

Abstract
Background
Endothelial microparticles are associated with chronic kidney disease (CKD) and complement activation. We hypothesized that the complement pathway is activated in patients with CKD via endothelial microparticles and that complement activation correlates with endothelial dysfunction in CKD.
Methods and Results
We analyzed complement data of 30 healthy subjects, 30 patients with stage III/IV CKD, and 30 renal transplant recipients with stage III/IV CKD, evaluating the potential correlation of complement fragments with brachial artery flow–mediated dilation, Chronic Kidney Disease Epidemiology Collaboration glomerular filtration rate, and urinary albumin/creatinine ratio. Endothelial microparticles were characterized via proteomic analysis and compared between study groups. Complement fragment Ba was significantly increased in CKD and post–kidney transplant CKD. Plasma Ba levels correlated significantly with lower brachial artery flow–mediated dilation, lower Chronic Kidney Disease Epidemiology Collaboration glomerular filtration rate, and higher urinary albumin/creatinine ratio. Factor D levels were significantly higher in the plasma microparticles of patients with CKD versus healthy controls. Plasma microparticles isolated from patients with CKD and containing factor D activated the alternative pathway in vitro.
Conclusion
The alternative complement pathway is activated in CKD and correlates with endothelial dysfunction and markers of CKD. Future studies are needed to evaluate whether endothelial microparticles with increased factor D play a pathologic role in CKD‐associated vascular disease.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT02230202.
Keywords: chronic kidney disease, Microparticles complement activation
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Inflammation
Clinical Perspective
What Is New?
In patients with chronic kidney disease, the alternative complement pathway is activated and correlates with endothelial dysfunction and with markers of kidney disease, and microparticles with increased factor D are more prevalent and may play an important role in the activation of the complement pathway.
What Are the Clinical Implications?
Future studies are needed to evaluate complement fragments and microparticles as markers of cardiovascular and kidney disease and as potential therapeutic targets.
Introduction
Chronic kidney disease (CKD) is a highly prevalent condition1 and an independent risk factor for cardiovascular disease (CVD). Approximately 50% of patients with CKD will die of cardiovascular complications.2 Indeed, patients with CKD are more likely to die of CVD than they are to reach end‐stage renal disease.3, 4 The increased risk of CVD in this patient population is only partially explained by traditional (Framingham)5, 6 risk factors, and “nontraditional” factors likely influence the risk of CVD.7 Systemic inflammation has been proposed to contribute to the development of CVD in patients with CKD,8 as inflammatory markers, such as C‐reactive protein and interleukin‐6, are elevated in CKD.9, 10, 11, 12 In some observational studies, cytokines prospectively predicted the development of CVD.12
Greater understanding of the underlying molecular causes of inflammation in CKD may lead to biomarkers that more accurately predict the risk of CVD than those currently available and may reveal new therapeutic targets for preventing cardiovascular complications. Immunosuppressive drugs are commonly used in CKD to treat concomitant diseases, but little is known about the direct effects of these drugs on systemic inflammation. Several drugs that target specific immune targets have been tested in CKD, but these studies have generated conflicting results.13, 14 Given the heterogeneity of CKD and the complexity of the immune system, the development of effective and safe therapies will require additional insights into pathogenesis and new mechanistic biomarkers of immune activation in this disease.
Microparticles (also called extracellular vesicles) are submicrometer‐sized membrane vesicles (0.05–1 μm diameter) that are actively shed from cells in response to activation, injury, and apoptosis.15, 16 A number of recent studies have shown that diseases of the vasculature and kidneys, including CKD, are associated with increased numbers of circulating endothelial microparticles.17, 18, 19, 20, 21, 22 We have previously reported that the microparticles released from endothelial cells can, under some conditions, activate the complement system, and that generation of complement‐activating microparticles is associated with vascular and renal injury in mice.23 The complement system is an important part of the body's defense against pathogens, but complement activation also contributes to the pathogenesis of a broad range of diseases, including CVD, ischemia/reperfusion injury, atypical hemolytic uremic syndrome, and renal allograft injury.24, 25, 26, 27, 28, 29 The production of complement‐activating microparticles might trigger acute inflammatory diseases (such as atypical hemolytic uremic syndrome) in susceptible patients, and the continuous production of complement‐activating microparticles might also be an important cause of chronic inflammation in CKD. As such, complement activation has been proposed to play a role in CKD.30 Consistent with this hypothesis, we previously observed elevated levels of complement activation fragments in a small number of patients with CKD.31
Based upon the observations discussed above, we hypothesized that microparticles generated in patients with CKD activate the complement system and are associated with vascular endothelial dysfunction. Endothelial dysfunction is common in CKD and has been shown to predict CVD prospectively in several groups, including healthy individuals,32, 33 individuals with peripheral vascular disease,34 and CKD.35 To explore this hypothesis we recruited healthy control subjects and patients with stage III and IV CKD. We also recruited renal transplant recipients with renal dysfunction in order to examine the effects of immunosuppression on the complement system in the setting of CKD. We measured vascular endothelial function and the levels of complement activation fragments, and we examined the molecular composition of microparticles in plasma from these patients. The overall goal of these experiments was to identify novel biomarkers of vascular injury in patients with CKD and to explore potential novel mechanisms of vascular and renal injury in these patients.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Patient Characteristics
This is a pilot study including 30 healthy subjects, 30 patients with stage III and IV CKD , and 30 renal transplant recipients with stage III and IV CKD. Healthy subjects were recruited by public advertisement. Patients with stage III and IV CKD and kidney transplant recipients receiving maintenance immunosuppression consisting of tacrolimus, mycophenolate mofetil, and corticosteroids were recruited from our CKD and transplant clinics, respectively, at the University of Colorado Hospital. Individuals with CKD and kidney transplant recipients were considered eligible for participation if they were at least 18 years of age, had stage III/IV CKD with a Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) estimated glomerular filtration equation (eGFR) of 20 to 59 mL/min per 1.73 m2,1 and were able to give informed consent. The only exclusion criteria for healthy subjects was pregnancy or breastfeeding. Exclusion criteria for the other groups included pregnancy or breastfeeding, uncontrolled hypertension, body mass index ≥40 kg/m2, life expectancy <1 year, history of significant liver disease or significant congestive heart failure (ejection fraction <20%), hospitalizations within the past 3 months, or active infection on antibiotic therapy. For individuals with CKD of their native kidneys, history of immunosuppressive therapy in the past year was an additional exclusion criteria. The study was approved by the Colorado Multiple Institutional Review Board. The nature, benefits, and risks of the study were explained to the volunteers, and their written informed consent was obtained before participation. All the study procedures were subsequently conducted during 1 visit at the Clinical and Translational Research Center at the University of Colorado Anschutz Medical Campus.
