

Abstract
Background
Desmin filament proteins interlink the contractile myofibrillar apparatus with mitochondria, nuclei and the sarcolemma. Mutations in the human desmin gene cause cardiac disease, remodeling, and heart failure but the pathophysiological mechanisms remain unknown.
Methods and Results
Cardiomyocyte‐specific overexpression of mutated desmin (a 7 amino acid deletion R172‐E178, D7‐Des Tg) causes accumulations of electron‐dense aggregates and myofibrillar degeneration associated with cardiac dysfunction. Though extensive studies demonstrated that these altered ultrastructural changes cause impairment of cardiac contractility, the molecular mechanism of cardiomyocyte death remains elusive. In the present study, we report that the D7‐Des Tg mouse hearts undergo aberrant mitochondrial fission associated with increased expression of mitochondrial fission regulatory proteins. Mitochondria isolated from D7‐Des Tg hearts showed decreased mitochondrial respiration and increased apoptotic cell death. Overexpression of mutant desmin by adenoviral infection in cultured cardiomyocytes led to increased mitochondrial fission, inhibition of mitochondrial respiration, and activation of cellular toxicity. Inhibition of mitochondrial fission by mitochondrial division inhibitor mdivi‐1 significantly improved mitochondrial respiration and inhibited cellular toxicity associated with D7‐Des overexpression in cardiomyocytes.
Conclusions
Aberrant mitochondrial fission results in mitochondrial respiratory defects and apoptotic cell death in D7‐Des Tg hearts. Inhibition of aberrant mitochondrial fission using mitochondrial division inhibitor significantly preserved mitochondrial function and decreased apoptotic cell death. Taken together, our study shows that maladaptive aberrant mitochondrial fission causes desminopathy‐associated cellular dysfunction.
Keywords: cardiomyopathy, desminopathy, mitochondrial fission, mitochondrial respiration
Subject Categories: Basic Science Research, Pathophysiology, Cardiomyopathy, Mechanisms, Genetically Altered and Transgenic Models
Clinical Perspective
What Is New?
Desminopathy is a myofibrillar protein aggregation disease whose proteotoxic sequelae affect regular mitochondrial positioning and mitochondrial function, which in turn may lead to cardiomyocyte dysfunction.
This study shows that a proteotoxic stimulus, D7‐Des expression, leads to aberrant mitochondrial fission and mitochondrial dysfunction, resulting in apoptotic cell death in a mouse model of desminopathy.
Inhibition of aberrant mitochondrial fission using mitochondrial division inhibitor significantly preserved mitochondrial function and decreased cell death.
What Are the Clinical Implications?
Desminopathies are associated with derangement of the mitochondrial spatial organization, and accumulation of electron‐dense aggregates, but the pathogenic molecular sequelae remain obscure.
Treatment with a small molecule inhibitor of mitochondrial fission significantly improved mitochondrial respiration and decreased cell death, suggesting a causal role for aberrant mitochondrial fission in desminopathy.
Our study showed that aberrant mitochondrial fission is maladaptive in desminopathy and sheds light on the molecular mechanism of desminopathy‐associated cell death.
Introduction
Desmin is the intermediate filament protein that interlinks neighboring myofibrils at the level of Z‐discs, connecting the entire myofibrillar apparatus to mitochondria and nuclei in striated muscle cells.1, 2 In a healthy heart, desmin maintains the proper mitochondrial positioning along the sarcomere and contributes to normal mitochondrial function by preserving mitochondrial spatial organization.3 Desmin disorganization can affect mitochondrial positioning, compromise mitochondrial function, and lead to cardiomyocyte dysfunction.3 A pivotal milestone in understanding the necessity of desmin's role was the generation of desmin‐null mice. Those mice developed a progressive and generalized myopathy that mainly affected the myocardium. The striated muscles that were examined all showed irregular mitochondrial shape and distribution, including aggregation of sarcolemmal mitochondria, which correlated with weakened muscles and increased fatigue.4, 5
Desmin's essential role was underscored by showing that mutations in the desmin gene (DES) (chromosome 2q35) cause myopathies and cardiomyopathies.6 To date, more than 67 disease‐causing DES mutations have been detected in human myofibrillar diseases.6, 7, 8 Desmin‐related cardiomyopathy (DRC) is a family of genetic disorders caused by mutations in desmin and other desmin‐related proteins that results in a spectrum of myofibrillar myopathies. Desminopathies are characterized by accumulations of electron‐dense aggregates and myofibrillar degeneration in the skeletal muscle and cardiomyocytes.7 Clinically, patients with DRCs exhibit slowly progressive muscle weakness, conduction‐system disease including cardiac arrhythmias, and cardiomyopathy.7 Clinical onsets of DRC range from the first to the eighth decades of life and disease manifestations comprise pure myopathy, cardiomyopathy, or both.7 The majority of desminopathies are caused by heterozygous mutations accounting for autosomal‐dominant cases, although some rare recessive cases and, even more rarely, cases with complete lack of desmin have been reported.3, 7
DRC represents a cardiac proteotoxicity disease that is prevalent in many forms of human heart failure. The mutant desmin transgenic mouse (D7‐Des Tg) carries a 7 amino acid deletion R172‐E178 in DES and provides a clinically relevant mouse model of cardiac proteotoxicity.9, 10, 11, 12 This model manifests a collapse of the desmin network, and accumulation of desmin aggregates, which contributes to cardiomyopathy.10 DRC‐causing mutations result in early perturbations in mitochondrial structure. For example, mitochondrial disorganization was observed in the mutant αB‐crystallin Tg (CryABR120G) mouse models of DRC.10, 13, 14 In DRC mice expressing either D7‐Des or CryABR120G, mitochondrial spatial organization was highly perturbed, and the myofibrils were interspersed with electron‐dense aggregates. Similarly, tissue biopsy samples from patients diagnosed with myofibrillar myopathies show abnormal mitochondrial enzyme staining and have reduced mitochondrial complex‐I activities.15 Despite extensive studies, the genetic pathologies for the DRCs are only partially defined and the pathogenic sequelae remain obscure.
Previous reports have noted that both mitochondrial morphology and positioning are altered in the muscles of DRCs.10, 16 It is unknown whether these alterations result in maladaptive changes in mitochondrial dynamics and directly contribute to mitochondrial dysfunction, resulting in cardiomyopathies in DRC. Here, we demonstrate that aberrant mitochondrial fission and mitochondrial respiratory dysfunction are associated with cardiac dysfunction and explore the functional consequences in the D7‐Des Tg mouse model of cardiac proteotoxicity. Using a mitochondrial division chemical inhibitor, we show that aberrant fission inhibition improved mitochondrial function, reduced aggregate accumulation, and reduced cellular toxicity.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. Other researchers can contact the corresponding authors about methodological questions.
Materials
Materials are as follows: AdEasy system (Agilent Technologies), OptiMEM (Gibco), DMEM (Gibco), fetal bovine serum (Gibco), Cell Lytic M (Sigma‐Aldrich), Protease Inhibitor Cocktail (Roche), pre‐cast 7.5% to 15% Criterion Gels (BioRad), 4′‐6‐diamidino‐2‐phenylindole (Invitrogen), mdivi‐1 (Sigma‐Aldrich), Oligomycin (Sigma‐Aldrich), carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone (FCCP) (Sigma‐Aldrich), Rotenone (Sigma‐Aldrich), Antimycin A (Sigma‐Aldrich), Ponceau S (Acros Organic), Calcium Green‐5N (Invitrogen), Vectashield Hardset (Vector Labs, H1400), and antimycin A (Sigma‐Aldrich) were used.
Animals
D7‐Des Tg mice have been described previously10 and are in an FVB/N background. All procedures for handling animals complied with the Guide for Care and Use of Laboratory Animals and were approved by the ACUC Committee of LSU Health Sciences Center‐Shreveport. All animals were cared for according to the National Institutes of Health guidelines for the care and use of laboratory animals. Timed pregnant Sprague‐Dawley rats were purchased from Charles River Laboratories International, Inc (Portage, MI).
Neonatal Rat Cardiomyocyte Isolation and Culture
Neonatal rat cardiomyocytes (NRCs) were isolated from the ventricles of 1‐ to 2‐day‐old Sprague–Dawley rat pups as previously described.17 Ventricular tissues of rat pups were digested with collagenase at 37°C overnight, followed by further digestion in trypsin. After a preplating step to remove cardiac fibroblasts, isolated cardiomyocytes were plated at 1.5×106 cells per 10‐cm2 plate and 1.0×105 cells per chamber in 2‐well glass chamber slides in αMEM containing 10% fetal bovine serum (Gibco) and 1% antibiotic‐antimycotic (Gibco). Cells underwent different treatments 24 hours after plating and were maintained in DMEM (Gibco) containing 2% fetal bovine serum and 1% antibiotic‐antimycotic. All cell culture treatments were repeated in 3 independent experiments.
Adenovirus Infection of NRCs
We prepared adenoviral constructs containing D7‐Des by cloning into a pShuttle‐CMV vector; replication‐deficient recombinant adenoviruses were generated using the AdEasy system (Agilent Technologies).17, 18, 19 NRCs were infected with D7‐Des adenovirus at different multiplicities of infection (1–10) for 2 hours and then replaced with fresh medium. Cells were harvested for 72 hours postinfection. Infection of parallel plates with adenovirus expressing β‐galactosidase served as controls for all the experiments.
mdivi‐1 Treatment
For the measurement of mdivi‐1's effects on mitochondrial respiration and cellular toxicity, we treated cardiomyocytes with mdivi‐1 72 hours after plating. NRCs were treated with different concentrations of mdivi‐1 (1, 5, and 10 μmol/L) or the same volume of vehicle (dimethyl sulfoxide) for 24 hours. To determine the effects of mdivi‐1 on D7 Des expression induced pathogenesis, NRCs were infected with D7‐Des adenovirus (10 multiplicities of infection) 24 hours after plating the cardiomyocytes. Cardiomyocytes were treated with 5 μmol/L mdivi‐1 at 72 hours postinfection. All biochemical and mitochondrial respiration experiments using an XF24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA) were performed 24 hours after mdivi‐1 treatment.
