells [10], ET-1 typically acts in an autocrine fashion signaling, but under conditions of marked endothelial activation, ET-1 can spill over into the systemic circulation and affect distant organ systems [11].
Once released, ET-1 acts largely on two cell-surface G-protein-coupled receptors, the ET-1 Type A (ETA), located primarily on vascular smooth muscle, and Type B (ETB) receptors on endothelial, vascular smooth muscle, and renal epithelial cells [9]. Binding of the ETA receptor elicits vasoconstriction via increased Ca2+ influx and generation of reactive oxygen species (ROS) [12,13]. In contrast, ETB receptors on the endothelium allow ET-1 to signal in an autocrine fashion and stimulate nitric oxide synthase (NOS) and NO production, which serves the vasodilatory arm of this system [14]. Therefore, it can be appreciated that there is a balance of ET-1-mediated control of vascular tone, with vasodilation promoting proper blood pressure regulation during pregnancy but aberrant signaling of these receptors encouraging vasoconstriction and hypertension in PE.
The ET System in Normal Pregnancy
Human studies have revealed changes in ET receptor expression during pregnancy, whereas most of our understanding for the functional role of ET-1 in blood pressure regulation comes from experimental animal studies. For example, ETA and ETB receptor expression is increased in the uterus in pregnant versus non-pregnant women and is thought to modulate contraction during normal labor [15–17]. Alternatively spliced forms of ETA in smooth muscle cells isolated from human placenta may control the amount of function ETA involved in constriction [18]. Unfortunately, these studies only yield correlative data and they do not provide functional information regarding the role of ET-1 in blood pressure regulation during pregnancy.
Pharmacological and genetic studies in rodents provide a better insight into the function of the ET system during normal pregnancy. ETA receptor control of blood pressure remains similar to non-pregnant levels. Some studies show that antagonism of this receptor reduces blood pressure similarly between groups [19] and others a minimal change in response to antagonism during pregnancy [20]. With regard to ETB, its expression is increased in decidua in humans [21] but is not altered by pregnancy in isolated blood vessels in rodents [22]. Yet, studies point to greater ETB receptor-mediated relaxation in human and rodent arteries during pregnancy [23, 24]. This is attributed to pregnancy-related factors, including relaxin and proteinases, to convert big-ET-1 to the alternate form of ET(1–32), which signals via ETB to stimulate NOS in endothelial cells [24, 25]. Whether ETB receptor control of blood pressure is greater during pregnancy is debatable. Some investigators suggest that it contributes to the increased vasodilation and reduced blood pressure during pregnancy [23, 26], where- as others do not [19]. Pharmacological blockade of ETB increases blood pressure during pregnancy, but whether this response is greater than non-pregnant conditions to implicate greater ETB signaling in control of blood pressure during pregnancy is lacking. This requires further investigation, and genetic ETB-deficient models have become available for this purpose.
Pregnancy-related factors allow big-ET-1 conversion to ET(1–32) that acts on ETB receptors within the kidney [25, 27, 28]. This is thought to increase GFR during pregnancy, which is likely due to greater relaxation of the renal afferent and efferent arterioles [29]. ETB receptors are shown to contribute to efferent arteriole dilation via NO signaling [30, 31]. It is unknown the extent to which this pathway modulates ETA-induced constriction in the renal circulation. Furthermore, the role of the ETA or ETB pathways on direct tubular handling of sodium has not been examined during pregnancy. It is possible that relaxin shunts big-ET-1 away from ET-1(1–21) and its sodium excretory ability and towards ET-1(1–32) having vasodilatory actions without direct signaling of sodium control in the tubules. This is unknown. Again, what is known is that under non-pregnant conditions from rodent studies, ET-1 promotes sodium excretion via both ET receptors in females via NO signaling, whereas pregnancy is associated with increased GFR and renal blood flow via NO signaling but reduced sodium excretion due to attenuated NO signaling. Overall, this allows for plasma volume expansion of normal pregnancy [32], but the complete understanding of ET-1 involvement in this process is not yet fully elucidated. Although urinary levels of ET-1 are higher in pregnant versus non-pregnant women and increases as pregnancy progresses [33], it is unclear the relative levels of the different ET-1 cleavage products and their actions on vascular and renal function in proper blood pressure regulation during normal pregnancy.
