

Abstract
Background
Exogenous testosterone decreases serum concentrations of high-density lipoprotein cholesterol (HDL-C) in men, but whether this alters cardiovascular risk is uncertain.
Objective
To investigate the effects of testosterone and estradiol on HDL particle concentration and metrics of HDL function.
Methods
We enrolled 53 healthy men, 19–55 years of age, in a double-blinded, placebo-controlled, randomized trial. Subjects were rendered medically castrate using the GnRH receptor antagonist acyline and administered either 1) placebo gel, 2) low-dose transdermal testosterone gel (1.62%, 1.25 g), 3) full replacement dose testosterone gel (1.62%, 5 g) or 4) full replacement dose testosterone gel together with an aromatase inhibitor for 4 weeks. At baseline and end-of-treatment, serum HDL total macrophage and ABCA1-specific cholesterol efflux capacity (CEC), HDL particle concentration and size, and HDL protein composition were determined.
Results
Significant differences in serum HDL-C were observed with treatment across groups (p=0.01 in overall repeated measures ANOVA), with increases in HDL-C seen after both complete and partial testosterone deprivation. Medical castration increased total HDL particle concentration (median[IQR] 19.1[1.8] nmol/L at baseline vs. 21.3[3.1] nmol/L at week 4, p=0.006). However, corresponding changes in total macrophage CEC and ABCA1-specific CEC were not observed. Change in serum 17β-estradiol concentration correlated with change in total macrophage CEC (β=0.33 per 10 pg/mL change in serum 17β-estradiol, p=0.03).
Conclusions
Testosterone deprivation in healthy men leads to a dissociation between changes in serum HDL-C and HDL CEC. Changes in serum HDL-C specifically due to testosterone exposure may not reflect changes in HDL function.
Keywords: sex steroids, HDL, cholesterol efflux, lipoproteins, cardiovascular disease
Introduction
Low circulating testosterone concentrations associate with increased cardiovascular disease (CVD) risk in men,1 but whether this association reflects a causal relationship remains uncertain. Similarly, the impact of testosterone replacement therapy (TRT) on CVD risk is unclear and particularly controversial with regard to its use in older men with late-onset hypogonadism.1,2 Concern stems in part from the observation that exogenous testosterone administration to men often modestly lowers serum concentrations of high-density lipoprotein cholesterol (HDL-C),3 a well-established negative risk factor for CVD.4
Measurement of HDL-C concentrations may have limited utility as a predictor of CVD risk. Despite the strong, inverse association between HDL-C and CVD risk in epidemiologic studies, recent clinical trials have demonstrated that pharmacologic treatments designed to increase HDL-C have failed to confer cardiovascular protection despite substantially raising HDL-C concentrations.5,6 Thus, the possibility exists that TRT also could result in a dissociation between HDL-C and CVD risk. Increasingly, alternative metrics of HDL composition and function have been employed as indices of the relationship between HDL and CVD.7 The use of these alternative metrics may offer key insights into HDL-associated mechanisms through which testosterone may influence CVD risk in men.
The best characterized function for HDL is its role in reverse cholesterol transport (RCT), the process by which cholesterol from peripheral cells is transferred by HDL to the liver for excretion in the bile. HDL mediates RCT both directly by delivering the cholesterol to liver via scavenger receptor class B type 1 (SR-BI), as well as indirectly, through transfer of cholesterol to apolipoprotein B-containing particles and delivery via LDL receptor.8 In addition to RCT, HDL can also deliver cholesterol to steroidogenic cells, adipocytes, and endothelial cells through interaction with SR-BI.9,10 HDL cholesterol efflux capacity (CEC), the ability of HDL to accept cholesterol from macrophages – the first critical step of RCT - can be quantified in clinical samples using cell-based assays.11 Importantly, serum HDL-C concentrations are dissociated from HDL CEC; HDL-C accounts for less than half of the variance in HDL CEC,11 underscoring the limitations of serum HDL-C as a measure of HDL function.12 Furthermore, HDL CEC has been shown to be a predictor of both prevalent and incident coronary artery disease independent of HDL-C concentrations.13,14 HDL particles of different sizes have been shown to preferentially mediate cholesterol efflux through discrete pathways,15,16 and large HDL subclasses in particular have been associated with lower CVD prevalence and severity although these findings have not been uniform across studies.17 Further, lower total HDL particle concentration has been associated with prevalent cerebrovascular disease and incident cardiovascular events.18–21 HDL particle concentration and size distribution have been shown to more closely associate with CVD than HDL-C.18,19 Thus, quantifying changes in these alternative HDL metrics may generate important new insights into the relationship between testosterone and HDL and thereby into the relationship between testosterone and CVD risk in men.
To investigate the relationship between testosterone and HDL, we measured pre-specified HDL-associated metrics using serum samples collected during a randomized, double-blind clinical intervention study in which healthy, eugonadal men underwent sex steroid manipulation for a 4-week treatment period. At baseline and end-of-treatment, serum HDL CEC, and HDL particle concentration, size distribution, and protein composition were quantified in order to comprehensively characterize the effects of testosterone on HDL in men.
