

Abstract
Background
Ischemia/reperfusion injury (IRI) is one of the most predominant complications of ischemic heart disease. Gastrin has emerged as a regulator of cardiovascular function, playing a key protective role in hypoxia. Serum gastrin levels are increased in patients with myocardial infarction, but the pathophysiogical significance of this finding is unknown. The purpose of this study was to determine whether and how gastrin protects cardiac myocytes from IRI.
Methods and Results
Adult male Sprague‐Dawley rats were used in the experiments. The hearts in living rats or isolated Langendorff‐perfused rat hearts were subjected to ischemia followed by reperfusion to induce myocardial IRI. Gastrin, alone or with an antagonist, was administered before the induction of myocardial IRI. We found that gastrin improved myocardial function and reduced the expression of myocardial injury markers, infarct size, and cardiomyocyte apoptosis induced by IRI. Gastrin increased the phosphorylation levels of ERK1/2 (extracellular signal‐regulated kinase 1/2), AKT (protein kinase B), and STAT3 (signal transducer and activator of transcription 3), indicating its ability to activate the RISK (reperfusion injury salvage kinase) and SAFE (survivor activating factor enhancement) pathways. The presence of inhibitors of ERK1/2, AKT, or STAT3 abrogated the gastrin‐mediated protection. The protective effect of gastrin was via CCK2R (cholecystokinin 2 receptor) because the CCK2R blocker CI988 prevented the gastrin‐mediated protection of the heart with IRI. Moreover, we found a negative correlation between serum levels of cardiac troponin I and gastrin in patients with unstable angina pectoris undergoing percutaneous coronary intervention, suggesting a protective effect of gastrin in human cardiomyocytes.
Conclusions
These results indicate that gastrin can reduce myocardial IRI by activation of the RISK and SAFE pathways.
Keywords: gastrin, ischemia/reperfusion injury, Langendorff, reperfusion injury salvage kinase, RISK (reperfusion injury salvage kinase), SAFE (survivor activating factor enhancement)
Subject Categories: Myocardial Infarction, Ischemia, Mechanisms
Clinical Perspective
What Is New?
Ischemia/reperfusion injury is an important factor that affects the treatment and prognosis of myocardial infarction.
Exploration for new intrinsic or exogenous methods and potential therapeutic agents aimed at ameliorating ischemia/reperfusion injury has become an area of intensive research.
Our present study showed that gastrin reduced cardiac ischemia/reperfusion injury in both in vivo and in vitro experiments.
We also found an inverse correlation between serum levels of cardiac troponin I and gastrin in unstable angina pectoris patients undergoing percutaneous coronary intervention.
What Are the Clinical Implications?
We believe that gastrin treatment may represent a potential therapeutic strategy for the alleviation of myocardial ischemia/reperfusion injury in the future.
Introduction
Ischemic heart diseases are major threats to human health worldwide.1 The probability of saving ischemic cardiomyocytes and limiting the size of the infarction is dependent on prompt and effective myocardial reperfusion.2 There is, however, an inherent risk in the process of reperfusion because it can produce more myocardial injury, called myocardial ischemia/reperfusion injury (IRI).3, 4 Consequently, exploring new therapeutic strategies to reduce IRI is an important research direction.
Previous studies have suggested that people in a “hungry” condition are more prone to myocardial injury than those people who are “full” at the time of the event.5 It is well known that gastrointestinal hormones increase significantly after a meal (especially a high‐protein meal); therefore, the relationship between the gastrointestinal hormones and IRI warrants serious attention. Numerous gastrointestinal hormones are secreted during a meal, such as gastrin and cholecystokinin. The blood level of gastrin markedly increases after a meal, ≈10‐ to 20‐fold more than cholecystokinin.6 The postprandial increase in serum gastrin level is associated with the postprandial decrease in blood pressure in hypertensive but not normotensive adults.7 Our previous study showed that a synergistic interaction between renal CCK2R (cholecystokinin 2 receptor) and D1‐like dopamine receptors plays a pivotal role in maintaining normal blood pressure.8 Plasma gastrin level is associated with decreased cardiovascular mortality risk, the opposite of that found with plasma cholecystokinin level.9 Gastrin and its receptor, CCK2R (also known as CCKBR), are highly and widely expressed in the heart.10, 11, 12 The intracoronary infusion of gastrin 17, the endogenous ligand of CCK2R, or the CCK2R agonist pentagastrin dose‐dependently increases coronary blood flow and myocardial contractility in anesthetized pigs.10 It is of interest that plasma serum gastrin levels are increased in patients with myocardial infarction.13 Moreover, previous studies showed that increased progastrin gene expression in the liver counterbalances hypoxia‐induced weight loss and weakness in mice.14 Nevertheless, the pathophysiological significance of these findings in the heart is not known. We wondered whether there gastrin has a protective effect on myocardial IRI. This study found that gastrin may provide protection from myocardial IRI in a Langendorff heart and in an in vivo heart IRI rat model. Furthermore, we found that RISK (reperfusion injury salvage kinase) and SAFE (survivor activating factor enhancement) pathways were involved in mediating the cardioprotective effects of gastrin. To show the clinical pathophysiological significance of this finding, we investigated the correlation between serum cardiac troponin I (cTnI) and serum gastrin levels in patients with unstable angina pectoris undergoing percutaneous coronary intervention (PCI).
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Animals
Eight‐week‐old male Sprague‐Dawley rats (200–250 g), obtained from the animal center of the Third Military Medical University, were fed with normal rat diet. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996) and approved by the animal care and use committee of the Third Military Medical University.
Isolated Rat Heart Experiments
Isolated hearts were perfused at constant temperature (37°C) and coronary flow (12 mL/min) using the Langendorff technique.15 Sprague‐Dawley rats were anaesthetized by the intraperitoneal injection of sodium pentobarbital (50 mg/kg body weight). Heparin (300 IU) was injected into the inferior vena cava. The hearts were quickly removed, the aortas cannulated, and the hearts retrogradely perfused with Krebs–Henseleit bicarbonate buffer (containing [in mmol/L] glucose 11.0, NaCl 118, KCl 4.8, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25.0, CaCl2 1.2 at pH 7.4), which was equilibrated in 95% O2–5% CO2. A latex balloon connected to a pressure transducer was inserted into the left ventricle through the left atrium. The mechanical activities of the hearts were continuously recorded with a PowerLab system (AD Instruments), filled with aqueous solution to achieve left ventricular end‐diastolic pressure of ≈5 to 10 mm Hg. The measurements included left ventricular‐developed pressure, heart rate, left ventricular end‐diastolic pressure, and the maximum rate of rise of the left ventricular pressure (+dp/dtmax). The treatment, timing, duration, and recovery from normothermic global ischemia, induced by the interruption of perfusion, are illustrated in Figure 1. The hearts were immediately stored at −80°C for subsequent analysis.
Figure 1.
