

Abstract
Background
The epithelial growth factor receptor family of tyrosine kinases modulates embryonic formation of semilunar valves. We hypothesized that mice heterozygous for a dominant loss‐of‐function mutation in epithelial growth factor receptor, which are Egfr Vel/+ mice, would develop anomalous aortic valves, valve dysfunction, and valvular cardiomyopathy.
Methods and Results
Aortic valves from Egfr Vel/+ mice and control mice were examined by light microscopy at 2.5 to 4 months of age. Additional Egfr Vel/+ and control mice underwent echocardiography at 2.5, 4.5, 8, and 12 months of age, followed by histologic examination. In young mice, microscopy revealed anatomic anomalies in 79% of Egfr Vel/+ aortic valves, which resembled human unicuspid aortic valves. Anomalies were not observed in control mice. At 12 months of age, histologic architecture was grossly distorted in Egfr Vel/+ aortic valves. Echocardiography detected moderate or severe aortic regurgitation, or aortic stenosis was present in 38% of Egfr Vel/+ mice at 2.5 months of age (N=24) and in 74% by 8 months of age. Left ventricular enlargement, hypertrophy, and reversion to a fetal myocardial gene expression program occurred in Egfr Vel/+ mice with aortic valve dysfunction, but not in Egfr Vel/+ mice with near‐normal aortic valve function. Myocardial fibrosis was minimal or absent in all groups.
Conclusions
A new mouse model uniquely recapitulates salient functional, structural, and histologic features of human unicuspid aortic valve disease, which are phenotypically distinct from other forms of congenital aortic valve disease. The new model may be useful for elucidating mechanisms by which congenitally anomalous aortic valves become critically dysfunctional.
Keywords: aortic valve, aortic valve regurgitation, aortic valve stenosis
Subject Categories: Valvular Heart Disease, Congenital Heart Disease
Clinical Perspective
What Is New?
Herein, we report discovery of a new mouse model that uniquely recapitulates salient functional, structural, and histologic features of human unicuspid aortic valve disease.
Mice heterozygous for a dominant loss‐of‐function mutation in the epithelial growth factor receptor have a high prevalence of unicuspid aortic valves.
The incidence of physiologically significant aortic valve dysfunction (regurgitation, stenosis, or both) approximately doubles between infancy and middle age in mice with unicuspid aortic valves.
What Are the Clinical Implications?
Congenitally anomalous aortic valves, of which ≈10% are unicuspid, account for about half of all aortic valve replacement surgeries.
In patients with congenitally anomalous aortic valves, symptomatic aortic valve dysfunction may be delayed until adulthood, suggesting a window for medical interventions that target progression of valve dysfunction.
This new mouse model has the potential to facilitate discovery of mechanisms by which congenitally anomalous aortic valves develop overt dysfunction and produce valvular cardiomyopathy.
Introduction
Malformations of the aortic valve are the most common form of congenital cardiac anomaly, accounting for more than half of aortic valve replacement procedures.1, 2 A bicuspid valve is the most common form of congenital aortic valve anomaly, but ≈10% of those with an anomaly leading to the requirement for valve replacement have a unicuspid valve.3 The natural history of unicuspid aortic valves is poorly understood, because unicuspid anatomic features are difficult to diagnose with most imaging methods, and thus are not differentiated from bicuspid anatomic features until the time of valve surgery or autopsy.4 Clinical manifestations of unicuspid aortic valves may arise during infancy but, more important, often appear only after several decades of life.2, 5 Mechanisms by which patients with clinically silent unicuspid aortic valves progress to overt valvular cardiomyopathy are not well understood, in part because of absence of relevant experimental models.
In this study, we pursued 3 lines of investigation. First, because elements of epithelial growth factor receptor (EGFR) signaling pathways regulate embryonic formation of the aortic valve in mice,6 and putatively in humans,7 we examined structural and histologic architecture of aortic valves from mice heterozygous for a dominant loss‐of‐function mutation in Egfr, the Velvet mutation (Vel).8 Second, because impairment of aortic valve function may begin to manifest clinically over a wide range of ages in humans with congenital anomalies, we hypothesized that the cumulative incidence of valve dysfunction would increase with age in Egfr Vel/+ (Velvet) mice. Third, because left ventricular (LV) responses to hemodynamic stress may differ importantly in patients with congenital aortic valve disease, compared with patients who acquire aortic valve disease in late adulthood, we studied structural, functional, histologic, and molecular responses to aortic valve dysfunction in myocardium from Velvet mice with congenital aortic valve anomalies.
Methods
The authors confirm that the data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure, on reasonable request addressed to the corresponding author. All studies were approved by the Institutional Animal Care and Use Committee at the University of Iowa (United States Public Health Service Animal Welfare Assurance No. A3021‐01).
Mice
Egfr +/+ (control) and littermate Egfr Vel/+ (Velvet) mice bred on a C57/BL6 background were maintained on normal chow diet (Harlan Teklad 7004 Rodent Diet). The breeding strategy used male Velvet mice × female control mice, to minimize complications of pregnancy related to maternal genotype. Because male Velvet mice, but not female Velvet mice, were used to maintain breeder stocks, the experimental group contained more female Velvet mice (70%) than male Velvet mice. Sexes of control mice were configured using approximately the same proportions (75% female). Genotype was ascertained on the basis of appearance of fur and vibrissae, which are wavy in Velvet mice.8 Accuracy of that method was confirmed in tissue samples from 6 Velvet mice and 3 control mice, using polymerase chain reaction. The Velvet mutation is a single‐nucleotide mutation (A to G) in the EGFR‐tk coding region.8 The wild‐type forward primer is 5′‐CACAGCATGTCAAGATCAGAGA‐3′. The Velvet forward primer is 5′‐CACAGCATGTCAAGATCAGAGG‐3′. The common reverse primer is 5′‐TACTCCCAGGACTTACTTTGCC‐3′. Under temperature‐optimized polymerase chain reaction conditions, Velvet mice produce a single band of the same size (99 bp) with both sets of primers, whereas wild‐type mice produce a single band only when the primer set for wild type is used. Primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Blood Pressure
Arterial systolic pressure was measured with a tail cuff, in unsedated mice, as previously described.9
Echocardiography
Midazolam (0.15 mg, SC) was used to produce light conscious sedation. Parasternal long‐ and short‐axis views were obtained using high‐frequency echocardiography (Vevo 2100; VisualSonics, Toronto, ON, Canada) to assess LV mass, volumes, and systolic function, using the biplane area‐length method previously validated in our laboratory.10 M‐mode images were then acquired to measure systolic aortic cusp separation (ACS), as described previously.11 Severity of aortic valve stenosis (AS) was classified as follows: none, ACS ≥1.00 mm; mild, 0.80 mm≤ACS<1.00 mm; moderate, 0.66 mm≤ACS<0.80 mm; and severe, ACS <0.66 mm. Presence and severity of AS were subsequently confirmed visually, using 2‐dimensional short‐axis images, as previously described.12 For group analysis, AS was deemed “significant” if moderate or severe valve stenosis was present. Color Doppler images were acquired in a parasternal long‐axis plane to determine the presence of aortic regurgitation (AR). Severity of aortic valve regurgitation was graded by measuring length of the maximum diastolic regurgitant jet and dividing by the end‐diastolic long‐axis length of the LV. Trace or mild AR was recorded when jet length was <25% of LV length; moderate, 25% to 50%; and severe, >50%. AR was deemed “significant” if moderate or severe valve regurgitation was present, as previously validated using cardiac magnetic resonance.13
Anatomic Features of the Aortic Valve
Hearts from Velvet mice (N=14) and control mice (N=6), at 2.5 to 4 months of age, were excised and placed in cold normal saline for immediate low‐power stereoscopic visualization of aortic valve anatomic features. Some examples demonstrating either normal or anomalous valve anatomic features were then selected for scanning electron microscopy, as previously described.12 Briefly, trimmed hearts containing aortic valves were excised; fixed with 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer for 1 hour; postfixed with 1% osmium tetroxide for 1 hour; dehydrated through sequential incubation in 25%, 50%, 75%, 95%, and 100% ethanol; and dried using the CO2 Sorvall Critical Point Drying System (DuPont, Wilmington, DE). Samples were mounted onto aluminum stubs, sputter coated with gold‐palladium, and examined with an S‐4800 scanning electron microscope (Hitachi High Technologies America Inc, Pleasanton, CA).