The ET System in PE
ET-1 Levels in PE
Multiple studies have examined circulating ET-1 levels in normal pregnant and preeclamptic women, and found elevated plasma ET-1 in the preeclamptic group, with some studies indicating that circulating ET-1 correlates with the severity of the disease symptoms, though this is not a universal finding [34–39]. Verdonk et al. recently investigated the relationship between disturbed angiogenic balance, arterial pressure, and ET-1 in pregnant women with a high (> 85; n = 38) or low (< 85) soluble Fms-like tyrosine kinase-1 (sFlt-1)/ placental growth factor (PlGF) ratio [40]. Plasma ET-1 levels were increased in women with a high ratio. In addition, plasma ET-1 correlated positively with sFlt-1. Aggarwal et al. also investigated the correlation between ET-1 and sFlt-1, PlGF, and soluble endoglin (sEng) levels during uncomplicated normotensive pregnancy and PE [41]. Their results also show an association between elevation of sFlt-1, sEng, and ET-1 in the maternal circulation in PE, which strengthens the possibility that ET-1 could be a mediator in pathogenesis of PE secondary to the anti-angiogenic factors, sFlt-1, and sEng released by the placenta [41].
A number of experimental models of PE are also associated with elevated tissue levels of the ET-1 precursor, prepro-ET-1 mRNA. Both the renal cortex and medulla of placental ischemic rats express significantly higher levels of prepro-ET-1 when compared to normal pregnant controls [20]. Furthermore, chronic elevation of sFlt-1 in pregnant rats directly increased prepro-ET-1 gene expression in the renal cortex [42]. It has also been shown that infusion of the pro-inflammatory cytokine, tumor necrosis factor-alpha (TNF-α), directly induced hypertension in pregnant rats and is associated with significant increases in the expression of prepro-ET-1 in the maternal vasculature, placenta, and kidney [43]. Finally, infusion of the AT1-AA into the pregnant rat results in moderate hypertension and is associated with increased prepro-ET-1 expression in both the renal cortex and placenta [44].
Another potential mechanism for increased ET-1 levels in PE is via matrix metalloproteinase (MMP) activation. MMPs are enzymes that cleave the ET-1 precursor, big-ET-1 to active ET-1. Interestingly, the expression levels of MMPs (particularly MMP-2 and MMP-1) have been shown to increase in women who subsequently develop PE. Abdalvand et al. recently hypothesized that the increased MMP-2 expression leads to increased big-ET-1 conversion, and therefore increasing vasoconstriction [45]. They reported increased vascular contractility to big-ET-1 in the reduced uterine perfusion pressure (RUPP) model of PE that was likely attributable to upstream enzymatic pathways. These data are consistent with the greater contribution of MMP to cleave big-ET-1 to ET-1 partially responsible for increased big-ET-1-induced contractility. Nugent et al. also recently examined the potential role of MMP-1 as an activator of protease-activated receptor 1 (PAR-1), which is known to mediate the release of ET-1 in endothelial cells [46]. They reported increased serum and vascular MMP-1 in women with PE and hypothesized that the action of MMP-1 on PAR-1 might have vasoconstrictive effects. They demonstrated that MMP-1 has potent vasoconstrictor effects and the ability to enhance vascular reactivity to vasoconstrictor hormones, which are mediated by an endothelial ET-1 pathway. The authors concluded that increased circulating levels of MMP-1, and increased expression in systemic vessels of women with PE, may contribute to the development of hypertension in the mother.
ETA Receptors in PE
A number of experimental animal models have been utilized to examine the etiology and development of PE [47–49]. One of which we and others have used with great success is the RUPP model, where mechanical restriction of blood flow to the placenta causes hypoxia and ischemia [49]. This model has been used in species ranging from rats to non-human primates and mimics a number of the pathological features of human preeclampsia, such as hypertension, angiogenic imbalance, renal injury, proteinuria, and endothelial dysfunction [20, 50••, 51]. The renal cortex and medulla of RUPP rats express significantly higher prepro-ET-1, when compared to normal pregnant controls [18]. We have also reported that serum from RUPP animals significantly induces production of ET-1 from the endothelial cells when compared to those exposed to serum from normal pregnant animals, suggesting that circulating factors produced by placental ischemia are responsible for increased vascular ET-1 production [52]. When an ETA receptor antagonist was administered to the RUPP rats, the associated hypertension was abolished, and there was a trend for improved renal function [20]. Tam Tam et al. also reported that pretreatment with ETA attenuates both the mean arterial pressure (MAP) and uterine artery resistance index (UARI) in the RUPP group without affecting these parameters in the normal pregnant group [53]. The improvement in UARI could be one potential mechanism for the reduction in MAP in response to ETA in pregnant dams with ischemic placentas. Collectively, these studies suggest that placental ischemia induces factors that activate the production of ET-1 in the vasculature, which acts through ETA receptors to mediate the maternal hypertension seen in RUPP model. Zhou et al. also demonstrated that chronic hypoxia during gestation triggers PE-like symptoms in pregnant rats via heightened ET-1- and ETA-mediated signaling, providing a molecular mechanism linking gestational hypoxia and increased risk of PE [54].