Echocardiography
Echocardiograms were performed on isoflurane‐anesthetized mice with a VisualSonics Vevo 2100 Imaging System (Toronto, Ontario, Canada) using a 40‐MHz transducer to assess cardiac functional parameters.20, 21 Briefly, 2‐dimensional directed M‐mode echocardiographic images along the parasternal short axis were recorded by investigators blinded to genotype to determine left ventricular (LV) size and systolic function. M‐mode measurements included the LV internal dimensions in systole and diastole (LVIDs and LVIDd, respectively) as well as the diastolic thickness of LV posterior wall and diastolic intraventricular septum thickness (IVSd). Percent fractional shortening was calculated using [(LVIDd−LVIDs)/LVIDd]×100. From these data, LV end‐systolic, LV end‐diastolic diameter, LV mass, and percent ejection fraction were calculated. Systolic shortening of the interventricular septum and posterior wall was calculated using: [(systolic thickness−diastolic thickness)/systolic thickness]×100.
Electron Microscopy
For electron microscopic examination, D7 Des Tg and non‐transgenic (Ntg) hearts were perfused with 1% paraformaldehyde/2% glutaraldehyde in the cardioplegic buffer, then with 1% paraformaldehyde/2% glutaraldehyde in 0.1 mol/L cacodylate buffer, pH 7.2, postfixed in 1% OsO4 and processed for thin sectioning as described previously.20, 21
Mitochondria Isolation
Mitochondria in D7 Des Tg and Ntg hearts were isolated as described previously.22, 23 Briefly, hearts were harvested, homogenized in MS‐EGTA buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 5 mmol/L HEPES, and 1 mmol/L EGTA, pH 7.4) and subjected to differential centrifugation. Finally, mitochondria were lysed with 1x cell lytic M (Sigma‐Aldrich) containing protease and phosphatase inhibitors.
Mitochondrial Respiration
Mitochondrial oxygen consumption rate was measured with an XF24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA) by methods as described previously.22, 23, 24 Heart mitochondria were isolated using MS‐EGTA buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 5 mmol/L HEPES, and 1 mmol/L EGTA, pH 7.4) by differential centrifugation as described above. Mitochondria (50 μg/well) were seeded in XF24 culture plates, and respiration was measured in mitochondrial assay buffer (220 mmol/L mannitol, 70 mmol/L sucrose, 10 mmol/L KH2PO4, 5 mmol/L MgCl2, 2 mmol/L HEPES, 1 mmol/L EGTA, 0.2% fatty acid‐free bovine serum albumin, pH 7.4) supplemented with 7 mmol/L pyruvate and 1 mmol/L malate. Mitochondrial oxygen consumption rate (OCR) was measured and plotted at basal conditions followed by sequential addition of 1 μg/mL oligomycin (ATP‐synthase inhibitor), 4 μmol/L FCCP (a mitochondrial uncoupler), and 0.5 μmol/L rotenone (complex I inhibitor) plus 0.5 μmol/L antimycin A (complex III inhibitor). The OCR values were normalized to total protein content in the corresponding wells and expressed as pmol/min per μg protein.
For intact cardiomyocytes, NRCs were seeded at a density of 8×104 cells/well into gelatin‐coated Seahorse Bioscience XF microplates and were grown in DMEM (Gibco) containing 2% fetal bovine serum (Gibco) and 1% antibiotic‐antimycotic (Gibco). Twenty‐four hours after plating the NRCs, cardiomyocytes were infected with D7‐Des adenovirus for 2 hours, and then the media was replaced with DMEM (Gibco) containing 2% fetal bovine serum and 1% antibiotic‐antimycotic. Infection of parallel plates with adenovirus expressing β‐galactosidase served as a control for all experiments. NRCs were incubated with DMEM (containing no glucose and pyruvate, Gibco) supplemented with 10 mmol/L glucose and 2 mmol/L pyruvate in a CO2‐free incubator at 37°C for 1 hour before loading the plate in the XF24 analyzer. The OCR was measured over a period of 86 minutes over which time oligomycin (1 μmol/L), FCCP (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L) were sequentially added to each well at specified time points.
Histological Analysis and Immunofluorescence Microscopy
Hearts were collected, fixed in 10% buffered formalin, and embedded in paraffin as described previously.20, 21 Serial 5‐μm heart sections from each group were stained with Masson's trichrome21 and Sirius Red.25 Fibrosis and collagen deposition within sections were quantitated using ImageJ software (NIH) as described previously. To quantitate fibrosis, blue‐stained areas and nonstained myocyte areas from each section were determined using color‐based thresholding. Collagen deposition was quantified by red‐stained areas and nonstained myocyte areas from each section using color‐based thresholding. The percentage of total fibrosis area was calculated as the blue‐stained areas divided by total surface area from each section. The relative amount of collagen deposition area was calculated as the red‐stained areas divided by total surface area from each section.
Paraffin‐embedded heart tissue sections (5 μm) were stained for immunofluorescent detection of desmin aggregates in D7‐Des Tg mice hearts according to the procedures described previously.10, 20 Briefly, heart tissue sections (5 μm) were deparaffinized through washes in xylene, and graded alcohol series following rehydration with deionized water and 0.1M phosphate‐buffered saline, pH 7.4 (PBS) for 10 minutes each. Heat‐induced antigen retrieval was carried out by boiling in 10 mmol/L citrate buffer for 30 minutes. Slides were then cooled down to room temperature (RT), washed with PBS and blocked with 1% BSA, 0.1% cold water fish skin gelatin, and 1% Tween 20 in PBS for 1 hour at RT. Then, tissue sections were immunolabeled by overnight incubation of the sections with rabbit anti‐desmin antibody (Biomeda) diluted at blocking buffer at 1:100 dilution at 4°C. Next, sections were washed with PBS and incubated with anti‐rabbit secondary antibody conjugated with Alexa 568 dye (Invitrogen) for 1 hour at RT. Sections were then washed and counterstained with 4′‐6‐diamidino‐2‐phenylindole (Invitrogen) following mounting with Vectashield Hard Set (Vector Laboratories). Stained tissue sections were assessed by investigators blinded to genotype for desmin aggregate staining on a Leica TCS SP5 spectral confocal microscope using a ×63 oil objective (NA=1.4), and images were acquired with Leica LAS (AF 2.6.3) software.
Immunocytochemistry
Immunocytochemistry was conducted in NRCs infected with D7‐Des adenovirus to evaluate mitochondrial morphology, staining for the mitochondrial outer membrane protein Tom20. Briefly, isolated NRCs were plated on Lab‐Tek II chamber slides (Thermo Scientific, 154461) at a density of 1×105 cells/well. Immunocytochemistry was then performed according to the procedures reported previously.17, 20, 21 Plated cells were first washed with 0.1 mol/L PBS followed by fixation and permeabilization in 4% paraformaldehyde with 0.5% Triton‐X 100 in PBS for 15 minutes. Antigen retrieval was carried out by incubating in 0.1 mol/L glycine solution, pH 3.5 for 30 minutes at RT. The cells were then incubated in blocking solution containing 1% BSA, 0.1% cold water fish skin gelatin, 0.1% Tween‐20, and 0.05% sodium azide in PBS for 1 hour at RT. Following blocking, cells were incubated with rabbit anti‐Tom20 antibody (Santa Cruz) at 1:500 dilution in blocking solution overnight at 4°C. The next morning, cells were washed and incubated with anti‐rabbit secondary antibody conjugated with Alexa 568 dye (Invitrogen) for 1 hour at RT. Next, cells were washed with PBS, counterstained with 4′‐6‐diamidino‐2‐phenylindole (Invitrogen), and mounted with Vectashield Hardest (Vector Laboratories). Stained cells were then observed on a Leica TCS SP5 spectral confocal microscope using a ×63 oil objective (NA=1.4) and imaged using Leica LAS (AF 2.6.3) software. All image analyses were performed in an investigator‐blinded manner.
Mitochondrial Ca2+ Retention Capacity and Swelling Assay
Mitochondrial Ca2+ retention capacity was measured with Calcium Green 5N (Molecular Probes) as previously described.22, 23 Cardiac mitochondria (100 μg) were suspended in KCl buffer containing 125 mmol/L KCl, 20 mmol/L HEPES, 2 mmol/L MgCl2, 2 mmol/L potassium phosphate, and 40 μmol/L EGTA, pH 7.2, 200 nmol/L Calcium Green‐5N (Molecular Probes), 7 mmol/L pyruvate, and 1 mmol/L malate. Mitochondria were challenged with additions of CaCl2 and Calcium Green‐5N fluorescence was monitored using the BioTek Synergy IV plate reader (BioTek). Ca2+ pulses of 20 nmol/mg of mitochondrial protein were added every 180 s using auto‐injector. The Ca2+ pulse induced a peak in the extramitochondrial Ca2+ concentration that returns to near baseline level as Ca2+ enters the mitochondrial matrix. With increasing mitochondrial Ca2+ loading, extramitochondrial Ca2+ starts accumulating until the addition of Ca2+ leads to a sustained Ca2+ increase indicated by a massive release of mitochondrial Ca2+ evident by a sustained increase in fluorescence. Mitochondrial Ca2+ retention capacity was quantitated as the amount of Ca2+ required to trigger a massive Ca2+ release by the mitochondria resulting from mitochondrial permeability transition pore (MPTP) opening.
For mitochondrial swelling assay, mitochondria isolated from hearts were suspended in swelling buffer containing 120 mmol/L KCl, 10 mmol/L Tris pH 7.4, 5 mmol/L KH2PO4, 7 mmol/L pyruvate, 1 mmol/L malate, and 10 μmol/L EDTA. Mitochondrial swelling was induced by challenging the mitochondria with 200 μmol/L CaCl2, and the absorbance at 540 nm was monitored.22, 23
ATP Measurements
Cellular and mitochondrial ATP contents were measured in freshly prepared whole cell lysates and mitochondrial suspensions from left ventricles (LVs) according to manufacturer's instructions using the ATP Bioluminescence Assay Kit HS II (Roche).26, 27 Freshly excised LVs were chopped and homogenized using a hand‐driven glass/Teflon potter Elvehjem homogenizer (Wheaton) in cell lysis reagent provided in the kit on ice. Fresh mitochondria suspensions were collected as described above and homogenized in cell lysis reagent (Roche) using this method on ice. Prepared whole cell and mitochondria lysates were then left at RT for 5 minutes following boiling at 100°C for 2 minutes to extract ATP according to manufacturer's instructions (Roche). The lysates were then centrifuged at 10 000g for 1 minute and supernatants were collected to measure the ATP level. ATP content was measured immediately after addition of luciferase reagent (Roche) by automated injection and luciferase‐driven bioluminescence was measured on a FLUOstar OPTIMA microplate reader (BMG Labtech). ATP levels in samples were then normalized to protein content as measured by Bio‐Rad protein assay reagent.26
mtDNA Quantification
Total DNA was isolated from mouse hearts using the Quick‐gDNA MiniPrep kit (Zymo Research) according to the manufacturer's protocol. The mtDNA content was measured by PCR of cytochrome B and β‐actin as described.28, 29 Primer sequences used for cytochrome B and β‐actin were as follows: 5′‐CCACTTCATCTTACCATTTATTATCGC‐3′ (forward primer) and 5′‐TTTTATCTGCATCTGAGTTTAA‐3′ (reverse primer) for mitochondrial cytochrome B, and 5′‐CTGCCTGACGGCCAGG‐3′ (forward primer) and 5′‐CTATGGCCTCAGGAGTTTTGTC‐3′ (reverse primer) for genomic β‐actin.