Aortic Valve Histologic Features
After euthanasia, thoracotomy was performed to remove the heart and proximal aorta, which were frozen in optimal cutting temperature compound (10.24% polyvinyl alcohol, 4.26% polyethylene glycol, and 85.5% nonreactive ingredients). The apical two thirds of the heart was removed, and remaining tissue was secured in a cryostat. Sections, 10 μm thick, were obtained from proximal, mid, and distal aortic valve from each mouse. Slides were stained with Alizarin Red or Picrosirius Red to quantitate the amount of calcium or collagen, respectively, as described previously.12 Movat's Pentachrome stain was used to visualize proteoglycans and collagen in the aortic valve, as described previously.13 Sources of primary antibodies for immunostaining were as follows: collagen‐1 (Abcam, Cambridge, MA), α‐smooth muscle actin (α‐SMA; Abcam), Runx2 (Santa Cruz Biotechnology, Dallas, TX), and CD31 (BD Biosciences, San Jose, CA).
To detect phosphorylated EGFR in aortic valve, hearts from control mice and Velvet mice, aged 2.5 to 4 months, were incubated in DMEM in the presence (N=5 control, and N=6 Velvet) or absence (N=5 control, and N=5 Velvet) of recombinant EGF (40 ng/mL; R&D Systems, Minneapolis, MN), for 12 minutes, then frozen in optimal cutting temperature compound, cut in 10‐μm sections, and stained with anti–phosphorylated EGFR antibody (Cell Signaling, Danvers, MA).
Definition of Aortic Valve Abnormalities
Valves were defined as abnormal on the basis of gross anatomic features (stereoscopic imaging and scanning electron microscopy), valve motion (echocardiography), and histologic features. A normal aortic valve was defined by 3 thin leaflets, with each leaflet fully extending to the wall of the aortic sinuses; and 3 valve commissures, with normal systolic opening and no evidence of stenosis or regurgitation. An abnormal valve was defined on the basis of failure of separation of leaflets. Histologically, anomalous valves were thick and demonstrated failure of demarcation between valve cusps and the aortic wall, with integration of valve‐like matrix (proteoglycan) into the collagenous valve annulus. A unicuspid valve was defined by one (or zero) commissures and a single leaflet that opened with a funnel or tear‐drop shape in systole.
Studies in Myocardial Tissue
The apical two thirds of the LV, which had been removed to facilitate sectioning of the aortic valve, was cut in 10‐μm transverse sections, and stained with Masson's Trichrome, as previously described.13 For studies of gene expression, myocardium was homogenized in Trizol, and RNA was prepared using RNeasy mini kit (Qiagen, Germantown, MD). cDNA was generated using Moloney Murine Leukemia Virus reverse transcriptase with random hexamers. We measured expression of skeletal actin, junctophilin‐2, brain‐type natriuretic peptide, and α‐myosin heavy chain, which reflect reversion to a fetal myocardial gene program. We also measured myocardial expression of collagen‐1a1 and collagen‐3a1, which are downstream reporters of a profibrotic milieu, using carboxyfluorescein. Expression of each gene was normalized to expression of β‐actin (real‐time quantitative reverse transcription–polymerase chain reaction with 2′‐chloro‐7′phenyl‐1,4‐dichloro‐6‐carboxy‐fluorescein), in the same well. Sources of TaqMan primers are shown in Table 1.
Table 1.
Sources of Primers for Myocardial Gene Expression Studies
| Gene | Vendor | Catalog No. |
|---|---|---|
| Acta1 | IDT | Mm.PT.58.7312945 |
| Jph2 | IDT | Mm.PT.58.5417675 |
| Col1a1 | IDT | Mm.PT.58.7562513 |
| Col3a1 | IDT | Mm.PT.58.13848686 |
| Nppb (BNP) | IDT | Mm.PT.58.8584045.g |
| Myh6 | IDT | Mm.PT.58.31314128 |
| Actb | Thermo Fisher | 4352341E |
IDT indicates Integrated DNA Technologies (Coralville, IA).
Blood Chemistries
Plasma cholesterol, inorganic phosphorus, and calcium were measured spectrophotometrically using commercial kits (C7510 and P7516 [Pointe Scientific Inc, Canton, MI]; and C503 [Teco Diagnostics, Anaheim, CA], respectively).
Statistical Analysis
Data source materials (echocardiograms, histologic specimens, and blood) were analyzed by observers (RMW, KAZ, MKK) blinded with respect to genotype. All continuous variables are reported as mean±SEM. Continuous variables were compared between groups using unpaired Student t tests. Presence or absence of a discrete condition (moderate/severe AS or moderate/severe AR) was compared between groups using χ2 analysis. Echocardiographic data from 3 Velvet mice with severe AR/AS and severe LV systolic dysfunction that were humanely euthanized before 12 months of age were grouped with data from mice that survived to 12 months of age, to report cumulative incidence of aortic valve dysfunction and LV remodeling over the duration of the study. Statistical significance was set at P<0.05, except where values for control mice were used as a basis for comparison for 2 distinct Velvet groups (those with AR/AS and those without significant AR/AS), where statistical significance was set at P<0.025.
Results
Body mass, plasma calcium and phosphorus, heart rate, and systolic blood pressure were normal in Velvet mice (Table 2). Plasma cholesterol was lower in Velvet mice, compared with control mice. Survival to 8 months of age was not different in Velvet mice (82%) compared with control mice (88%; P=not significant).
Table 2.