Several studies have investigated the role of the ET-1 system in models of sFlt-1 excess in pregnancy. In support of the role of ET-1 in mediating sFlt-1-induced hypertension, our group has recently reported that continuous infusion of sFlt-1 in pregnant rats directly increased the level of ET-1 in the renal cortex and caused an increase in the MAP of ~ 20 mmHg [42]. Administration of an ETA receptor antagonist abolished this hypertension, which strongly supports that ET-1 is an important mediator of sFlt-1-induced hypertension. Kappers et al. also reported that the administration of Sunitinib, a tyrosine kinase inhibitor of the vascular endothelial growth factor (VEGF) receptor, induces a reversible rise in blood pressure in patients, and also in rats, associated with activation of the ET-1 system and generalized microvascular dysfunction [55]. Moreover, Sunitinib in swine results in ET-mediated hypertension [56]. Thus, another potential mechanism whereby VEGF blockade could increase blood pressure is by enhancing ET-1 synthesis.
The innate immune system has also been implicated in PE. For example, increased production of TNF-α is seen in both preeclamptic women and rodents undergoing chronic placental ischemia [43, 57, 58]. Studies in vitro demonstrated that 301 production of ET-1 by endothelial cells could be mediated by 302 exposure to TNF-α [59]. Studies from our group have demonstrated that administration of the soluble TNF-α receptor, Etanercept, reduces hypertension associated with placental ischemia in pregnant rats [43]. This treatment is accompanied by reduced expression of prepro-ET-1 in the renal cortex and medulla as well as the placenta itself. It has also been shown that infusion of TNF-α alone induces hypertension in pregnant rats, producing an approximate 20 mmHg increase in MAP in late gestation [60]. This is also associated with a significant increase in the expression of prepro-ET-1 in the maternal vasculature, placenta, and kidney. As seen in the RUPP model, administration of an ETA receptor antagonist in these animals completely abolished the associated hypertension [43]. LIGHT, a novel tumor necrosis factor superfamily member, is also significantly elevated in the circulation and placentas of preeclamptic women [61]. Injection of LIGHT into pregnant mice causes placental apoptosis, small fetuses, and key features of PE, with hypertension and proteinuria. The work reported in this study was the first to show that elevated LIGHT, coupled with enhanced lymphotoxin β and herpes virus entry mediator receptor activation, promotes placental damage and triggers release of potent vasoactive factors, namely sFlt-1 and ET-1. Together, these data suggest that ET-1, acting via the ETA receptor signaling, is a crucial mediator of TNF-α-induced hypertension in pregnancy.
AT1-AA is elevated in PE women as well as in RUPP rats. Studies show that infusion of the AT1-AA directly into the pregnant rat results in moderate hypertension that is associated with increased prepro-ET-1 expression in both the renal cortex and the placenta. Administration of an ETa receptor antagonist abrogated the hypertensive response to AT1-AA, highlighting the importance of the ET-1 system in the manifestation of AT1-AA-induced hypertension [44]. Recent studies also report that IgG from women with PE increase preproET-1 mRNA expression via angiotensin II type 1 receptor activation in kidneys and placentas in pregnant mice [62]. The ETA receptor-specific antagonist, BQ123, significantly reduced autoantibody-induced hypertension, proteinuria, and renal damage in these mice, suggesting that autoantibody-induced ET-1 production contributes to the pathogenesis of PE.
ETB Receptors in PE
Less is known about the regulation of ETB receptors in PE and their role in blood pressure regulation in this maternal disorder [63•]. In the RUPP model, there is reduced vascular expression of ETB receptors compared to their normal pregnant counterparts [64], which implicates reduced ETb receptor signaling in the vasculature during the pathogenesis of PE. Most studies have only examined this in systemic vessels and not in the kidney vasculature. Nevertheless, administration of an ETB agonist was still able to reduce blood pressure in RUPP rats [64], and ETB-deficient pregnant rats have exaggerated placental ischemia-induced hypertension [26]. It is hypothesized that the exaggerated placental ischemia-induced hypertension in ETB-deficient rats is due to defective NO signaling, which suggests that ETB are still present and act to buffer hypertension in wild-type controls. Thus, ETb receptors could be an important target in PE, and upregulation of endothelial ETB receptors, by using pharmacological agonists or genetic approaches, may represent novel strategies to manipulate this system in treatment of this maternal disorder. Foremost, it would be interesting to determine if ETa blockade is capable of completely reducing hypertension in ETB-deficient pregnant rats to determine if the latter receptor is important for the efficacy of ETA blockade. Moreover, a novel therapeutic option to increase NO signaling in PE and downstream of defective ETB signaling are activators and stimulators of the NO receptor, soluble guanylate cyclase (sGC) [59], or agents to increase bioavailability of the product 3 of NO-sGC signaling, cGMP. Sildenafil, an inhibitor of phosphodiesterase enzyme 5 (PDE5) that quenches bioavailable 372 cGMP, has been reported to attenuate hypertension and intra- 373 uterine growth restriction in a model of superimposed PE [65•]. These targeted strategies are highlighted in Fig. 1.
