

Abstract
Objective
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal inherited disease characterized by ventricular arrhythmias induced by physical exercise or emotional stress. The major cause of CPVT is mutations in RYR2, which encodes the cardiac ryanodine receptor channel. Recent advances in sequencing technology have yielded incidental findings of RYR2 variants in other cardiac diseases. Analyzing the characteristics of RYR2 variants related to CPVT will be useful for differentiation from those related to other cardiac diseases. We examined the phenotypic characteristics of patients with RYR2 variants.
Methods
Seventy-nine probands carrying RYR2 variantswhose diagnoses were either CPVT (n=68) or long QT syndrome (LQTS; n=11) were enrolled. We compared the characteristics of the electrocardiogram (ECG) and the location of the RYR2 mutations-N-terminal (NT), central region (CR) or C-terminal (CT)-between the two patient groups.
Results
Using the ECGs available from 53 probands before β-blocker therapies, we analyzed the heart rates (HRs). CPVT probands showed bradycardia more frequently (25/44; 57%) than LQTS probands (1/9; 11%; p=0.024). In CPVT patients, 20 mutations were located in NT, 25 in CR and 23 in CT. In LQTS patients, 5 mutations were located in NT, 2 in CR and 4 in CT. There were no significant differences in the locations of the RYR2 mutations between the phenotypes.
Conclusion
Bradycardia was highly correlated with the phenotype of CPVT. When a clinically-diagnosed LQTS patient with bradycardia carries an RYR2 mutation, we should be careful to avoid making a misdiagnosis, as the patient may actually have CPVT.
Keywords: catecholaminergic polymorphic ventricular tachycardia, long QT syndrome, bradycardia, RYR2 mutation
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal inherited primary electrical disease characterized by bidirectional or polymorphic ventricular arrhythmias caused by exercise or emotional stress, particularly in young patients. The diagnosis includes the absence of the other primary electrical disease, such as long QT syndrome (LQTS) (1). CPVT shares clinical features with LQTS in terms of causing faintness or sudden death by exercise or emotional stress. It is therefore often difficult to make differential diagnosis between these two clinical situations (2,3).
Pathogenic RYR2 mutations were identified in nearly 60% of patients with CPVT (4). The cardiac ryanodine receptor channel (RyR2) encoded by RYR2 is a calcium release channel localized on the sarcoplasmic reticulum (SR) membrane. Gain-of-function type RYR2 mutations cause abnormal Ca2+ leak from the SR, resulting in delayed depolarization and ventricular arrhythmias observed in CPVT (1,5). Mutations in RYR2 are causative for inherited primary arrhythmia syndromes (IPASs), such as CPVT (1,5), LQTS (6), short-coupled polymorphic ventricular tachycardia (7,8) and idiopathic ventricular fibrillation (9).
One issue regarding the genetic screening of RYR2 is associated with its large size, as the gene consists of 105 exons. The recent clinical introduction of next-generation sequencing (NGS) (10) has allowed us to detect disease-causing mutations not only in RYR2 but in other candidate genes for IPASs. Using a targeted panel gene analysis with NGS, we extensively identified RYR2 variants in our cohort of clinically-diagnosed CPVT (n=68) and clinically-diagnosed LQTS (n=11) probands.
Therefore, the present study aimed (1) to examine the phenotypic characteristics of carriers with RYR2 variants and (2) to clarify the relationships among mutation locations in RYR2. These data will offer important insights into the contribution of RYR2 mutations to IPASs.
Materials and Methods
Patients
Seventy-nine probands carrying RYR2 variantswhose diagnoses were either CPVT (n=68) or LQTS (n=11) were enrolled. Table 1 shows the demographic characteristics of the study population. The diagnosis of CPVT or LQTS was made based on clinical symptoms, electrocardiogram (ECG) findings and modified Schwartz scores (2,11). When a patient had a Schwartz score of ≥3.5 and showed neither bidirectional ventricular tachycardia (bVT) nor polymorphic ventricular tachycardia (pVT) on ECG, we diagnosed the patient with LQTS. In contrast, when a patient had a Schwartz score of <3.5 or had a Schwartz score of ≥3.5 but showed bVT or pVT on ECG, we diagnosed the patient with CPVT.
Table 1.
Definition of Bradycardia in This Study.
| Age (years) | Heart rate (bpm) |
|---|---|
| 0 | <100 |
| 1-3 | <95 |
| 4-5 | <75 |
| 6-8 | <65 |
| ≥ 9 | <60 |
We excluded patients being treated with β-blockers from the analysis of the heart rate. Because the normal range of the heart rate varies with age, bradycardia was defined as follows: in children aged <9 years, a heart rate below the second percentile of the established age- and sex-appropriate norms; in adults or children ≥ 9 years, <60 beats/min on resting ECG without taking β-blockers (12,13) (Table 1). We compared the ECG findings, such as the heart rate and QTc interval, between patients with each disease.