FMD Measurements
Brachial artery flow–mediated dilation (BA‐FMD) was measured at the Clinical and Translational Research Center by a trained technician using high‐resolution ultrasonography (GE Vivid 7 Dimension) as described originally by Celermajer et al36 and subsequently by our group.37, 38 ECG gated end‐diastolic ultrasound images and Doppler flow of the artery were acquired during baseline and FMD conditions. Reactive hyperemia was produced by inflating a pediatric forearm cuff around the forearm to 250 mm Hg for 5 minutes followed by rapid deflation to measure FMD. A commercially available software package (Vascular Analysis Tools 5.8.1, Medical Imaging Applications) was used to concurrently acquire ECG gated brachial artery diameters. BA‐FMD was determined and reported as the percent change from baseline. Doppler flow of the brachial artery was also measured, and peak shear rate was calculated as a potential covariate. The images were analyzed by an independent research assistant who was blinded to the study groups.
Clinical Variables
Race/ethnicity were evaluated by questionnaire. Diabetes mellitus status was defined as history of diabetes mellitus according to the medical record, current treatment with oral hypoglycemic agents or with insulin, or fasting glucose ≥126 mg/dL. Weight and height were measured, and body mass index was calculated and expressed as kilograms per square meter. Blood pressure was measured via automated cuff after 10 minutes of rest at the beginning of each visit. We measured clinical labs including serum creatinine and albumin‐to‐creatinine ratio (ACR) at the University of Colorado Hospital clinical lab. eGFR was calculated based on the CKD‐EPI formula.1 Urinary ACR was reported as milligrams per gram.
Materials
Complement ELISAs were purchased from Quidel (San Diego, CA). For flow cytometry, sizing beads (Life Technologies, Carlsbad, CA), counting beads (Life Technologies) and compensation beads (Invitrogen) were used for gating and counting. Antibodies to identify endothelial microparticles included anti‐CD41a (eBiosciences, Thermo Fisher, Waltham, MA), anti‐CD105 (Novus Bio, Littleton, CO), anti–immunoglobulin G (ABcam, Cambridge, UK), and anti‐C3b/iC3b (mAb 3E7, generated as previously described).39 Factor D–depleted serum and purified factor D were purchased from Complement Technologies (Tyler, TX).
Complement Fragment Measurements
Plasma was collected into EDTA‐containing tubes, which were inverted several times and placed immediately on ice. Within 10 minutes the samples were centrifuged at 1000g at 4°C, and the layer of plasma was removed with a Pasteur pipette. For urine samples, 9 mL of freshly voided urine was immediately mixed with 1 mL of 10 mmol/L Tris buffer, pH 8.6, with 0.05% Tween 20, 0.01% NaN3, and protease inhibitors (10 mmol/L benzamidine, 10 mmol/L ε‐aminocaproic acid, 20 mmol/L EDTA and 100 kallikrein inhibitor units of aprotinin) to prevent protein degradation after collection.40, 41 The sample was centrifuged at 1000g for 10 minutes at 4°C, and the supernatant was removed. Plasma and urine samples were stored at −80°C until use. Complement activation fragments (Ba, C4a, C3a, C5a, sC5b‐9) in plasma and urine were measured using commercial ELISAs according to the manufacturer's instructions (Quidel). The plasma and urine samples were diluted 1:10 and 1:15, respectively.
Microparticle Isolation
Plasma microparticles were isolated as previously published.31 The samples were thawed in a 37°C water bath and then centrifuged at 400g for 15 minutes at 4°C. The supernatants were collected and the volume recorded. Samples were then centrifuged at 20 000g for 2.5 hours. The pellets were resuspended in MP buffer (Hank's buffered saline solution containing 20 mmol/L HEPES and 5 mmol/L glucose) at a volume of 40% of the initial plasma volume.
Flow Cytometry
Similar to work previously published by our group,31 after microparticles were isolated and resuspended, they were stained using the antibodies to CD105, CD41a, immunoglobulin G, and C3b/iC3b. Bound antibodies were detected at the University of Colorado Flow Cytometry Shared Resource using a Moflo Astrios EQ flow cytometer (Beckman Coulter, Brea, CO). Sizing beads were used to identify particles in the 0.2 to 1 μm range. Equivalent numbers (0.6×105) of counting beads were added to the microparticles before antibody staining to determine the number of microparticles in a fixed volume of plasma.
Proteomic Analysis of Microparticles
EDTA plasma samples (≈200 μL) were centrifuged as previously described to isolate microparticles. The supernatant was removed and the pellet was washed gently. Next, the microparticles were resuspended in SB17 lysis buffer (40 mmol/L HEPES, 101 mmol/L NaCl, 5 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L EDTA, 0.05% Tween 20, pH 7.5) by pipetting up and down 6 times. Samples were incubated in a 37°C water bath for 15 minutes with occasional agitation. The samples were centrifuged at room temperature for 5 minutes at 14 000g. Supernatants were collected, and protein concentrations were determined on a Nanodrop 2000 with secondary confirmation of concentration on select samples by BioRad (Hercules, CA) bicinchoninic acid protein concentration analysis. The proteome in each sample was analyzed at the University of Colorado Genomics and Microarray Core using the Somalogic SOMAscan assay. This assay uses aptamers (single‐stranded DNA that binds to specific proteins) to quantify >1100 different proteins. The assay can measure proteins in concentrations down to the femtomolar range and is useful for analysis of small samples. The aptamers bind to proteins in their native conformation. The assay detects several different complement activation fragments and is specific for the tertiary structure of the fragments specified.