Materials and Methods
Study design and subjects
A total of 53 subjects with normal baseline serum testosterone concentrations were enrolled. Of these, 45 completed all study visits and study-related procedures and were included in the analyses. As described elsewhere,22 enrolled subjects were healthy, eugonadal men between 19–55 years of age. Exclusion criteria included a history of diabetes, uncontrolled hypertension, body mass index >33 kg/m2, and current or recent use of testosterone, glucocorticoids, or lipid-lowering medications including statins.22
The GnRH receptor antagonist acyline (300 μg/kg body weight) 23 was administered by subcutaneous injection to all subjects at baseline and study week 2. Subjects were randomized to receive one of the following regimens administered daily over the 4-week treatment period: 1) placebo transdermal gel and placebo tablets (Castrate group), 2) low-dose testosterone gel (1.25g of 1.62% gel; AndroGel, Abbvie Inc; Chicago, IL) and placebo tablets (Low T/E group), 3) full replacement dose testosterone gel (5g of 1.62% gel) and placebo tablets (Normal T/E group), or 4) full replacement dose testosterone gel and the aromatase inhibitor letrozole to confer selective estrogen deprivation (Apotex Corporation; Ontario, Canada) (Normal T/Low E group). Double-blinded randomization to treatment groups was performed with a block size of 4. At baseline and end-of-treatment (study week 4), serum HDL CEC was measured by 2 methods with serum depleted of apolipoprotein B (apoB)-containing particles. Total HDL particle concentration (HDL-Pima) and particle size distribution were quantified by calibrated ion mobility analysis.18 This study was approved by the University of Washington Institutional Review Board and registered at ClinicalTrials.gov (ID NCT01686828). Written informed consent was obtained from all subjects before any study procedures were performed.
Sample collection and processing
Serum sex steroids were measured by liquid chromatography-tandem mass spectrometry as previously described.22 For measurement of fasting lipid concentrations and HDL analyses, whole blood was collected and allowed to clot for 15 minutes on ice. Samples were then centrifuged at 3000 rpm at 4°C for 10 minutes and immediately stored at −80°C. Samples were kept at −80°C until batched and analyzed together after a single thaw. Fasting lipid concentrations were measured at the Northwest Lipid Metabolism and Diabetes Research Center (NWLMDRC; Seattle, WA) using established, validated methods.24
Serum HDL CEC measurement
Serum HDL capacity to efflux cholesterol from macrophages was determined using the methods developed by Rothblat and colleagues,13 which we have employed previously.25,26 Briefly, plasma was converted to serum by addition of Ca2+ and depleted of apoB-containing particles by polyethylene glycol precipitation (serum HDL). Serum HDL in DMEM medium (1.1% serum) was incubated for 4 hours with cells loaded with radiolabeled [3H]-cholesterol. Total serum HDL CEC (total macrophage CEC) was determined using cAMP-stimulated J774 cells treated with an acyl–coenzyme A:cholesterol acyltransferase inhibitor.13 CEC mediated specifically by ABCA1 (ABCA1 CEC) was quantified using BHK-1 cells transfected with mifepristone-inducible human ABCA1 transporter.27,28 CEC was quantified as the % of [3H]-cholesterol in the medium relative to all labeled cholesterol in the culture system (cells and medium) after subtraction of values obtained after serum-free incubation. ABCA1 CEC was determined by subtracting basal CEC (BHK-1 cells not stimulated with mifepristone) from mifepristone-stimulated CEC in BHK-1 cells. All samples were assayed in duplicate, and mean values are presented. Assays for each cell type were done on the same day, and the coefficient of variation for macrophage CEC was 7.9%, and ABCA1 CEC 6.7%.
Quantification of HDL particle size and concentration
HDL particle concentration (HDL-Pima) and size distribution were measured using calibrated ion mobility analysis (cIMA).18,28 This method yields a stoichiometry of apoA-I and sizes and relative abundances of HDL subclasses that are in excellent agreement with those determined by non-denaturing gradient gel electrophoresis and analytical ultracentrifugation.18,29 Particle concentration was calibrated using glucose oxidase and validated using external, well characterized plasma samples. Four species of HDL particles were detected by cIMA analysis and classified as extra small (xs-HDL-Pima, average diameter 7.8 nm), small (s-HDL-Pima, average diameter 8.4 nm), medium (m-HDL-Pima, average diameter 9.5 nm), or large (l-HDL-Pima, average diameter 10.8 nm). Total HDL particle (t-HDL-Pima) concentration was calculated as the sum total of all 4 particle sizes. Intra-assay coefficients of variation averaged 11.2% (6.7–18.2%), and inter-assay values averaged 11.1% (3.8–18.8%).
Quantification of HDL-associated proteins
The abundance of HDL-associated proteins was quantified by MS using a targeted proteomics strategy to measure 34 proteins as described previously.30 HDL proteins were spiked with 15N-apoA-I as an internal standard, denatured, reduced and alkylated, and digested with trypsin. Tryptic digests of HDL were chromatographed using a nanoAquity UPLC (Waters, MA) with a C18 trapping column (Reprosil C18 100 Å, 5 μm, 0.1 × 40 mm; Dr. Maisch, Germany) (trapping flow rate 4 μL/min), a capillary analytical column (Reprosil C18 100 Å, 5 μm, 100×0.075 mm; Dr. Maisch, Germany), and a 30 min linear gradient of acetonitrile, 0.1% formic acid (7–35%) in 0.1% formic acid in water at a flow rate of 0.6 μL/min. The NanoAquity UPLC was connected to a Thermo TSQ Vantage triple-quadrupole mass spectrometer with electrospray ionization. The instrument was operated in SRM mode with 10 ms dwell time and the peptides were monitored with collision energies optimized to maximize the signal. Peak areas were integrated using Skyline software.31
Statistical analyses
All variables were tested for normality using the Shapiro-Wilk test, and non-normally distributed variables were log-transformed. Repeated measures ANOVA (RM-ANOVA) was performed to determine whether time-by-treatment group interactions existed for each of the endpoints except for HDL-associated proteins. Within-group differences over the 4-week treatment period were assessed with Wilcoxon signed rank tests using non-log transformed values. Baseline differences among treatment groups were determined by ANOVA. For HDL-associated proteins, peak area values for each protein were log-transformed and standardized using Z-scores. Differences in the change in protein abundance across treatment groups were determined using one-way ANOVA. To examine associations between changes in serum sex steroid concentrations and HDL-associated endpoints, multistep linear regression analyses were used, with all study subjects analyzed as a single cohort. For fasting lipoprotein concentrations, dependent variables were changes in serum testosterone and 17β-estradiol concentrations, which were treated as ordinal variables (per 3.47 nmol/L [1 ng/mL] change in serum testosterone or per 36.7 pmol/L [10 pg/mL] change in serum 17β-estradiol). For HDL CEC and HDL particle concentrations, dependent variables included changes in serum testosterone and 17β-estradiol concentrations as well as change in serum HDL-C concentration, which was treated as a continuous variable. For HDL-associated proteins, dependent variables included changes in serum testosterone and 17β-estradiol concentrations, total macrophage CEC, and the fractions of large and small HDL particles. Only significant variables were included in the final models for all regression analyses. A p-value threshold of <0.05 defined statistical significance except for analysis of HDL-associated protein abundance for which an adjusted p-value threshold of <0.0015 (Bonferroni adjustment) was used to correct for multiple comparisons. Data are presented as median[interquartile range (IQR)] or mean±SD as indicated, and standardized β-coefficients are shown. Statistical analyses were performed using SPSS 25 (IBM Corporation, Armonk, New York), and graphs were generated using GraphPad Prism 5 (GraphPad Inc., La Jolla, CA).
