

Abstract
Background
Carboxyl‐terminal modulator protein (CTMP) has been implicated in cancer, brain injury, and obesity. However, the role of CTMP in pathological cardiac hypertrophy has not been identified.
Methods and Results
In this study, decreased expression of CTMP was observed in both human failing hearts and murine hypertrophied hearts. To further explore the potential involvement of CTMP in pathological cardiac hypertrophy, cardiac‐specific CTMP knockout and overexpression mice were generated. In vivo experiments revealed that CTMP deficiency exacerbated the cardiac hypertrophy, fibrosis, and function induced by pressure overload, whereas CTMP overexpression alleviated the response to hypertrophic stimuli. Consistent with the in vivo results, adenovirus‐mediated gain‐of‐function or loss‐of‐function experiments showed that CTMP also exerted a protective effect against hypertrophic responses to angiotensin II in vitro. Mechanistically, CTMP ameliorated pathological cardiac hypertrophy through the blockade of the protein kinase B signaling pathway. Moreover, inhibition of protein kinase B activation with LY294002 rescued the deteriorated effect in aortic banding–treated cardiac‐specific CTMP knockout mice.
Conclusions
Taken together, these findings imply, for the first time, that increasing the cardiac expression of CTMP may be a novel therapeutic strategy for pathological cardiac hypertrophy.
Keywords: angiotensin II, aortic banding, carboxyl‐terminal modulator protein, pathological cardiac hypertrophy, signal transduction
Subject Categories: Heart Failure, Hypertrophy, Remodeling
Clinical Perspective
What Is New?
The expression of carboxyl‐terminal modulator protein is decreased in human failing hearts, murine hypertrophied hearts, and neonatal rat cardiomyocytes treated with angiotensin II.
Carboxyl‐terminal modulator protein is a negative regulator of pressure overload–induced cardiac hypertrophy, fibrosis, and cardiac dysfunction in vivo and in vitro.
Carboxyl‐terminal modulator protein exerts its antihypertrophic effect by inhibiting the protein kinase B signaling pathway.
What Are the Clinical Implications?
These findings elaborate a new mechanism underlying the pathogenesis of cardiac hypertrophy, which indicates that increasing the cardiac expression of carboxyl‐terminal modulator protein may be a novel therapeutic strategy for this disease.
This study is instructive for cardiologists and other related physicians to understand the development of pathological cardiac hypertrophy.
Introduction
Pathological cardiac hypertrophy is initially an adaptive response of the heart to a variety of common cardiovascular stresses, such as uncontrolled hypertension, myocardial ischemia, and valvular stenosis.1 However, prolonged stimulation induces a shift from compensated hypertrophy to decompensated hypertrophy, which directly leads to cardiac dysfunction, heart failure, and even sudden death.2 Thus, pathological cardiac hypertrophy is a crucial predisposing factor for cardiovascular events and is the leading cause of cardiac morbidity and mortality globally.3, 4 Multiple signaling pathways, including the mitogen‐activated protein kinase (MAPK) pathway, the phosphatidylinositol 3‐kinase/protein kinase B (AKT) pathway, and the protein kinase C pathway, have been identified in the initiation and development of pathological cardiac hypertrophy.5, 6, 7 Nevertheless, the molecular mechanisms that mediate these pathways remain incompletely understood. A better understanding of these mechanisms should shed light on novel therapeutic targets for ameliorating pathological cardiac hypertrophy.