Generation of an Inhibitory Antibody to Factor D
To produce inhibitory monoclonal antibodies to factor D, mice with targeted deletion of the gene for factor D were immunized with purified human factor D (Complement Technology). The mice were screened for the development of inhibitory antibodies to factor D by testing their sera in an ELISA using factor D–coated plates and an in vitro assay of alternative complement pathway inhibition (described below). Spleen cells from one of the mice were fused to a myeloma cell line in the University of Colorado Monoclonal Antibody Center. Candidate hybridomas were cloned by limiting dilution, and clones capable of recognizing factor D by ELISA and inhibiting alternative pathway activity were identified. One of the hybridomas, designated 1679, was identified as an effective inhibitor of the alternative pathway in human serum.
Alternative Pathway Activation Assay
To measure alternative pathway activity, zymosan A particles (Sigma‐Aldrich, St. Louis, MO) were incubated with human serum, and C3 deposition on the particles was measured as previously described.42 Briefly, 10 μL of serum was incubated with 109 zymosan particles at 37°C for 30 minutes in a master mix containing 5 mmol/L MgCl2 and 10 mmol/L EGTA. In some experiments, factor D–depleted serum was used. Purified factor D, microparticles from patient samples, and inhibitory antibody to factor D were added to some samples as described in the text.
Statistical Analysis
Descriptive statistics are reported according to study group as n (%) for categorical variables and mean (standard deviation) or median (interquartile range) for the continuous variables. The potential correlation between complement activation fragments and kidney function (including CKD‐EPI eGFR and urinary ACR) and BA‐FMD was evaluated by Spearman correlation coefficients. To account for the difference in age between the healthy controls and the subjects in both CKD stage III/IV and posttransplant recipients we applied linear regression models to the relation between complement activation fragments and markers of kidney function and BA‐FMD. Comparisons between multiple groups were performed using 1‐way ANOVA with a Tukey's multiple comparison test or Wilcoxon test with Critchlow‐Fligner (DSCF) multiple comparison analysis as appropriate. A P value of <0.05 was considered statistically significant. Statistical analysis was carried out using SAS version 9.4 (SAS Institute, Cary, NC).
Results
Clinical Characteristics
Compared with CKD stage III/IV patients and posttransplant recipients, healthy controls were younger, mostly women, and had lower body mass index and blood pressure and higher BA‐FMD (Table 1). Patients with CKD had significantly lower BA‐FMD (6.4±5.5%) compared with healthy controls (9.9±7.1%; P=0.05) but not compared with posttransplant subjects (6.95±3.8%). When evaluating the whole group of participants, BA‐FMD correlated with higher eGFR and lower urinary ACR, although this did not achieve statistical significance (data not shown).
Table 1.
Clinical Characteristics According to Study Group
| Healthy Control (n=30) | CKD Stage III & IV (n=30) | Posttransplant CKD Stage III & IV (n=29) | P Value (ANOVA) | |
|---|---|---|---|---|
| Age, y | 38±13 | 59±15 | 52±14 | <0.001 |
| Sex (Male) | 6 (20%) | 18 (60%) | 17 (57%) | 0.003 |
| Race (White) | 27 (90%) | 25 (76%) | 30 (100%) | 0.01 |
| History of DM | 0 (0%) | 12 (36%) | 6 (20%) | <0.0001 |
| BMI, kg/m2 | 26.3±5.5 | 29.2±5.2 | 25.9±5.0 | 0.035 |
| SBP, mm Hg | 116±10 | 130±12 | 136±14 | <0.001 |
| DBP, mm Hg | 72±8 | 77±10 | 82±11 | 0.0004 |
| CKD‐EPI eGFR, mL/min per 1.73 m2 | 82±17 | 37±8 | 44±10 | <0.0001 |
| ACR, mg/g | 0.08±0.09 | 5±14 | 3±6 | 0.11 |
| FMD % Δ | 9.9±7.1 | 6.4±5.5 | 6.95±3.8 | 0.046 |
Healthy controls were younger, mostly women, had lower BMI and blood pressure and higher BA‐FMD compared with those with CKD and posttransplant. Patients with CKD had significantly lower BA‐FMD compared with healthy. Values are expressed as means±standard deviation or %=percent of patients. ACR indicates urinary albumin/creatinine ratio; BMI, body mass index; CKD, chronic kidney disease; CKD‐EPI eGFR, Chronic Kidney Disease Epidemiology Collaboration estimated glomerular filtration rate; DBP, diastolic blood pressure; DM, diabetes mellitus; FMD % Δ, percent change in brachial artery flow mediated dilation; SBP, systolic blood pressure.
Complement Activation Fragments Are Elevated in Patients With CKD
Complement activation fragments were elevated in patients with stage III/IV CKD, but this achieved statistical significance only for fragment Ba and C5b‐9 (Figure 1). Ba, a marker of activation of the alternative pathway of complement, was also increased in posttransplant recipients compared with healthy controls but not with individuals with stage III/IV CKD. Complement fragments C4a and C5a were mildly higher in patients with stage III/IV CKD compared with healthy controls and posttransplant subjects, but this was not statistically significant. Importantly, as shown in Table 2, there was significant correlation between the plasma levels of the different measured complement activation fragments, suggesting that elevation of these fragments was attributable to increased complement activation and not simply decreased renal clearance of any one fragment. This is additionally supported by the finding of higher Ba levels in the urine of those with stage III/IV CKD versus healthy controls.
Figure 1.

Levels of complement activation fragments are higher in patients with stage III/IV CKD compared with healthy controls. A, Plasma Ba and C5b‐9 levels were significantly higher in stage III/IV CKD and transplant patients compared with healthy controls. There was also a trend toward higher levels of the other complement fragments in CKD patients, but these levels were not significantly higher than in healthy controls. B, Urine Ba levels were higher in patients with stage III/IV CKD compared with healthy controls. ***P<0.001, *P<0.05.
Table 2.