Results
Study subjects and serum sex steroid concentrations
A total of 45 subjects completed all study visits and procedures and met criteria for study drug adherence established a priori and consequently were included in the current analyses. Subjects were healthy men with normal baseline serum testosterone concentrations, and detailed enrollment data and baseline characteristics have been reported previously.22 On-treatment serum sex steroid concentrations were achieved as intended for all 4 treatment groups (Figure 1), with highly significant differences in serum testosterone and 17β-estradiol concentrations seen across groups during treatment (p=0.0002 for both in overall RM-ANOVA).
Figure 1.

Baseline and end-of-treatment serum total testosterone (A) and 17β-estradiol (B) concentrations in study subjects classified by treatment group. Data points are shown as median and standard error.
Testosterone deprivation increases serum concentrations of HDL-C
At baseline, none of the lipid parameters differed significantly among groups. No significant treatment effects were found for any of the serum lipid parameters except HDL-C, for which a significant effect was observed (p=0.01 in overall RM-ANOVA, Figure 2). A single subject in the Normal T/Low E had a very high baseline serum HDL-C concentration (3.81 mmol/L). Exclusion of this subject in sensitivity analysis did not affect the findings, and the time-by-group interaction for serum HDL-C concentration remained significant and unchanged (p=0.01).
Figure 2.

Changes in serum lipids across treatment groups during the 4-week treatment period. No significant differences across groups nor within-group changes were seen for total cholesterol (A), triglycerides (B), very low-density lipoprotein cholesterol (C), or low-density lipoprotein cholesterol (D). A significant difference across groups was seen over the treatment period for high-density lipoprotein cholesterol (HDL-C) (E), with significant increases in HDL-C concentrations evident in both the Castrate and Low T/E groups. Whiskers = minimum to maximum values, **p<0.01 for within-group change
Significant treatment-induced increases in serum HDL-C concentration were evident for both groups deficient in testosterone, the Castrate group (1.19[0.19] mmol/L at baseline vs. 1.35[0.22] mmol/L at week 4, p=0.006) and the Low T/E group (1.09[0.49] mmol/L at baseline vs. 1.30[0.60] mmol/L at week 4, p=0.007). In contrast, no changes in serum HDL-C were observed in the 2 treatment groups that received full-dose testosterone (Normal T/E and Normal T/Low E groups). A significant inverse correlation was found between change in serum testosterone concentration and change in serum HDL- C across all study subjects (β=−0.39 per 3.47 nmol/L change in serum testosterone, p=0.009). Change in serum 17β-estradiol concentration did not associate with changes in HDL-C nor any of the other serum lipid parameters (data not shown).
Testosterone deprivation does not increase serum HDL CEC
The CEC of serum HDL (apoB-depleted serum) was measured to determine whether increases in HDL-C consequent to testosterone deprivation showed attendant changes in HDL efflux capacity. Changes in total macrophage HDL CEC did not differ among groups over the treatment period (p=0.65 in overall RM-ANOVA, Figure 3A). Despite a significant increase in serum HDL-C concentrations among subjects in the Castrate group, total macrophage CEC remained unchanged (14.1[3.4]% at baseline vs. 15.7[5.3]% at week 4, p=0.13). No changes in total macrophage CEC were found in the other 3 treatment groups. These findings were unchanged by adjustment for the serum HDL-C concentration in the RM-ANOVA models (data not shown).
Figure 3.

Changes in CEC across treatment groups during the 4-week treatment period. Neither differences across groups nor within-group changes were seen for total macrophage CEC (A). A significant increase in ABCA1 CEC was evident over the 4-week treatment period among subjects who underwent selective estrogen deprivation (B). Whiskers = minimum to maximum values, *p<0.05 for within-group change
Whereas change in total macrophage CEC exhibited a modest, positive correlation with change in serum HDL-C (β=0.30, p=0.05), no association was found between change in total macrophage CEC and serum testosterone concentration. However, total macrophage CEC exhibited a positive correlation with change in serum 17β-estradiol (β=0.37 per 36.7 pmol/L change in 17β-estradiol, p=0.02).
Similar to total macrophage CEC, no differences across treatment groups were observed for ABCA1 CEC (p=0.34 in overall RM-ANOVA, Figure 3B), regardless of correction for changes in serum HDL-C concentration (data not shown). End-of-treatment ABCA1 CEC also remained similar to baseline values among subjects in the Castrate group (5.8[1.7]% at baseline vs. 6.2[1.6]% at week 4, p=0.38). However, selective estrogen deprivation led to a small but significant increase in ABCA1 CEC (Normal T/Low E group, 5.3[2.0]% at baseline vs. 6.4[1.9]% at week 4, p=0.047). No other significant within-group changes in ABCA1 CEC were found over the 4-week treatment period.