Basic Characteristics
| Characteristics | Control Group | Velvet Groupa |
|---|---|---|
| Body mass, g | 28±5 | 24±1 |
| Heart rate, min−1 | 671±28 | 638±18 |
| Systolic blood pressure, mm Hg | 101±2 | 105±2 |
| Plasma calcium, mg/dL | 8.5±0.1 | 8.3±0.1 |
| Plasma phosphorus, mg/dL | 9.1±0.9 | 8.8±0.6 |
| Plasma cholesterol, mg/dL | 144±7 | 105±3b |
Data are given as mean±SEM. N=4 per group.
Velvet mice without significant aortic regurgitation or aortic stenosis.
P<0.05 vs control.
Prevalence of Anomalous Aortic Valves
At 2.5 to 4 months of age, stereoscopic imaging in 14 Velvet (Egfr Vel/+) aortic valves revealed gross congenital anomalies in 11, versus no anomalies in 6 control (Egfr +/+) valves (χ2=7.54, P=0.006; Figure 1). Aortic valve abnormalities in Velvet mice with significant AR and AS were characterized by a single leaflet with ≤1 commissure; anatomic features and systolic opening were similar to human unicuspid aortic valves5, 14 (Figures 1 through 2, 3, Videos S1 and S2).
Figure 1.
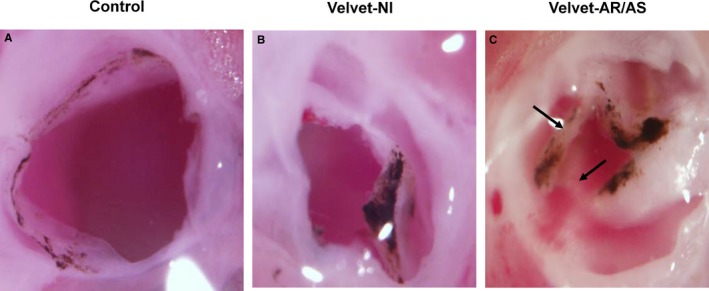
Light micrographs of aortic valves at 2.5 to 4 months of age. A, Control valve. B, Velvet valve without echocardiographic evidence of significant aortic regurgitation (AR) or aortic stenosis (AS), designated as functionally normal (Nl). C, Velvet valve with severe AR and severe AS. Arrows indicate noncoplanar architecture of valve tissue. Differences in pigmentation between control valves and Velvet valves are not consistently observed.
Figure 2.
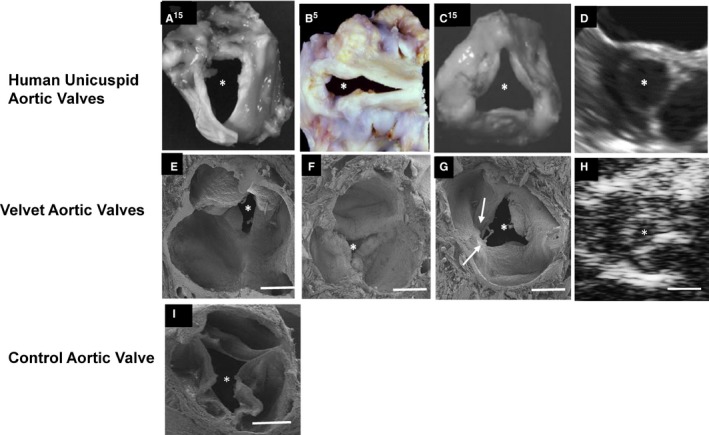
Anomalies in Velvet mice resemble human unicuspid aortic valves. A through C, Photographs of explanted human unicuspid aortic valves. D, Short‐axis midsystolic transesophageal echocardiogram from an adult patient with unicuspid aortic valve. E through G, Scanning electron micrographs (SEMs) of aortic valves from Velvet mice. H, Short‐axis midsystolic echocardiogram from a Velvet mouse. I, SEM of aortic valve from a control mouse. The asterisk indicates valve orifice. Arrows indicate noncoplanar architecture of valve tissue. White bar=400 μm. A and C, Reproduced from Roberts and Ko14 with permission. Copyright ©2005, Wolters Kluwer Health, Inc. B, Reproduced from Fealey et al5 with permission. Copyright ©2012, Elsevier.
Figure 3.
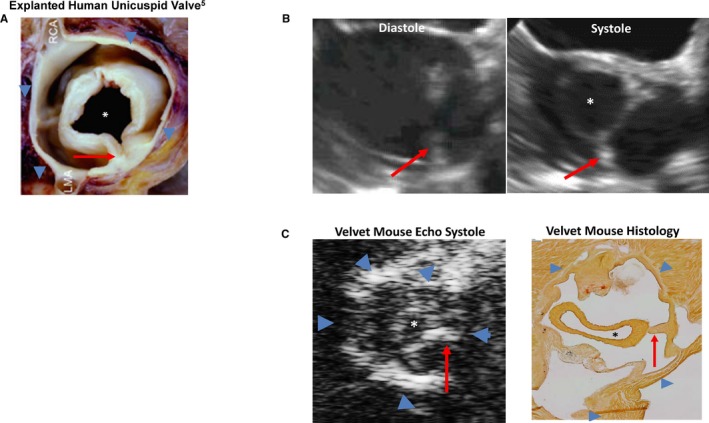
Stenotic unicuspid aortic valves. A circular orifice (asterisk) attaches to valve annulus (blue arrowheads) via a unicommissural stalk (red arrow). A, Explanted valve. B, Transesophageal echocardiogram from a 19‐year‐old man with unicuspid aortic valve (see Video S1). C, Echocardiogram and histologic features from a Velvet mouse with unicuspid aortic valve and severe aortic stenosis (see Video S2). LMA indicates left main coronary artery; RCA, right coronary artery. A, Reproduced from Fealey et al5 with permission. Copyright ©2012, Elsevier.
Incidence of Aortic Valve Dysfunction Increased With Age in Velvet Mice
At 2.5 months of age, incidence of hemodynamically significant AR, AS, or both was 38% in Velvet mice and 0% in control mice (Figure 4). By 8 months of age, cumulative incidence of significant aortic valve dysfunction increased to 74% in Velvet mice (odds ratio, 1.97 [95% confidence range, 1.11–3.49]; χ2=4.91; P=0.03 versus 2.5 months of age), and remained 0% in control mice. Increase in cumulative incidence of valve dysfunction between 2.5 and 8 months of age was explained largely by development of significant AS (with or without AR), which increased from 23% to 60% (P<0.05). The cumulative incidence of aortic valve dysfunction did not increase further in either Velvet mice or control mice, between 8 and 12 months of age. At 12 months of age, 5 of 7 male Velvet mice versus 12 of 16 female Velvet mice demonstrated moderate or severe aortic valve dysfunction (χ2<0.11; P=0.74).
Figure 4.