Plasma Levels of Complement Activation Fragments Correlate With Each Other
| LN_C3aa | C4a | C5a | sC5b_9 | |
|---|---|---|---|---|
| LN_Baa |
0.20 0.06 |
0.22 0.036 |
0.15 0.17 |
0.29 0.006 |
| LN_C3aa |
0.27 0.01 |
0.02 0.9 |
0.06 0.6 |
|
| C4a |
0.29 0.007 |
0.28 0.008 |
||
| C5a |
0.24 0.02 |
Complement activation fragments shown: Ba, C3a, C4a, C5a, and sC5b_9. Data are shown as Spearman correlation coefficient followed by the P value.
Variable not normally distributed and presented as natural log (LN).
Complement Activation Fragment Ba Correlates With Biomarkers of CKD
Higher plasma Ba levels correlated inversely with CKD‐EPI eGFR (Spearman correlation coefficients −0.82, P<0.0001) and positively with urinary ACR (correlation coefficients 0.5, P<0.0001). Results of this unadjusted analysis are shown in Table 3. After adjusting for age using linear regression analysis, higher plasma Ba levels were still associated with lower CKD‐EPI eGFR with a β estimate of −0.01 (95% confidence interval, −0.013, −0.007; P<0.0001) and with higher urinary ACR with a β estimate of 0.003 (95% confidence interval, 0.002, 0.004; P<0.0001).
Table 3.
Plasma Levels of Complement Activation Fragment Ba Correlate With Biomarkers of CKD
| LN_Baa | |
|---|---|
| CKD‐EPI eGFR, mL/min per 1.73 m2 |
−0.82 <0.0001 |
| Urinary ACR, mg/g |
0.5 <0.0001 |
Data are shown as Spearman correlation coefficient followed by the P value. ACR indicates urinary albumin/creatinine ratio; CKD‐EPI eGFR, Chronic Kidney Disease Epidemiology Collaboration estimated glomerular filtration rate.
Variable not normally distributed and presented as natural log (LN).
Plasma Levels of Complement Fragment Ba Correlate With BA‐FMD
Higher plasma Ba levels correlated inversely and significantly with BA‐FMD in all the participants (correlation coefficient=−0.26 with a P value of 0.016) suggesting that alternative pathway activation correlates with endothelial dysfunction (Figure 2). We found no significant correlation between plasma levels of Ba and BA‐FMD in the separate groups. In addition, after adjusting for age, the association between complement fragment Ba and BA‐FMD was no longer significant (P=0.16). This is likely attributable to the small number of participants. We did not find a significant correlation between sC5b‐9 and BA‐FMD.
Figure 2.
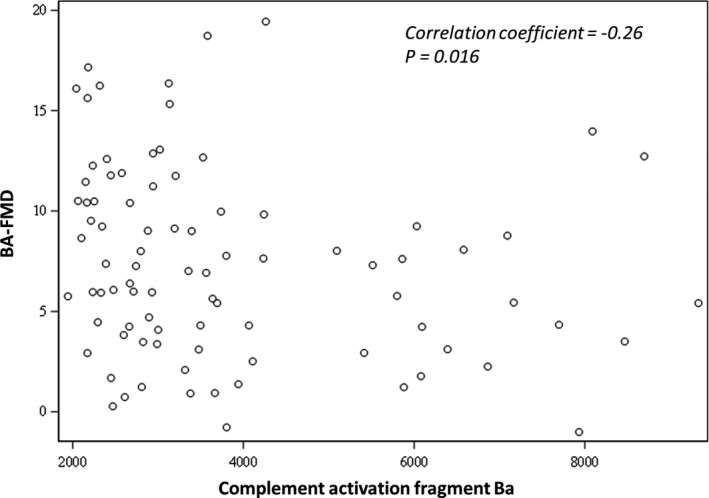
Plasma levels of complement fragment Ba are significantly and inversely correlated with BA‐FMD in all of the recruited participants (correlation coefficient −0.26, P=0.016) suggesting that complement activation is related to endothelial dysfunction.
The Alternative Pathway Activating Protein Factor D Is Increased in Microparticles of Patients With CKD
We previously found that endothelial microparticles can activate the alternative pathway, but we did not identify the molecular cause of this activation.23 To analyze the microparticle proteome, protein was extracted from microparticle pellets and analyzed by the SOMAscan assay (which measures >1100 proteins and can measure the abundance of most proteins in the fmol/L to pmol/L range).
We next examined those proteins whose abundance was different among the different patient groups. One of the strongest differences between the microparticle proteome of healthy controls and patients with CKD was the level of factor D, the rate‐limiting protease of the alternative pathway. Factor D was significantly more abundant in the microparticles of CKD and transplant patients compared with healthy controls (Figure 3A), whereas the level of factor B was lower in the microparticles of patients with CKD versus healthy controls. Factor B is cleaved during alternative pathway activation, and lower levels of the protein may reflect its consumption during this process.
Figure 3.

Complement proteins in microparticles from patients with CKD are altered compared with microparticles in healthy controls. We used an aptamer‐based assay (SOMAscan) to measure microparticle proteins with high sensitivity. A, Factor D levels were significantly higher in the microparticles of patients with CKD and patients with transplants with CKD compared with healthy controls. Factor B levels were lower in these groups compared with healthy controls. B, Levels of C3, iC3b, and C3d were not significantly different among the patient groups. C, Levels of the complement inhibitory protein CD59 was higher in patients with CKD than in healthy controls. Levels of factor H and decay accelerating factor were not statistically different among the 3 groups. ***P<0.001, *P<0.05.
The SOMAscan can distinguish C3, iC3b, and C3d and measure their concentrations. The levels of all 3 forms of C3 were similar in microparticle samples from patients with CKD and transplants to levels in healthy controls (Figure 3B). Levels of DAF and CD59 were both higher in microparticles from patients with CKD and patients with CKD with transplants compared with healthy controls. DAF and CD59 are complement regulatory proteins, and higher levels of these proteins may explain why elevated factor D levels in the microparticles of patients with CKD is not associated with greater C3 deposition on the surface of the microparticles.