Change in ABCA1 CEC did not correlate with changes in serum testosterone or 17β-estradiol concentrations or with change in serum HDL-C. In sensitivity analyses, exclusion of the subject with very high HDL-C concentrations did not change the absence of significant time-by-group interactions for either total macrophage or ABCA1 CEC (p=0.72 and p=0.39, respectively).
Testosterone and 17β-estradiol exposure both contribute to changes in HDL particle subclass distribution
Across the groups there was no significant difference in total HDL particle concentration (t-HDL-Pima) (p=0.42 in overall RM-ANOVA, Figure 4). However, a significant, ~10% increase in t-HDL-Pima was found in the Castrate group (19.1[1.8] nmol/L at baseline vs. 21.3[3.1] nmol/L at week 4, p=0.006). Further supporting a relationship between serum testosterone and t-HDL-Pima, a significant inverse correlation was seen between changes in serum testosterone concentration and t-HDL-Pima (β=−0.36 per 3.47 nmol/L change in serum testosterone, p=0.01). However, when serum HDL-C concentration was included in the model as a covariate, the inverse correlation between testosterone and t-HDL-Pima was lost due to association of change in serum HDL-C with change in t-HDL-Pima (β=0.58, p<0.0001).
Figure 4.
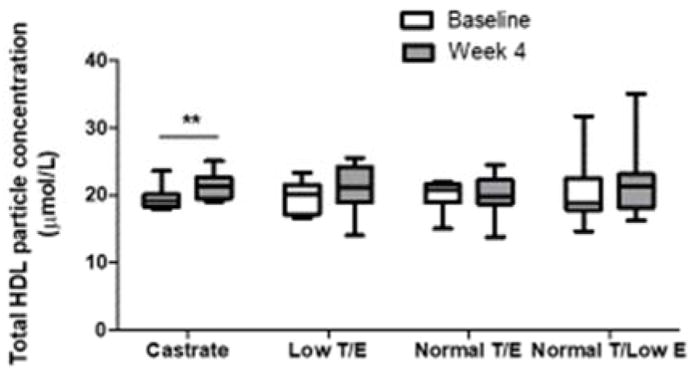
Change in total HDL particle concentration across treatment groups during the 4-week treatment period. A time-by-group interaction was not found for total HDL particle concentration, but a significant within-group change was noted for subjects in the Castrate group over the 4-week treatment period. Whiskers = minimum to maximum values. **p<0.01 for within-group change
Relative fractions of HDL particle subclasses were calculated, with extra-small, small, medium, and large particles expressed as a % of t-HDL-Pima. No differences were observed across the groups for extra small HDL particles (xs-HDL-Pima), nor were within-group differences observed over the treatment period (Figure 5A). In contrast, significant changes in the fraction of small HDL particles (s-HDL-Pima) were found among the groups during treatment (p=0.009 in overall RM-ANOVA, Figure 5B). In the 2 treatment groups rendered testosterone deficient, s-HDL-Pima decreased, whereas s-HDL-Pima increased in the other 2 groups, with a statistically significant within-group decrease in s-HDL-Pima for the Low T/E group (p=0.002, Figure 5B). The treatment-induced change in s-HDL-Pima exhibited an inverse correlation with change in serum HDL-C concentration (β=−0.37, p=0.01) but did not correlate with changes in serum sex steroid concentrations.
Figure 5.

Baseline and end-of-treatment HDL particle size distribution in all study subjects classified by treatment group. No differences across groups were observed for extra small HDL particle fraction (xs-HDL-Pima) (A), but a significant treatment effect was seen for small HDL particles (s-HDL-Pima) (B) with a significant decrease in small HDL particle fraction in the Low T/E group. No differences across groups were evident for medium HDL particle fraction (m-HDL-Pima) (C), but change in the fraction of large HDL particles (l-HDL-Pima) did differ across groups, with a significant decrease in large HDL particle fraction among subjects in the Normal E/Low E group. Data are presented as a % of total HDL particle concentration (total HDL-Pima). Whiskers = minimum to maximum values. *p<0.05 for within-group change
No differences in the fraction of medium HDL particles (m-HDL-Pima), were found among the treatment groups, nor were within-group changes observed for any group (Figure 5C). However, change in m-HDL-Pima exhibited a significant inverse correlation with change in serum 17β-estradiol concentration (β=−0.32 per 36.7 pmol/L change in 17β-estradiol, p=0.045). Changes in the fraction of large HDL particles (l-HDL-Pima) differed among groups over the treatment period (p=0.03 in overall RM-ANOVA, Figure 5D), with a significant within-group decrease evident in the Normal T/Low E group (p=0.005). Interestingly, the changes in l-HDL-Pima correlated significantly with change in both serum testosterone and 17β-estradiol concentrations but with opposite relationships. Thus, change in serum testosterone correlated negatively with change in l-HDL-Pima (β=−0.51 per 3.47 nmol/L change in testosterone, p=0.003, Figure 6A), whereas change in serum 17β-estradiol showed a positive correlation with change in l-HDL-Pima (β=0.49 per 36.7 pmol/L change in 17β-estradiol, p=0.004, Figure 6B). In sensitivity analyses, significant treatment effects persisted for s-HDL-Pima and l-HDL-Pima with exclusion of the subject with a very high serum HDL-C concentration (p=0.004 and p=0.03 in overall RM-ANOVA, respectively). Further, a subject in the Normal T/E group exhibited a very high increment in serum 17β-estradiol concentration during the treatment period. However, the correlation between changes in serum 17β-estradiol concentration and l-HDL-Pima remained significant though attenuated when this subject was excluded (β=0.36 per 36.7 pmol/L change in 17β-estradiol, p=0.02).
Figure 6.