Genetic analyses
The protocol for the genetic analyses was approved by our Institutional Ethics Committee and carried out under the guidance of this committee. All patients and their guardians provided their written informed consent before undergoing a genetic analysis. Genomic DNA was isolated from peripheral white blood cells. RYR2 variants were screened by the Sanger method using an ABI PRISM-3130 (Applied Biosystems, Foster City, USA) or by targeted gene-sequencing methods using MiSeq (Illumina, San Diego, USA) and confirmed by the Sanger method. NGS screening was also used to search for variants of LQTS-related genes. We excluded from the analysis variants for which the minor allele frequencies (MAFs) in ethnicity-matched controls were greater than 0.005.
All variants were evaluated by several in silico predicting software programs: Polymorphism Phenotyping (PolyPhen)-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), PROVEAN (http://provean.jcvi.org/index.php) and CADD (http://cadd.gs.washington.edu/score). Scores for each variant are summarized in the Supplementary material. When the variants were reported in the ExAC database (http://exac.broadinstitute.org/), dbSNP database (https://www.ncbi.nlm.nih.gov/snp/) or in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), we added the information to the Supplementary material.
RyR2 mutation locations
According to the previous report (14), we categorized the RYR2 mutation sites as N-terminal (NT: 1-2177), central region (CR: 2178-4075) and C-terminal (CT: 4076-4959).
Statistical analyses
All statistical analyses were conducted using the Stata version 14.0 software program (LightStone, Tokyo, Japan). Continuous variables with normal distribution are shown as the mean ± standard deviation (SD), and variables without a normal distribution are shown as the median [interquartile range (IQR)]. Levene's tests were conducted to assess the equality of variance for comparable groups. Differences between groups were evaluated by Fisher's exact test, Student's t-test and the Mann-Whitney U test when necessary. All p values <0.05 were considered statistically significant.
Results
Clinical characteristics of the study population
The demographic characteristics of the study patients are summarized in Table 2. The clinical features of each patient and variant information are summarized in the Supplementary material. Forty-three (54.4%) patients were men, of whom 36 had CPVT and 7 LQTS. The mean age at the time of the genetic analysis showed no significant differences between CPVT and LQTS (p=0.51). However, patients with LQTS (n=11) had a significantly longer QTc interval and higher Schwartz score than those with CPVT. The median QTc interval in patients with LQTS was 480.0 (460.0-515.0), and the mean Schwartz score was 4.7±1.5. In contrast, in CPVT patients, the median QTc interval was 411.5 (392.8-433.3), and the mean Schwartz score was 2.3±0.9.
Table 2.
Demographic Characteristics of the Study Population.
| CPVT | LQTS | p value | |
|---|---|---|---|
| Number | 68 | 11 | |
| Male | 36 (53%) | 7 (64%) | 0.75# |
| Age (IQR) | 13.0 (10.0-16.3) | 16.0 (8.5-32.0) | 0.51* |
| QTc (IQR) | 411.5 (392.8-433.3) | 480.0 (460.0-515.0) | <0.001* |
| Schwartz score | 2.3±0.9 | 4.7±1.5 | <0.001** |
| Patients with CPA | 29 (43%) | 3 (27%) | 0.51# |
Values for Age and QTc are represented by median (IQR: inter-quartile range).
Values for Schwartz score is mean±SD.
CPA: cardiopulmonary arrest
# Fisher’s exact test, *Mann-Whitney U test, **Student t-test
Among the 79 probands, 29 CPVT patients (43%) and 3 LQTS patients (27%) experienced fatal cardiac events that required cardiopulmonary resuscitation (CPR), showing no significant differences in the disease severity between the 2 phenotypes (p=0.51).
Heart rate in RYR2 mutation carriers
We excluded 26 patients (CPVT, n=24; LQTS, n=2) from the analysis of the heart rate because we could not obtain their ECGs before the administration of β-blockers. Fig. 1 shows the relationship between the heart rate at rest and the age of ECG recording in patients with CPVT and LQTS. The heart rates in CPVT patients (open circles) were lower than those of LQTS patients (filled circles), especially at older ages. Fig. 2A depicts the frequency distribution of patients showing bradycardia: 25 out of 44 CPVT patients (57%) vs. 1 out of 9 LQTS patients (11%). Thus, the proportion of patients with CPVT having bradycardia was significantly higher than that of patients with LQTS (p=0.024).
Figure 1.