Lactate Dehydrogenase Release Assay
Lactate dehydrogenase release from NRCs was measured using the Cytotoxicity Detection Kit (Roche) according to the manufacturer's instructions. Color development was measured using a microplate reader at an absorbance of 492 nm (Bio‐Rad).17
Protein Extraction and Western Blot Analyses
Soluble and insoluble fractions were prepared from hearts harvested in cold PBS, pH 7.4 containing 1% Triton‐X100, 2.5 mmol/L EDTA, 0.5 mmol/L PMSF, and a complete protease inhibitor mixture, and then vortexed for 30 s as described previously.21 The heart homogenates were centrifuged at 12 000g for 15 minutes and the supernatants were collected (soluble fraction). The pellets were dissolved in DNAase (1 mg/mL in 10 mmol/L Tris, 15 mmol/L MgCl2) (Roche), sonicated on ice, and the protein was quantitated with a modified Bradford assay. The insoluble protein was then diluted in RIPA buffer and 3 μg of resuspended protein used for subsequent immunoblotting with appropriate antibodies.
Total proteins were prepared from NRCs washed with PBS and the cells were lysed with Cell Lytic M (Sigma‐Aldrich) lysis buffer, supplemented with Complete Protease Inhibitor Cocktail (Roche).17, 18 The lysed cells were homogenized by sonication and centrifuged at 14 000g for 15 minutes to sediment any insoluble material. The protein content of the soluble lysates was measured using the modified Bradford protocol/reagent relative to a BSA standard curve (BioRad). Protein lysates were separated on SDS‐PAGE using pre‐cast 7.5% to 15% Criterion Gels (BioRad) and transferred to PVDF membranes (BioRad). Membranes were blocked for 1 hour in 5% nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: Anti‐Drp1 (1:1000, Cell Signaling Technology), Anti‐pDrp‐Ser616 (1:1000, Cell Signaling Technology), Anti‐Mfn2 (1:1000, Cell Signaling Technology), anti‐OPA1 (1:1000, Cell Signaling Technology), anti‐OMA1 (1:200. Santa Cruz Biotechnology), Anti‐Fis1 (1:200, Santa Cruz Biotechnology), Anti‐mitochondrial fission factor (1:200, Santa Cruz Biotechnology), Anti‐Tom20 (1:200, Santa Cruz Biotechnology), Anti‐Tim23 (1:200, Santa Cruz Biotechnology), Anti‐PGC1α (1:200, Santa Cruz Biotechnology), Anti‐β‐Actin (1:1000, Santa Cruz Biotechnology), Anti‐OXPHOS (1:1000, Abcam), Anti–pyruvate dehydrogenase (1:1000, Abcam), Anti‐Bax (1:1000, Cell Signaling Technology), Anti‐COXIV (1:1000, Cell Signaling Technology), Anti‐cleaved PARP1 (1:200, Santa Cruz Biotechnology), Anti‐CHOP (1:1000, Cell Signaling Technology), Anti‐P62 (Progen Biotechnik GmbH), Anti‐Desmin (1:2000, Biomeda), Anti‐CryAB (1:10 000, Enzo Life Sciences), Anti‐GRP75 (1:200, Santa Cruz Biotechnology), Anti‐Periostin (1:500, Santa Cruz Biotechnology), and β‐Tubulin (1:500) (Cell Signaling Technology). Membranes were then washed, incubated with alkaline‐phosphatase‐conjugated secondary antibodies (Santa Cruz Biotechnology), exposed with ECF reagent (Amersham) and finally, detected on a ChemiDoc Touch Imaging System (BioRad). Ponceau S protein stain of the transfer membrane was used as a loading control. Densitometry on scanned membranes was done using ImageJ software (National Institutes of Health, Bethesda, MD).
Statistical Analysis
Data are expressed as mean±SEM. All statistical tests were done with GraphPad Prism software. Data were analyzed using Student t test (P<0.05) for 2 groups and groups of 4 or more with 1‐way ANOVA, followed by Tukey's post hoc test. For certain data sets with smaller sample size (n=3), the Kruskal–Wallis test was applied (using R software, 64 bit version 3.4.3), and data were presented in graphs showing median and interquartile ranges. A value of P<0.05 was considered statistically significant.
Results
Desmin Aggregation, Hypertrophic Remodeling, and Cardiac Dysfunction in D7‐Des Tg Hearts
Hearts derived from the D7‐Des Tg mice showed mutant protein aggregate formation in the pellet (insoluble) fraction without alteration of desmin in the soluble fraction (Figure 1A). CryAB protein levels were also significantly increased in both pools in the D7‐Des Tg hearts (Figure 1A). Immunostaining of the D7‐Des Tg hearts showed abnormal desmin arrangements, confirming that the abnormal aggregates were desmin positive. The aberrant desmin aggregates disrupted the continuity and overall organization of the desmin network (Figure 1B). The D7‐Des Tg hearts showed significant interstitial fibrosis as quantified by Masson's trichrome staining (Figure 2A) and collagen deposition detected using Sirius Red staining compared with Ntg mice (Figure 2B). The D7‐Des Tg hearts also showed an increased level of periostin expression that is consistent with the increased fibrosis (Figure 2C). Heart‐to‐body‐weight ratio confirmed cardiac hypertrophy (Figure 2D). Consistent with these data, echocardiography showed a significant increase in LV mass index, septal (interventricular septum) and posterior wall thicknesses as well as significant changes in systolic and diastolic LV chamber dimensions in the D7‐Des Tg hearts (Figure 3A through 3L). Systolic shortening of the interventricular septum of Ntg mice showed ≈31% compared with only ≈18% in D7‐Des mice. Systolic shortening of the posterior wall showed ≈32% for Ntg and ≈20% for D7‐Des Tg, suggesting that the D7‐Des Tg hearts were not only thicker at end diastole, but they also failed to contract to the same degree as Ntg (thicken up) in systole. Interestingly, the degree of cardiac dysfunction in the 6‐month‐old D7‐Des Tg mice used in this study was significantly worse than reported in a previous study,10 likely because of the older age of our cohorts. Overall, the D7‐Des Tg mice showed desmin aggregate formation, adverse cardiac hypertrophic remodeling, and defective cardiac contractile function recapitulating aspects of human desminopathies.
Figure 1.
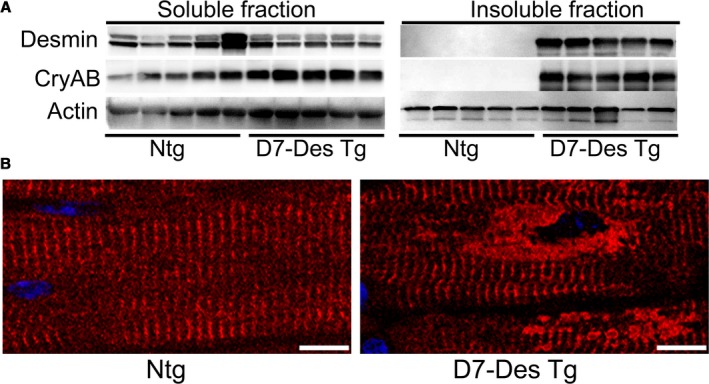
Desmin expression in D7‐Des Tg mice hearts. A, Western blot analysis of desmin and αB‐crystallin protein expression in the soluble and insoluble protein fractions derived from 6‐month‐old D7‐Des Tg and Ntg hearts. n=5 mice per group. B, Confocal microscopy images of immunostaining of desmin (red) in heart sections showing aberrant desmin aggregates in D7‐Des Tg cardiomyocytes (Scale bars: 5 μm). CryAB indicates αB‐Crystallin; D7‐Des Tg, mutant desmin transgenic mouse; Ntg, non‐transgenic.
Figure 2.
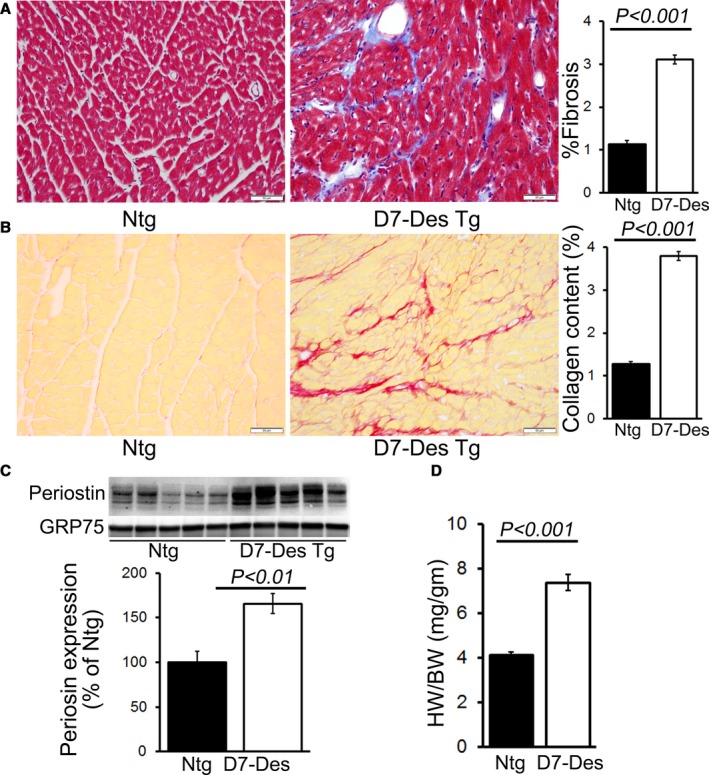
D7‐Des expression results in cardiac hypertrophy and fibrosis. A, Representative micrographs of Masson's trichrome‐stained LV myocardium and quantification of the cardiac fibrosis in Ntg and D7‐Des Tg hearts (n=5 mice per group). B, Representative micrographs of Sirius Red–stained LV myocardium and quantification of the myocardial collagen deposition in Ntg and D7‐Des Tg hearts (n=5 mice per group). C, Representative Western blot and densitometric quantification showing significantly increased expression of periostin in D7‐Des Tg hearts (n=5 mice per group). D, Heart‐weight‐to‐body‐weight ratios (HW/BW) showing LV hypertrophy in the D7‐Des Tg hearts (n≥6 or more mice per group). Bars represent mean±SEM. P values were determined by Tukey's post hoc test. Scale bars: 50 μm. D7‐Des Tg indicates mutant desmin transgenic mouse; LV, left ventricular; Ntg, non‐transgenic.