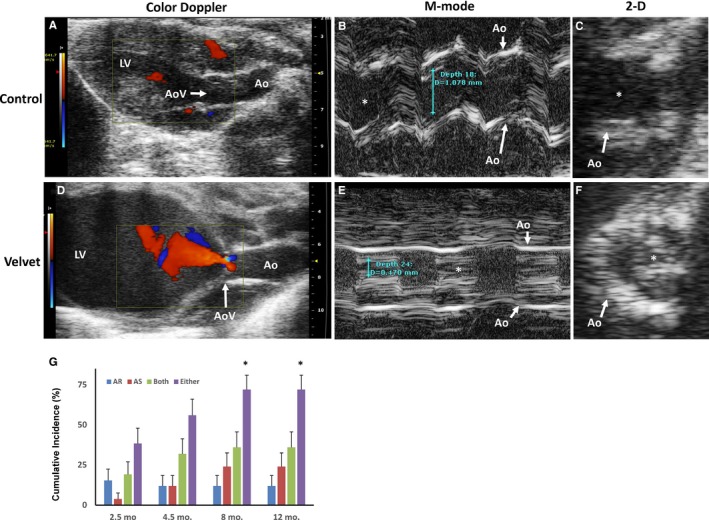
Aortic valve (AoV) function. Echocardiographic images from a control mouse (A through C) and from a Velvet mouse (D through F). Early diastolic color Doppler depicts moderate aortic regurgitation (AR; orange) in the Velvet mouse. M‐mode tracings show normal systolic orifice (white asterisk) dimension in the control mouse and severe aortic stenosis (AS) in the Velvet mouse. Two‐dimensional (2‐D) image of the AoV during systole confirms severe stenosis in the Velvet mouse. G, Cumulative incidence of moderate or severe AoV dysfunction in Velvet mice. N=24, N=24, N=23, and N=23 for ages 2.5, 4.5, 8, and 12 months, respectively. Ao indicates aorta; LV, left ventricle. *P<0.05 vs 2.5 months.
Disordered Extracellular Matrix in Velvet Aortic Valves
In control aortic valves, sites of cusp attachment to aorta consisted of fibrillar collagen oriented predominantly parallel to or perpendicular to valve cusps (Figure 5A, C), as previously reported.12 In Velvet valves, histologic architecture was grossly distorted. Annulus attachment sites at the base of the valve often were amorphous, with fusion (or failure of separation) of cusp and annulus (Figure 5B). Although collagen‐1, a principal structural isoform, was present in equal proportions in Velvet valves versus control valves (Figure 5E–G), regions were observed in Velvet valves where collagen fibers did not demonstrate coherent organization, or were present in a nonfibrillar state (Figure 5D).
Figure 5.
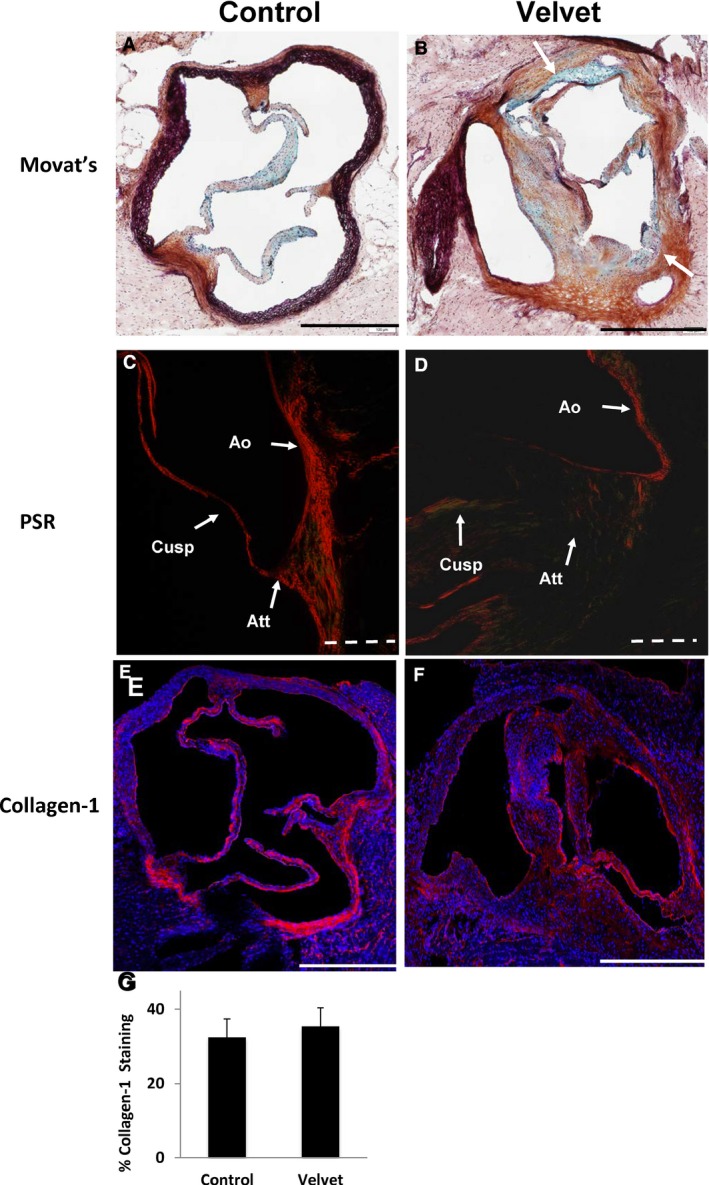
Extracellular matrix in aortic valves. A and B, Movat's Pentachrome staining shows discrete cusp attachment sites, which are composed mostly of collagen (brown), in the control valve. Control valve cusps contain mostly collagen and proteoglycan (blue). Demarcation between valve cusps and aortic wall is less distinct in the Velvet valve with both aortic regurgitation and aortic stenosis (white arrows). C and D, Picrosirius Red (PSR) staining viewed under polarized light depicts collagen fibers at site of cusp attachment (Att) oriented roughly parallel to the aortic wall (Ao) in the control valve. There is minimal transmission of polarized light, indicating lack of organization of collagen fibers, at the site of cusp attachment in the Velvet valve. E and F, Immunostaining for collagen‐1 (red) and cell nuclei (blue). G, Group data indicate similar proportion of valve tissue staining positive for collagen‐1 in Velvet vs control (N=6 each). Images and data were obtained from mice at 12 months of age. Solid bar=500 μm; dashed bar=100 μm. Data are given as mean±SEM.
Minimal Transdifferentiation of Valve Interstitial Cells in Velvet Mice
Calcification, which can occur as a result of transdifferentiation of valve interstitial cells,15 was minimal or absent in Velvet valves and control valves, at 12 months of age (Figure 6A through 6C). Calcification was not greater in Velvet valves with significant dysfunction versus Velvet valves without dysfunction (Figure 6D). Immunostaining for the procalcific transcription factor Runx2 was minimal or absent in Velvet valves and control valves. α‐SMA, which is a marker for valve interstitial cell transdifferentiation to myofibroblasts,16 was minimal or absent in Velvet aortic valves and control valves (Figure 6E through 6H).