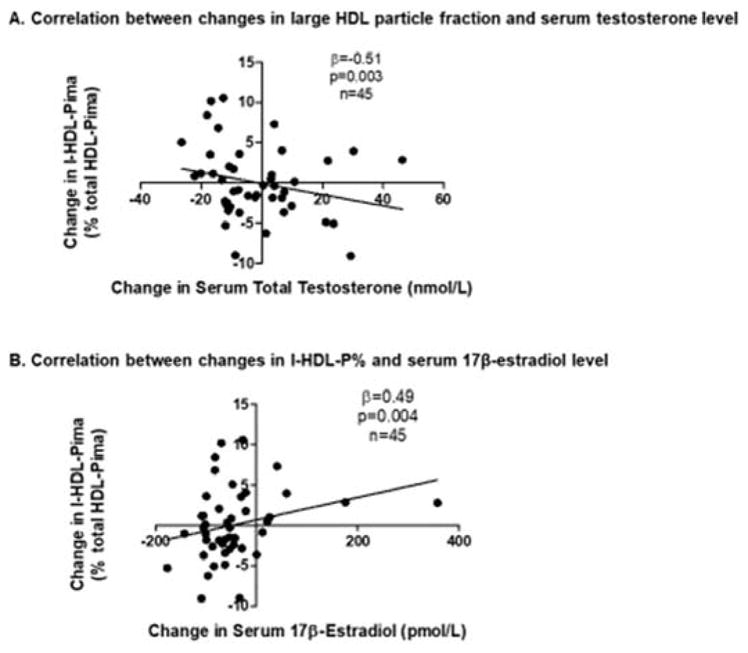
Correlations between changes in serum sex steroid concentrations and change in the fraction of large HDL particles. When all study subjects were grouped as a single cohort, a significant inverse association was found between change in serum testosterone concentration and change in the fraction of large HDL particles (A), whereas a positive association was found between change in serum 17β-estradiol concentration and change in the fraction of large HDL particles (B).
Change in the fraction of small HDL particles is the primary determinant of changes in the abundance of HDL-associated proteins
The abundance of 34 HDL-associated proteins was determined at baseline and at the end of the 4-week treatment period using targeted proteomics. Across treatment groups, no significant differences were found for changes in the abundance of any HDL-associated proteins after correcting for multiple comparisons (Supplementary Table 1). Consistent with these negative findings in the primary analysis, changes in serum sex steroid concentrations correlated with changes in the abundance of only 3 HDL-associated proteins. Change in serum testosterone concentration showed a significant inverse correlation with change in HDL-associated apolipoprotein L1 (APOL1) abundance (β=−0.36 per 3.47 nmol/L change in testosterone, p=0.01). Change in serum 17β-estradiol concentration exhibited a strong inverse correlation with change in abundance of HDL-associated apolipoprotein C4 (APOC4) (β=−0.41 per 36.7 pmol/L change in 17β-estradiol, p=0.004) and a weaker, inverse correlation with change in abundance of HDL-associated apolipoprotein A-I (APOA1) (β=−0.34 per 36.7 pmol/L change in 17β-estradiol, p=0.03). Of the HDL-Pima metrics, the change in s-HDL-Pima was significantly, positively correlated with changes in the abundance of 13 of 34 measured proteins, whereas change in l-HDL-Pima correlated only with the change in abundance of prenylcysteine oxidase 1 (PCYOX1) (Supplementary Table 2). Change in total macrophage CEC correlated significantly with changes in the abundance of 3 HDL-associated proteins [clusterin, APOA1, and paraoxonase 3 (PON3)].
Discussion
We demonstrate that short-term testosterone deprivation increases serum concentrations of HDL-C in healthy men but that these increases in HDL-C are not reflected by enhanced HDL CEC. Change in serum testosterone did not associate with changes in total macrophage or ABCA1 CEC. In contrast, change in serum 17β-estradiol concentration correlated positively with the change in total macrophage but not ABCA1 CEC. Changes in both serum testosterone and 17β-estradiol concentrations were strongly associated with changes in HDL particle concentration and subclasses. Medical castration but not partial testosterone deprivation led to increases in total HDL particle concentration. Further, testosterone deprivation associated with an increase in the fraction of large HDL particles, whereas selective estrogen deprivation led to a decrease in the fraction of large HDL particles. Collectively, these findings indicate discrete roles for androgens and estrogens in the regulation of HDL particle size distribution and HDL function and highlight the dissociation between HDL-C concentrations and HDL function (serum HDL CEC) with sex steroid manipulation.
Serum HDL CEC is strongly associated with both prevalent and incident CVD,13,14 suggesting it could prove both an important therapeutic target and a clinical metric of CVD risk.32 However, the positive association between CEC and CVD risk has not been a universal finding. For example, pharmacologic inhibition of cholesteryl ester transfer protein (CETP) markedly increased HDL CEC but failed to reduce incident CVD.33 Higher serum HDL CEC also has been found in subjects with familial hypertriglyceridemia and metabolic syndrome relative to healthy controls,34,35 findings that may result from the positive association between serum triglyceride and pre-β1-HDL concentrations.36
To date, only a few studies have examined the relationship between sex steroids and alternative HDL-associated metrics in men. Whereas we previously found that short-term medical castration in men led to increased total macrophage CEC,25 in both the previous and the present studies, we consistently observed an increase in HDL-C with medical castration that was not reflected by proportionate increases in CEC. Conversely, in an earlier study of hypogonadal older men, testosterone replacement did not significantly change serum HDL-C concentrations or total macrophage CEC.26 These negative findings are most likely due to the fact that subjects had only mild, age-associated hypogonadism, and serum concentrations of both testosterone and estradiol rose only modestly with testosterone replacement.