Carboxyl‐terminal modulator protein (CTMP), also known as thioesterase superfamily member 4, contains 2 functional domains, including a C‐terminal thioesterase domain and a conserved mitochondrial‐related N‐terminal domain.8, 9 CTMP is ubiquitously expressed in various tissues, including the heart.10 As a nuclear encoded mitochondrial protein, CTMP is synthesized and translocated by mitochondrial localization signals.11 On cell death stimulation, CTMP is released to the cytosol and makes cells sensitive to apoptosis.11 Meanwhile, accumulating evidence suggests that CTMP plays vital roles in numerous pathological processes, such as tumor,8, 12, 13 neuroprotection,14 and metabolism.15 Initially, CTMP has been determined to exert a complex influence in multiple cancers by regulating cell survival, proliferation, and angiogenesis.12, 13, 16 Moreover, mounting studies manifest that inhibition of CTMP would be beneficial in the neuroprotection in response to brain injury, no matter whether the subjects combined with diabetes mellitus or not.9, 14, 17 In addition, a recent study makes clear that the expression of CTMP is higher in the adipose tissue of both high‐fat diet–induced diabetic mice and genetically modified obese mice compared with their controls, which suggests CTMP is a key player in obesity.15 Cardiometabolic disorders share certain common pathophysiological process and are orchestrated by similar signaling pathways, such as insulin resistance and insulin signaling,18, 19 indicating a potential involvement of CTMP in pathological cardiac hypertrophy. Nevertheless, the role of CTMP in pathological cardiac hypertrophy remains to be elucidated.
Given that aortic banding (AB) provides a more reproducible model of cardiac hypertrophy and a more gradual time course in the development of heart failure,20 we perform AB model instead of conventional left anterior descending ligation. In the current study, we first observed that CTMP expression was downregulated in the hearts of patients with dilated cardiomyopathy or hypertrophic cardiomyopathy and in mice after AB. Next, by using gain‐of‐function and loss‐of‐function approaches in vivo and in vitro, we demonstrated, for the first time, that CTMP ameliorated pathological cardiac hypertrophy by inhibiting the AKT signaling pathway. Moreover, restraining the AKT signaling pathway with an AKT inhibitor rescued the deleterious effects of CTMP knockout induced by AB. Taken together, our results indicate that CTMP is a novel negative regulator of pathological cardiac hypertrophy via its inhibition of the AKT signaling pathway.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. The data are available from the authors on request.
Reagents
Antibodies against the following proteins were purchased from Cell Signaling Technology: MAPK/extracellular signal–regulated kinase (ERK) kinase 1/2 (9122, 1:1000 dilution), ERK1/2 (4695, 1:1000 dilution), c‐Jun N‐terminal kinase 1/2 (9252, 1:1000 dilution), P38 (9212, 1:1000 dilution), phosphorylated MAPK/ERK kinase 1/2Ser217/221 (9154, 1:1000 dilution), phosphorylated ERK1/2Thr202/Tyr204 (4370, 1:1000 dilution), phosphorylated c‐Jun N‐terminal kinase 1/2Thr183/Tyr185 (4668, 1:1000 dilution), phosphorylated P38Thr180/Tyr182 (4511, 1:1000 dilution), AKT (4691, 1:1000 dilution), mammalian target of rapamycin (mTOR; 2983, 1:1000 dilution), glycogen synthase kinase 3β (GSK3β; 9315, 1:1000 dilution), ribosomal protein S6 kinase β‐1 (P70S6) (2708, 1:1000 dilution), phosphorylated AKTSer473 (4060, 1:1000 dilution), phosphorylated mTORSer2448 (2971, 1:1000 dilution), phosphorylated GSK3βSer9 (9322, 1:1000 dilution), phosphorylated P70S6Ser371 (9208, 1:1000 dilution), CTMP (4612, 1:500 dilution), and GAPDH (2118, 1:1000 dilution). Antibodies against atrial natriuretic peptide (sc20158, 1:200 dilution) were purchased from Santa Cruz Biotechnology. Peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, 1:10 000 dilution) were used for visualization of Western blot bands. The bicinchoninic acid protein assay kit was purchased from Pierce. Fetal bovine serum was purchased from Gibco (10099141). The cell culture reagents and other reagents were purchased from Sigma.