Figure 3.
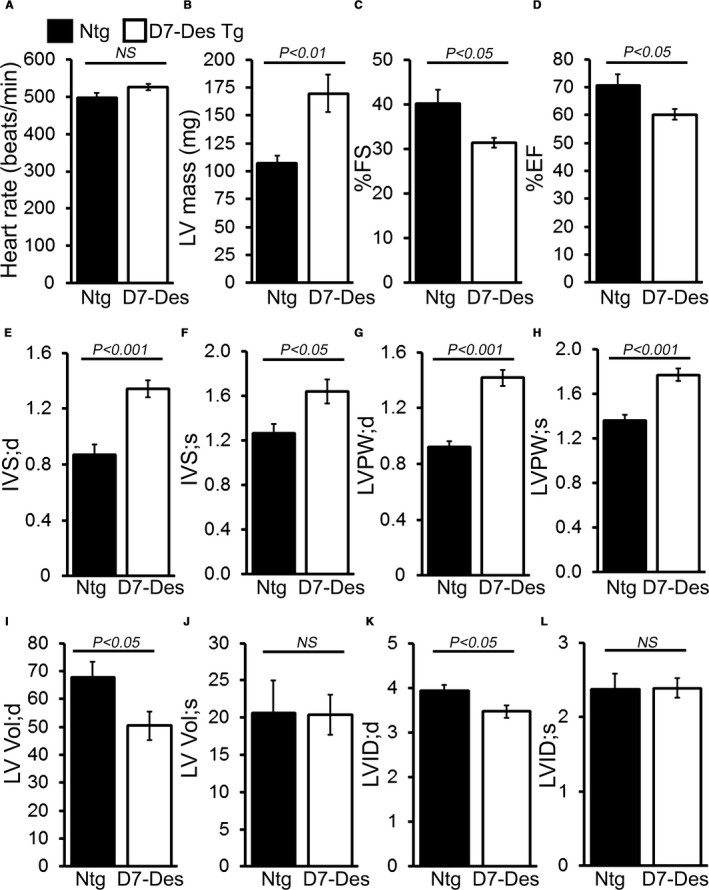
M‐mode echocardiography indices of cardiac structure and function in 6‐month‐old D7‐Des Tg mice. A, Heart rate (beats/min), (B) Left ventricular (LV) mass. (C) Percent fractional shortening (%FS). D, Percent ejection fraction (%EF). E and F, LV interventricular septum diastolic (IVS;d) and systolic (IVS;s) thickness. G and H, LV posterior wall diastolic (LVPW;d) and systolic (LVPW;s) thickness. I and J, LV diastolic (LV Vol;d) and systolic volume (LV Vol;s). K and L, LV internal dimension in end‐diastole (LVID;d) and end‐systole (LVIDs). Bars represent mean±SEM. n=10 mice per group. P value vs Ntg mice by Tukey's post hoc test. D7‐Des Tg indicates mutant desmin transgenic mouse; NS, not significant; Ntg, non‐transgenic.
Aberrant Mitochondrial Fission in D7‐Des Tg Hearts
Ultrastructural analysis by electron microscopy showed that mitochondria are well organized and in a regular pattern along the sarcomeres in Ntg hearts. In contrast, D7‐Des Tg mice showed highly perturbed mitochondrial spatial organization (Figure 4A). Relative mitochondrial DNA (mtDNA) content measured as a function of total genomic DNA (nDNA) in hearts of D7‐Des Tg was significantly increased compared with Ntg (Figure 4B). Given the alterations in mitochondrial morphology and mtDNA copy number, we hypothesized an impaired balance of fission/fusion and investigated the effect of D7‐Des overexpression on mitochondrial fission (ie, DRP1 and Fis1) and fusion proteins (ie, OPA1, and Mfn2). Immunoblot analysis revealed that DRP1, Phospho‐DRP1, FIS1, Mff, and MFN2 were significantly increased in whole cell lysates from D7‐Des Tg hearts compared with Ntg (Figure 4C). There were no significant changes in OPA1 and Tim 23 levels. We also observed significantly increased expression of DRP1, MFN2, Mff, and Tim 23 in the mitochondrial fraction in D7‐Des Tg hearts compared with Ntg (Figure 4D). There were no significant changes in OPA1, OMA1, Fis1, and Tom20 levels in the mitochondrial fraction. Ponceau‐S staining of the Western blots was used to confirm equivalent loading. The increased number of mitochondria indicated by TOM20 and mtDNA is likely because of aberrant mitochondrial fission without any effect on expression of the mitochondrial biogenesis factor PGC‐1α in the D7‐Des Tg hearts (Figure 4C).
Figure 4.
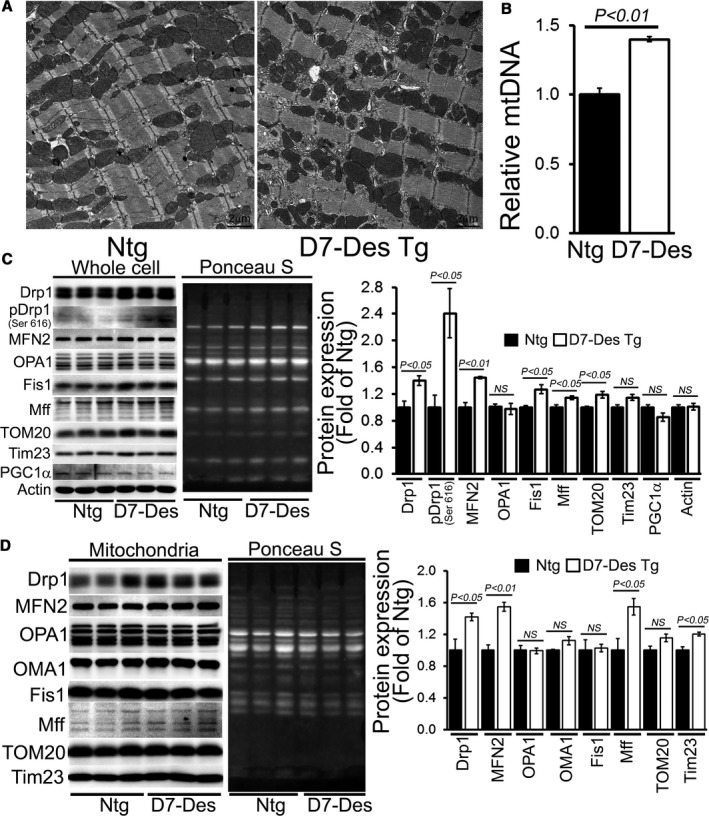
Aberrant mitochondrial fission and altered expression of fission regulatory proteins in D7‐Des Tg hearts. A, Representative transmission electron micrographs of hearts from D7‐Des Tg mice (Scale bars: 2 μm). B, Relative mtDNA content expressed compared with total genomic DNA (nDNA) in hearts of D7‐Des Tg was significantly increased compared with Ntg. Representative Western blot and densitometric quantification of the (C) whole cell fraction and (D) mitochondrial fraction showing expression of mitochondrial dynamic regulatory proteins in the D7‐Des Tg hearts: Drp1, pDrp1 (Ser 616), Fis 1, OPA1, OMA1, MFN2, Mff, Tom20, Tim23, and PGC1α. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading across the gel. Bars represent mean±SEM. n=6 mice per group. P values were determined by Tukey's post hoc test. D7‐Des Tg indicates mutant desmin transgenic mouse; mtDNA, mitochondrial DNA; NS, not significant; Ntg, non‐transgenic.
Mitochondrial Respiratory Dysfunction in the D7‐Des Tg Mouse
The abnormal mitochondrial morphology and distribution in D7‐Des Tg mice pointed to potential mitochondrial dysfunction. To ascertain this, we isolated mitochondria from both D7‐Des Tg and Ntg mouse hearts and measured mitochondrial respiration. Real‐time OCRs in isolated mitochondria show that basal respiration, representing the sum of all physiological mitochondrial oxygen consumption, was decreased in the D7‐Des Tg samples, indicating lower respiratory function compared with Ntg hearts (Figure 5A and 5B). The injection of oligomycin, an ATP synthase inhibitor, leads to a decrease in basal respiration that is reflective of oxygen consumption used to generate ATP (Figure 5C). In addition to low basal respiration, D7‐Des Tg mitochondria presented a very low ATP‐linked OCR (Figure 5C), indicating only a small contribution of mitochondria toward ATP generation in these cells. The addition of FCCP uncouples respiration from oxidative phosphorylation and allows for the measurement of maximal OCR, which was lower in D7‐Des Tg mitochondria (Figure 5D), indicating lower overall mitochondrial activity. The extent of nonmitochondrial oxygen‐consuming processes was estimated by inhibiting the respiratory chain with rotenone and antimycin A and therefore, the non‐mitochondrial respirationwas also significantly lower D7‐Des Tg mitochondria (Figure 5E). The respiratory reserve capacity, calculated by subtracting basal OCR from FCCP‐stimulated OCR, was significantly lower in D7‐Des Tg mitochondria (Figure 5F), and ATP turnover measured by ATP‐linked respiration subtracted from the basal OCR was significantly decreased in D7‐Des Tg mitochondria (Figure 5G). The maximum respiration calculated by nonmitochondrial respiration subtracted from FCCP‐stimulated OCR was also significantly lower in D7‐Des Tg mitochondria (Figure 5H). We conclude that mitochondria isolated from D7‐Des hearts are functionally compromised.
Figure 5.