Figure 6.
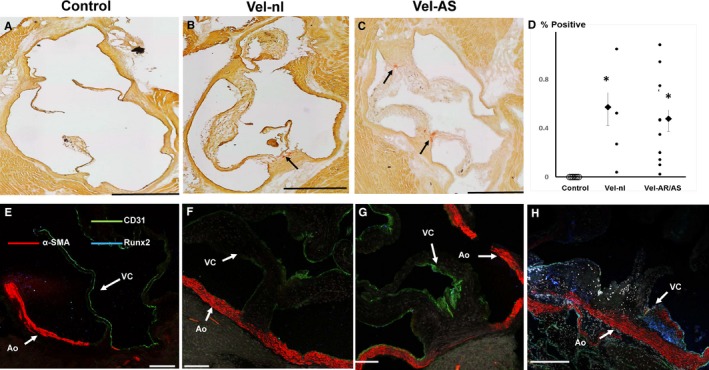
Valve interstitial cell transdifferentiation and calcification at 12 months of age. A through C, Alizarin Red staining shows modest calcification in Velvet valves (arrows) and none in control valve. D, Quantitation of Alizarin Red staining. Calcification was not greater in valves with echocardiographic evidence of moderate or severe aortic regurgitation (AR) or aortic stenosis (AS) than in Velvet valves with minimal valve dysfunction (Vel‐nl). Smaller points represent individual valves. Larger points and bars represent mean±SEM. E through G, Immunostaining for CD31, α‐smooth muscle actin (α‐SMA), and Runx2 in control and Velvet valves; α‐SMA is abundant in aortic wall (Ao), but not in valve cusps (VCs). Runx2 staining is minimal or absent. H, Immunostains from a Reversa valve, which is known to undergo valve interstitial cell transdifferentiation to myofibroblasts (α‐SMA) and osteogenic transdifferentiation (Runx2),16 serve as “positive control.” Black bar=500 μm. White bar=100 μm. *P<0.025 vs control.
EGFR Activity in the Aortic Valve
Ligand binding to EGFR signaling initiates homodimerization, or heterodimerization with another member of the human epidermal growth factor receptor family, followed by phosphorylation of EGFR, which activates downstream signaling. The Velvet mutation inhibits coordination of ATP to EGFR, with consequent loss of kinase activity and downstream signaling.8 We reasoned that phosphorylation of EGFR might still occur in Velvet valves, after formation of chimeric wild‐type/Velvet dimers, or by heterodimerization with other tyrosine kinase moieties. We detected phosphorylated EGFR in Velvet aortic valves, of the same order of magnitude as in control valves under basal conditions (Figure 7). After incubation with EGF, we observed greater immunostaining for phosphorylated EGFR in control mice, but not Velvet mice, compared with samples that were not incubated with EGF.
Figure 7.
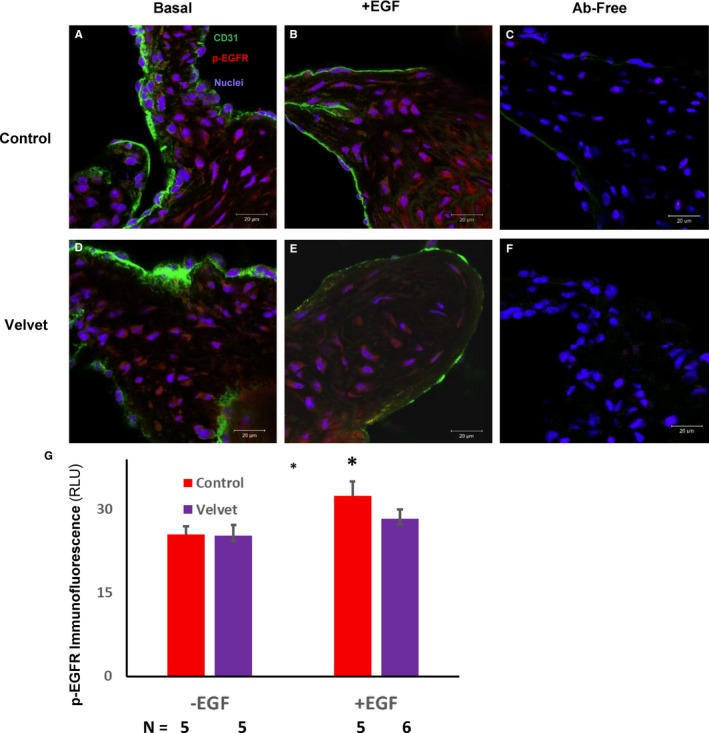
Immunostaining for phosphorylated epithelial growth factor receptor (p‐EGFR) in the aortic valve. A and D, Under basal conditions, p‐EGFR (red) is present in both control and Velvet valves. B and E, After incubation with EGF, level of p‐EGFR may increase somewhat less in the Velvet valve than in the control valve. C and F, In the absence of anti‐CD31 and anti–p‐EGFR antibodies (Abs), there is negligible autofluorescence or secondary antibody fluorescence. G, Group data. Original magnification ×63. Bar=20 μm. RLU indicates relative light unit. *P<0.05 vs EGF.
LV Remodeling in Velvet Mice
At all ages, Velvet mice with significant AS or AR demonstrated ≈2‐fold increases in LV end‐diastolic volume and LV mass (Figure 8). LV end‐diastolic volume and mass were normal in Velvet mice without significant valve dysfunction. In younger Velvet mice with aortic valve dysfunction, LV ejection fraction was normal. Of 17 Velvet mice with severe AR/AS, 3 developed overt LV dysfunction (ejection fraction, 0.15, 0.16, and 0.06, respectively) by 8 months of age. LV ejection fraction was >0.65 in all other Velvet mice. Thus, LV ejection fraction for the whole group of Velvet mice with severe AR/AS was not statistically significantly different from LV ejection fraction in Velvet mice without AR/AS or control mice.
Figure 8.

Left ventricular (LV) remodeling in Velvet mice with aortic valve dysfunction. Short‐axis end‐diastolic images from a control mouse (A) and from a Velvet mouse with severe aortic regurgitation (AR) and severe aortic stenosis (AS) (B). LV enlargement (C) and hypertrophy (D) were present at all ages in Velvet mice with moderate/severe AR or AS (Vel‐AR/AS), but not in Velvet mice with normal or mildly impaired valve function (Vel‐nl). LV ejection fraction (EF) remained normal in Velvet mice until 8 months of age, when ≈18% demonstrated severely decreased LVEF (P=not significant for all Vel‐AR/AS vs control, at all ages; E). Data are given as mean±SEM. EDV indicates end‐diastolic volume; RV, right ventricle. *P<0.025 vs age‐matched control.