Human Heart Samples
Samples were collected from the left ventricles (LVs) of failing human hearts with dilated cardiomyopathy and hypertrophic cardiomyopathy in patients undergoing heart transplantation. The control samples were collected from the LVs of normal heart donors with brain death or those who died from accidents but whose hearts were inappropriate for transplantation because of technical reasons. We obtained written informed consent from the patients and the relatives of the donors. All the experiments involving human samples were conducted in accordance with the principles of the Declaration of Helsinki and the ethical regulations of the Human Research Ethics Committee of Renmin Hospital of Wuhan University (Wuhan, China). Detailed information of human heart samples is shown in Table S1.
Animals
All of the animal procedures were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University.
Production of cardiac‐specific CTMP knockout mice
CTMPloxP/loxP (CTMP‐Flox) mice were created using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR‐associated (CRISPR/Cas9) system. The second exon of CTMP was flanked by loxP sites; thus, 2 single‐guide RNAs (single‐guide RNA1 and single‐guide RNA2) targeting CTMP introns 1 and 2 were designed. The donor plasmid containing exon 2, flanked by 2 loxP sites and the 2 homologous arms, was used as the template. The donor plasmid, single‐guide RNA1 and single‐guide RNA2, and Cas9 mRNAs were coinjected into 1‐cell‐stage embryos. The obtained mice with exon 2 flanked by 2 loxP sites on 1 allele were used to establish homozygous CTMP‐Flox mice. A simple schematic diagram is shown in Figure S1A. The polymerase chain reaction (PCR) primers CTMP‐P1 to CTMP‐P6, which were used for identification, are listed in Table S2. CTMP‐Flox mice were bred with ɑ‐major histocompatibility complex (MHC)–MerCreMer (MCM) (Jackson Laboratory, stock No. 005650) transgenic mice to generate CTMP‐Flox/α‐MHC‐MCM mice. For the cardiac‐specific knockout of CTMP (CTMP‐CKO), 6‐week‐old CTMP‐Flox/α‐MHC‐MCM mice were injected with tamoxifen (Sigma‐Aldrich, T5648, 25 mg/kg per day) for 5 consecutive days. The controls, α‐MHC‐MCM mice and CTMP‐Flox mice, were treated with equal dose of tamoxifen injection. No mice died or showed any morbidity after tamoxifen injection.
Generation of cardiac‐specific CTMP transgenic mice
Conditional CTMP transgenic mice were generated by microinjecting CAG‐loxP‐CAT‐loxP‐CTMP into fertilized C57BL/6 embryos. The obtained mice were identified using PCR analysis of tail genomic DNA. The primers used for identification are as follows: 5′‐CATGTCTGGATCGATCCCCG‐3′ and 5′‐CAGTAGTGACATCAATGATGG‐3′. The founder mice were then crossed with α‐MHC‐MCM mice to generate CAG‐CAT‐CTMP/α‐MHC‐MCM mice. To commence cardiac‐specific CTMP overexpression, 6‐week‐old CAG‐CAT‐CTMP/α‐MHC‐MCM mice were intraperitoneally injected with tamoxifen for 5 consecutive days to induce Cre‐mediated CAT gene excision. The α‐MHC‐MCM mice (nontransgenic) with the same administration of tamoxifen served as controls.
Animal Model of AB
AB was performed to induce cardiac hypertrophy by pressure overload, as previously described.4, 21 In brief, the mice (males; aged 8–10 weeks; weight, 24–27 g) were anesthetized with sodium pentobarbital via an intraperitoneal injection until the toe pinch reflex disappeared. The left chest of each mouse was opened to visualize the thoracic aorta at the second intercostal space. A 7‐0 silk suture was used to band the thoracic aorta against a 26/27‐gauge needle. After ligation, the needle was gently removed, and the thoracic cavity was closed. Next, the adequacy of the constriction was assessed with Doppler analysis. The sham‐operated group underwent a similar procedure with no aorta constriction.