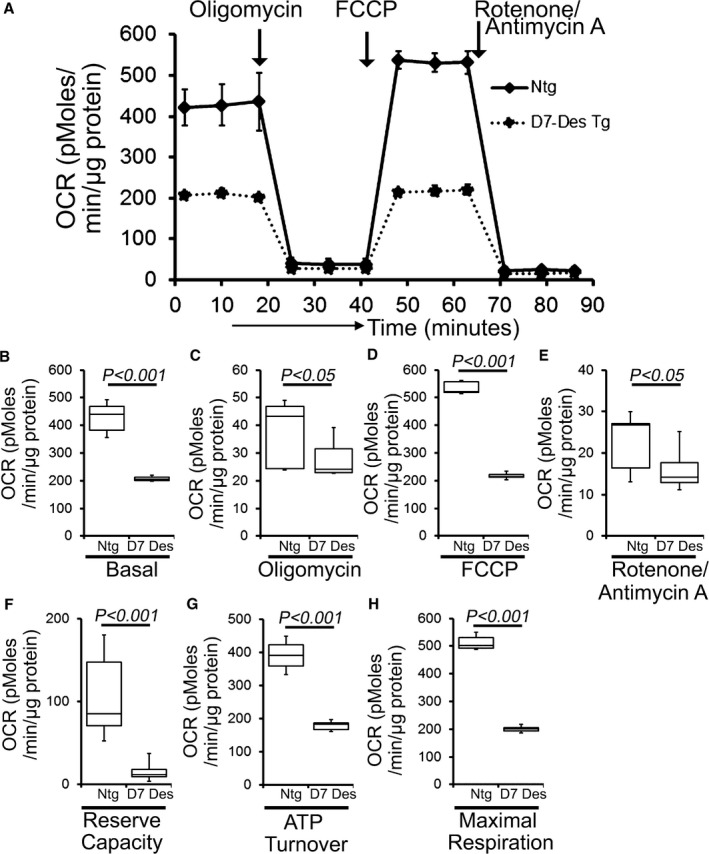
Suppression of mitochondrial respiration in D7‐Des Tg mice hearts. A, Mitochondrial oxygen consumption rate (OCR) profiles in isolated mitochondria from 6‐month‐old Ntg and D7‐Des Tg hearts. Arrow indicates the sequential addition of oligomycin (1 μmol/L), FCCP (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L). OCR profiles are expressed as pmol O2/min per μg of protein. Graph shows OCR under (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Key parameters of mitochondrial function, including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration were significantly decreased in D7‐Des Tg mice. n=3 mice per group. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. D7‐Des Tg indicates mutant desmin transgenic mouse; FCCP, carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone; Ntg, non‐transgenic.
Because the D7‐Des Tg hearts showed defective mitochondrial respiration, we compared OXPHOS protein levels in whole cell lysates and the mitochondrial fractions using 6‐month‐old D7‐Des Tg and Ntg mice hearts of mixed sex. D7‐Des Tg hearts showed significantly increased levels of Complex III and II in the whole cell lysates, and Complex V and III subunits in mitochondrial fractions (Figure 6A and 6B). No significant changes were observed in the expression of pyruvate dehydrogenase complex proteins except E3 bp in the mitochondrial fractions (Figure 6A and 6B). Actin was used to confirm the purity of the mitochondrial fraction and Cox IV was used as a marker for the mitochondrial fraction. Ponceau S staining of proteins was used to confirm equal loading (Figure 6A). Measurement of mitochondrial ATP production and cellular ATP levels demonstrated a significantly lower ATP pool in the D7‐Des Tg hearts (Figure 6C). Collectively, we find that the defective mitochondrial respiration in the D7‐Des Tg hearts is associated with altered expression of OXPHOS proteins.
Figure 6.

Impaired OXPHOS protein expression and ATP content in the D7‐Des Tg hearts. A, Representative Western blot showing expression of Complex I, Complex II, Complex III, Complex V, and PDH complex protein derived from the mitochondrial fractions isolated from 6‐month‐old D7‐Des Tg hearts. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading. Actin was used as a whole cell lysate and COX IV as a mitochondrial marker to indicate the purity of the fractionation. B, Densitometric quantification of OXPHOS complex and PDH complex protein. C, Mitochondrial and whole cellular ATP content in the D7‐Des Tg hearts. n=3 mice per group. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. D7‐Des Tg indicates mutant desmin transgenic mouse; NS, not significant; Ntg, non‐transgenic; PDH, pyruvate dehydrogenase.
Activation of Apoptosis in the D7‐Des Tg Heart
Previous data showed that mitochondrial dysfunction in mutant αB‐Crystallin mouse models of DRC correlated with opening of the MPTP and activation of apoptotic cell death.13, 30 MPTP opening results in mitochondrial swelling, eventually rupturing the outer mitochondrial membrane, releasing potent apoptogens into the cytoplasm and triggering apoptotic cell death. Mitochondrial swelling is classically determined by challenging isolated mitochondria with calcium and measuring the decrease in light scattering.31 We isolated mitochondria from the hearts of D7‐Des Tg as well as Ntg mice at 6 months of age, and examined their ability to undergo calcium‐induced swelling. Mitochondria isolated from D7‐Des Tg hearts showed significantly decreased baseline absorption, indicating increased swelling under basal conditions. In addition, D7‐Des Tg mitochondria showed a dramatically attenuated response after addition of calcium compared with Ntg hearts (Figure 7A). We performed parallel experiments using Calcium Green‐5N to measure mitochondrial calcium retention capacity to test the calcium threshold needed to trigger MPTP opening.32 Calcium pulses of 20 nmol/mg of mitochondrial protein resulted in a rapid increase in Calcium Green‐5N fluorescence followed by a decline in the fluorescence intensity of the calcium sensor, and this decline in fluorescence corresponded to mitochondrial calcium uptake (Figure 7B). With increasing mitochondrial Ca2+ loading, extramitochondrial Ca2+ starts accumulating until the addition of Ca2+ leads to a sustained Ca2+ increase, indicating a massive release of mitochondrial Ca2+. Mitochondrial Ca2+ retention capacity was quantitated as the amount of Ca2+ required to trigger a massive Ca2+ release by the mitochondria resulting from MPTP opening. The D7‐Des heart mitochondria showed significantly lower mitochondrial calcium retention capacity compared with Ntg mitochondria (Figure 7B). Mitochondrial swelling and MPTP opening can result in apoptosis via the mitochondrial localization of Bax. We measured Bax in both the whole cell and mitochondrial fractions and observed increased Bax levels in both fractions in the D7‐Des Tg hearts (Figure 7C). We also observed increased expression of cleaved PARP1, CHOP, and p62 in the D7‐Des Tg hearts compared with Ntg (Figure 7D). Thus, the cellular toxicity in D7‐Des Tg hearts is associated with mitochondria‐mediated activation of apoptotic pathways.
Figure 7.
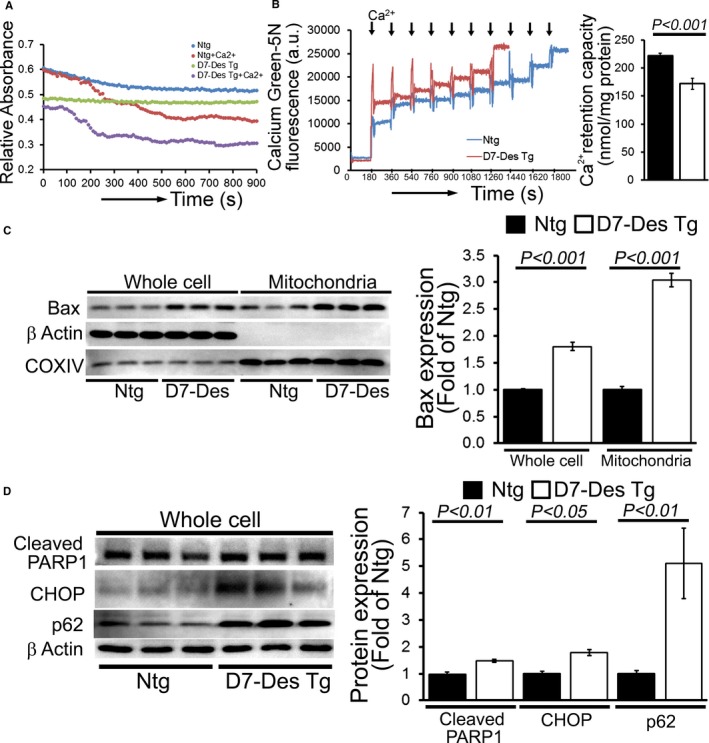
Altered mitochondrial swelling and calcium retention capacity in D7‐Des Tg hearts. A, Representative images of calcium‐induced mitochondrial swelling isolated from the D7‐Des Tg and Ntg hearts at 6 months of age. Mitochondrial swelling was induced by the addition of 200 μmol/L CaCl2. The assay was performed in 3 independent experiments, and representative tracings are shown. B, Representative traces and quantification of mitochondrial Ca2+‐retention capacity of D7‐Des Tg and Ntg hearts at 6 months of age. Fluorescence reading of Ca2+ measured with Calcium Green‐5N indicator in solution with subsequent addition of Ca2+ pulses of 20 nmol/mg of mitochondrial protein. Cumulative Ca2+ additions are shown at each arrowhead (n=5–6 mice per group). C, Representative Western blot and densitometric quantification shows significantly increased Bax expression and mitochondrial localization in the D7‐Des Tg mice hearts. D, Representative Western blot and densitometric quantification showing increased expression of cleaved PARP1, CHOP, and p62 protein isolated from the D7‐Des Tg mice heart (n=6 mice per group). Bars represent mean±SEM. P values were determined by Tukey's post hoc test. D7‐Des Tg indicates mutant desmin transgenic mouse; Ntg, non‐transgenic.
D7‐Des Overexpression in Cardiomyocytes Induces Mitochondrial Fission and Respiratory Dysfunction
To determine whether D7‐Des expression directly results in altered mitochondrial fission and respiratory dysfunction, we overexpressed D7‐Des in NRCs via adenoviral‐mediated infections (0–10 multiplicities of infection) (Figure 8A and 8B). Similar to D7‐Des Tg hearts, adenoviral‐mediated D7‐Des overexpression in NRCs increased desmin aggregate formation (Figure 8C). D7‐Des overexpression significantly increased Drp1 expression (Figure 8D), Bax expression (Figure 8E), and lactate dehydrogenase release, a measure of cell death, in NRCs (Figure 8F). Immunostaining with TOM20 indicated increased fragmented mitochondria in NRCs with D7‐Des overexpression (Figure 8G). These data are consistent with the in vivo data obtained with the Tg mice.