Endothelial Microparticle Numbers Are Not Significantly Increased in CKD
Microparticle numbers and surface C3 fragment deposition were also analyzed by flow cytometry. Microparticles in the 0.2 to 1 μm size range were identified using sizing beads, and microparticles derived from endothelial cells were identified by detection of surface CD105 (Figure 4A). Using forward scatter to compare microparticles before and after purification, the average size was slightly greater after purification in both healthy control samples and CKD samples (Figure 4B and Figure S1). The size of the purified microparticles was the same in all 3 groups of patients, however (Figure 4C). There was a trend toward higher numbers of endothelial microparticles in the plasma of CKD and transplant patients compared with healthy controls, but the difference was not statistically significant (Figure 4D). Similarly, deposited C3 fragments on the microparticle surface did not differ significantly among the 3 groups (Figure 4E).
Figure 4.
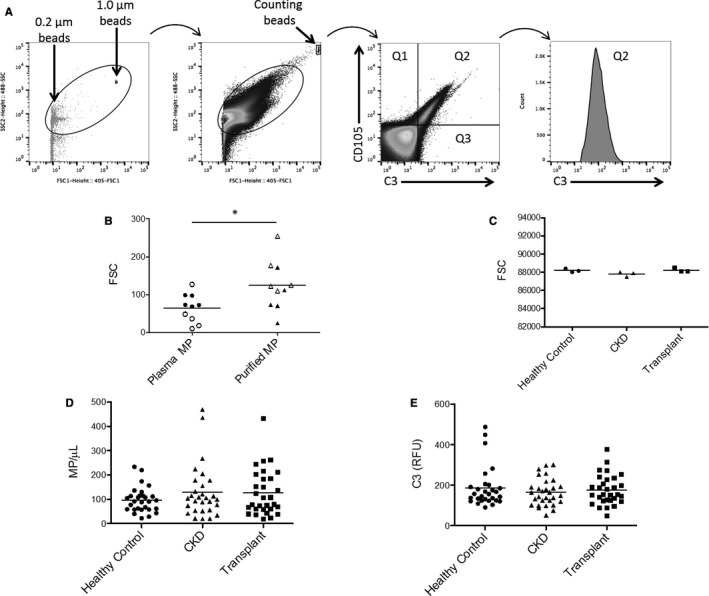
Endothelial microparticle number and bound C3 fragments are not significantly increased in patients with CKD. A, We used sizing beads to identify 0.2 to 1.0 μm by forward scatter and size scatter. Counting beads were added to the plasma before microparticle purification, and were used to compare the number of microparticles isolated from each sample. Endothelial microparticles were analyzed by gating on the CD105+CD41a‐ microparticle population. We also analyzed C3 fragment deposition on the endothelial microparticles. B, The size of the microparticles purified from plasma was slightly larger than those in the original plasma sample when compared by forward scatter (FSC). *P<0.05. Data points from healthy control samples are white, and data points from CKD samples are solid black. C, The size of the purified microparticles was the same in the 3 different patient groups studied. D, The number of microparticles in patients with CKD and patients with transplants was not significantly higher than in healthy controls. E, The amounts of C3 fragments deposited on the endothelial microparticles in patients with CKD and patients with transplants was not significantly different than in healthy controls.
Factor D in Microparticles Activates the Alternative Pathway in Serum
Because we identified greater levels of factor D in the microparticles of patients with CKD and elevated alternative pathway activation fragments in the plasma of these patients, we next sought to determine whether factor D in plasma microparticles is functionally active. To do this we tested the ability of purified microparticles to restore alternative pathway activity to factor D depleted serum. Zymosan particles activate the alternative pathway when incubated with serum.42 As expected, no alternative pathway activity was seen when factor D–depleted serum was used in this assay (Figure 5A). Alternative pathway activity was restored, however, when purified factor D protein was added to the reaction. We also generated a novel inhibitory monoclonal antibody to factor D (Figure S2). Addition of the anti–factor D antibody reduced alternative pathway activity in factor D–depleted serum that was reconstituted with purified factor D. We next isolated microparticles from 250 μL of plasma from a patient with CKD. When the microparticle pellet was added to factor D–deficient serum, alternative pathway activity increased (Figure 5B). We did not see a similar increase in alternative pathway activity when microparticles from a healthy control subject were added to the reaction. The addition of the anti–factor D antibody to the reaction mix blocked the increase in alternative pathway activity when the microparticles were added, confirming that complement activation was caused by factor D in the microparticle pellet.
Figure 5.
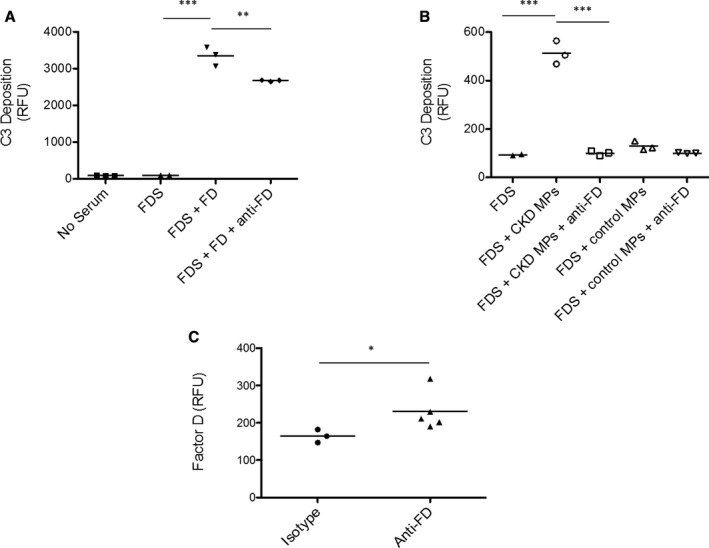
Microparticle‐associated factor D activates plasma complement proteins. To test whether FD in plasma microparticles is catalytically active, we examined whether it could restore complement activity to (FDS. A, Using an alternative pathway assay, we confirmed that purified FD restored activity to FDS. We also confirmed that an anti‐FD reduced complement activity in the FDS reconstituted with FD. B, Similarly, (CKD MPs restored activity to the FDS. The addition of an inhibitory antibody to factor D prevented this response, however, confirming that the effect was due to factor D protein contained within the isolated microparticles. C, Staining of the microparticles for factor D confirmed that it was present on the surface. Anti‐FD indicates anti‐factor D antibody; CKD MPs, microparticles purified from the plasma of a patient with CKD; FD, factor D; and FDS, factor D–depleted serum. ***P<0.001, **P<0.01, *P<0.05.