Echocardiographic Evaluation
After animals were anesthetized with 2% inhaled isoflurane, transthoracic echocardiography was performed to measure cardiac function at the indicated time point. Using a Mylab 30CV machine (ESAOTE) with a 15‐MHz transducer, LV structure and function were measured. The LV end‐diastolic diameter and LV end‐systolic diameter were determined using the LV M‐mode with a sweep speed of 50 mm/s at the midpapillary muscle level. In addition, LV fractional shortening was calculated, as previously described.21, 22
Histological Analysis
When the hearts were excised, they were placed in a 10% potassium chloride solution (50 g potassium chloride was dissolved into pure water, and the solution was fixed to 500 mL) to arrest them in diastole. After fixation in 10% formalin, the hearts were dehydrated and embedded in paraffin. Then, 5‐μm‐thick sections of the hearts were stained with hematoxylin and eosin and picrosirius red for morphological examinations and collagen deposition assessments, respectively.22 Subsequently, quantitative digital analysis imaging (Image‐Pro Plus 6.0) was performed to measure the cross‐sectional area of cardiomyocytes and collagen volume using captured images.
Cultured Neonatal Rat Cardiac Myocytes and Adenovirus Infection
PBS containing 0.03% trypsin and 0.04% collagenase type II was used to isolate primary neonatal rat cardiomyocytes (NRCMs) from the hearts of 1‐ to 2‐day‐old Sprague‐Dawley rats, as described.4, 21 After removing fibroblasts by a differential attachment technique, NRCMs were seeded at a density of 2×105 cells per well onto 6‐well culture plates coated with gelatin in plating medium consisting of DMEM/F12 (C11330, Gibco) supplemented with 10% fetal bovine serum, bromodeoxyuridine (0.1 mmol/L, to inhibit the proliferation of fibroblasts), and penicillin/streptomycin. After 48 hours, the culture medium was replaced with serum‐free DMEM/F12 for 12 hours before stimulation with angiotensin II (AngII). Adenovirus harboring CTMP short‐hairpin RNA was used to silence CTMP expression. Adenoviral short‐hairpin RNA was used as a control. To overexpress CTMP, full‐length rat CTMP cDNA under the control of the cytomegalovirus promoter was inserted into replication‐defective adenoviral vectors (adenoviral CTMP). An adenoviral vector encoding the green fluorescent protein gene was used as a control (adenoviral green fluorescent protein). The NRCMs were infected with adenoviral short‐hairpin CTMP, adenoviral short‐hairpin RNA, adenoviral CTMP, and adenoviral green fluorescent protein at a multiplicity of infection of 100 for 24 hours.
Immunofluorescence Analysis
Immunofluorescence staining was used to assess the cell surface area of the NRCMs. The NRCMs were infected with the corresponding adenoviruses for 24 hours and then treated with AngII (1 μmol/L) or PBS for 48 hours. The NRCMs were fixed with 3.7% formaldehyde, permeabilized with 0.2% Triton X‐100 in PBS for 45 minutes, and stained with α‐actinin (05‐384, Merck Millipore, 1:100 dilution) using standard immunofluorescence staining techniques. Image‐Pro Plus 6.0 software was used to analyze and measure the cell size.
Quantitative Real‐Time PCR and Western Blotting
Total mRNA was extracted from mouse LV tissues and NRCMs with TRIzol (15596‐026, Invitrogen), and cDNA was synthesized by using the Transcriptor First Strand cDNA Synthesis Kit (04896866001, Roche). Quantitative real‐time PCR amplification was used by using SYBR Green (04887352001, Roche), and the results were normalized against the corresponding GAPDH gene expression. The primers used for real‐time PCR are presented in Table S3.