Figure 8.
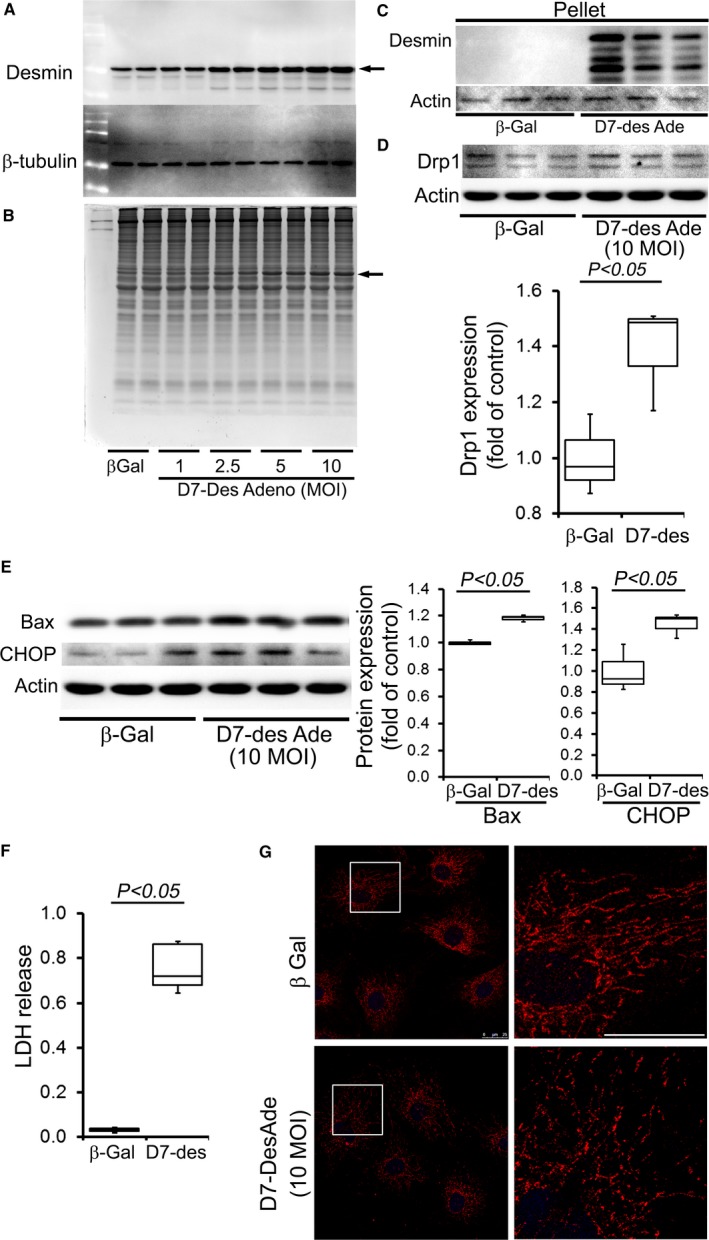
D7‐Des overexpression in NRCs. A, Overexpression of D7‐Des (indicated by arrow) in NRCs by adenoviral‐mediated infection at 1.0, 2.5, 5.0, and 10.0 MOI. B, Coomassie staining shows the D7‐Des protein (indicated by arrow). C, Pellet (insoluble) fraction shows accumulated desmin indicating aggregation. D, Drp1 expression in the NRCs is consistent with increased mitochondrial fission. E, Expression of Bax and CHOP in NRCs by D7‐Des overexpression. F, LDH release. n=3 replicates per group. Boxes represents interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. G, NRCs were infected with D7‐Des adenovirus at 10 MOI and mitochondrial morphology was monitored by staining with mitochondrial marker Tom 20 (Red). Tom 20 staining shows fragmented mitochondria in the D7‐Des adeno‐infected NRCs (scale bar: 25 μm). D7‐Des Ade indicates mutant desmin adenovirus; LDH, lactate dehydrogenase; MOI, multiplicity of infection; NRCs, neonatal rat cardiomyocytes.
To ensure that the decreased OCR we observed in D7‐Des hearts (Figure 5A) was not an artifact caused by the mitochondrial isolation process, we measured OCR in intact NRCs expressing either D7‐Des or β‐galactosidase (Figure 9A). NRCs expressing D7‐Des showed a dose‐dependent decrease in OCR at baseline (Figure 9B) as well as with the addition of oligomycin, FCCP, and rotenone plus antimycin A (Figure 9C–9E). Critical parameters of mitochondrial function including reserve capacity, ATP turnover, and maximal respiration (Figure 9F–9H) were also dose‐dependently decreased in D7‐Des expressing NRCs. We are thus able to demonstrate that adenoviral‐mediated D7‐Des expression in NRCs significantly increased mitochondrial fission and respiratory dysfunction, recapitulating the D7‐Des Tg heart data.
Figure 9.
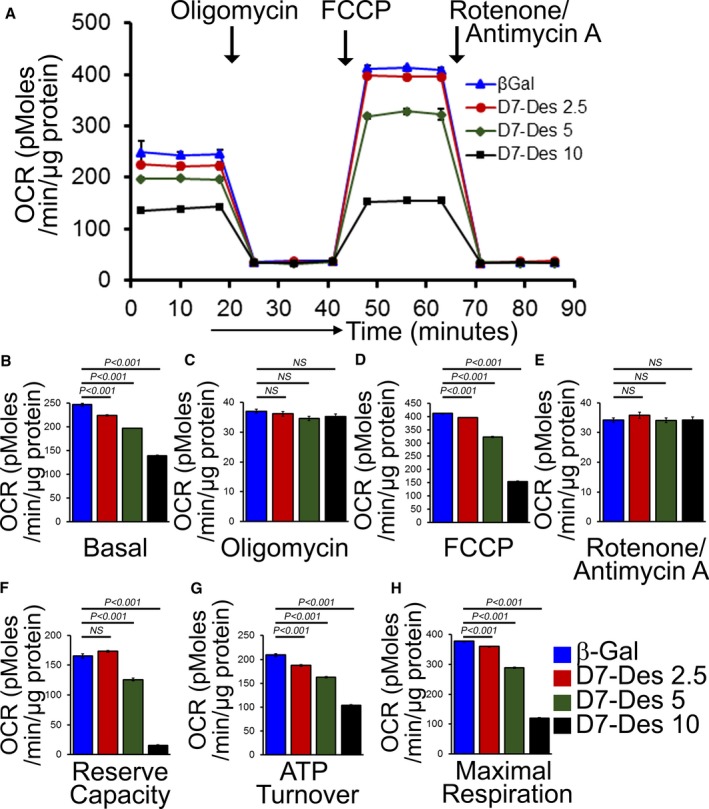
Suppression of mitochondrial respiration by adenovirus overexpression of D7‐Des in NRCs. A, Mitochondrial OCR profile in NRCs infected with D7‐Des adenovirus at 2.5, 5.0, and 10.0 multiplicities of infection or control adenovirus (β‐Gal). Arrows indicate the sequential addition of oligomycin (1 μmol/L), FCCP (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L). OCR profile is expressed as pmol O2/min per μg of protein. Graph showing OCR at (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Key parameters of mitochondrial function include (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration. Bars represent mean±SEM. n=5 wells per group. P values were determined by Tukey's post hoc test. D7‐Des Tg indicates mutant desmin transgenic mouse; FCCP, carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone; NRCs, neonatal rat cardiomyocytes; NS, not significant; OCR, oxygen consumption rate.
Inhibition of Mitochondrial Fission in Cardiomyocytes Rescues D7‐Des Overexpression‐Induced Mitochondrial Dysfunction
To test whether aberrant mitochondrial fission in the D7‐Des Tg hearts is responsible for mitochondrial dysfunction and cellular toxicity, we used the mitochondrial fission inhibitor, mdivi‐133, in NRCs. Earlier studies using mdivi‐1 at a concentration of 10 μmol/L reported no toxic effects in cultured neurons.34, 35 Therefore, we treated the NRCs with different concentrations of mdivi‐1 (1, 5, and 10 μmol/L) or the same volume of vehicle (dimethyl sulfoxide) to determine whether mitochondrial fission inhibition by mdivi‐1 affects mitochondrial respiration and cellular toxicity. We did not detect any toxic effects of midivi‐1 in uninfected NRCs on the mitochondrial respiratory profile (Figure 10A–10H) or cellular toxicity as measured by lactate dehydrogenase release (Figure 10I) at doses of 1 to 10 μmol/L. We then treated the D7‐Des expressing NRCs with 5 μmol/L mdivi‐1 or vehicle (dimethyl sulfoxide) for 24 hours. Treatments with mdivi‐1 significantly decreased Drp1 expression, indicating inhibition of mitochondrial fission (Figure 11A). We also observed decreased expression of Bax and CHOP (Figure 11B) as well as lactate dehydrogenase release (Figure 11C) indicating cellular protection by mdivi‐1 treatment. Desmin protein in the pellet (aggregated protein) fraction was also significantly decreased by mdivi‐1 treatment (Figure 11D). Desmin expression in the whole cell fraction was not significantly different with or without mdivi‐1 treatment, suggesting that mdivi‐1 did not affect D7‐Des adenovirus infection or the virus ability to drive protein expression, and Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (Figure 11E).
Figure 10.

Effects of mitochondrial fission inhibitor mdivi‐1 on mitochondrial respiration in NRCs. A, Mitochondrial oxygen consumption rate (OCR) profile in NRCs treated with 1, 5, and 10 μmol/L mdivi‐1 treatment for 24 hours in NRCs. Arrows indicate the sequential addition of oligomycin (1 μmol/L), FCCP (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L). OCR profile is expressed as pmol O2/min per μg of protein. Graph shows OCR at (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Key parameters of mitochondrial function including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration were not significantly changed by mdivi‐1 treatment. I, LDH release in the medium with increasing dose of mdivi‐1. Bars represent mean±SEM. n=5 wells per group. P values were determined by Tukey's post hoc test. FCCP indicates carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone; LDH, lactate dehydrogenase; NRC, neonatal rat cardiomyocyte.
Figure 11.