Fig. 1.
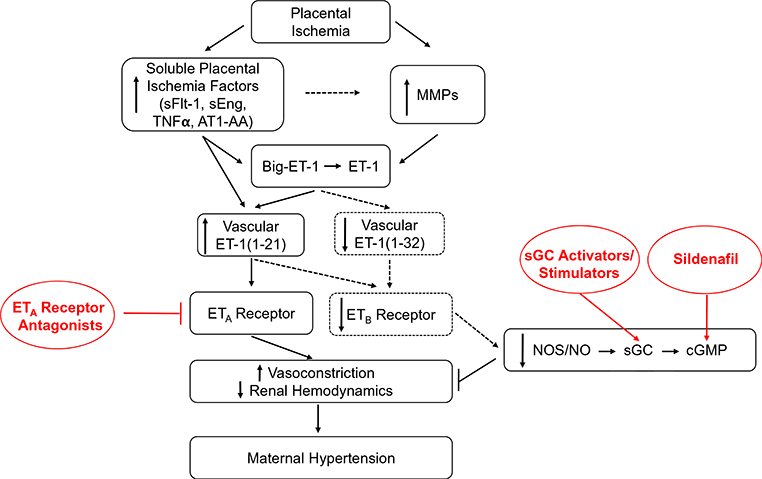
Hypothetical scheme highlighting the importance of the ET-1 system in blood pressure regulation during placental ischemia and PE. Placental ischemia induces the release of soluble factors for the placenta that target the vasculature. There, they activate the endothelium to produce ET-1(1–21). This acts on the ETA receptor on underlying vascular smooth muscle to elicit vasoconstriction and reduced renal hemodynamics to result in maternal hypertension. It has been shown that placental ischemia-induced hypertension in RUPP rats is linked to increased activity of matrix metalloproteinases (MMPs) in the blood vessel wall, which cleave big-ET-1 to active ET-1(1–21). The dashed line indicates that it is unknown if the placental factors mediate increased MMP Furthermore, it is unknown the extent that ET-1(1–21) or its alterative form, ET-1(1–32), is produced or activates any remaining ETB receptors during hypertensive pregnancies. Knowing the vasoprotective mechanisms that are still available for targeting in the mother will allow for a full understanding of how upcoming therapeutic approaches are beneficial in PE. These include the therapies illustrated in red, including ETA antagonists, activators and stimulators of sGC, or compounds to increase cGMP bioavailability, like the PDE5 inhibitor, sildenafil. This understanding could allow for optimization of these pharmacological strategies in PE
Targeting ETA Receptor Signalingas a Potential Therapeutic Target in PE
ETA receptor antagonists are used in the treatment of numerous cardiovascular diseases including pulmonary and systemic hypertension, congestive heart failure, myocardial infarction, vascular restenosis and atherosclerosis, renal failure, cerebrovascular disease, and cancer. Given the myriad of experimental models of PE, which have proven susceptible to ETA antagonism, could the ET-1 system be a therapeutic approach in managing the hypertension associated with PE? The clinical relevance of this potential therapeutic approach is tempered by work showing that genetic knockout of the ETA receptor leads to birth defects and eventual embryonic lethality in mice [66]. As a result, administration of ET receptor antagonists is contraindicated in pregnancy [67]. However, specific windows during early to mid-gestation have been identified in animal studies, where pharmacological antagonism of ETA caused phenotypes similar to that seen in the knockout. Administration of the ETA antagonist only during late gestation was not performed, and it is entirely plausible that ETA receptor antagonists might prove safe and efficacious in later pregnancy, when the symptoms of PE are most severe [68]. Furthermore, development of ETA receptor antagonists that do not cross the placental barrier would circumvent these problems altogether. Indeed, Thaete and colleagues reported of an ET receptor antagonist that had limited access to the fetal compartment during chronic maternal administration late in pregnancy [69]. Moreover, linking ETA receptor antagonists with elastin-like peptides that do not cross the placental barrier could also be an approach to limit the effects of ETA receptor antagonism within the fetal circulation but allow beneficial effects localized to the maternal circulation in PE [70].
Conclusions
Although numerous studies highlight the importance of the ET system in the pathogenesis of PE, further work is needed to determine whether ET receptor antagonists could provide an effective therapy for the management of this disease.
Acknowledgments
This work was supported by funds in a grant awarded to JPG from the NHLBI (5P01HL051971, 5R01HL108618, 5T32HL105324) and to FTS from NHLBI (4R00HL130577–02).