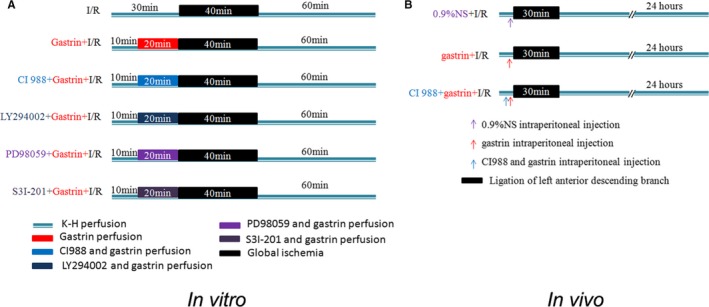
Schematic illustration of the protocol used for ischemia/reperfusion‐injured (I/R‐injured) rat heart. A, Schematic illustration of the protocol for I/R‐injured rat heart in vitro using the Langendorff perfusion system. After 30 minutes of stabilization, the isolated Langendorff‐perfused hearts were subjected to 40 minutes of global ischemia followed by 60 minutes of reperfusion. Gastrin: infusion of gastrin before ischemia for 20 minutes. CI988+gastrin: infusion of CI988 and gastrin before ischemia for 20 minutes. LY294002+gastrin: infusion of LY294002 and gastrin before ischemia for 20 minutes. PD98059+gastrin: infusion of PD98059 and gastrin before ischemia for 20 minutes. S3I‐201+gastrin: infusion of S3I‐201 and gastrin before ischemia for 20 minutes. B, Schematic illustration of the protocol used for I/R‐injured rat heart in in vivo experiment. I/R‐injury was induced by occlusion of the left anterior descending coronary artery for 30 minutes with a small microvascular clamp and then released for 24 hours. 0.9%NS+I/R indicates intraperitoneal injection of normal saline before ischemia/reperfusion; CI988+gastrin+I/R, intraperitoneal injection of CI988 and gastrin before ischemia/reperfusion; gastrin+I/R, intraperitoneal injection of gastrin before ischemia/reperfusion.
Myocardial In Vivo IRI
Myocardial IRI in vivo was induced, as described previously.16 Briefly, the rats were anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal injection). Adequate anesthesia was ensured by monitoring the absence of a withdrawal response to a paw pinch. After anesthesia, the rats were mechanically ventilated, and then the hearts were subjected to myocardial ischemia by ligation of the left anterior descending coronary artery for 30 minutes, and then followed by reperfusion for 24 hours. Sham‐operated rats were subjected to the same operation without left anterior descending artery ligation (according to the protocol described in Figure 1B). For tissue collection, the rats were euthanized by an overdose of pentobarbital sodium (150 mg/kg intraperitoneal injection) and cervical dislocation.
Establishment of Hypoxia/Reoxygenation Models
Primary cultures of neonatal rat cardiomyocytes from 1‐ to 3‐day‐old Sprague‐Dawley rats (Animal Center of the Third Military Medical University, Chongqing, China) were prepared using methods detailed previously.17 The in vitro hypoxia/reoxygenation (H/R) model was used according to methods described previously.17 Briefly, for hypoxia of cardiomyocytes, the culture medium was replaced with DMEM without glucose and serum, placed in a modular incubator chamber, and subsequently flushed with a gas mixture of 5% CO2 and 95% N2 for 60 minutes. Next, the sealed chamber was placed in an incubator (37°C). After 4 hours of hypoxemic incubation, the cultured cardiomyocytes were supplemented with fresh normal medium and then exposed to 95% air and 5% CO2 for 8 hours.
Myocardial Necrosis Determination
The degree of myocardial damage was assessed by measuring the level of cTnI and lactate dehydrogenase (LDH) in the coronary effluents before ischemia perfusion, at the end of reperfusion in vitro, and postoperatively at 24 hours in vivo. The TnI concentration was measured using a high‐sensitivity autoanalyzer assay (Cobas e411; Hoffmann‐La Roche AG). LDH activity was measured using an LDH enzymatic assay kit. The myocardial infarct size was determined by triphenyltetrazolium chloride staining. The isolated, perfused rat hearts were cut into 6 transverse slices (2 mm thick), along the long axis of the left ventricle, from apex to base. The slices were then incubated in sodium phosphate buffer containing 1% triphenyltetrazolium chloride for 15 minutes to visualize the unstained infarcted region. Infarction areas were enhanced by immersion in 4% paraformaldehyde solution for 48 hours before measurement. The infarct and heart areas were quantified by planimetry with ImageJ software (National Institutes of Health). The infarct size was expressed as a percentage of the total area of the heart.
Cell Viability
Cell viability was detected using a Cell Counting Kit (CCK‐8; Dojindo). In brief, primary cultures of cardiomyocytes, inoculated in 96‐well plates, were washed with PBS. Thereafter, 10 μL of the CCK‐8 solution mixed with 90 μL of medium were added to each well. After incubating the cells at 37°C for 1 hour, optical densities at 450 nm were read with a Varioskan Flash microplate reader (Thermo Scientific). Cell viability was expressed as the percentage of control.
Apoptosis Detection
Cardiomyocyte apoptosis in paraffin‐embedded sections was detected using an in situ apoptotic cell death detection kit based on the TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) assay, following the manufacturer's instructions (Roche).18, 19 Cardiomyocytes were labeled with antisarcomeric tropomyosin antibody at 4°C overnight, and nuclei were identified with DAPI (4′,6‐diamidino‐2‐phenylindole) staining. The number of TUNEL‐positive cardiomyocytes was detected in 5 randomly selected nonadjacent images of peri‐infarcted areas. The index of apoptosis is presented as the percentage of TUNEL‐positive cardiomyocytes. Apoptosis in primary cultures of cardiomyocytes was determined as described in our previous report.17
Western Blot
The expression of tissue protein was measured by immunoblotting, as described previously.19 The total protein concentration was measured using Coomassie brilliant blue. Approximately 30 μg of tissue protein lysates were loaded onto SDS‐polyacrylamide gel, electrophoresed, and then transferred onto a polyvinylidene difluoride membrane. Nonspecific antigen was blocked with nonfat milk for 1 hour at room temperature. The membranes were incubated in primary antibodies at 4°C overnight and in a secondary antibody (1:10 000) for 2 hours. Finally, the membranes were visualized by an enhanced chemiluminescence method. The following antibodies were used for immunoblotting: total AKT (protein kinase B; 1:800); phosphorylated AKT (1:800); cleaved caspase 3 (1:200); total STAT3 (signal transducer and activator of transcription 3; 1:800); phosphorylated STAT3 (1:800); total ERK1/2 (extracellular signal‐regulated kinase 1/2; 1:800), phosphorylated ERK1/2 (1:800); and caspase 3 (1:200). GAPDH was used for normalization.
Patients
The participants were recruited between December 2016 and April 2017 from the Department of Cardiology of Daping Hospital in the Third Military Medical University. These patients presented with unstable angina pectoris and ST‐segment depression but with cardiac ischemic marker levels in the normal range. They underwent interventional therapy within 72 hours of admission. To minimize bias, only patients who underwent implantation of a single stent were included in this study. Blood levels of cTnI (ng/mL) were measured in blood drawn immediately before and 24 hours after PCI. In addition, blood was drawn during PCI to measure gastrin levels (pg/mL), using an ELISA kit. This study was approved by the ethics committee of the Medical Faculty of Daping Hospital. Written informed consent was obtained from all patients or their families, in accordance with the Declaration of Helsinki.