Mitochondrial fission inhibition by mdivi‐1 in D7‐Des overexpressed NRCs. A, Representative Western blot and densitometric quantification showing Drp1, OPA1, and PGC1α expression after 5 μmol/L mdivi‐1 or vehicle for 24 hours in D7‐Des overexpressed cardiomyocytes. B, Expression of Bax and CHOP in the whole cell lysate, and (C) LDH release in the medium after 5 μmol/L mdivi‐1 or vehicle treatment. D, Desmin expression in the pellet fraction was significantly decreased after mdivi‐1 treatment. E, Desmin expression in the whole cell fraction was not significantly changed after mdivi‐1 treatment. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading. n=3 replicates per group. Boxes represents interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal–Wallis test. LDH indicates lactate dehydrogenase; MOI, multiplicities of infection; NRCs, neonatal rat cardiomyocytes; NS, not significant.
To test whether inhibition of mitochondrial fission by mdivi‐1 could rescue the mitochondrial respiratory defects in D7‐Des expressing NRCs, we measured mitochondrial respiration in the presence of 5 μmol/L mdivi‐1 or vehicle (dimethyl sulfoxide) for 24 hours. mdivi‐1‐mediated inhibition of mitochondrial fission significantly increased OCR at baseline (Figure 12A and 12B) as well as with the addition of oligomycin, FCCP, and rotenone plus antimycin A (Figure 12C through 12E). Critical parameters of mitochondrial function including reserve capacity, ATP turnover, and maximal respiration (Figure 12F–12H) were significantly increased in the mdivi‐1‐treated group. Therefore, inhibition of mitochondrial fission by mdivi‐1 treatment rescued D7‐Des expression‐induced aberrant mitochondrial fission and respiratory dysfunction.
Figure 12.
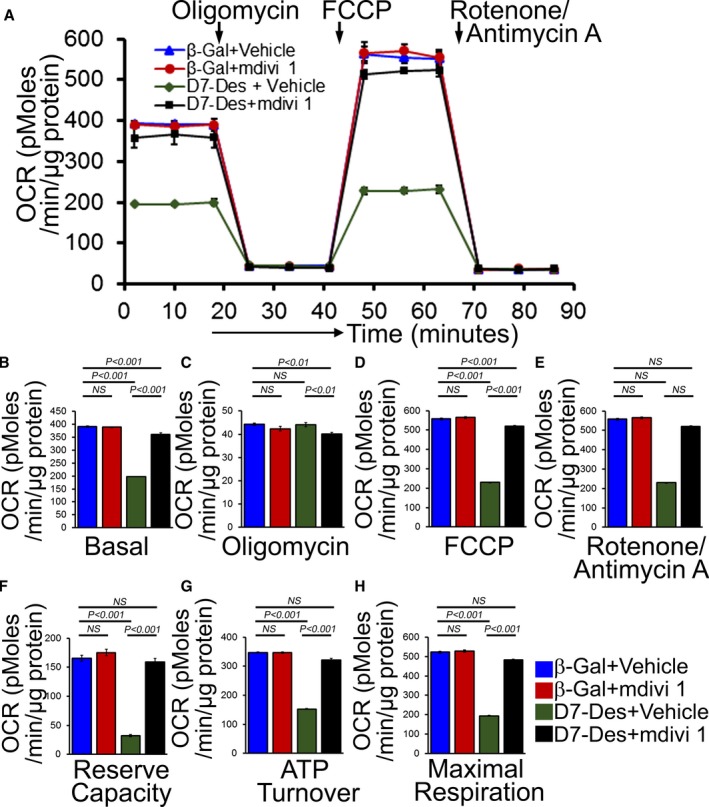
Mitochondrial fission inhibition by mdivi‐1 preserved mitochondrial respiration in D7‐Des overexpressed NRCs. A, Mitochondrial oxygen consumption rate (OCR) profile in NRCs treated with 5 μmol/L mdivi‐1 or vehicle for 24 hours in D7‐Des overexpressed cardiomyocytes. Arrow indicates the sequential addition of oligomycin (1 μmol/L), FCCP (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L). OCR profile is expressed as pmol O2/min per μg of protein. Graph shows OCR under (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Critical parameters of mitochondrial function including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration were significantly rescued by mdivi‐1 treatment in D7‐Des overexpressed NRCs. Bars represent mean±SEM. n=5 wells per group. P values were determined by Tukey's post hoc test. FCCP indicates carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone; NRCs, neonatal rat cardiomyocytes; NS, not significant.
Discussion
Desmin is the main intermediate filament protein in cardiomyocytes, accounting for 2% of total cellular protein, forming a 3‐dimensional scaffold around the myofibrillar Z‐disc, and interconnecting the contractile apparatus with the subsarcolemmal cytoskeleton, the nuclei, and other organelles. Desmin mutations can result in various myopathies that are characterized by desmin network disorganization, accumulation of insoluble desmin‐containing aggregates, and sarcomere disarray.9, 36, 37 Expression of the 7 amino acid deletion (R173–E179) mutation in desmin leads to a desminopathy characterized by defects in skeletal, cardiac, and smooth muscle.38, 39 These patients are at risk for cerebrovascular accidents and may develop generalized muscle weakness, respiratory failure, and intestinal pseudo‐obstruction that can lead to death.38, 39 Ultrastructural analysis of postmortem muscles show characteristic myofibrillar disruption and aggregation of desmin filaments in the myocardium.38, 39 In addition, mitochondrial dysfunction occurs early and acts causally in a wide variety of human protein aggregate disease pathogenesis affecting the central nervous system40, 41 and striated muscle tissue.16, 42 Muscle biopsy specimens from human desminopathies and myofibrillar myopathies showed evidence of mitochondrial pathology.43, 44 Moreover, patients with mutations in desmin, αB‐crystallin, filamin‐C, myotilin, ZASP, FHL1, and plectin, which are all causative of myofibrillar myopathies, showed mitochondrial dysfunction.36 The functional relationship between altered mitochondrial dynamics and function caused by these disease‐causing mutations remains mostly unknown in the majority of these diseases.
The link between mitochondrial dysfunction and desminopathy is reflected by defects in mitochondrial morphology, positioning, and respiratory enzyme function in the desmin knockout mouse.45, 46, 47 In the present study, we show that desminopathy is associated with aberrant mitochondrial fission and increased expression of mitochondrial fission regulatory proteins both in vivo and in vitro using D7‐Des expression systems. D7‐Des expression also caused a reduction in mitochondrial respiration in both isolated mitochondria and intact cardiomyocytes. Neurodegenerative protein aggregation diseases such as Alzheimer disease, Huntington disease, amyotrophic lateral sclerosis, and Parkinson disease also exhibit excessive mitochondrial fission, producing increased levels of reactive oxygen species and defective mitochondrial function.48, 49, 50 Biochemical studies of the causative mutant proteins including amyloid β, phosphorylated Tau, mutant Htt, mutant LRRK2, or mutant DJ1 document interactions with mitochondria in affected neurons. These interactions are primarily responsible for increased free‐radical levels, ultimately causing an impairment of mitochondrial dynamics, dysfunction, and neuronal damage. Future studies will be directed towards understanding whether D7‐Des acts in a similar fashion.
Mitochondrial fission is regulated and maintained by Drp1 GTPase. Drp1 is a cytosolic protein that is translocated to the outer membrane of the mitochondria where it interacts with multiple proteins, including Fis1 and Mff. We found that DRP1, phospho‐DRP1, FIS1, and Mff were significantly increased in D7‐Des Tg hearts compared with Ntg. Aberrant mitochondrial fission may involve several possible mechanisms such as excessive production of reactive oxygen species and activation of fission regulatory proteins. Studies on Huntington disease pathogenesis showed altered mitochondrial dynamics, fragmentation of mitochondria, subsequent mitochondrial dysfunction, and cellular death in Huntington disease–affected neurons. Studies also showed that Huntington disease interacts with the mitochondrial fission protein Drp1, and elevates levels of GTPase Drp1, resulting in increased fission and reduced fusion in Huntington disease–affected neurons. Future studies are needed to define whether D7‐Des directly interacts with Drp1 and regulates GTPase Drp1 enzymatic activity, which in turn increases mitochondrial fission and creates an imbalance in mitochondrial dynamics.
Mitochondrial translocation of Bax is a crucial step for inducing apoptotic cell death via the intrinsic apoptotic pathway. We found that D7‐Des overexpression is associated with mitochondrial localization of Bax along with the mitochondrial fission protein Drp1. Studies showed that Bax and Drp1 are binding partners, and Drp1 promotes mitochondrial translocation of Bax.51 Though both Bax and Drp1 translocate to discrete foci on the mitochondria, studies showed that mitochondrial Bax participates in apoptotic fragmentation of mitochondria by stabilizing Drp1.52, 53, 54 Mitochondrial Drp1 promotes Bax oligomerization and leads to cytochrome C release from mitochondria.55 Therefore, we used the mitochondrial division inhibitor mdivi‐1, which is known to selectively inhibit mitochondrial fission by Drp1 (18). Mdivi‐1 was reported to inhibit the activity of the mitochondrial fission regulator Drp1 and also impedes apoptosis early in the intrinsic pathway by blocking Bax/Bak‐dependent mitochondrial outer membrane permeabilization.33 Moreover, studies also showed that mdivi‐1 also blocks pro‐apoptotic Bax‐dependent cytochrome C release from isolated mitochondria33 and attenuates neural cell death in vitro and in vivo.56, 57 In the current study, we found that mdivi‐1 inhibited Drp1‐mediated mitochondrial fission, decreased Bax expression, and significantly improved mitochondrial respiration, resulting in decreased cell death induced by D7‐Des in cardiomyocytes.
The degree of cardiac dysfunction observed in D7‐Des Tg mice used in this study was significantly worse than reported in previous study, likely because of the older age of our cohorts.10 Reduction in mitochondrial OCR in D7‐Des heart mitochondria could result from defects in mitochondrial substrate uptake, pyruvate dehydrogenase activity, the activity of the entire TCA cycle, or the flux through the electron transport chain. Therefore, further studies are needed to define the role of D7‐Des proteotoxicity on cardiac mitochondrial substrate preference. A potential technical limitation of the ATP measurements carried out in this study is that increased fibrosis in D7‐Des hearts may result in differential shearing forces during the homogenization step, thereby affecting measured ATP levels. An additional limitation of our study is that while there has been no report of midivi‐1 exerting a direct effect on protein aggregation and disaggregation, our experiments were not able to differentiate the potential for the protein directly affecting these processes in a mitochondria‐independent way. Similarly, the reduction in aggregated desmin in mdivi‐1‐treated cardiomyocytes also suggests a possible role of Drp1 activity in regulating aggregate formation. Future studies will be directed towards understanding the direct role of Drp1 on D7‐Des aggregate formation and the subsequent conservation of cardiac contractile function.