Myocardial Adaptation to Aortic Valve Dysfunction
Velvet mice with significant valve dysfunction demonstrated increased expression of skeletal actin and brain‐type natriuretic peptide, consistent with reversion toward a fetal gene program in myocardium (Figure 9A and 9B).17 Myocardial expression of junctophilin‐2 and expression of α‐myosin heavy chain (Myh6) were decreased in Velvet mice with significant valve dysfunction, findings that are also consistent with reversion to a fetal gene program (Figure 9C and 9D).18 There was no significant increase in expression of collagen‐1a1 or collagen‐3a1 in myocardium from Velvet mice with valve dysfunction (Figure 9E and 9F). In Velvet mice without significant valve dysfunction, myocardial gene expression did not differ significantly from control mice. Interstitial fibrosis was minimal or absent in myocardium from both control mice and Velvet mice with valve dysfunction (Figure 9G through 9I).
Figure 9.
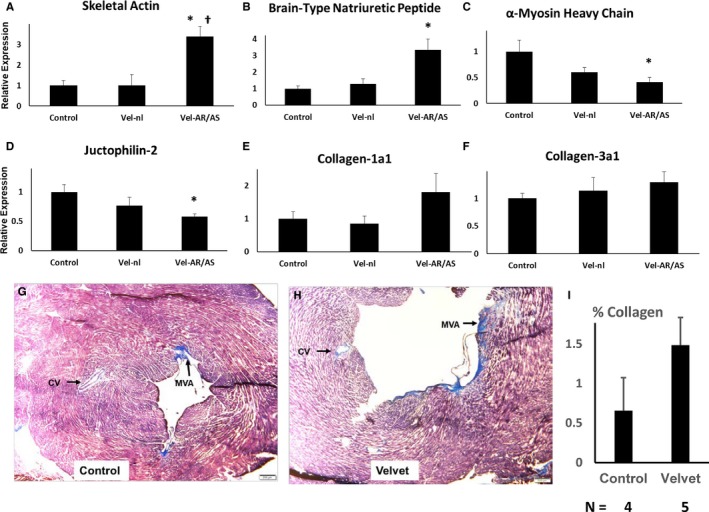
Myocardial adaptation to hemodynamic stress at 12 months of age. A through F, Gene expression was first normalized to β‐actin expression, then normalized to expression in control myocardium. Results are pooled from 4 hearts from each group. G through I, Masson's Trichrome staining of left ventricular myocardium from a control mouse (left ventricular ejection fraction [LVEF], 0.79) and from a Velvet mouse (LVEF, 0.15). Staining in mitral valve annulus (MVA) served as positive control for collagen. Staining around coronary vessels (CVs) was seen occasionally in Velvet myocardium, but not in control myocardium. Small clear bar at lower right=200 μm. Vel‐nl indicates Velvet mice with normal or mildly impaired valve function; Vel‐AR/AS, Velvet mice with moderate/severe aortic regurgitation or aortic stenosis. *P<0.025 vs control; † P<0.05 vs Vel‐nl.
Discussion
Novel findings of this study are as follows: (1) discovery of a new experimental model, the Velvet mouse, that uniquely recapitulates morphologic features of unicuspid aortic valve disease in humans; (2) progressive increase in cumulative incidence of overt aortic valve dysfunction, in the setting of congenitally anomalous valve anatomic features; and (3) structural and molecular adaptation in stressed myocardium, with minimal interstitial fibrosis.
Clinical Relevance of Congenitally Anomalous Aortic Valves in Velvet Mice
Aortic valves that are categorized as “unicuspid” comprise a spectrum of structural and histologic anomalies, including noncommissural and unicommissural varieties.5, 14 Thus, a unicuspid aortic valve does not function as a simple flap, or “trap door,” but has a variety of complex features, resulting in an orifice in the shape of a funnel or tear drop (Figure 10, Video S3). Valve dysfunction resulting in morbidity and mortality occurs across a wide range of ages, from infancy to middle age, with typical presentation at a younger age than bicuspid aortic valve disease or calcific disease of trileaflet valves. Surprisingly perhaps, a few humans with unicuspid aortic valves never develop symptoms or significant LV remodeling.19 Similarly, overt aortic valve dysfunction appeared over a broad range of ages in Velvet mice, but some maintained near‐normal valve function up to 12 months of age.
Figure 10.
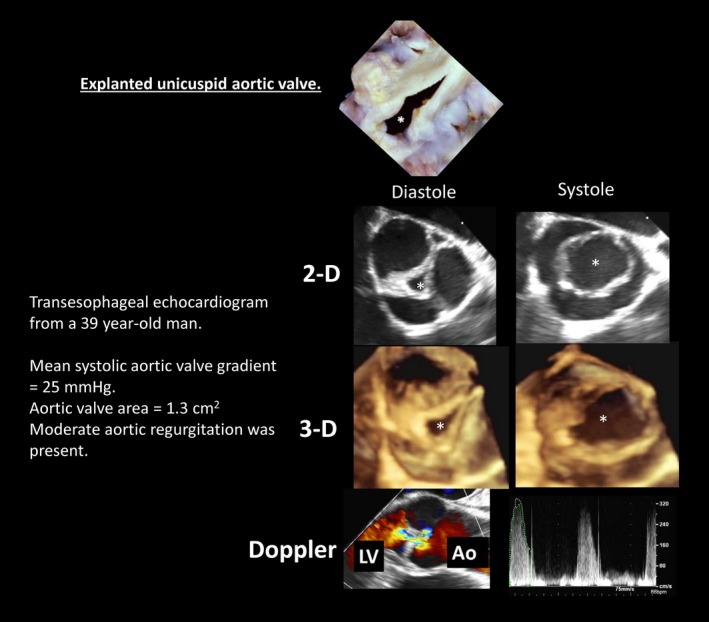
Human unicuspid aortic valves. The asterisk indicates valve orifice. Three‐dimensional (3‐D) images may be viewed in cine format in Video S3. Ao indicates aorta; 2‐D, 2 dimensional; LV, left ventricle. Top panel is reproduced from Fealey et al5 with permission. Copyright ©2012, Elsevier.
The unicuspid valve phenotype is distinct from the bicuspid valve phenotype, which typically has 2 leaflets and 2 commissures, with valve dysfunction occurring later in life because of superimposed fibrocalcific changes. In humans20 and mice9 with bicuspid aortic valves, early postnatal architecture of each individual cusp is usually near normal, after which progressive matrix remodeling and calcification occur at variable rates.
“Enlarged” aortic valves have been reported in mice homozygous for a different hypomorphic Egfr allele.6 Those mice, which are known as waved‐2 mice, however, are born with 3 discrete aortic valve cusps and rarely develop AS; the incidence of aortic valve dysfunction does not increase with age.13 Thus, to our knowledge, aortic valves in Velvet mice are unique among experimental models, in their resemblance to unicuspid valves in humans.