Statistical Analysis
All data are shown as mean±SEM. Student unpaired t test was used to compare 2 independent samples. Repeated measures ANOVA was used to compare different data at multiple time points from the same rat or patient. The Kolmogorov–Smirnov test was used to test the normality of the cTnI and gastrin distributions at a significance level of 0.05. When the index was normally distributed, a 2‐sample t test or 1‐way ANOVA and Holm–Sidak test were used to compare ≥2 groups. Otherwise, a nonparametric Mann–Whitney test or a Kruskal–Wallis test was used. Linear regression analysis with variable selection was used to assess the relationship between cTnI changes (differences in log‐transformed cTnI levels after and before PCI) and gastrin levels in patients’ blood after accounting for confounding factors.
Results
Gastrin Improves the Reperfusion Recovery of Cardiac Function
To determine the effects of gastrin on IRI, we used an isolated perfused‐heart preparation. Both ischemia/reperfusion‐injured (I/R‐injured) and gastrin‐treated (gastrin+I/R) hearts were subjected to 40 minutes of global no‐flow ischemia followed by 60 minutes of reperfusion. The gastrin+I/R hearts were pretreated before ischemia with gastrin (10−9 mol/L) for 20 minutes. Gastrin+I/R hearts exhibited significantly better functional recovery than IRI hearts, as determined by the developed pressure. Figure 2A shows that the developed pressure in the gastrin+I/R group was higher than in the vehicle (0.9% NS)‐treated I/R group. Figure 2B shows that +dp/dtmax was higher in the gastrin+I/R group than the vehicle‐treated I/R group. In addition, the end‐diastolic pressure in gastrin+I/R hearts during reperfusion was lower than that in I/R hearts (Figure 2C). Collectively, these data suggest that gastrin pretreatment improves cardiac functional recovery in the in vitro experiments.
Figure 2.
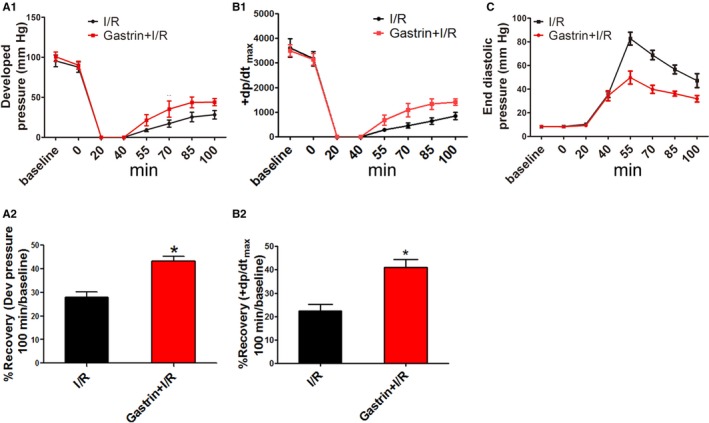
The protective effect of gastrin on cardiac function in a Langendorff‐perfused system. Cardiac function was assessed by developed (Dev) pressure, the rate of rise of left ventricular pressure (+dp/dtmax), and end‐diastolic pressure. Significant differences in data in panels A1, B1, and C were tested by repeated‐measures ANOVA and the Holm–Sidak test. Significant differences in data in panels A2 and B2 was assessed by the Student unpaired t test. *P<0.01 vs I/R, n=12). Gastrin+I/R indicates intraperitoneal injection of gastrin before ischemia/reperfusion; I/R indicates ischemia/reperfusion.
Gastrin Ameliorates I/R‐Induced Cardiac Injury by Alleviation of Cellular Apoptosis
It is well appreciated that maintaining adequate numbers of myocytes is critical to the overall preservation of cardiac structural integrity and function after IRI.20 Given the markedly improved recovery of myocardial function in gastrin‐pretreated hearts, we next determined the effects of gastrin on I/R‐related cellular damage. The extent of necrotic and apoptotic cell death was examined after ischemia for 40 minutes followed by 60 minutes of reperfusion. Before the induction of ischemia (preischemia), the release of cTnI and LDH—biochemical markers for necrotic cell death—was not different between gastrin+I/R and untreated I/R‐injured hearts. After IRI, however, cTnI and LDH release was markedly increased but suppressed by gastrin pretreatment (cTnI shown in Figure 3A, and LDH shown in Figure 3B; 40% and 51%, respectively). The protection may be ascribed to cellular apoptosis, which was reduced by gastrin treatment, as shown by the TUNEL assay (Figure 3C) and immunoblotting of cleaved caspase 3, an index of caspase 3 activity (Figure 3D). Moreover, the ratio of infarct to risk region, determined by triphenyltetrazolium chloride staining, was markedly reduced in the gastrin pretreated group relative to the vehicle‐treated I/R group (Figure 3E1 and 3E2).
Figure 3.
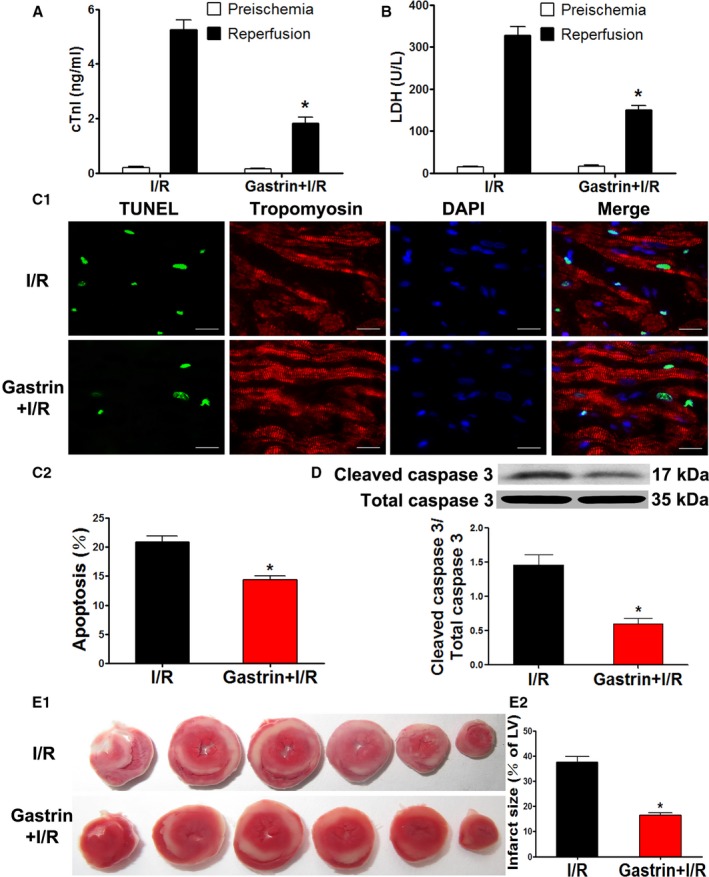
Gastrin attenuated I/R‐induced cardiac injury in a Langendorff‐perfused system. A and B, Coronary effluent was collected at the end of reperfusion, and total cTnI (A) and LDH (B) concentrations were measured. *P<0.05 vs I/R, Student t test, n=6 per group. C and D, Apoptosis of cardiomyocytes was determined by TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) assay (C) and caspase 3 activity (D). E, Infarct size was determined by triphenyltetrazolium chloride staining. *P<0.01 vs I/R, Student t test, n=12. cTnI indicates cardiac troponin I; DAPI, 4′,6‐diamidino‐2‐phenylindole; gastrin+I/R, intraperitoneal injection of gastrin before ischemia/reperfusion; I/R indicates ischemia/reperfusion; LDH, lactate dehydrogenase; LV, left ventricle.