For Western blotting, the LV tissues and NRCMs were used to extract total proteins in a lysis buffer (720 μL of radioimmunoprecipitation assay, 20 μL of phenylmethyl sulphonyl fluoride, 100 μL of complete protease inhibitor cocktail, 100 μL of Phos‐stop, 50 μL of NaF, and 10 μL of Na3VO4), and the bicinchoninic acid protein assay kit (Pierce) was used to measure protein concentrations. The loading buffer was composed of 40% glycerol, 4% lithium dodecyl sulfate, 0.8 mol/L Tris‐HCl, 0.025% Phenol Red, 0.025% Coomassie Blue, 2 mmol/L EDTA disodium, and 50 mmol/L dithiothreitol. The samples were boiled before loading onto the gel. Protein (50 μg) was used for SDS‐PAGE (Invitrogen), and the proteins were then transferred to a polyvinylidene difluoride membrane (Millipore). After blocking with 5% fat‐free milk at room temperature for 1 hour, the membranes were probed with specific primary antibodies at 4°C overnight. Subsequently, the membranes were incubated with peroxidase‐conjugated secondary antibodies at room temperature for 1 hour. The membranes were incubated with electrochemiluminescence reagents (Bio‐Rad) before visualization with the Bio‐Rad ChemiDoc XRS+ system. The expression levels of specific proteins were normalized to GAPDH.
Treatment of Mice With LY294002
LY294002 (Sigma, L9908), an AKT inhibitor, was dissolved in dimethyl sulfoxide. Then, the mixture was prepared and injected into the animal at a dose of 50 mg/kg for 4 weeks after AB. The control group for this experiment was injected with the same volume of dimethyl sulfoxide without LY294002.
Statistical Analysis
Results are presented as the mean±SD. No significant deviations from gaussian distributions were seen for any group using the Shapiro‐Wilk test. However, by Levene's test, there were deviations from homogeneity of variance (heteroscedasticity) for some group comparisons. For group comparisons showing no significant heteroscedasticity, the 2‐tailed Student t test or 1‐way ANOVA was performed; for group comparisons showing significant heteroscedasticity, the Welch test and the Brown‐Forsythe test were performed. For comparisons of ≥3 groups, post hoc pairwise comparisons used either T tests with Bonferroni correction or Tamhane's T2 test when there was significant heteroscedasticity. P<0.05 was considered to be significant. Statistical analyses were performed with SPSS for Windows software, version 21.0 (IBM Corp).
Results
Reduced Expression of CTMP in Hypertrophic Hearts
The expression level of CTMP in pathological cardiac hypertrophy was first detected by using Western blotting to evaluate its potential involvement in the development of pathological cardiac hypertrophy. We found that CTMP expression was significantly lower in the tissues of cases of both dilated cardiomyopathy (Figure 1A) and hypertrophic cardiomyopathy (Figure 1B) compared with normal heart tissues, indicating the clinical relevance of CTMP to the development of pathological cardiac hypertrophy. When hypertrophy was induced by AB, the mRNA and protein levels of CTMP in the mouse heart tissues were also remarkably decreased in a time‐dependent manner compared with those in the sham hearts (Figure S2 and Figure 1C). Consistently, in the NRCMs, time‐dependent reduction of CTMP expression could be induced by AngII treatment (Figure 1D). Collectively, this evidence suggests that CTMP may play an important role in pathological cardiac hypertrophy.
Figure 1.