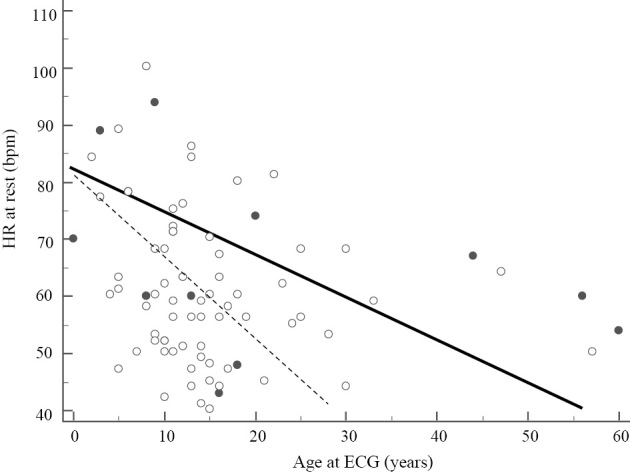
Relationship between the HR at rest and the age of ECG recordings. The HRs in CPVT patients (open circle) were lower than those of LQTS (filled circle). Open circles were fitted to the dotted line and filled circles were to the black line.
Figure 2.
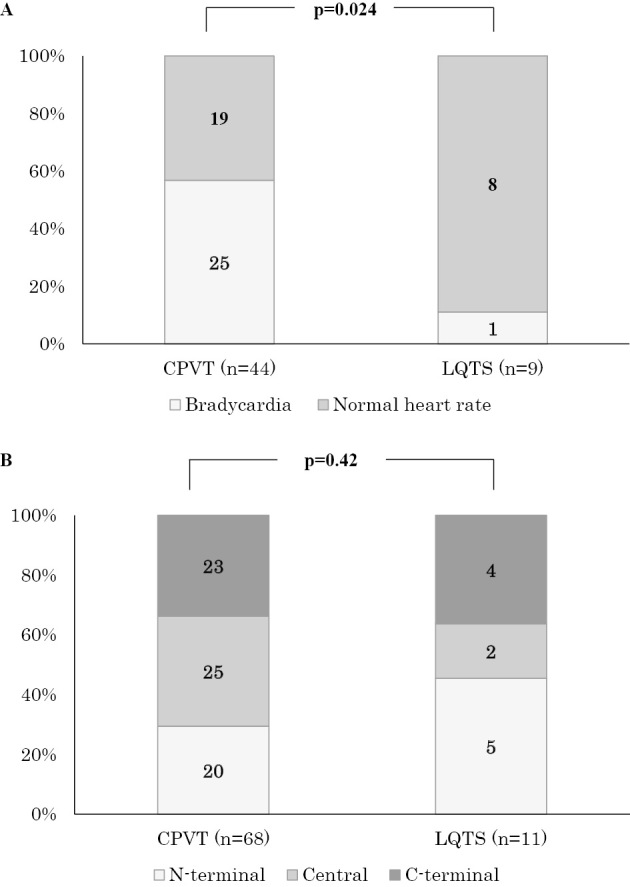
Distribution of bradycardia and RYR2 mutation locations in patients with CPVT and LQTS. A) Distribution of the bradycardia. The proportion of patients with CPVT having bradycardia was significantly higher than that of patients with LQTS. B) Mutation locations in RYR2. There were no significant differences in the locations of RYR2 mutations between CPVT and LQTS.
Location of mutations
Fig. 2B shows the frequency of the three different locations of RYR2 mutations described above. Regarding how the distribution of RYR2 mutation sites differed between the two different phenotypes, no significant differences were found in the mutation sites.
Discussion
For several years, RYR2 mutations have been regarded as specific for CPVT. However, NGS has enabled us to identify RYR2 mutations in other phenotypes as well, such as LQTS. Therefore, we can no longer diagnose a patient with CPVT simply because they carry an RYR2 mutation. In this study, we analyzed the phenotypical and genotypical characteristics of 79 probands with RYR2 variants whose diagnoses were either CPVT or LQTS. To differentiate CPVT from LQTS, we used modified Schwartz scores (11). Regarding the Schwartz score definitions, patients with a score ≥3.5 were deemed likely to have LQTS. However, we detected 8 CPVT patients with a score ≥3.5 and with bVT at ECG. Our data suggest that some CPVT patients with higher Schwartz scores might be misdiagnosed as LQTS despite showing bVT, which is specific for CPVT.