Footnotes
Compliance with Ethical Standards
Conflict of Interest The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
This article is part of the Topical Collection on Preeclampsia
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.American College of O, Gynecologists, Task Force on Hypertension in P. Hypertension in pregnancy. report of the American College of Obstetricians and Gynecologists’ Task Force on hypertension in pregnancy. Obstet Gynecol. 2013;122(5):1122–31. 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 2.Roberts JM, Taylor RN, Goldfien A. Clinical and biochemical evidence of endothelial cell dysfunction in the pregnancy syndrome preeclampsia. Am J Hypertens. 1991;4(8):700–8. [DOI] [PubMed] [Google Scholar]
- 3.Palei AC, Spradley FT, Warrington JP, George EM, Granger JP. Pathophysiology of hypertension in pre-eclampsia: a lesson in integrative physiology. Acta Physiol (Oxf). 2013;208(3):224–33. 10.1111/apha.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salam RA, Das JK, Ali A, Bhaumik S, Lassi ZS. Diagnosis and management of preeclampsia in community settings in low and middle-income countries. J Fam Med Prim Care. 2015;4(4):501–6. 10.4103/2249-4863.174265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Khaja KA, Sequeira RP, Alkhaja AK, Damanhori AH. Drug treatment of hypertension in pregnancy: a critical review of adult guideline recommendations. J Hypertens. 2014;32(3):454–63. 10.1097/HJH.0000000000000069. [DOI] [PubMed] [Google Scholar]
- 6.In: Bale JR, Stoll BJ, Lucas AO, editors. Improving birth outcomes: meeting the challenge in the developing world. Washington (DC); 2003. [PubMed] [Google Scholar]
- 7.Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972;1:177–91. [PubMed] [Google Scholar]
- 8.Warrington JP, George EM, Palei AC, Spradley FT, Granger JP. Recent advances in the understanding of the pathophysiology of preeclampsia. Hypertension. 2013;62(4):666–73. https://doi.org/45710.1161/HYPERTENSIONAHA.113.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev 2016;68(2):357–418. 10.1124/pr.115.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner OF, Christ G, Wojta J, Vierhapper H, Parzer S, Nowotny PJ, et al. Polar secretion of endothelin-1 by cultured endothelial cells. J Biol Chem. 1992;267(23):16066–8. [PubMed] [Google Scholar]
- 11.Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004; 61(2):227–37. [DOI] [PubMed] [Google Scholar]
- 12.Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther. 2005;315(3):1058–64 10.1124/jpet.105.091728. [DOI] [PubMed] [Google Scholar]
- 13.Schroeder AC, Imig JD, LeBlanc EA, Pham BT, Pollock DM, Inscho EW. Endothelin-mediated calcium signaling in preglomerular smooth muscle cells. Hypertension. 2000;35(1 Pt 474 2):280–6. [DOI] [PubMed] [Google Scholar]
- 14.Taylor TA, Gariepy CE, Pollock DM, Pollock JS. Gender differences in ET and NOS systems in ETB receptor-deficient rats: effect of a high salt diet. Hypertension. 2003;41(3 Pt 2):657–62. 10.1161/01.HYP.0000048193.85814.78. [DOI] [PubMed] [Google Scholar]
- 15.Wolff K, Faxen M, Lunell NO, Nisell H, Lindblom B. Endothelin receptor type A and B gene expression in human nonpregnant, term pregnant, and preeclamptic uterus. Am J Obstet Gynecol. 1996;175(5):1295–300. [DOI] [PubMed] [Google Scholar]
- 16.Wolff K, Nisell H, Modin A, Lundberg JM, Lunell NO, Lindblom B. Contractile effects of endothelin 1 and endothelin 3 on 485 myometrium and small intramyometrial arteries of pregnant women at term. Gynecol Obstet Investig. 1993;36(3): 166–71. [DOI] [PubMed] [Google Scholar]
- 17.Osada K, Tsunoda H, Miyauchi T, Sugishita Y, Kubo T, Goto K. Pregnancy increases ET-1-induced contraction and changes receptor subtypes in uterine smooth muscle in humans. Am J Phys. 1997;272(2 Pt 2):R541–8. 10.1152/ajpregu.1997.272.2.R541. [DOI] [PubMed] [Google Scholar]