Interestingly, change in serum 17β-estradiol concentration exhibited a positive association with the change in total macrophage but not ABCA1-mediated CEC. Previous work demonstrated that in the J774 model system, total macrophage CEC comprises pathways mediated by ABCA1, ABCG1, and SR-BI and passive diffusion.37 Thus, our findings suggest that estradiol may increase cholesterol efflux via ABCG1- and/or SR-BI-mediated pathways, possibly by changing the distribution of HDL particle subclasses. In this study, estradiol exposure associated strongly with an increase in large HDL particles, and a recent clinical study identified a strong, positive association between large HDL particles and SR-BI-mediated efflux.36 Finally, a significant increase in ABCA1 CEC was observed exclusively among subjects rendered estrogen deficient. Mechanisms underlying this observation are unknown, but this finding underscores the importance of measuring pre-β1-HDL and plasminogen in future studies, as these may be significant contributors to ABCA1-specific efflux by serum HDL.11,16
HDL particle size has been shown to influence HDL function and to associate with CVD risk. The smaller HDL particles predominantly mediate cholesterol efflux through the ABCA1-dependent pathway, whereas larger HDL particles preferentially mediate efflux through ABCG1- and SR-BI-dependent pathways.15,38 Our finding of a positive correlation between changes in serum 17β-estradiol concentration and the fraction of large HDL particles is consistent with previous studies in women that have shown positive correlations between serum estradiol and large HDL subclasses.39,40 Further, an increase in the larger, more buoyant, HDL2 subclass was found after 8 weeks of transdermal estrogen therapy in a small cohort of men undergoing androgen deprivation for treatment of prostate cancer.41 In contrast to estrogen-mediated effects, change in serum testosterone exhibited a negative correlation with change in the relative fraction of large HDL particles. These opposite associations of testosterone and estradiol with large HDL particle concentration likely explain why no significant within-group changes in the fraction of large HDL particles were found for the Castrate or Low T/E groups, which underwent simultaneous androgen and estrogen deprivation. The opposing effects of testosterone and estradiol on large HDL particles may also explain the variable findings from past studies that have examined changes in HDL particle size consequent to testosterone replacement therapy.42,43 Both testosterone and estradiol play roles in the regulation of hepatic lipase, 43,44 an effect that may prove a key mechanism whereby sex steroids contribute to the remodeling of HDL particles. Testosterone and estrogen also have been implicated in the regulation of SR-BI expression 45,46 and therefore could influence hepatic uptake of HDL-associated cholesterol as well as HDL remodeling. Recent work also suggested that HDL particles of varying sizes may be secreted from the liver concurrently and remain unchanged in circulation,47 raising the possibility that sex steroid signaling within hepatocytes could influence the size distribution of secreted HDL particles.
Additional work is needed to better establish the mechanisms whereby sex steroids influence HDL particle size, and, moreover, to determine how this remodeling affects HDL particle function. Across groups, no significant differences were found in the changes in abundance of HDL-associated proteins over the 4-week treatment period. In contrast, change in the fraction of small HDL particles showed significant correlations with nearly 40% of the quantified HDL-associated proteins. Notably, all of these correlations were positive, suggesting that HDL particles may become enriched in these proteins as their size and lipid cargo decrease. A critical future area of research therefore will be the simultaneous quantification of HDL phospholipid and neutral lipid cargo after sex steroid manipulation in men. In our prior pilot study of experimental testosterone deprivation in healthy men, medical castration resulted in an increase in abundance of HDL-associated clusterin.25 The absence of this finding in our current study is most likely due to the fact that macrophage CEC increased significantly after medical castration in our prior study, as macrophage CEC was the only significant correlate of change in HDL-associated clusterin abundance in the present study. Changes in serum sex steroid concentrations did exhibit correlations with changes in the abundance of 3 HDL-associated proteins, the strongest of which was an inverse correlation between changes in serum 17β-estradiol and HDL-associated APOC4 abundance. The absence of any significant findings in the primary analysis could be explained by the fact that subjects in 3 of the 4 treatment groups were rendered estrogen deficient. In addition, our treatment duration was fairly short, and a longer treatment period may be necessary to induce greater sex steroid-mediated changes in HDL protein cargo. Nonetheless, our findings overall suggest that although sex steroids may indirectly influence the abundance of HDL-associated proteins through conferring changes in HDL particle size, they do not appear to be significant regulators of HDL protein composition.
Our study was limited by a relatively small sample size as it was not designed specifically for the HDL-associated endpoints. Although all subjects in our study cohort were healthy, eugonadal men, subjects spanned a broad range of ages and serum lipid concentrations. Thus, follow up studies will be necessary to clearly determine interactions between sex steroid exposure and age and other indices of cardiometabolic risk in the regulation of HDL composition and function. Importantly, too, the utility of quantifying changes in HDL CEC after a clinical intervention for predicting cardiovascular outcomes remains to be established. To date, aggregate findings underscore the limited clinical utility of any single HDL-associated metric in isolation for predicting CVD risk on an individual basis and highlight the need to identify novel metrics that specifically reflect HDL-mediated cholesterol efflux from vascular macrophages, a critical determinant of HDL’s atheroprotective effects.48 Nonetheless, our study benefited from use of well validated assays to quantify serum HDL CEC and HDL particle concentration and size.
Conclusion
Our findings support roles for both testosterone and estradiol in regulating HDL particle size and function in men. They suggest that changes in HDL-C consequent to testosterone deprivation may not be a reliable predictor of changes in HDL CEC, and perhaps, therefore, may not impact CVD risk through this mechanism. Additional work is essential to delineate pathways by which sex steroids may regulate HDL remodeling and function and thereby help clarify the respective roles of androgens and estrogens in influencing cardiovascular risk in men. Critically, too, clinical studies are needed to determine whether the present findings are similarly evident with sex steroid manipulation in clinical settings, including exogenous testosterone replacement therapy for male hypogonadism, male hormonal contraceptive regimens, or androgen deprivation therapy for treatment of prostate cancer.
Supplementary Material
Supplementary Table 1. Differences in the change in abundance of HDL-associated proteins across treatment groups
Highlights.