Staining of the microparticles with the anti–factor D antibody confirmed that factor D protein is detectable on the microparticle surface (Figure 5C). Proteins can adhere to the surface of microparticles in a calcium‐dependent fashion or by binding of the proteins to phospholipids on the microparticle.43 We tested whether factor D levels on the microparticles are reduced by the addition of 20 mmol/L EDTA to chelate calcium or 50 U/mL heparin to compete with phospholipids on the microparticle surface. We did not see a reduction of factor D levels in either of these conditions (data not shown).
Discussion
In this study, we show evidence of systemic complement activation in patients with stage III/IV CKD. Notably, we have found that complement activation fragment Ba and C5b‐9 are significantly increased in the plasma of patients with stage III/IV CKD and that plasma levels of Ba correlated inversely with CKD‐EPI GFR. Urinary levels of Ba were mildly increased, suggesting that the higher levels of plasma Ba are related to increased alternative pathway activation and are not simply due to reduced renal clearance in the setting of reduced GFR. The conclusion that elevated levels of Ba are caused by complement activation is further supported by the correlation with levels of multiple complement activation fragments. In addition, plasma levels of Ba were correlated with urinary ACR, an important marker of kidney damage and endothelial dysfunction. Importantly, the significant correlation between plasma levels of Ba and BA‐FMD is consistent with a functional role of the alternative complement pathway in vascular and kidney disease in this patient population.
Using an unbiased proteomics approach, we found that levels of factor D in plasma microparticles of patients with CKD were significantly higher than levels in microparticles from control patients. Factor D is a serine protease synthesized in the adipose tissue in humans, and it is the rate‐limiting catalytic enzyme of alternative pathway activation.44 It was previously shown that factor D accumulates in CKD,45 but our data indicate that factor D is specifically increased in microparticles of patients with stage III/IV CKD. Our in vitro data show, for the first time, that plasma microparticles containing factor D cause alternative pathway activation. An inhibitory monoclonal antibody to factor D that we developed blocked alternative pathway activation by the microparticles in vitro, confirming that factor D in the microparticles is functional and that it can be pharmacologically blocked.
Collectively, these data indicate that microparticle‐associated factor D increases in CKD and is associated with systemic activation of the alternative complement pathway. Alternative pathway activation, in turn, correlates with vascular endothelial dysfunction. Another study in patients who did not have CKD also recently linked factor D levels with CVD, further supporting a functional link between alternative pathway activity and vascular injury.46 Thus, microparticle‐associated factor D and plasma Ba may be biomarkers and/or mediators of vascular injury in CKD.
Plasma Ba and microparticle‐associated factor D levels of patients with transplants were similar to that in patients with CKD, indicating that standard immunosuppressive medications (mycophenolate mofetil, tacrolimus, and corticosteroids) do not affect factor D levels or alternative pathway activation. These drugs are therefore unlikely to prevent complement‐mediated vascular injury in these patients. Multiple complement inhibitory drugs are currently in clinical development, however, including several alternative pathway inhibitors.47 It is possible that therapeutic complement inhibitors could block some of the systemic inflammatory effects of CKD and could provide a novel approach for preventing CVD in these patients.
Alternative pathway activation contributes to a wide range of different inflammatory and autoimmune diseases.48 Even when complement activation is initiated by immune complexes or immunoglobulin bound to self antigens, amplification through the alternative pathway is an important cause of inflammation and tissue damage. Increased levels of microparticle‐associated factor D in patients with CKD could exacerbate the inflammatory process.49 Conversely, alternative pathway–mediated diseases, such as atypical hemolytic uremic syndrome, are frequently associated with defects in regulation of the alternative pathway that could further amplify factor D–mediated complement activation in patients with CKD.50 This could lead to a vicious cycle whereby complement dysregulation causes kidney injury, which then causes further alternative pathway activation.
Limitations of our study include the small number of patients and the cross‐sectional design of the study. CKD is caused by many different primary diseases and is associated with older age and a large number of comorbidities. Of note, the association between complement fragment Ba and BA‐FMD was attenuated after adjusting for age most likely because of the small number of participants. Although the association between complement fragment Ba and markers of kidney disease remained significant, the estimate was small. It is important to examine these findings in future studies that are adequately powered to evaluate potential confounders including age and CKD‐associated comorbidities. Other limitations include the inability to examine whether complement activation predicts future hard outcomes such as cardiovascular events due to the cross‐sectional nature of the study. Importantly, this data cannot distinguish whether increased alternative pathway activation is a cause of disease or simply a marker of inflammation in CKD. Another potential limitation is the absence of standardized methodology for the measurement of microparticles in humans.51 Notwithstanding these limitations, data such as ours may lead to greater interest in microparticles, and future studies should focus on evaluating the methodology and whether in fact microparticles may be a valid marker of CVD.
In conclusion, we have found that the alternative pathway of complement is activated in patients with CKD. Ba levels in plasma correlate with vascular dysfunction, suggesting that plasma Ba is a biomarker of CVD in these patients. Microparticles in the plasma of patients with CKD also have increased levels of factor D, the activating enzyme of the alternative pathway. The micro‐particle associated protein is functional and activates the alternative pathway in the plasma, demonstrating that altered factor D homeostasis may be an important mechanism of alternative pathway activation and systemic inflammation. These complement perturbations were similar in patients with kidney transplants with CKD, indicating that the immunosuppressive drugs routinely used in patients with transplants do not attenuate complement activation. Future longitudinal studies can confirm an association of alternative pathway activation with clinical outcomes and test whether use of complement inhibitory drugs provides an effective means of preventing CVD in this patient population.
Sources of Funding
This work was supported by a grant from the American Heart Association (14GRNT20120018) and National Institutes of Health Grant R01 DK076690 (Thurman). It was also supported by NIH T32 DK007135.
Disclosures
Thurman and Holers receive royalties from Alexion Pharmaceuticals, Inc, and are also consultants for AdMIRx, Inc, a company that develops complement inhibitors. They also hold stock and will receive royalty income from AdMIRx. The remaining authors have no disclosures to report.