To further determine whether the protective effect of gastrin was caused by targeting of cardiomyocytes, we isolated cardiomyocytes from neonatal rats and established an in vitro model of H/R, which resembles I/R in vivo, and investigated the possible protective effects of gastrin on H/R injury in these cells. The cardiomyocytes were pretreated with vehicle or gastrin (10−10, 10−9, 10−8, 10−7, and 10−6 mol/L) before H/R treatment. We found that pretreatment with 10−8, 10−7, and 10−6 mol/L gastrin concentration‐dependently attenuated the decrease in cell viability caused by H/R treatment, achieving significance at the concentration of 10−8 mol/L (Figure 4A). The results suggest a protective effect of gastrin against H/R injury in primary cultures of rat cardiomyocytes. Cellular apoptosis, measured by TUNEL assay (Figure 4B1 and 4B2) and cleaved caspase 3 expression (Figure 4C), was markedly increased by H/R in the vehicle‐treated group and ameliorated by gastrin treatment.
Figure 4.
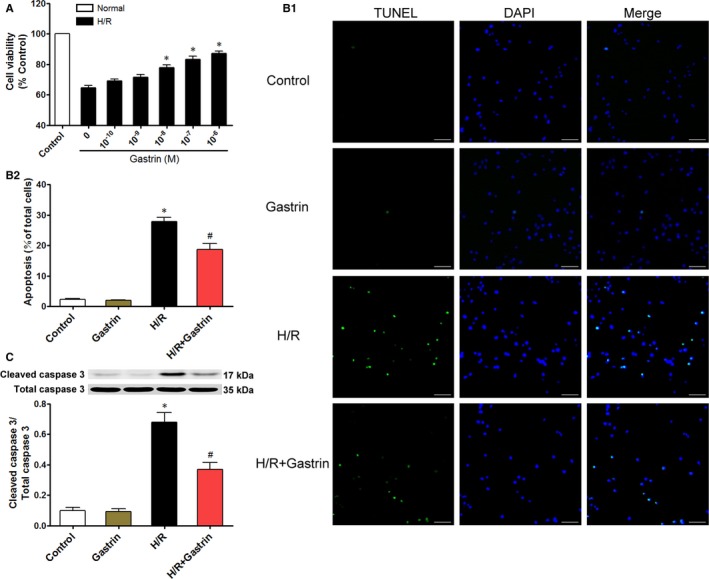
Protective effects of gastrin on H/R‐injury: Primary cultured cardiomyocytes were pretreated with gastrin (0, 10−10, 10−9, 10−8, 10−7, and 10−6 mol/L) before H/R treatment (A). *P<0.007 vs 0 mol/L group. Data are presented as mean±SEM, n=6. Gastrin attenuated I/R‐induced cardiomyocyte apoptosis: Representative images (B1) and quantification (B2) of in situ TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) assay of primary cultured cardiomyocytes. TUNEL‐positive is green and DAPI is blue. Scale bar=400 μm. Cardiomyocyte apoptosis was also analyzed by Western blotting of cleaved and total caspase 3 (C). *P<0.01 vs control; # P<0.01 vs H/R, 1‐way ANOVA, Holm–Sidak test. Data are presented as mean±SEM, n=6 per group. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; H/R, hypoxia/reoxygenation; 0 mol/L group, group without gastrin.
RISK and SAFE Pathways Are Involved in Mediating the Protective Effect of Gastrin on the I/R‐Injured Heart
The RISK and SAFE pathways are known as major protective signaling cascades in both ischemic pre‐ and postconditioning.21 Phosphorylation of ERK1/2, AKT, and STAT3 represents RISK and SAFE activation.22 In the present study, the phosphorylation of ERK1/2 (Figure 5A), AKT (Figure 5B), and STAT3 (Figure 5C) was found to be increased in I/R‐injured hearts after treatment with gastrin. The involvement of the RISK and SAFE pathways was further confirmed by the use of their corresponding inhibitors. In the presence of PD98095 (10 μmol/L),23 an ERK1/2 inhibitor, the protective effects of gastrin on the I/R‐injured hearts were decreased, as determined by measurement of cTnI (Figure 6A), LDH release (Figure 6B), apoptosis (TUNEL assay, Figure 6C), cleaved caspase 3 protein (Figure 6D), and cardiac infarct size (Figure 6E). Correspondingly, the protective effects of gastrin were blocked in the presence of LY294002 (50 μmol/L; AKT inhibitor)22 and S3I‐201 (100 μmol/L; STAT3 inhibitor; Figure 6),24 which proves that the RISK and SAFE pathways are involved in mediating the cardioprotective effects of gastrin.
Figure 5.
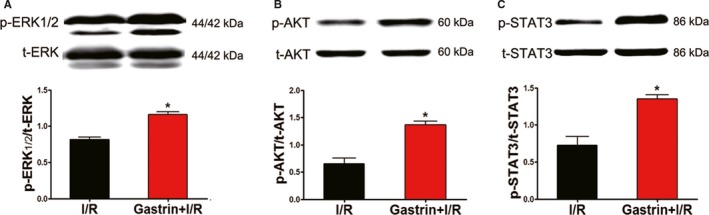
Gastrin promoted phosphorylation of ERK1/2 (A), AKT (B), and STAT3 (C) in a Langendorff‐perfused system. ERK1/2, AKT and STAT3 activation is represented by corresponding protein phosphorylation. The results are expressed as a ratio of phosphorylation (p) and total (t) corresponding protein. *P<0.01 vs I/R, Student t test, n=8 per group. AKT, protein kinase B; ERK1/2, extracellular signal‐regulated kinase 1/2; I/R, ischemia/reperfusion; STAT3, signal transducer and activator of transcription 3.
Figure 6.
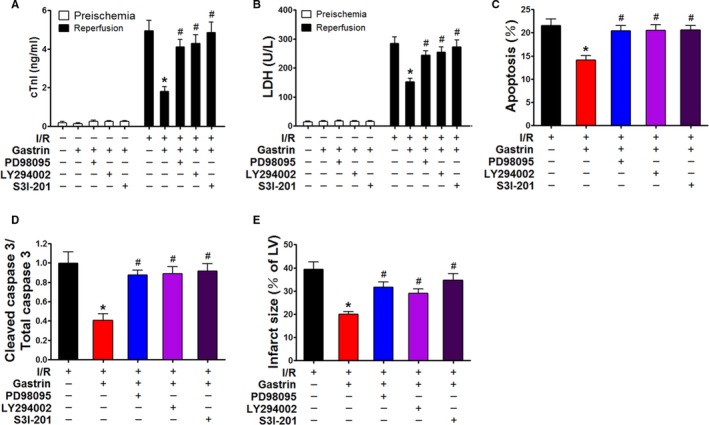
Role of RISK and SAFE pathways in mediating the protective effects of gastrin on I/R‐injured heart in a Langendorff perfusion system. The protective effect of gastrin on I/R‐injured heart was investigated with or without the presence of specific inhibitors. These inhibitors included PD98095 (ERK1/2 inhibitor), LY294002 (AKT inhibitor), and S3I‐201 (STAT3 inhibitor). The concentrations of cTnI (A) and LDH (B) in coronary effluent, number of TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling)–positive cells (C), caspase 3 activity (D), and infarct size of heart (E) were detected in all groups at the end of reperfusion. *P<0.01 vs I/R, # P<0.01 vs gastrin+I/R, 1‐way ANOVA, Holm–Sidak test, n=8 per group. AKT indicates protein kinase B; cTnI indicates cardiac troponin I; ERK1/2, extracellular signal‐regulated kinase 1/2; gastrin+I/R, intraperitoneal injection of gastrin before ischemia/reperfusion; I/R indicates ischemia/reperfusion; LDH, lactate dehydrogenase; LV, left ventricle; RISK, reperfusion injury salvage kinase; SAFE, survivor activating factor enhancement; STAT3, signal transducer and activator of transcription 3.