Testosterone deprivation in healthy men increased HDL-C and HDL particle number.
A corresponding increase in HDL cholesterol efflux capacity was not observed.
Estrogen deprivation increased ABCA1-specific cholesterol efflux capacity.
HDL cholesterol may not reflect sex steroid-mediated changes in HDL function.
Acknowledgments
This work was supported by an American Heart Association Grant-in-Aid Award; the National Institutes of Health [grant numbers 6K12 HD053984, 30 DK017047, T32DK007247-37, HD042454, and RO1AG037603]; and the University of Washington Robert McMillen Professorship in Lipid Research. Testosterone and placebo gel were supplied by AbbVie Inc. (Chicago, IL).
Footnotes
Disclosure summary: The authors report no conflicts of interest in this work.
This study was registered at ClinicalTrials.gov (ID NCT01686828).
Authors’ Contributions
KBR performed the study, analyzed resultant data, and drafted the manuscript; TV contributed to the study design, performed HDL-based assays, analyzed resultant data, and revised the manuscript; JHC performed the study and revised the manuscript; JWH contributed to the study design, performed HDL-based assays, and revised the manuscript; STP contributed to the study design, provided study funding, and revised the manuscript. All authors approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corona G, Rastrelli G, Monami M, et al. Hypogonadism as a risk factor for cardiovascular mortality in men: a meta-analytic study. Eur J Endocrinol. 2011;165(5):687–701. doi: 10.1530/EJE-11-0447. [DOI] [PubMed] [Google Scholar]
- 2.Spitzer M, Huang G, Basaria S, Travison TG, Bhasin S. Risks and benefits of testosterone therapy in older men. Nat Rev Endocrinol. 2013;9(7):414–424. doi: 10.1038/nrendo.2013.73. [DOI] [PubMed] [Google Scholar]
- 3.Wu FC, von Eckardstein A. Androgens and coronary artery disease. Endocr Rev. 2003;24(2):183–217. doi: 10.1210/er.2001-0025. [DOI] [PubMed] [Google Scholar]
- 4.Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79(1):8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 6.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 7.Toth PP, Barter PJ, Rosenson RS, et al. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7(5):484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Karathanasis SK, Freeman LA, Gordon SM, Remaley AT. The Changing Face of HDL and the Best Way to Measure It. Clin Chem. 2017;63(1):196–210. doi: 10.1373/clinchem.2016.257725. [DOI] [PubMed] [Google Scholar]
- 9.Hoekstra M. SR-BI as target in atherosclerosis and cardiovascular disease - A comprehensive appraisal of the cellular functions of SR-BI in physiology and disease. Atherosclerosis. 2017;258:153–161. doi: 10.1016/j.atherosclerosis.2017.01.034. [DOI] [PubMed] [Google Scholar]
- 10.Connelly MA, Williams DL. SR-BI and HDL cholesteryl ester metabolism. Endocr Res. 2004;30(4):697–703. doi: 10.1081/erc-200043979. [DOI] [PubMed] [Google Scholar]
- 11.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30(4):796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenson RS, Brewer HB, Ansell B, et al. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation. 2013;128(11):1256–1267. doi: 10.1161/CIRCULATIONAHA.113.000962. [DOI] [PubMed] [Google Scholar]
- 13.Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371(25):2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinecke JW. Small HDL promotes cholesterol efflux by the ABCA1 pathway in macrophages: implications for therapies targeted to HDL. Circ Res. 2015;116(7):1101–1103. doi: 10.1161/CIRCRESAHA.115.306052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pamir N, Hutchins PM, Ronsein GE, et al. Plasminogen promotes cholesterol efflux by the ABCA1 pathway. JCI Insight. 2017;2(15) doi: 10.1172/jci.insight.92176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Superko HR, Pendyala L, Williams PT, Momary KM, King SB, Garrett BC. High-density lipoprotein subclasses and their relationship to cardiovascular disease. J Clin Lipidol. 2012;6(6):496–523. doi: 10.1016/j.jacl.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Hutchins PM, Ronsein GE, Monette JS, et al. Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin Chem. 2014;60(11):1393–1401. doi: 10.1373/clinchem.2014.228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khera AV, Demler OV, Adelman SJ, et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin) Circulation. 2017;135(25):2494–2504. doi: 10.1161/CIRCULATIONAHA.116.025678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otvos JD, Collins D, Freedman DS, et al. Low-density lipoprotein and high-density lipoprotein particle subclasses predict coronary events and are favorably changed by gemfibrozil therapy in the Veterans Affairs High-Density Lipoprotein Intervention Trial. Circulation. 2006;113(12):1556–1563. doi: 10.1161/CIRCULATIONAHA.105.565135. [DOI] [PubMed] [Google Scholar]
- 21.Parish S, Offer A, Clarke R, et al. Lipids and lipoproteins and risk of different vascular events in the MRC/BHF Heart Protection Study. Circulation. 2012;125(20):2469–2478. doi: 10.1161/CIRCULATIONAHA.111.073684. [DOI] [PubMed] [Google Scholar]
- 22.Chao J, Rubinow KB, Kratz M, Amory JK, Matsumoto AM, Page ST. Short-Term Estrogen Withdrawal Increases Adiposity in Healthy Men. J Clin Endocrinol Metab. 2016;101(10):3724–3731. doi: 10.1210/jc.2016-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbst KL, Coviello AD, Page S, Amory JK, Anawalt BD, Bremner WJ. A single dose of the potent gonadotropin-releasing hormone antagonist acyline suppresses gonadotropins and testosterone for 2 weeks in healthy young men. J Clin Endocrinol Metab. 2004;89(12):5959–5965. doi: 10.1210/jc.2003-032123. [DOI] [PubMed] [Google Scholar]