Aortic Valve Dysfunction
Mechanistically, processes leading to development of end‐stage valve dysfunction have not been characterized in humans with unicuspid aortic valves, but the variable time course suggests that cumulative tissue responses to disturbed flow, integrated over time, may contribute to progressive valve dysfunction.21, 22, 23
Anatomic aortic valve anomalies were present in about three quarters of Velvet mice. At young ages, however, significant aortic valve dysfunction is present in only about half that number. By 8 months of age, fully 74% of Velvet mice developed significant valve dysfunction. Together, the findings suggest that prolonged exposure to disturbed flow across anatomically distorted valves may be a key mechanism leading to valve dysfunction as age increases.
Characterization of specific mechanisms by which disturbed flow invokes tissue responses leading to overt valve dysfunction is beyond the scope of this study. But, at this preliminary stage, we are able to discount some mechanisms that are present in other forms of aortic valve disease. In an established mouse model of acquired AS, valve interstitial cells undergo transdifferentiation into myofibroblasts, which express α‐SMA and produce interstitial collagen.16 Myofibroblasts may then transdifferentiate further into osteoblast‐like cells, which produce matrix calcification.24 Similar processes occur in diseased human valves, in which α‐SMA and calcification colocalize.25 In aortic valves from Velvet mice, α‐SMA and calcification are minimal, even when significant valve dysfunction is present. The findings are analogous to observations in some children and young adults with unicuspid aortic valves who require aortic valve surgery, although valve calcification is often minimal or absent.5
LV Remodeling and Valvular Cardiomyopathy
Adult mice that are subjected to acute severe pressure overload,18 and mice that acquire severe AS during adult life, develop LV systolic dysfunction or die shortly thereafter.11, 12 Humans who acquire severe AS during adult life usually develop symptoms within 2 years.26 The clinical course is more diverse in patients with congenital AS, in whom symptom onset may be delayed for decades.27, 28 In Velvet mice with significant AR/AS, LV mass and end‐diastolic volume were approximately double that of mice without valve dysfunction. LV remodeling was accompanied by reversion to key features of the fetal myocardial gene program. We found remarkably little myocardial fibrosis, however, despite prolonged and extensive LV remodeling. Similar findings have been reported in children and young adults with congenital AS.29 LV systolic dysfunction was not observed in Velvet mice until 8 months of age, when only ≈18% manifest severely decreased LV ejection fraction.
Together, findings in humans and new findings in Velvet mice indicate that LV responses to initiation of hemodynamic stress during early life differ dramatically from LV responses to hemodynamic stress that are first encountered during adult life. Discovery of mechanisms by which LV systolic function is preserved during hemodynamic stress in young humans and young Velvet mice, and discovery of pathways that produce exhaustion of those compensatory mechanisms, could lead to therapies that ameliorate development of valvular cardiomyopathy, which arises later in life.
Limitations
Our understanding of specific cellular and molecular processes that produce increased cumulative incidence of aortic valve dysfunction is incomplete at this early stage of investigation, in this new experimental model of congenital aortic valve disease. Virtual absence of pro‐osteogenic signaling and valve calcification in Velvet valves may focus future investigations on processes involved in remodeling of valve extracellular matrix, which is dysregulated in Velvet mice.
The extent to which postnatal impairment of EGFR signaling contributed to unique phenotypic features of Velvet mice is not clear. Our finding of constitutive phosphorylation (activation) of EGFR, and the suggestion of diminished augmentation by exogenous EGF in Velvet aortic valves, is qualitatively concordant with results reported previously in neonatal liver.8 Those findings suggest an opportunity for therapeutic intervention, because EGFR activity can be modulated pharmacologically.30
We report congenital valve anomalies in mice with a single genotype (Velvet mutation). Although an association between intronic mutations in loci influencing EGFR signaling and human congenital AS has been reported,7 the genetic basis for congenital aortic valve disease in humans is unknown in most cases. For that reason, and others, translation of findings in Velvet mice to the clinical setting will require meticulous independent confirmation in humans.
Summary
Congenital aortic valve disease comprises a range of abnormalities affecting valve leaflets, commissures, and the aorta, resulting in valve dysfunction and valvular cardiomyopathy. We report a novel mouse model with congenital unicuspid aortic valve anomalies that progress to overt valvular cardiomyopathy. Pathogenic processes in Velvet mice differ importantly from other mouse models of aortic valve disease, but recapitulate salient features of humans with unicuspid aortic valves. These findings suggest that the unicuspid aortic valve phenotype is distinct from bicuspid aortic disease; future animal and human studies of aortic valve disease should include precise phenotypic descriptors.
Sources of Funding
This study was supported by grant OD019941 from the National Institutes of Health, by Grant‐In‐Aid 16GRNT29720002 from the American Heart Association, and by a generous gift to the University of Iowa Research Foundation by Gerald and Marlene Manatt.
Disclosures
None.
Supporting information
Video S1. Transesophageal echocardiogram from a 19‐year‐old man with unicuspid aortic valve. For anatomical annotation, see Figure 3.
Video S2. Echocardiogram from a Velvet mouse with unicuspid aortic valve. For anatomical annotation, see Figure 3.
Video S3. Transesophageal 3D echocardiogram from a 39‐year‐old man with unicuspid aortic valve. For anatomical annotation, see Figure 10.
Acknowledgments
We thank Jeffrey Harless of the University of Iowa College of Dentistry for expert assistance with polarized light microscopy; William Kutschke, for assistance with blood pressure measurement; and Allyn L. Mark, MD, for thoughtful review of the article.
(J Am Heart Assoc. 2018;7:e006908 DOI: 10.1161/JAHA.117.006908.)
This article was handled independently by Hossein Ardehali, MD, PhD, as a guest editor. The editors had no role in the evaluation of the manuscript or in the decision about its acceptance.