- 18.Bourgeois C, Robert B, Rebourcet R, Mondon F, Mignot TM, Duc Goiran P, et al. Endothelin-1 and ETA receptor expression in vascular smooth muscle cells from human placenta: a new ETA recetor messenger ribonucleic acid is generated by alternative splicing of exon 3. J Clin Endocrinol Metab. 1997;82(9):3116–23. 10.1210/jcem.82.9.4209. [DOI] [PubMed] [Google Scholar]
- 19.Thaete LG, Neerhof MG. Endothelin and blood pressure regulation in the female rat: studies in normal pregnancy and with nitric oxide synthase inhibition-induced hypertension. Hypertens Pregnancy. 2000;19(2):233–47. [DOI] [PubMed] [Google Scholar]
- 20.Alexander BT, Rinewalt AN, Cockrell KL, Massey MB, Bennett WA, Granger JP. Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension. 2001;37(2 Pt 2):485–9. [DOI] [PubMed] [Google Scholar]
- 21.Kohnen G, Campbell S, Irvine GA, Church HJ, MacLachlan F, Titterington M, et al. Endothelin receptor expression in human decidua. Mol Hum Reprod. 1998;4(2):185–93. [DOI] [PubMed] [Google Scholar]
- 22.Kerchner LJ, Novak J, Hanley-Yanez K, Doty KD, Danielson LA, Conrad KP. Evidence against the hypothesis that endothelialendothelin B receptor expression is regulated by relaxin and pregnancy. Endocrinology. 2005;146(6):2791–7. 10.1210/en.2004-1602. [DOI] [PubMed] [Google Scholar]
- 23.Mazzuca MQ, Dang Y, Khalil RA. Enhanced endothelin receptor type B-mediated vasodilation and underlying [Ca(2)(+)]i in mesenteric microvessels of pregnant rats. Br J Pharmacol. 2013;169(6):1335–51. 10.1111/bph.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP. Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension. 2011;57(6): 1151–60. 10.1161/HYPERTENSIONAHA.110.165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad KP. Maternal vasodilation in pregnancy: the emerging role of relaxin. Am J Physiol Regul Integr Comp Physiol. 2011;301(2): R267–75. 10.1152/ajpregu.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dent EA, Spradley FT, Granger JP. Endothelin receptor type B (ETB)-deficient pregnant rats have exaggerated placental ischemia-induced hypertension. FASEB J. 2017;31(1 Supplement):851–8.. [Google Scholar]
- 27.Jeyabalan A, Novak J, Doty KD, Matthews J, Fisher MC, Kerchner LJ, et al. Vascular matrix metalloproteinase-9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short-term administration of relaxin. Endocrinology. 2007;148(1): 189–97. 10.1210/en.2006-0989. [DOI] [PubMed] [Google Scholar]
- 28.Conrad KP, Davison JM. The renal circulation in normal pregnancy and preeclampsia: is there a place for relaxin? Am J Physiol Renal Physiol. 2014;306(10):F1121–35. 10.1152/ajprenal.00042.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng A, Conrad KP, Baylis C. Relaxin mediated renal vasodilation in the rat is associated with falls in glomerular blood pressure. Am J Physiol Regul Integr Comp Physiol. 2017;314:R147–52. 10.1152/ajpregu.00148.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inscho EW, Imig JD, Cook AK, Pollock DM. ETA and ETB receptors differentially modulate afferent and efferent arteriolar responses to endothelin. Br J Pharmacol. 2005;146(7):1019–26. 10.1038/sj.bjp.0706412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schildroth J, Rettig-Zimmermann J, Kalk P, Steege A, Fahling M, Sendeski M, et al. Endothelin type A and B receptors in the control of afferent and efferent arterioles in mice. Nephrol Dial Transplant. 2011;26(3):779–89. 10.1093/ndt/gfq534. [DOI] [PubMed] [Google Scholar]
- 32.West CA, Sasser JM, Baylis C. The enigma of continual plasma volume expansion in pregnancy: critical role of the renin-angiotensin- aldosterone system. Am J Physiol Renal Physiol. 2016;311(6): F1125–F34. 10.1152/ajprenal.00129.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamas P, Worgall S, Sulyok E, Rascher W. Renal electrolyte and water handling in normal pregnancy: possible role of endothelin-1. Eur J Obstet Gynecol Reprod Biol. 1994;55(2):89–95. [DOI] [PubMed] [Google Scholar]