Gastrin, via CCK2R, Activates the RISK and SAFE Pathways
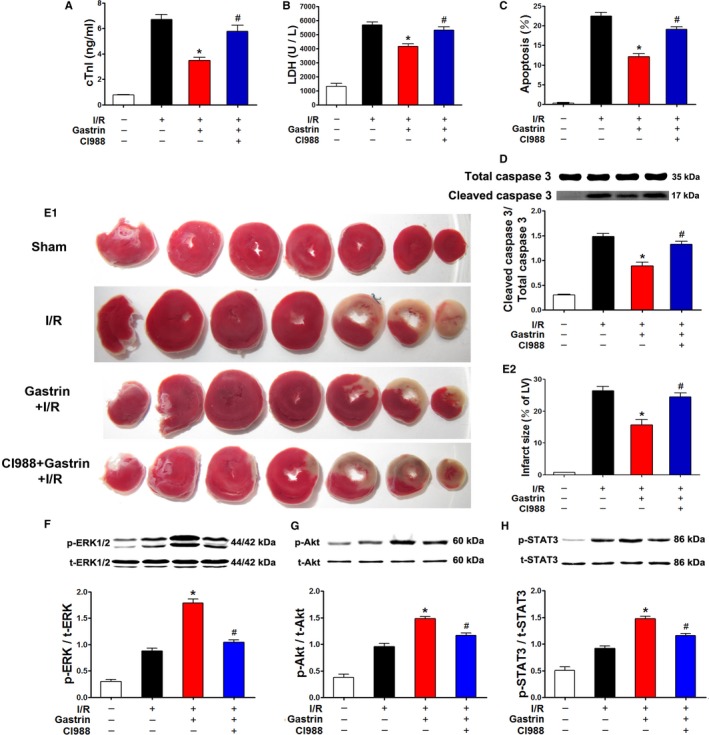
Gastrin shares the same receptor with cholecystokinin. It is known that the cholecystokinin receptor has 2 subtypes, type 1 and type 2. The expression of CCK2R (CCKBR) is greater than CCK1R in both myocardium and coronary artery10 and has a 500‐ to 1000‐fold higher affinity for gastrin than CCK1R.25, 26 In the present study, the protective effect of gastrin was abolished in the presence of a CCK2R inhibitor, CI988 (Figure 7A–7E). Furthermore, the gastrin‐mediated up‐regulation of the phosphorylation of AKT, ERK1/2, and STAT3 was also blocked in the presence of CI988 (10−8 mol/L) (Figure 7F–7H), which indicates that CCK2R is involved in mediating the protective effects of gastrin.
Figure 7.

CCK2R antagonist CI988 abolished the gastrin‐mediated protective effect against I/R‐injury. The protective effect of gastrin was assessed in a Langendorff‐perfused system in the presence and absence of CI988. The cTnI concentration (A) was measured using an autoanalyzer, and LDH concentration (B) was measured by ELISA. Cardiomyocyte apoptosis was determined by TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) assay (C) and caspase 3 activity (D). Cardiac infarct size was measured by triphenyltetrazolium chloride staining (E). Phosphorylation of ERK1/2 (F), AKT (G), and STAT3 (H) was determined by immunoblotting. *P<0.01 vs I/R, # P<0.01 vs gastrin+I/R, 1‐way ANOVA, Holm–Sidak test, n=8 per group. AKT indicates protein kinase B; CCK2R, cholecystokinin 2 receptor; cTnI indicates cardiac troponin I; ERK1/2, extracellular signal‐regulated kinase 1/2; gastrin+I/R, intraperitoneal injection of gastrin before ischemia/reperfusion; I/R indicates ischemia/reperfusion; LDH, lactate dehydrogenase; LV, left ventricle; p, phosphorylated; STAT3, signal transducer and activator of transcription 3; t, total.
Confirmation of the Protective Effect of Gastrin Against IRI In Vivo
The above‐mentioned results were replicated in in vivo experiments. To further confirm the gastrin‐mediated protective effects, we performed studies using an in vivo I/R model. The results were consistent with those found in the in vitro experiments: IRI increased infarct size and increased cTnI and LDH concentrations, accompanied by increased cardiac apoptosis. Pretreatment with gastrin attenuated the infarct size and cTnI and LDH concentrations and reduced cardiomyocyte apoptosis (Figure 8A–8E). The protective effect is probably mediated by the RISK and SAFE signaling pathways because phosphorylation of ERK1/2, AKT, and STAT3 was increased by gastrin. In the presence of CI988, the above‐mentioned protective effects of gastrin were blocked (Figure 8F–8H), further confirming the role of CCK2R in mediating the cardioprotective effects of gastrin.
Figure 8.
Gastrin attenuated I/R‐induced myocardial damage in in vivo experiments. The protective effect of gastrin was confirmed in an in vivo I/R model. These parameters included plasma cTnI (A) and LDH (B) concentrations, cardiac apoptosis determined by TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) assay (C) and caspase 3 activity (D), and infarct size (E). The protective effects of gastrin against I/R injury were assessed in the presence and absence of CI988, a CCK2R antagonist. CI988 blocked the gastrin‐induced RISK and SAFE activation in I/R‐treated rat heart. The protective effect of gastrin was assessed in I/R‐treated rat heart in the presence and absence of CI988. RISK and SAFE activation is shown as phosphorylation of ERK1/2 (F), AKT (G), and STAT3 (H) in I/R‐treated rat heart. *P<0.01 vs I/R, # P<0.01 vs gastrin+I/R, 1‐way ANOVA, Holm–Sidak test, n=8 per group. AKT indicates protein kinase B; CCK2R, cholecystokinin 2 receptor; cTnI indicates cardiac troponin I; ERK1/2, extracellular signal‐regulated kinase 1/2; gastrin+I/R, intraperitoneal injection of gastrin before ischemia/reperfusion; I/R indicates ischemia/reperfusion; LDH, lactate dehydrogenase; LV, left ventricle; RISK, reperfusion injury salvage kinase; SAFE, survivor activating factor enhancement; STAT3, signal transducer and activator of transcription 3.
Association Between Serum cTnI and Gastrin Levels in Patients With Unstable Angina Pectoris Undergoing PCI
PCI is often companied by cardiac injury, and cTnI is a marker of myocardial injury.27, 28 Consequently, we studied the relationship between the serum levels of cTnI and gastrin in patients with unstable angina pectoris undergoing PCI to determine whether gastrin has a protective effect on cardiomyocytes in patients, after accounting for the confounding factors summarized in Table.
Table 1.