References
- 1. Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL; American Heart Association Council on Cardiovascular Disease in the Young . Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995–3014. [DOI] [PubMed] [Google Scholar]
- 2. Williams RG, Pearson GD, Barst RJ, Child JS, del Nido P, Gersony WM, Kuehl KS, Landzberg MJ, Myerson M, Neish SR, Sahn DJ, Verstappen A, Warnes CA, Webb CL; National Heart, Lung, and Blood Institute Working Group on research in adult congenital heart disease . Report of the National Heart, Lung, and Blood Institute Working Group on research in adult congenital heart disease. J Am Coll Cardiol. 2006;47:701–707. [DOI] [PubMed] [Google Scholar]
- 3. Roberts WC, Ko JM, Hamilton C. Comparison of valve structure, valve weight, and severity of the valve obstruction in 1849 patients having isolated aortic valve replacement for aortic valve stenosis (with or without associated aortic regurgitation) studied at 3 different medical centers in 2 different time periods. Circulation. 2005;112:3919–3929. [DOI] [PubMed] [Google Scholar]
- 4. Mookadam F, Thota VR, Garcia‐Lopez AM, Emani UR, Alharthi MS, Zamorano J, Khandheria BK. Unicuspid aortic valve in adults: a systematic review. J Heart Valve Dis. 2010;19:79–85. [PubMed] [Google Scholar]
- 5. Fealey ME, Edwards WD, Miller DV, Maleszewski JJ. Unicommissural aortic valves: gross, histological, and immunohistochemical analysis of 52 cases (1978–2008). Cardiovasc Pathol. 2012;21:324–333. [DOI] [PubMed] [Google Scholar]
- 6. Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet. 2000;24:296–299. [DOI] [PubMed] [Google Scholar]
- 7. McBride KL, Zender GA, Fitzgerald‐Butt SM, Seagraves NJ, Fernbach SD, Zapata G, Lewin M, Towbin JA, Belmont JW. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res A Clin Mol Teratol. 2011;91:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du X, Tabeta K, Hoebe K, Liu H, Mann N, Mudd S, Crozat K, Sovath S, Gong X, Beutler B. Velvet, a dominant Egfr mutation that causes wavy hair and defective eyelid development in mice. Genetics. 2004;166:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El Accaoui RN, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, Lund DD, Brooks RM, Doshi H, Zimmerman KA, Kutschke W, Anseth KS, Heistad DD, Weiss RM. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2014;306:H1302–H1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short‐term pressure overload. Circulation. 2000;101:2863–2869. [DOI] [PubMed] [Google Scholar]
- 11. Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. [DOI] [PubMed] [Google Scholar]
- 12. Chu Y, Lund DD, Doshi H, Keen HL, Knudtson KL, Funk ND, Shao JQ, Cheng J, Hajj GP, Zimmerman KA, Davis MK, Brooks RM, Chapleau MW, Sigmund CD, Weiss RM, Heistad DD. Fibrotic aortic valve stenosis in hypercholesterolemic/hypertensive mice. Arterioscler Thromb Vasc Biol. 2016;36:466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hajj GP, Chu Y, Lund DD, Magida JA, Funk ND, Brooks RM, Baumbach GL, Zimmerman KA, Davis MK, El Accaoui RN, Hameed T, Doshi H, Chen B, Leinwand LA, Song LS, Heistad DD, Weiss RM. Spontaneous aortic regurgitation and valvular cardiomyopathy in mice. Arterioscler Thromb Vasc Biol. 2015;35:1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111:920–925. [DOI] [PubMed] [Google Scholar]
- 15. Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller JD, Weiss RM, Serrano KM, Brooks RM II, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buttrick PM, Kaplan M, Leinwand LA, Scheuer J. Alterations in gene expression in the rat heart after chronic pathological and physiological loads. J Mol Cell Cardiol. 1994;26:61–67. [DOI] [PubMed] [Google Scholar]
- 18. Chen B, Guo A, Zhang C, Chen R, Zhu Y, Hong J, Kutschke W, Zimmerman K, Weiss RM, Zingman L, Anderson ME, Wehrens XH, Song LS. Critical roles of junctophilin‐2 in T‐tubule and excitation‐contraction coupling maturation during postnatal development. Cardiovasc Res. 2013;100:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts WC, Vowels TJ, Ko JM. Natural history of adults with congenitally malformed aortic valves (unicuspid or bicuspid). Medicine (Baltimore). 2012;91:287–308. [DOI] [PubMed] [Google Scholar]
- 20. Michelena HI, Prakash SK, Della Corte A, Bissell MM, Anavekar N, Mathieu P, Bossé Y, Limongelli G, Bossone E, Benson DW, Lancellotti P, Isselbacher EM, Enriquez‐Sarano M, Sundt TM III, Pibarot P, Evangelista A, Milewicz DM, Body SC; BAVCon Investigators . Bicuspid aortic valve: identifying knowledge gaps and rising to the challenge from the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation. 2014;129:2691–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conti CA, Della Corte A, Votta E, Del Viscovo L, Bancone C, De Santo LS, Redaelli A. Biomechanical implications of the congenital bicuspid aortic valve: a finite element study of aortic root function from in vivo data. J Thorac Cardiovasc Surg. 2010;140:890–896, 896.e891–e892. [DOI] [PubMed] [Google Scholar]
- 22. Hope MD, Hope TA, Meadows AK, Ordovas KG, Urbania TH, Alley MT, Higgins CB. Bicuspid aortic valve: four‐dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology. 2010;255:53–61. [DOI] [PubMed] [Google Scholar]
- 23. Ohura N, Yamamoto K, Ichioka S, Sokabe T, Nakatsuka H, Baba A, Shibata M, Nakatsuka T, Harii K, Wada Y, Kohro T, Kodama T, Ando J. Global analysis of shear stress‐responsive genes in vascular endothelial cells. J Atheroscler Thromb. 2003;10:304–313. [DOI] [PubMed] [Google Scholar]
- 24. Hsu JJ, Lim J, Tintut Y, Demer LL. Cell‐matrix mechanics and pattern formation in inflammatory cardiovascular calcification. Heart. 2016;102:1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña‐Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC); European Association for Cardio‐Thoracic Surgery (EACTS) , Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón‐Esquivias G, Baumgartner H, Borger MA, Carrel TP, De Bonis M, Evangelista A, Falk V, Iung B, Lancellotti P, Pierard L, Price S, Schäfers HJ, Schuler G, Stepinska J, Swedberg K, Takkenberg J, Von Oppell UO, Windecker S, Zamorano JL, Zembala M. Guidelines on the management of valvular heart disease (version 2012). Eur Heart J. 2012;33:2451–2496. [DOI] [PubMed] [Google Scholar]
- 27. Jashari H, Rydberg A, Ibrahimi P, Bajraktari G, Henein MY. Left ventricular response to pressure afterload in children: aortic stenosis and coarctation: a systematic review of the current evidence. Int J Cardiol. 2015;178:203–209. [DOI] [PubMed] [Google Scholar]
- 28. Orwat S, Diller GP, van Hagen IM, Schmidt R, Tobler D, Greutmann M, Jonkaitiene R, Elnagar A, Johnson MR, Hall R, Roos‐Hesselink JW, Baumgartner H; ROPAC Investigators . Risk of pregnancy in moderate and severe aortic stenosis: from the multinational ROPAC Registry. J Am Coll Cardiol. 2016;68:1727–1737. [DOI] [PubMed] [Google Scholar]
- 29. Dusenbery SM, Lunze FI, Jerosch‐Herold M, Geva T, Newburger JW, Colan SD, Powell AJ. Left ventricular strain and myocardial fibrosis in congenital aortic stenosis. Am J Cardiol. 2015;116:1257–1262. [DOI] [PubMed] [Google Scholar]
- 30. Tönisen F, Perrin L, Bayarmagnai B, van den Dries K, Cambi A, Gligorijevic B. EP4 receptor promotes invadopodia and invasion in human breast cancer. Eur J Cell Biol. 2017;96:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Transesophageal echocardiogram from a 19‐year‐old man with unicuspid aortic valve. For anatomical annotation, see Figure 3.
Video S2. Echocardiogram from a Velvet mouse with unicuspid aortic valve. For anatomical annotation, see Figure 3.
Video S3. Transesophageal 3D echocardiogram from a 39‐year‐old man with unicuspid aortic valve. For anatomical annotation, see Figure 10.