- 34.George EM, Granger JP. Endothelin: key mediator of hypertension in preeclampsia. Am J Hypertens. 2011;24(9):964–9. 10.1038/ajh.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George EM, Granger JP. Linking placental ischemia and hypertension in preeclampsia: role of endothelin 1. Hypertension. 2012;60(2):507–11. 10.1161/HYPERTENSIONAHA.112.194845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George EM, Palei AC, Granger JP. Endothelin as a final common pathway in the pathophysiology of preeclampsia: therapeutic implications. Curr Opin Nephrol Hypertens. 2012;21(2):157–62. 10.1097/MNH.0b013e328350094b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benigni A, Orisio S, Gaspari F, Frusca T, Amuso G, Remuzzi G. Evidence against a pathogenetic role for endothelin in pre-eclampsia. Br J Obstet Gynaecol. 1992;99(10):798–802. [DOI] [PubMed] [Google Scholar]
- 38.Mastrogiannis DS, O’Brien WF, Krammer J, Benoit R. Potential role of endothelin-1 in normal and hypertensive pregnancies. Am J Obstet Gynecol. 1991;165(6 Pt 1):1711–6. [DOI] [PubMed] [Google Scholar]
- 39.Taylor RN, Varma M, Teng NN, Roberts JM. Women with preeclampsia have higher plasma endothelin levels than women with normal pregnancies. J Clin Endocrinol Metab. 1990;71(6): 1675–7. 10.1210/jcem-71-6-1675. [DOI] [PubMed] [Google Scholar]
- 40.Verdonk K, Saleh L, Lankhorst S, Smilde JE, van Ingen MM, Garrelds IM, et al. Association studies suggest a key role for endothelin-1 in the pathogenesis of preeclampsia and the accompanying renin-angiotensin-aldosterone system suppression. Hypertension. 2015;65(6):1316–23. 10.1161/HYPERTENSIONAHA.115.05267. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal PK, Chandel N, Jain V, Jha V. The relationship between circulating endothelin-1, soluble fms-like tyrosine kinase-1 and soluble endoglin in preeclampsia. J Hum Hypertens. 2012;26(4): 236–41. 10.1038/jhh.2011.29. [DOI] [PubMed] [Google Scholar]
- 42.Murphy SR, LaMarca BB, Cockrell K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010;55(2): 394–8. 10.1161/HYPERTENSIONAHA.109.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep. 2007;9(6):480–5. [DOI] [PubMed] [Google Scholar]
- 44.LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension. 2009;54(4):905–9. 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdalvand A, Morton JS, Bourque SL, Quon AL, Davidge ST. Matrix metalloproteinase enhances big-endothelin-1 constriction in mesenteric vessels of pregnant rats with reduced uterine blood 604 flow. Hypertension. 2013;61(2):488–93. 10.1161/HYPERTENSIONAHA.111.00055. [DOI] [PubMed] [Google Scholar]
- 46.Nugent WH, Mishra N, Strauss JF 3rd, Walsh SW. Matrix metalloproteinase 1 causes vasoconstriction and enhances vessel reactivityto angiotensin II via protease-activated receptor 1. Reprod Sci. 609 2016;23(4):542–8. 10.1177/1933719115607998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexander BT, Llinas MT, Kruckeberg WC, Granger JP. L-Arginine attenuates hypertension in pregnant rats with reduced uterine perfusion pressure. Hypertension. 2004;43(4):832–6. 10.1161/01.HYP.0000119192.32360.a9. [DOI] [PubMed] [Google Scholar]
- 48.Taylor RN, Roberts JM, Cunningham FG, Lindheimer MD, Chesley LC. Chesley’s hypertensive disorders in pregnancy. 4th 616 ed. Amsterdam: Elsevier/AP, Academic Press is an imprint of 617 Elsevier; 2015. [Google Scholar]
- 49.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, et al. Reduced uterine perfusion pressure (RUPP) model for 620 studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med. 2006;122:383–92. [DOI] [PubMed] [Google Scholar]
- 50.••.Spradley FT, Tan AY, Joo WS, Daniels G, Kussie P, Karumanchi SA, et al. Placental growth factor administration abolishes placental ischemia-induced hypertension. Hypertension. 2016;67(4):740–7. 10.1161/HYPERTENSIONAHA.115.06783.This was the first study to implicate recombinant human placental growth factor as a potential treatment for preeclampsia in a model of placental ischemia-induced hypertension.