General Characteristics of the Participants
| Characteristic | Result |
|---|---|
| Age, y | 59.55±10.7 |
| Body mass index, kg/m2 | 23.2±2.77 |
| Smoking, % (yes/no) | 56/92 (35.6/64.4) |
| TC, mmol/L | 4.35±1.19 |
| TG, mmol/L | 1.67±1.9 |
| HDL‐C, mmol/L | 1.15±0.25 |
| LDL‐C, mmol/L | 2.64±0.81 |
| DM, % (yes/no) | 136/12 (91.9/8.1) |
| Sex, % (male/female) | 76/72 (51.4/48.6) |
Data presented as mean±SEM except as noted. DM indicates diabetes mellitus; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; TC, total cholesterol; TG, triglycerides.
Because the cTnI change is heavily skewed to the right, we performed a log transformation and defined y as the change (after versus before PCI) in the log‐transformed cTnI. The scatter plot in Figure 9 shows that the cTnI change tends to be negatively associated with gastrin. To account for potential confounding factors, we considered a linear regression by regressing y on gastrin together with the other 9 variables in Table. We then performed stepwise model selection using the Akaike information criterion, and the result shows that the model with age and gastrin variables gives the smallest value. The results suggest that age has a significant positive effect, whereas gastrin has a significant negative effect on the cTnI change, that is, after adjusting for the age effect, significant inverse dependence still exists between gastrin and the changes in cTnI levels after PCI.
Figure 9.
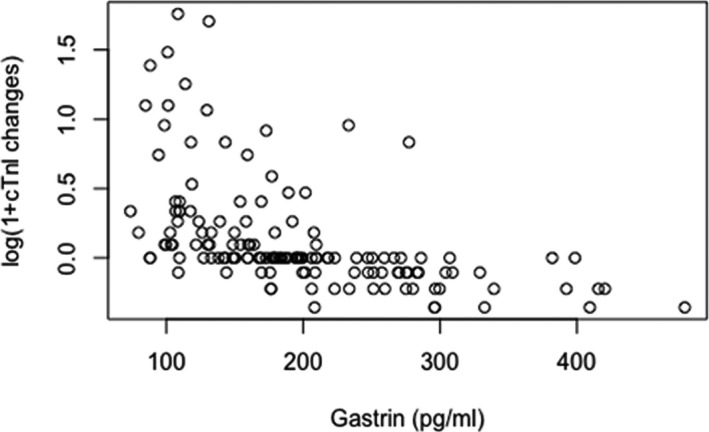
Scatter plot of the changes in log‐transformed serum cardiac troponin I (cTnI) and gastrin levels of unstable angina pectoris patients undergoing percutaneous coronary intervention (PCI). The y‐axis is log(post‐PCI)−log(pre‐PCI).
Discussion
In this study, we demonstrated (1) that gastrin attenuated IRI in the heart in both in vivo and in vitro experiments; (2) that both RISK and SAFE pathways played a crucial role in mediating the cardioprotective effects of gastrin in the I/R‐injured heart; and (3) that in unstable angina pectoris patients undergoing PCI, an inverse correlation was observed between the serum levels of cTnI and gastrin. As far as we know, this report is the first to elucidate the myocardial protective effects of gastrin in IRI and to link causally the protective effects of gastrin with the RISK and SAFE pathways.
From the gut to the heart, a wide variety of cardiovascular actions of gastrointestinal hormones have been reported, including hemodynamics and cardioprotection. Distension of the stomach with gastrin balloons has been associated with increased heart rate and arterial pressure in anesthetized pigs,29 and gastric reflex relaxation following colonic distention increased the secretion of gastrointestinal hormones, such as vasoactive intestinal polypeptide.30 Furthermore, several studies have demonstrated the cardioprotective effects of gastrointestinal hormones. Some gastrointestinal hormones, for example, have been shown to mitigate myocardial IRI through antiapoptotic, antioxidant, and anti‐inflammatory effects (eg, GLP‐1 [glucagon‐like peptide 1]31). There are also reports that gastrointestinal hormones may be important in controlling blood pressure (eg, gastrin8, 32) and improving the efficacy of mesenchymal stem cell–based therapy for ischemic heart disease (eg, ghrelin33). These results point toward the existence of a gastro–heart axis that regulates cardiovascular function.
Gastrin is one of the main gastrointestinal hormones, normally produced by G cells in response to food intake. That the physiological function of gastrin in regulating gastric acid secretion occurs via the CCK2R is now well established.25, 34 In recent years, however, several lines of evidence have emerged to suggest that there is much more to the biology of gastrin than is indicated by earlier work.25, 35 Gastrin has been shown to display proliferative,36, 37 antiapoptotic,38 and cell migration39 properties. In previous reports, the concentration of gastrin in serum was found to be significantly increased during myocardial infarction.13 A previous report also showed a significant correlation between serum gastrin concentration and incidence of myocardial infarction.40 Gastrin is, indeed, increased during myocardial infarction and may play a beneficial or harmful role; however, no direct evidence proves a harmful or protective role for gastrin in myocardial infarction. In this study, we found that gastrin promotes recovery of cardiac function and reduces cardiac necrosis‐induced IRI.
There are 2 subtype receptors in the gastrin peptide family, namely, CCK1R and CCK2R. Gastrin has a 500‐ to 1000‐fold higher affinity for CCK2R than CCK1R.25, 26 CCK2R is highly expressed not only in the stomach and the gastrointestinal tract but also in the heart, brain, and kidney.8, 10, 41 Consequently, gastrin is thought to mediate its biological effects primarily through CCK2R, which is a 7‐transmembrane domain G protein–coupled receptor.42 A study suggested that CCK2R antagonists have a protective effect against cerebral ischemia.18 Another study, however, demonstrated that the neuroprotective effect of gastrin on brain ischemia was attributable to a CCK2R agonist effect.43 The role of CCK2R in protecting against IRI is controversial. In the heart, gastrin 17 increases cardiac perfusion and function through CCK2R in anesthetized pigs.10 In the present study, we found that C1988, a selective inhibitor of CCK2R, abolished the gastrin‐induced cardioprotective effects; therefore, the potent cardioprotective role of gastrin in the I/R‐injured heart is likely mediated through CCK2R.
Many studies have shown that the cardioprotective effect against IRI is associated with activation of cell survival signaling pathways, including the RISK and SAFE pathways.44, 45 The prosurvival kinases are activated in the first few minutes of reperfusion to attenuate reperfusion‐related cell injury by antiapoptotic mechanisms.46 The RISK pathway involves PI3K (phosphatidylinositol 3‐kinase)/AKT and ERK1/2,47 whereas the SAFE pathway involves NF‐κB and STAT3.48 The PI3K/AKT pathway has been shown to play a role in the antiapoptotic effects of gastrin.49 Sustained gastrin stimulation enhances cell survival, in part by decreasing apoptosis via the ERK1/2 pathway.38 In addition, activation of JAK2 (janus kinase 2)/STAT3 pathway by glycine‐extended gastrin has been shown to increase the survival of gastrointestinal tumor cells.50 In the present study, the cardioprotective effect of gastrin before treatment was abolished by inhibitors of PI3K/AKT, ERK1/2, and STAT3, which indicates that the RISK and SAFE pathways play a crucial role in mediating the cardioprotective effect of gastrin against IRI of rat hearts.