- 24.Guy J, Ogden L, Wadwa RP, et al. Lipid and lipoprotein profiles in youth with and without type 1 diabetes: the SEARCH for Diabetes in Youth case-control study. Diabetes Care. 2009;32(3):416–420. doi: 10.2337/dc08-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinow KB, Tang C, Hoofnagle AN, et al. Acute sex steroid withdrawal increases cholesterol efflux capacity and HDL-associated clusterin in men. Steroids. 2012;77(5):454–460. doi: 10.1016/j.steroids.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubinow KB, Vaisar T, Tang C, Matsumoto AM, Heinecke JW, Page ST. Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity. J Lipid Res. 2012;53(7):1376–1383. doi: 10.1194/jlr.P026005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergt C, Pennathur S, Fu X, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101(35):13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Khoudary SR, Hutchins PM, Matthews KA, et al. Cholesterol Efflux Capacity and Subclasses of HDL Particles in Healthy Women Transitioning Through Menopause. J Clin Endocrinol Metab. 2016;101(9):3419–3428. doi: 10.1210/jc.2016-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenson RS, Brewer HB, Chapman MJ, et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. 2011;57(3):392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- 30.Hoofnagle AN, Becker JO, Oda MN, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58(4):777–781. doi: 10.1373/clinchem.2011.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt A, Rohatgi A. HDL Cholesterol Efflux Capacity: Cardiovascular Risk Factor and Potential Therapeutic Target. Curr Atheroscler Rep. 2016;18(1):2. doi: 10.1007/s11883-015-0554-1. [DOI] [PubMed] [Google Scholar]
- 33.Nicholls SJ, Ruotolo G, Brewer HB, et al. Cholesterol Efflux Capacity and Pre-Beta-1 HDL Concentrations Are Increased in Dyslipidemic Patients Treated With Evacetrapib. J Am Coll Cardiol. 2015;66(20):2201–2210. doi: 10.1016/j.jacc.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Lucero D, Sviridov D, Svidirov D, et al. Increased cholesterol efflux capacity in metabolic syndrome: Relation with qualitative alterations in HDL and LCAT. Atherosclerosis. 2015;242(1):236–242. doi: 10.1016/j.atherosclerosis.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 35.Attia N, Ramaharo A, Paul JL, et al. Enhanced removal of cholesterol from macrophage foam cells to serum from type IV hypertriglyceridemic subjects. Atherosclerosis. 2008;198(1):49–56. doi: 10.1016/j.atherosclerosis.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 36.Asztalos BF, Horvath KV, Mehan M, Yokota Y, Schaefer EJ. Influence of HDL particles on cell-cholesterol efflux under various pathological conditions. J Lipid Res. 2017;58(6):1238–1246. doi: 10.1194/jlr.M075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adorni MP, Zimetti F, Billheimer JT, et al. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48(11):2453–2462. doi: 10.1194/jlr.M700274-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Du XM, Kim MJ, Hou L, et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ Res. 2015;116(7):1133–1142. doi: 10.1161/CIRCRESAHA.116.305485. [DOI] [PubMed] [Google Scholar]
- 39.Kuller LH, Gutai JP, Meilahn E, Matthews KA, Plantinga P. Relationship of endogenous sex steroid hormones to lipids and apoproteins in postmenopausal women. Arteriosclerosis. 1990;10(6):1058–1066. doi: 10.1161/01.atv.10.6.1058. [DOI] [PubMed] [Google Scholar]
- 40.El Khoudary SR, Brooks MM, Thurston RC, Matthews KA. Lipoprotein subclasses and endogenous sex hormones in women at midlife. J Lipid Res. 2014;55(7):1498–1504. doi: 10.1194/jlr.P049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purnell JQ, Bland LB, Garzotto M, et al. Effects of transdermal estrogen on levels of lipids, lipase activity, and inflammatory markers in men with prostate cancer. J Lipid Res. 2006;47(2):349–355. doi: 10.1194/jlr.M500276-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Bolu E, Sonmez A, Tapan S, et al. HDL cholesterol subfractions and the effect of testosterone replacement in hypogonadism. Horm Metab Res. 2013;45(6):443–448. doi: 10.1055/s-0033-1343447. [DOI] [PubMed] [Google Scholar]
- 43.Tan KC, Shiu SW, Kung AW. Alterations in hepatic lipase and lipoprotein subfractions with transdermal testosterone replacement therapy. Clin Endocrinol (Oxf) 1999;51(6):765–769. doi: 10.1046/j.1365-2265.1999.00882.x. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava N, Chowdhury PR, Averna M, Srivastava RA. Estrogen increases hepatic lipase levels in inbred strains of mice: a possible mechanism for estrogen-dependent lowering of high density lipoprotein. Mol Cell Biochem. 2001;220(1–2):87–93. doi: 10.1023/a:1010845032399. [DOI] [PubMed] [Google Scholar]
- 45.Ansari KI, Kasiri S, Hussain I, Bobzean SA, Perrotti LI, Mandal SS. MLL histone methylases regulate expression of HDLR-SR-B1 in presence of estrogen and control plasma cholesterol in vivo. Mol Endocrinol. 2013;27(1):92–105. doi: 10.1210/me.2012-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langer C, Gansz B, Goepfert C, et al. Testosterone up-regulates scavenger receptor BI and stimulates cholesterol efflux from macrophages. Biochem Biophys Res Commun. 2002;296(5):1051–1057. doi: 10.1016/s0006-291x(02)02038-7. [DOI] [PubMed] [Google Scholar]
- 47.Mendivil CO, Furtado J, Morton AM, Wang L, Sacks FM. Novel Pathways of Apolipoprotein A-I Metabolism in High-Density Lipoprotein of Different Sizes in Humans. Arterioscler Thromb Vasc Biol. 2016;36(1):156–165. doi: 10.1161/ATVBAHA.115.306138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenson RS, Brewer HB, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125(15):1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Differences in the change in abundance of HDL-associated proteins across treatment groups