- 51.Makris A, Yeung KR, Lim SM, Sunderland N, Heffernan S, Thompson JF, et al. Placental growth factor reduces blood pressure in a uteroplacental ischemia model of preeclampsia in nonhuman primates. Hypertension. 2016;67(6):1263–72. 10.1161/HYPERTENSIONAHA.116.07286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts L, LaMarca BB, Fournier L, Bain J, Cockrell K, Granger JP. Enhanced endothelin synthesis by endothelial cells exposed tosera from pregnant rats with decreased uterine perfusion. Hypertension. 2006;47(3):615–8. 10.1161/01.HYP.0000197950.42301.dd. [DOI] [PubMed] [Google Scholar]
- 53.Tam Tam KB, George E, Cockrell K, Arany M, Speed J, Martin JN Jr, et al. Endothelin type A receptor antagonist attenuates placental 641 ischemia-induced hypertension and uterine vascular resistance. Am 642 J Obstet Gynecol. 2011;204(4):330 e1–4. 10.1016/j.ajog.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou J, Xiao D, Hu Y, Wang Z, Paradis A, Mata-Greenwood E, et al. Gestational hypoxia induces preeclampsia-like symptoms via height-enedendothelin-1 signaling in pregnant rats. Hypertension. 2013;62(3):599–607. 10.1161/HYPERTENSIONAHA.113.01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kappers MH, Smedts FM, Horn T, van Esch JH, Sleijfer S, Leijten F, et al. The vascular endothelial growth factor receptor inhibitorsunitinib causes a preeclampsia-like syndrome with activation of the endothelin system. Hypertension. 2011;58(2):295–302. 10.1161/HYPERTENSIONAHA.111.173559. [DOI] [PubMed] [Google Scholar]
- 56.Kappers MH, de Beer VJ, Zhou Z, Danser AH, Sleijfer S, Duncker DJ, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59(1):151–7. 10.1161/HYPERTENSIONAHA.111.182220. [DOI] [PubMed] [Google Scholar]
- 57.Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. 2010;63(6):534–43. 10.1111/j.1600-0897.2010.00831.x. [DOI] [PubMed] [Google Scholar]
- 58.Redman CW, Tannetta DS, Dragovic RA, Gardiner C, Southcombe JH, Collett GP, et al. Review: does size matter? Placental debris and the pathophysiology of pre-eclampsia. Placenta. 2012;33(Suppl): S48–54. 10.1016/j.placenta.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Bakrania BA, Spradley FT, Satchell SC, Stec DE, Rimoldi JM, Gadepalli RSV, et al. Heme oxygenase-1 is a potent inhibitor of placental ischemia-mediated endothelin-1 production in cultured human glomerular endothelial cells. Am J Physiol Regul Integr Comp Physiol. 2017; 10.1152/ajpregu.00370.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension. 2005;46(1):82–6. 10.1161/01.HYP.0000169152.59854.36. [DOI] [PubMed] [Google Scholar]
- 61.Wang W, Parchim NF, Iriyama T, Luo R, Zhao C, Liu C, et al. Excess LIGHT contributes to placental impairment, increased secretion of vasoactive factors, hypertension, and proteinuria in preeclampsia. Hypertension. 2014;63(3):595–606. 10.1161/HYPERTENSIONAHA.113.02458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou CC, Irani RA, Dai Y, Blackwell SC, Hicks MJ, Ramin SM, et al. Autoantibody-mediated IL-6-dependent endothelin-1 elevation underlies pathogenesis in a mouse model of preeclampsia. J Immunol. 2011; 186(10):6024–34. 10.4049/jimmunol.1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.•.Mirabito Colafella KM Endothelin type B (ETB) receptors: friend or foe in the pathogenesis of pre-eclampsia and future cardiovascular disease (CVD) risk? Clin Sci (Lond). 2018;132(1):33–6. 10.1042/CS20171366.This review highlights the importance of studying Endothelin type B receptors in the pathogenesis of preeclampsia, and the therapeutic potential of these receptors.
- 64.Mazzuca MQ, Li W, Reslan OM, Yu P, Mata KM, Khalil RA. Downregulation of microvascular endothelial type B endothelin receptor is a central vascular mechanism in hypertensive pregnancy. Hypertension. 2014;64(3):632–43. 10.1161/HYPERTENSIONAHA.114.03315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.•.Gillis EE, Mooney JN, Garrett MR, Granger JP, Sasser JM. Sildenafil treatment ameliorates the maternal syndrome of preeclampsia and rescues fetal growth in the dahl salt-sensitive rat. Hypertension. 2016;67(3):647–53. 10.1161/HYPERTENSIONAHA.115.06071.This study characterizes a novel pre-clinical model of super-imposed preeclampsia in the Dahl Salt-Sensitive Rat.
- 66.Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, et al. Cranial and cardiac neural crest defects in endothelin—a receptor-deficient mice. Development. 1998;125(5):813–24. [DOI] [PubMed] [Google Scholar]
- 67.Kingman M, Ruggiero R, Torres F. Ambrisentan, an endothelin receptor type A-selective endothelin receptor antagonist, for the treatment of pulmonary arterial hypertension. Expert Opin Pharmacother. 2009;10(11):1847–58. 10.1517/14656560903061275. [DOI] [PubMed] [Google Scholar]
- 68.Taniguchi T, Muramatsu I. Pharmacological knockout of endothelin ET(A) receptors. Life Sci. 2003;74(2–3):405–9. [DOI] [PubMed] [Google Scholar]
- 69.Thaete LG, Khan S, Synowiec S, Dayton BD, Bauch J, Neerhof MG. Endothelin receptor antagonist has limited access to the fetal compartment during chronic maternal administration late in pregnancy. Life Sci. 2012;91(13–14):583–6. 10.1016/j.lfs.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 70.Bidwell GL 3rd, George EM. Maternally sequestered therapeutic polypeptides - a new approach for the management of preeclampsia. Front Pharmacol. 2014;5:201 10.3389/fphar.2014.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]