Increased postprandial gastrin levels have been associated with a postprandial decrease of blood pressure in hypertensive patients.7 Moreover, in hypertensive patients, fasting serum gastrin levels were inversely correlated with blood pressure, suggesting that gastrin is involved in the regulation of blood pressure.7, 32 Our laboratory found that the intrarenal infusion of gastrin induces natriuresis and diuresis.8 Similarly, increased plasma gastrin concentrations have also been reported in patients with acute myocardial infarction13; however, the pathophysiological significance of the phenomenon is not well characterized. Some studies have suggested that the increased gastrin level might represent an adaptive response to hypoxic conditions,14 because similar adaptive responses have been detected in gut cancer.51, 52 The results of the present study confirm the protective effect of elevated blood levels of gastrin against IRI. To make sure that the effect of gastrin on myocardial IRI is not limited to only animal experiments, we surveyed unstable angina pectoris patients after PCI who may have suffered myocardial IRI during balloon dilatation. We found that patients with higher gastrin levels had lower cardiac injury, which indicates a potential protective effect of gastrin on cardiomyocytes in PCI patients.
Collectively, our findings showed that increased levels of plasma gastrin in patients with myocardial infarction may play a protective effect against IRI. The activation of the RISK and SAFE pathways could trigger prosurvival and antiapoptotic effects to prevent myocardial cell death from the onset of acute myocardial infarction.
Sources of Funding
This study was supported by the National Natural Science Foundation of China (31430043, 31730043), Program of Innovative Research Team by National Natural Science Foundation (81721001), and the National Institutes of Health (R37HL023081‐37, R01DK039308‐30, and P01HL074940‐12).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e005171 DOI: 10.1161/JAHA.116.005171.)
Contributor Information
Hongyong Wang, Email: whysir@aliyun.com.
Chunyu Zeng, Email: chunyuzeng01@163.com.
References
- 1. Perricone AJ, Vander Heide RS. Novel therapeutic strategies for ischemic heart disease. Pharmacol Res. 2014;89:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kennedy JW. Limiting the size of myocardial infarction by early coronary artery reperfusion. Heart Dis Stroke. 1993;2:93–97. [PubMed] [Google Scholar]
- 3. Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. 2016;7:113–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liepinsh E, Makrecka M, Kuka J, Makarova E, Vilskersts R, Cirule H, Sevostjanovs E, Grinberga S, Pugovics O, Dambrova M. The heart is better protected against myocardial infarction in the fed state compared to the fasted state. Metabolism. 2014;63:127–136. [DOI] [PubMed] [Google Scholar]
- 6. Rehfeld JF, Friis‐Hansen L, Goetze JP, Hansen TV. The biology of cholecystokinin and gastrin peptides. Curr Top Med Chem. 2007;7:1154–1165. [DOI] [PubMed] [Google Scholar]
- 7. Jiang X, Wang W, Ning B, Liu X, Gong J, Gan F, Gao X, Zhang L, Jose PA, Qin C, Yang Z. Basal and postprandial serum levels of gastrin in normotensive and hypertensive adults. Clin Exp Hypertens. 2013;35:74–78. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y, Asico LD, Zheng S, Villar VA, He D, Zhou L, Zeng C, Jose PA. Gastrin and D1 dopamine receptor interact to induce natriuresis and diuresis. Hypertension. 2013;62:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goetze JP, Rehfeld JF, Alehagen U. Cholecystokinin in plasma predicts cardiovascular mortality in elderly females. Int J Cardiol. 2016;209:37–41. [DOI] [PubMed] [Google Scholar]
- 10. Grossini E, Caimmi P, Molinari C, Uberti F, Mary D, Vacca G. Intracoronary gastrin 17 increases cardiac perfusion and function through autonomic nervous system, CCK receptors, and nitric oxide in anesthetized pigs. J Appl Physiol. 2011;110:95–108. [DOI] [PubMed] [Google Scholar]
- 11. Gersl V, Goiny M, Uvnas‐Wallensten K. The occurrence of gastrin‐17 in the heart. Acta Physiol Scand. 1981;111:307–310. [DOI] [PubMed] [Google Scholar]
- 12. Jiang Y, Luo L, Gustafson EL, Yadav D, Laverty M, Murgolo N, Vassileva G, Zeng M, Laz TM, Behan J, Qiu P, Wang L, Wang S, Bayne M, Greene J, Monsma F Jr, Zhang FL. Identification and characterization of a novel RF‐amide peptide ligand for orphan G‐protein‐coupled receptor SP9155. J Biol Chem. 2003;278:27652–27657. [DOI] [PubMed] [Google Scholar]
- 13. Tansey MJ, Opie LH, Vinik A, Kennelly BM. Plasma pancreatic polypeptide and gastrin in the assessment of autonomic activity in acute myocardial infarction. Eur J Cardiol. 1981;12:243–259. [PubMed] [Google Scholar]
- 14. Laval M, Baldwin GS, Shulkes A, Marshall KM. Increased gastrin gene expression provides a physiological advantage to mice under hypoxic conditions. Am J Physiol Gastrointest Liver Physiol. 2015;308:G76–G84. [DOI] [PubMed] [Google Scholar]
- 15. Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. Hexokinase II and reperfusion injury: TAT‐HK2 peptide impairs vascular function in Langendorff‐perfused rat hearts. Circ Res. 2013;112:e3–e7. [DOI] [PubMed] [Google Scholar]
- 16. Yue R, Xia X, Jiang J, Yang D, Han Y, Chen X, Cai Y, Li L, Wang WE, Zeng C. Mitochondrial DNA oxidative damage contributes to cardiomyocyte ischemia/reperfusion‐injury in rats: cardioprotective role of lycopene. J Cell Physiol. 2015;230:2128–2141. [DOI] [PubMed] [Google Scholar]
- 17. Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L, Zhang S, Li K, Yu Z. Lycopene protects against hypoxia/reoxygenation‐induced apoptosis by preventing mitochondrial dysfunction in primary neonatal mouse cardiomyocytes. PLoS One. 2012;7:e50778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minamoto Y, Tanaka T, Shibata S, Watanabe S. Neuroprotective effect of cholecystokininB receptor antagonist on ischemia‐induced decrease in CA1 presynaptic fiber spikes in rat hippocampal slices. Neurosci Lett. 1994;167:81–84. [DOI] [PubMed] [Google Scholar]
- 19. Wang WE, Yang D, Li L, Wang W, Peng Y, Chen C, Chen P, Xia X, Wang H, Jiang J, Liao Q, Li Y, Xie G, Huang H, Guo Y, Ye L, Duan DD, Chen X, Houser SR, Zeng C. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose‐derived stem cells in infarcted myocardium. Circ Res. 2013;113:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia‐Dorado D, Hausenloy DJ, Heusch G, Vinten‐Johansen J, Yellon DM, Schulz R; Working Group of Cellular Biology of Heart of European Society of Cardiology . Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406–423. [DOI] [PubMed] [Google Scholar]