Supporting information
Figure S1. The size of microparticles was analyzed in patient plasma before and after purification.
Figure S2. Generation of an inhibitory antibody to factor D.
Acknowledgments
We would like to acknowledge the assistance of the University of Colorado Flow Cytometry Shared Resource. We would also like to acknowledge Dr Kenneth Jones for his input on analysis and data interpretation.
(J Am Heart Assoc. 2018;7:e007818 DOI: 10.1161/JAHA.117.007818.)
This article was handled independently by David M. Pollock, PhD, as a guest editor. The editors had no role in the evaluation of the manuscript or in the decision about its acceptance.
References
- 1. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF III, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD EPI . A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 3. Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow‐up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663. [DOI] [PubMed] [Google Scholar]
- 4. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW; American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention . Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154–2169. [DOI] [PubMed] [Google Scholar]
- 5. Whaley‐Connell AT, Sowers JR, Stevens LA, McFarlane SI, Shlipak MG, Norris KC, Chen SC, Qiu Y, Wang C, Li S, Vassalotti JA, Collins AJ; Kidney Early Evaluation Program Investigators . CKD in the United States: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis. 2008;51:S13–S20. [DOI] [PubMed] [Google Scholar]
- 6. Rao MV, Qiu Y, Wang C, Bakris G. Hypertension and CKD: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES), 1999–2004. Am J Kidney Dis. 2008;51:S30–S37. [DOI] [PubMed] [Google Scholar]
- 7. Sarnak MJ, Coronado BE, Greene T, Wang SR, Kusek JW, Beck GJ, Levey AS. Cardiovascular disease risk factors in chronic renal insufficiency. Clin Nephrol. 2002;57:327–335. [DOI] [PubMed] [Google Scholar]
- 8. Silverstein DM. Inflammation in chronic kidney disease: role in the progression of renal and cardiovascular disease. Pediatr Nephrol. 2009;24:1445–1452. [DOI] [PubMed] [Google Scholar]
- 9. Shlipak MG, Fried LF, Crump C, Bleyer AJ, Manolio TA, Tracy RP, Furberg CD, Psaty BM. Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation. 2003;107:87–92. [DOI] [PubMed] [Google Scholar]
- 10. Landray MJ, Wheeler DC, Lip GY, Newman DJ, Blann AD, McGlynn FJ, Ball S, Townend JN, Baigent C. Inflammation, endothelial dysfunction, and platelet activation in patients with chronic kidney disease: the chronic renal impairment in Birmingham (CRIB) study. Am J Kidney Dis. 2004;43:244–253. [DOI] [PubMed] [Google Scholar]
- 11. Oberg BP, McMenamin E, Lucas FL, McMonagle E, Morrow J, Ikizler TA, Himmelfarb J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004;65:1009–1016. [DOI] [PubMed] [Google Scholar]
- 12. Jalal D, Chonchol M, Etgen T, Sander D. C‐reactive protein as a predictor of cardiovascular events in elderly patients with chronic kidney disease. J Nephrol. 2012;25:719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nowak KL, Chonchol M, Ikizler TA, Farmer‐Bailey H, Salas N, Chaudhry R, Wang W, Smits G, Tengesdal I, Dinarello CA, Hung AM. IL‐1 inhibition and vascular function in CKD. J Am Soc Nephrol. 2017;28:971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ‐Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM; Investigators BT . Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest. 2010;90:1549–1557. [DOI] [PubMed] [Google Scholar]
- 16. Beyer C, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol. 2010;6:21–29. [DOI] [PubMed] [Google Scholar]
- 17. Hsu CY, Huang PH, Chiang CH, Leu HB, Huang CC, Chen JW, Lin SJ. Increased circulating endothelial apoptotic microparticle to endothelial progenitor cell ratio is associated with subsequent decline in glomerular filtration rate in hypertensive patients. PLoS One. 2013;8:e68644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang PH, Huang SS, Chen YH, Lin CP, Chiang KH, Chen JS, Tsai HY, Lin FY, Chen JW, Lin SJ. Increased circulating CD31+/annexin V+ apoptotic microparticles and decreased circulating endothelial progenitor cell levels in hypertensive patients with microalbuminuria. J Hypertens. 2010;28:1655–1665. [DOI] [PubMed] [Google Scholar]
- 19. Jourde‐Chiche N, Dou L, Sabatier F, Calaf R, Cerini C, Robert S, Camoin‐Jau L, Charpiot P, Argiles A, Dignat‐George F, Brunet P. Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J Thromb Haemost. 2009;7:1576–1584. [DOI] [PubMed] [Google Scholar]
- 20. Trappenburg MC, van Schilfgaarde M, Frerichs FC, Spronk HM, ten Cate H, de Fijter CW, Terpstra WE, Leyte A. Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol Dial Transplant. 2012;27:1446–1453. [DOI] [PubMed] [Google Scholar]
- 21. Chironi GN, Boulanger CM, Simon A, Dignat‐George F, Freyssinet JM, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–151. [DOI] [PubMed] [Google Scholar]
- 22. Boulanger CM, Amabile N, Guerin AP, Pannier B, Leroyer AS, Mallat CN, Tedgui A, London GM. In vivo shear stress determines circulating levels of endothelial microparticles in end‐stage renal disease. Hypertension. 2007;49:902–908. [DOI] [PubMed] [Google Scholar]
- 23. Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, Christians U, Thurman JM. Cyclosporine induces endothelial cell release of complement‐activating microparticles. J Am Soc Nephrol. 2013;24:1849–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown KM, Sacks SH, Sheerin NS. Mechanisms of disease: the complement system in renal injury—new ways of looking at an old foe. Nat Clin Pract Nephrol. 2007;3:277–286. [DOI] [PubMed] [Google Scholar]
- 25. McCullough JW, Renner B, Thurman JM. The role of the complement system in acute kidney injury. Semin Nephrol. 2013;33:543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noris M, Remuzzi G. Atypical hemolytic‐uremic syndrome. N Engl J Med. 2009;361:1676–1687. [DOI] [PubMed] [Google Scholar]