Taken together, our data suggest that aberrant mitochondrial fission is maladaptive in cardiac muscle in protein aggregation disease. Earlier studies of DRC animal models showed that the desmin aggregates and sarcomere disarray accompany and are partially responsible for impairment of cardiomyocyte contractility,58, 59 but these ultrastructural changes do not lead to cardiomyocyte cell death by themselves.58 Our data support the hypothesis that aberrant mitochondrial fission is likely involved in mitochondrial dysfunction and the apoptotic cell death associated with desminopathy. Small molecule inhibition of mitochondrial fission significantly improved mitochondrial respiration and decreased cell death, suggesting a causal role for aberrant mitochondrial fission in desminopathy. Given the critical role of mitochondria in heart, it is very likely that aberrant mitochondrial fission may be a common pathway leading to cellular dysfunction critical to various myofibrillar diseases. Overall, our data indicate that inhibition of mitochondrial fission may be a valuable therapeutic target of DRCs.
Author Contributions
Alam, Abdullah, Aishwarya, Peretik, and Bhuiyan performed experiments. James, Miriyala, and Panchatcharam analyzed data. Orr contributed to analytic tools. Robbins contributed to reagents. Alam, Abdullah, Aishwarya, and Bhuiyan designed experiments. Alam, Abdullah, and Bhuiyan wrote the manuscript and all authors contributed to the preparation of the manuscript.
Sources of Funding
This work was supported by the National Institutes of Health grants: K99 HL122354 and R00 HL122354 to Bhuiyan; R01 HL098435 and R01 HL133497 to Orr; PGM121307A to Bhuiyan, Orr, and Panchatcharam; and LSUHSC‐S Malcolm Feist Cardiovascular Postdoctoral Fellowship to Abdullah.
Disclosures
None.
Acknowledgments
The authors thank Mohammad Alfrad Nobel Bhuiyan from the Division of Biostatistics and Epidemiology at Cincinnati Children's Hospital for providing statistical assistance.
(J Am Heart Assoc. 2018;7:e009289 DOI: 10.1161/JAHA.118.009289.)
References
- 1. Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res. 1999;252:479–491. [DOI] [PubMed] [Google Scholar]
- 2. Wang K, Ramirez‐Mitchell R. A network of transverse and longitudinal intermediate filaments is associated with sarcomeres of adult vertebrate skeletal muscle. J Cell Biol. 1983;96:562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bar H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. [DOI] [PubMed] [Google Scholar]
- 4. Li Z, Colucci‐Guyon E, Pincon‐Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 1996;175:362–366. [DOI] [PubMed] [Google Scholar]
- 5. Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Azzimato V, Genneback N, Tabish AM, Buyandelger B, Knoll R. Desmin, desminopathy and the complexity of genetics. J Mol Cell Cardiol. 2016;92:93–95. [DOI] [PubMed] [Google Scholar]
- 8. Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dalakas MC, Park KY, Semino‐Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780. [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Osinska H, Dorn GW II, Nieman M, Lorenz JN, Gerdes AM, Witt S, Kimball T, Gulick J, Robbins J. Mouse model of desmin‐related cardiomyopathy. Circulation. 2001;103:2402–2407. [DOI] [PubMed] [Google Scholar]
- 11. Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino‐Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403. [DOI] [PubMed] [Google Scholar]
- 13. Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha‐B‐crystallin desmin‐related cardiomyopathy. Circulation. 2005;112:3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G‐alphaB‐crystallin causes aberrant desmin and alphaB‐crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. [DOI] [PubMed] [Google Scholar]
- 15. Reimann J, Kunz WS, Vielhaber S, Kappes‐Horn K, Schroder R. Mitochondrial dysfunction in myofibrillar myopathy. Neuropathol Appl Neurobiol. 2003;29:45–51. [DOI] [PubMed] [Google Scholar]
- 16. Winter L, Wittig I, Peeva V, Eggers B, Heidler J, Chevessier F, Kley RA, Barkovits K, Strecker V, Berwanger C, Herrmann H, Marcus K, Kornblum C, Kunz WS, Schroder R, Clemen CS. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol. 2016;132:453–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alam S, Abdullah CS, Aishwarya R, Orr AW, Traylor J, Miriyala S, Panchatcharam M, Pattillo CB, Bhuiyan MS. Sigmar1 regulates endoplasmic reticulum stress‐induced C/EBP‐homologous protein expression in cardiomyocytes. Biosci Rep. 2017;37:BSR20170898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhuiyan MS, Gulick J, Osinska H, Gupta M, Robbins J. Determination of the critical residues responsible for cardiac myosin binding protein C's interactions. J Mol Cell Cardiol. 2012;53:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhuiyan MS, McLendon P, James J, Osinska H, Gulick J, Bhandary B, Lorenz JN, Robbins J. In vivo definition of cardiac myosin‐binding protein C's critical interactions with myosin. Pflugers Arch. 2016;468:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez‐Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2:e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chandra M, Escalante‐Alcalde D, Bhuiyan MS, Orr AW, Kevil C, Morris AJ, Nam H, Dominic P, McCarthy KJ, Miriyala S, Panchatcharam M. Cardiac‐specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol. 2018;14:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, Hajjar RJ, Lebeche D. Therapeutic cardiac‐targeted delivery of miR‐1 reverses pressure overload‐induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2:e000078 DOI: 10.1161/JAHA.113.000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- 27. Yaniv Y, Spurgeon HA, Ziman BD, Lyashkov AE, Lakatta EG. Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am J Physiol Heart Circ Physiol. 2013;304:H1428–H1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Konokhova Y, Spendiff S, Jagoe RT, Aare S, Kapchinsky S, MacMillan NJ, Rozakis P, Picard M, Aubertin‐Leheudre M, Pion CH, Bourbeau J, Hepple RT, Taivassalo T. Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle. Skelet Muscle. 2016;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, Meyer JN. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015;1241:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maloyan A, Sayegh J, Osinska H, Chua BH, Robbins J. Manipulation of death pathways in desmin‐related cardiomyopathy. Circ Res. 2010;106:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gutierrez‐Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol. 2014;72:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cassidy‐Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak‐dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu JM, Yi Z, Liu SZ, Chang JH, Dang XB, Li QY, Zhang YL. The mitochondrial division inhibitor mdivi‐1 attenuates spinal cord ischemia‐reperfusion injury both in vitro and in vivo: involvement of BK channels. Brain Res. 2015;1619:155–165. [DOI] [PubMed] [Google Scholar]
- 35. Valenti D, Rossi L, Marzulli D, Bellomo F, De Rasmo D, Signorile A, Vacca RA. Inhibition of Drp1‐mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta. 2017;1863:3117–3127. [DOI] [PubMed] [Google Scholar]
- 36. Paulin D, Huet A, Khanamyrian L, Xue Z. Desminopathies in muscle disease. J Pathol. 2004;204:418–427. [DOI] [PubMed] [Google Scholar]
- 37. Schroder R, Vrabie A, Goebel HH. Primary desminopathies. J Cell Mol Med. 2007;11:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ariza A, Coll J, Fernandez‐Figueras MT, Lopez MD, Mate JL, Garcia O, Fernandez‐Vasalo A, Navas‐Palacios JJ. Desmin myopathy: a multisystem disorder involving skeletal, cardiac, and smooth muscle. Hum Pathol. 1995;26:1032–1037. [DOI] [PubMed] [Google Scholar]
- 39. Munoz‐Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez‐Figueras MT, Navas‐Palacios JJ, Ariza A, Fuchs E. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci USA. 1998;95:11312–11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. [DOI] [PubMed] [Google Scholar]
- 41. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. [DOI] [PubMed] [Google Scholar]
- 42. Nardin RA, Johns DR. Mitochondrial dysfunction and neuromuscular disease. Muscle Nerve. 2001;24:170–191. [DOI] [PubMed] [Google Scholar]
- 43. Jackson S, Schaefer J, Meinhardt M, Reichmann H. Mitochondrial abnormalities in the myofibrillar myopathies. Eur J Neurol. 2015;22:1429–1435. [DOI] [PubMed] [Google Scholar]
- 44. Joshi PR, Hauburger A, Kley R, Claeys KG, Schneider I, Kress W, Stoltenburg G, Weis J, Vorgerd M, Deschauer M, Hanisch F. Mitochondrial abnormalities in myofibrillar myopathies. Clin Neuropathol. 2014;33:134–142. [DOI] [PubMed] [Google Scholar]
- 45. Fountoulakis M, Soumaka E, Rapti K, Mavroidis M, Tsangaris G, Maris A, Weisleder N, Capetanaki Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J Mol Cell Cardiol. 2005;38:461–474. [DOI] [PubMed] [Google Scholar]
- 46. Linden M, Li Z, Paulin D, Gotow T, Leterrier JF. Effects of desmin gene knockout on mice heart mitochondria. J Bioenerg Biomembr. 2001;33:333–341. [DOI] [PubMed] [Google Scholar]
- 47. Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reddy PH. Misfolded proteins, mitochondrial dysfunction, and neurodegenerative diseases. Biochim Biophys Acta. 2014;1842:1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington's disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today. 2014;19:951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reddy PH. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J Alzheimers Dis. 2014;40:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang P, Wang P, Liu B, Zhao J, Pang Q, Agrawal SG, Jia L, Liu FT. Dynamin‐related protein Drp1 is required for Bax translocation to mitochondria in response to irradiation‐induced apoptosis. Oncotarget. 2015;6:22598–22612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy‐Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. [DOI] [PubMed] [Google Scholar]
- 55. Montessuit S, Somasekharan SP, Terrones O, Lucken‐Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy‐Wetzel E, Basanez G, Meda P, Martinou JC. Membrane remodeling induced by the dynamin‐related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy‐Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xie N, Wang C, Lian Y, Zhang H, Wu C, Zhang Q. A selective inhibitor of Drp1, mdivi‐1, protects against cell death of hippocampal neurons in pilocarpine‐induced seizures in rats. Neurosci Lett. 2013;545:64–68. [DOI] [PubMed] [Google Scholar]
- 58. Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid‐induced heart disease in desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2005;102:13592–13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J. Biochemical and mechanical dysfunction in a mouse model of desmin‐related myopathy. Circ Res. 2009;104:1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]