A previous (12) stated that one characteristic finding of CPVT on resting ECG is bradycardia, which may aid in the diagnosis of CPVT. In the present study, we found that bradycardia was more frequent in patients with CPVT than in those with LQTS. We also hypothesized that the locations of RYR2 mutations might be useful for distinguishing CPVT from LQTS, but the mutation locations were not associated with the phenotype in this study. Taken together, our findings suggested that the detection of RYR2 mutations during the genetic analysis may indicate the presence of CPVT, even in patients with a modified Schwartz score ≥3.5, particularly when the patient exhibits bVT or pVT and bradycardia on ECG.
There are no international diagnostic criteria for distinguishing CPVT from LQTS, and it is difficult to conclusively diagnose patients who have both QT prolongation and bVT or pVT with either of these diseases. There may even be a combined arrhythmic disease with a phenotype of both CPVT and LQTS. In the present study, however, we differentiated our patients' diagnoses based on the definition shown in the Methods section. Further studies will be needed to establish diagnostic criteria for combined arrhythmic diseases.
Immediately after recovery from a critical cardiac event, the QTc intervals are often prolonged. Therefore, we cannot decide whether a patient has CPVT or LQTS based solely on the QTc interval immediately after a cardiac event,. Although the exercise stress test (EST) may offer another reliable tool for a differential diagnosis, exercise itself also significantly prolongs the QTc intervals in RYR2-positive cases, as reported by Medeiros-Domingo et al. (15). In addition, the EST is not suitable for certain patients, such as those with severe brain injury. For these reasons, CPVT patients may be misdiagnosed with LQTS (2).
Study limitations
In this study, we included all of the RYR2 variants with MAFs <0.005, even if the variants were predicted to be benign by an in silico evaluation. Thus, some of the variants might not have affected the phenotype of the patients in this study. In addition, our population was relatively small, and further studies in larger populations will be needed to confirm the phenotypical differences between CPVT and LQTS patients with RYR2 mutations.
Conclusion
Among 79 RYR2 mutation carriers, bradycardia was more frequent in patients with CPVT than in those with LQTS. This difference may be useful for distinguishing CPVT from LQTS induced by RYR2 mutations. With the advent of NGS, we should be careful when diagnosing carriers of RYR2 mutations, especially those who show bradycardia.
The authors state that they have no Conflict of Interest (COI).
Financial Support
This work was supported by The Research Grant (KAKENHI) for Cardiovascular Diseases from the Ministry of Education, Culture, Sports, Science and Technology of Japan for Research on Hereditary Arrhythmias, the Health Sciences Research Grants for Clinical Research on Measures for Intractable Disease from the Ministry of Health, Labor, and Welfare of Japan, and by a Research Grant from Takeda Science foundation.
Supplementary Material
Clinical features and variant information of the patients
Acknowledgement
We thank all of the patients and their family members for participating in this study. We are also grateful to Ms. Arisa Ikeda, Ms. Madoka Tanimoto and Ms. Kazu Toyooka for their excellent technical assistance.
References
- 1.Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103: 196-200, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106: 69-74, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation 110: 2119-2124, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 119: 2426-2434, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Blayney LM, Lai FA. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther 123: 151-177, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taniguchi Y, Miyazaki A, Sakaguchi H, et al. Prominent QTc prolongation in a patient with a rare variant in the cardiac ryanodine receptor gene. Heart Vessels 32: 229-233, 2017. [DOI] [PubMed] [Google Scholar]
- 7.Fujii Y, Itoh H, Ohno S, et al. A type 2 ryanodine receptor variant associated with reduced Ca2+ release and short-coupled torsades de pointes ventricular arrhythmia. Heart Rhythm 14: 98-107, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Cheung JW, Meli AC, Xie W, et al. Short-coupled polymorphic ventricular tachycardia at rest linked to a novel ryanodine receptor (RyR2) mutation: leaky RyR2 channels under non-stress conditions. Int J Cardiol 180: 228-236, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paech C, Gebauer RA, Karstedt J, Marschall C, Bollmann A, Husser D. Ryanodine receptor mutations presenting as idiopathic ventricular fibrillation: a report on two novel familial compound mutations, c.6224T>C and c.13781A>G, with the clinical presentation of idiopathic ventricular fibrillation. Pediatr Cardiol 35: 1437-1441, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Buckton AJ, Wilkinson SL, et al. Towards clinical molecular diagnosis of inherited cardiac conditions: a comparison of bench-top genome DNA sequencers. PLoS One 8: e67744, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 124: 2181-2184, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Kawamura M, Ohno S, Naiki N, et al. Genetic background of catecholaminergic polymorphic ventricular tachycardia in Japan. Circ J 77: 1705-1713, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Rijnbeek PR, Witsenburg M, Schrama E, Hess J, Kors JA. New normal limits for the paediatric electrocardiogram. Eur Heart J 22: 702-711, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med 3: 43-52, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol 54: 2065-2074, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical features and variant information of the patients