- 27. Pickering MC, Cook HT. Translational mini‐review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. 2008;151:210–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pavlov VI, Skjoedt MO, Siow Tan Y, Rosbjerg A, Garred P, Stahl GL. Endogenous and natural complement inhibitor attenuates myocardial injury and arterial thrombogenesis. Circulation. 2012;126:2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leung VW, Yun S, Botto M, Mason JC, Malik TH, Song W, Paixao‐Cavalcante D, Pickering MC, Boyle JJ, Haskard DO. Decay‐accelerating factor suppresses complement C3 activation and retards atherosclerosis in low‐density lipoprotein receptor‐deficient mice. Am J Pathol. 2009;175:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fearn A, Sheerin NS. Complement activation in progressive renal disease. World J Nephrol. 2015;4:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thurman JM, Wong M, Renner B, Frazer‐Abel A, Giclas PC, Joy MS, Jalal D, Radeva MK, Gassman J, Gipson DS, Kaskel F, Friedman A, Trachtman H. Complement activation in patients with focal segmental glomerulosclerosis. PLoS One. 2015;10:e0136558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shechter M, Shechter A, Koren‐Morag N, Feinberg MS, Hiersch L. Usefulness of brachial artery flow‐mediated dilation to predict long‐term cardiovascular events in subjects without heart disease. Am J Cardiol. 2014;113:162–167. [DOI] [PubMed] [Google Scholar]
- 33. Shechter M, Issachar A, Marai I, Koren‐Morag N, Freinark D, Shahar Y, Shechter A, Feinberg MS. Long‐term association of brachial artery flow‐mediated vasodilation and cardiovascular events in middle‐aged subjects with no apparent heart disease. Int J Cardiol. 2009;134:52–58. [DOI] [PubMed] [Google Scholar]
- 34. Brevetti G, Silvestro A, Schiano V, Chiariello M. Endothelial dysfunction and cardiovascular risk prediction in peripheral arterial disease: additive value of flow‐mediated dilation to ankle‐brachial pressure index. Circulation. 2003;108:2093–2098. [DOI] [PubMed] [Google Scholar]
- 35. Yilmaz MI, Stenvinkel P, Sonmez A, Saglam M, Yaman H, Kilic S, Eyileten T, Caglar K, Oguz Y, Vural A, Cakar M, Altun B, Yenicesu M, Carrero JJ. Vascular health, systemic inflammation and progressive reduction in kidney function; clinical determinants and impact on cardiovascular outcomes. Nephrol Dial Transplant. 2011;26:3537–3543. [DOI] [PubMed] [Google Scholar]
- 36. Celermajer DS, Sorensen K, Ryalls M, Robinson J, Thomas O, Leonard JV, Deanfield JE. Impaired endothelial function occurs in the systemic arteries of children with homozygous homocystinuria but not in their heterozygous parents. J Am Coll Cardiol. 1993;22:854–858. [DOI] [PubMed] [Google Scholar]
- 37. Jalal DI, Decker E, Perrenoud L, Nowak KL, Bispham N, Mehta T, Smits G, You Z, Seals D, Chonchol M, Johnson RJ. Vascular function and uric acid‐lowering in stage 3 CKD. J Am Soc Nephrol. 2017;28:943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jalal DI, Jablonski KL, McFann K, Chonchol MB, Seals DR. Vascular endothelial function is not related to serum uric acid in healthy adults. Am J Hypertens. 2012;25:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DiLillo DJ, Pawluczkowycz AW, Peng W, Kennedy AD, Beum PV, Lindorfer MA, Taylor RP. Selective and efficient inhibition of the alternative pathway of complement by a mAb that recognizes C3b/iC3b. Mol Immunol. 2006;43:1010–1019. [DOI] [PubMed] [Google Scholar]
- 40. Ogrodowski JL, Hebert LA, Sedmak D, Cosio FG, Tamerius J, Kolb W. Measurement of SC5b‐9 in urine in patients with the nephrotic syndrome. Kidney Int. 1991;40:1141–1147. [DOI] [PubMed] [Google Scholar]
- 41. Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol. 2000;11:700–707. [DOI] [PubMed] [Google Scholar]
- 42. Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, Mitchell LM, Giclas PC, Salmon J, Gilkeson G, Holers VM. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody‐induced pregnancy loss in mice. Mol Immunol. 2005;42:87–97. [DOI] [PubMed] [Google Scholar]
- 43. Einfinger K, Badrnya S, Furtmuller M, Handschuh D, Lindner H, Geiger M. Phospholipid binding protein C inhibitor (PCI) is present on microparticles generated in vitro and in vivo. PLoS One. 2015;10:e0143137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. White RT, Damm D, Hancock N, Rosen BS, Lowell BB, Usher P, Flier JS, Spiegelman BM. Human adipsin is identical to complement factor‐D and is expressed at high‐levels in adipose‐tissue. J Biol Chem. 1992;267:9210–9213. [PubMed] [Google Scholar]
- 45. Volanakis JE, Barnum SR, Giddens M, Galla JH. Renal filtration and catabolism of complement protein D. N Engl J Med. 1985;312:395–399. [DOI] [PubMed] [Google Scholar]
- 46. Hertle E, Arts IC, van der Kallen CJ, Feskens EJ, Schalkwijk CG, Stehouwer CD, van Greevenbroek MM. The alternative complement pathway is longitudinally associated with adverse cardiovascular outcomes. The CODAM study. Thromb Haemost. 2016;115:446–457. [DOI] [PubMed] [Google Scholar]
- 47. Thurman JM, Le Quintrec M. Targeting the complement cascade: novel treatments coming down the pike. Kidney Int. 2016;90:746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–1310. [DOI] [PubMed] [Google Scholar]
- 49. Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD, Huang Y, Huang M, Brodsky RA. Small‐molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017;102:466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33:508–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu R, Greening DW, Zhu HJ, Takahashi N, Simpson RJ. Extracellular vesicle isolation and characterization: toward clinical application. J Clin Invest. 2016;126:1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The size of microparticles was analyzed in patient plasma before and after purification.
Figure S2. Generation of an inhibitory antibody to factor D.