- 22. Rahman S, Li J, Bopassa JC, Umar S, Iorga A, Partownavid P, Eghbali M. Phosphorylation of GSK‐3beta mediates intralipid‐induced cardioprotection against ischemia/reperfusion injury. Anesthesiology. 2011;115:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhagatte Y, Lodwick D, Storey N. Mitochondrial ROS production and subsequent ERK phosphorylation are necessary for temperature preconditioning of isolated ventricular myocytes. Cell Death Dis. 2012;3:e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choy MK, Movassagh M, Siggens L, Vujic A, Goddard M, Sanchez A, Perkins N, Figg N, Bennett M, Carroll J, Foo R. High‐throughput sequencing identifies STAT3 as the DNA‐associated factor for p53‐NF‐kappaB‐complex‐dependent gene expression in human heart failure. Genome Med. 2010;2:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dockray G, Dimaline R, Varro A. Gastrin: old hormone, new functions. Pflugers Arch. 2005;449:344–355. [DOI] [PubMed] [Google Scholar]
- 26. Guilloteau P, Le Meuth‐Metzinger V, Morisset J, Zabielski R. Gastrin, cholecystokinin and gastrointestinal tract functions in mammals. Nutr Res Rev. 2006;19:254–283. [DOI] [PubMed] [Google Scholar]
- 27. Isono T, Kamihata H, Sutani Y, Motohiro M, Yamamoto S, Kyoui S, Iharada Y, Kurimoto K, Hara K, Takahashi H, Iwasaka T. Nicorandil suppressed myocardial injury after percutaneous coronary intervention. Int J Cardiol. 2008;123:123–128. [DOI] [PubMed] [Google Scholar]
- 28. Zhou FZ, Song W, Yin LH, Song ZF, Yang S, Yang FB, Liu JF, Song YG, Zhang HY, Zhang ZM. Effects of remote ischemic preconditioning on myocardial injury and endothelial function and prognosis after percutaneous coronary intervention in patienta with acute coronary syndrome. Eur Rev Med Pharmacol Sci. 2017;21:4642–4648. [PubMed] [Google Scholar]
- 29. Vacca G, Vono P. The primary reflex effects of distension of the stomach on heart rate, arterial pressure and left ventricular contractility in the anaesthetized pig. Pflugers Arch. 1993;425:248–255. [DOI] [PubMed] [Google Scholar]
- 30. Bojo L, Lefebvre RA, Nellgard P, Cassuto J. Involvement of vasoactive intestinal polypeptide in gastric reflex relaxation. Eur J Pharmacol. 1993;236:443–448. [DOI] [PubMed] [Google Scholar]
- 31. Ametov AS, Kamynina LL, Akhmedova ZG. Cardioprotective effects of glucagon‐like peptide 1 receptor agonists. Kardiologiia. 2014;54:92–96. [DOI] [PubMed] [Google Scholar]
- 32. Wang YY, He WW, Liu YC, Lin YF, Hong LF. The effect of salt intake and potassium supplementation on serum gastrin levels in Chinese adults: a randomized trial. Nutrients. 2017;9:E389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han D, Huang W, Ma S, Chen J, Gao L, Liu T, Zhang R, Li X, Li C, Fan M, Chen Y, Cao F. Ghrelin improves functional survival of engrafted adipose‐derived mesenchymal stem cells in ischemic heart through PI3K/Akt signaling pathway. Biomed Res Int. 2015;2015:858349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindstrom E, Chen D, Norlen P, Andersson K, Hakanson R. Control of gastric acid secretion: the gastrin‐ECL cell‐parietal cell axis. Comp Biochem Physiol A Mol Integr Physiol. 2001;128:505–514. [DOI] [PubMed] [Google Scholar]
- 35. Dimaline R, Varro A. Novel roles of gastrin. J Physiol. 2014;592:2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng R, Aihara E, Kenny S, Yang L, Li J, Varro A, Montrose MH, Shroyer NF, Wang TC, Shivdasani RA, Zavros Y. Indian Hedgehog mediates gastrin‐induced proliferation in stomach of adult mice. Gastroenterology. 2014;147:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lavine JA, Attie AD. Gastrointestinal hormones and the regulation of beta‐cell mass. Ann N Y Acad Sci. 2010;1212:41–58. [DOI] [PubMed] [Google Scholar]
- 38. Selvik LK, Fjeldbo CS, Flatberg A, Steigedal TS, Misund K, Anderssen E, Doseth B, Langaas M, Tripathi S, Beisvag V, Laegreid A, Thommesen L, Bruland T. The duration of gastrin treatment affects global gene expression and molecular responses involved in ER stress and anti‐apoptosis. BMC Genomics. 2013;14:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mishra P, Senthivinayagam S, Rana A, Rana B. Glycogen synthase kinase‐3beta regulates Snail and beta‐catenin during gastrin‐induced migration of gastric cancer cells. J Mol Signal. 2010;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindstedt G, Bengtsson C, Lapidus L, Nystrom E. Hypergastrinemia—a risk factor for myocardial infarction? Clin Chem. 1985;31:585–590. [PubMed] [Google Scholar]
- 41. Sánchez‐Fernández C, González C, Mercer LD, Beart PM, Ruiz‐Gayo M, Fernández‐Alfonso MS. Cholecystokinin induces cerebral vasodilatation via presynaptic CCK2 receptors: new implications for the pathophysiology of panic. J Cereb Blood Flow Metab. 2003;23:364–370. [DOI] [PubMed] [Google Scholar]
- 42. Foucaud M, Archer‐Lahlou E, Marco E, Tikhonova IG, Maigret B, Escrieut C, Langer I, Fourmy D. Insights into the binding and activation sites of the receptors for cholecystokinin and gastrin. Regul Pept. 2008;145:17–23. [DOI] [PubMed] [Google Scholar]
- 43. Yasui M, Kawasaki K. CCKB receptor activation protects CA1 neurons from ischemia‐induced dysfunction in stroke‐prone spontaneously hypertensive rats hippocampal slices. Neurosci Lett. 1995;191:99–102. [DOI] [PubMed] [Google Scholar]
- 44. Wagner M, Siddiqui MA. Signaling networks regulating cardiac myocyte survival and death. Curr Opin Investig Drugs. 2009;10:928–937. [PubMed] [Google Scholar]
- 45. Lochner A, Huisamen B, Nduhirabandi F. Cardioprotective effect of melatonin against ischaemia/reperfusion damage. Front Biosci (Elite Ed). 2013;5:305–315. [DOI] [PubMed] [Google Scholar]
- 46. Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–419. [DOI] [PubMed] [Google Scholar]
- 47. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre‐, post‐, and remote conditioning. Circ Res. 2015;116:674–699. [DOI] [PubMed] [Google Scholar]
- 48. Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47:32–40. [DOI] [PubMed] [Google Scholar]
- 49. Ramamoorthy S, Stepan V, Todisco A. Intracellular mechanisms mediating the anti‐apoptotic action of gastrin. Biochem Biophys Res Commun. 2004;323:44–48. [DOI] [PubMed] [Google Scholar]
- 50. Beales IL, Ogunwobi O. Glycine‐extended gastrin inhibits apoptosis in colon cancer cells via separate activation of AKT and JNK pathways. Mol Cell Endocrinol. 2006;247:140–149. [DOI] [PubMed] [Google Scholar]
- 51. Westwood DA, Patel O, Baldwin GS. Gastrin mediates resistance to hypoxia‐induced cell death in xenografts of the human colorectal cancer cell line LoVo. Biochim Biophys Acta. 2014;1843:2471–2480. [DOI] [PubMed] [Google Scholar]
- 52. Xiao L, Kovac S, Chang M, Shulkes A, Baldwin GS, Patel O. Induction of gastrin expression in gastrointestinal cells by hypoxia or cobalt is independent of hypoxia‐inducible factor (HIF). Endocrinology. 2012;153:3006–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]