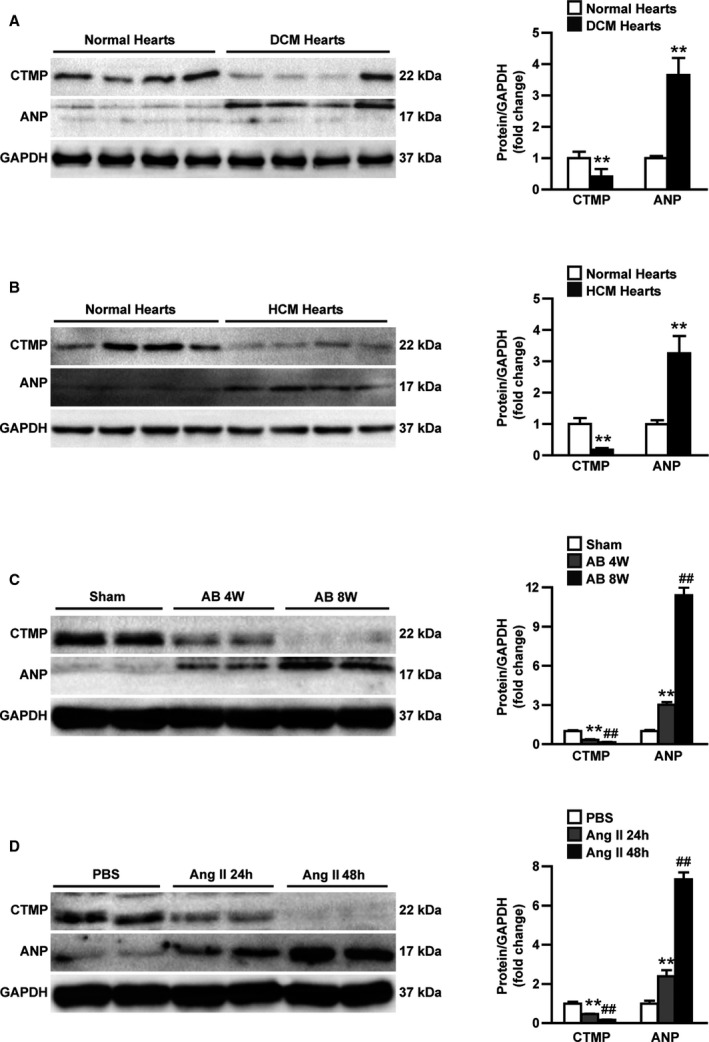
Carboxyl‐terminal modulator protein (CTMP) is downregulated in human failing hearts and murine hypertrophic hearts. A and B, Western blot analysis and quantitative results of CTMP and atrial natriuretic peptide (ANP) in left ventricular tissues from normal and dilated cardiomyopathy (DCM) human hearts (n=4 per group, **P<0.01 vs normal hearts; A) and normal and hypertrophic cardiomyopathy (HCM) human hearts (n=4 per group, **P<0.01 vs normal hearts; B). C, Western blot analysis and quantitative results of CTMP and ANP in the left ventricular tissues of mice subjected to sham operation, 4 weeks (4W) after aortic banding (AB) and 8 weeks (8W) after AB (n=6 per group, **P<0.01 vs sham, ## P<0.01 vs AB 4W). D, Western blot analysis and quantitative results of CTMP and ANP in neonatal rat cardiomyocytes treated with PBS or angiotensin II (AngII) for 24 hours (24h) and 48 hours (48h; n=3 independent experiments, **P<0.01 vs PBS, ## P<0.01 vs AngII 24h). The data are presented as the mean±SD by 2‐tailed Student t test (A [CTMP]), Welch test (A [ANP] and B), Bonferroni post hoc analysis (C [CTMP]), or Tamhane's T2 analysis (C [ANP] and D).
Ablation of CTMP in Cardiomyocytes Accelerates the Development of Cardiac Hypertrophy
We subsequently generated cardiac‐specific CTMP‐deficient mice using the CRISPR/Cas9 system to investigate the role of CTMP in the development of cardiac hypertrophy in vivo (Figure S1A through S1C). The deficiency of CTMP specifically in the heart was confirmed via Western blotting (Figure S1D). Cardiac‐specific CTMP deficiency (CTMP‐CKO) did not cause any defects in the hearts in terms of the structure and function in the natural condition (Figure 2A through 2C) compared with the ɑ‐MHC‐MCM or CTMP‐Flox control groups. After 4 weeks of AB induction, the ratios of heart weight/body weight (HW/BW), lung weight/BW, and HW/tibia length were markedly increased compared with the sham group (Figure 2A). Further detailed comparisons indicated that HW/BW, lung weight/BW, and HW/tibia length were significantly higher in the CTMP‐CKO mice compared with the α‐MHC‐MCM or CTMP‐Flox control mice after AB induction (Figure 2A). Histological examination indicated significantly larger heart volumes, greater thickness of the heart wall, and increased cardiomyocyte size in the CTMP‐CKO mice compared with those in the control mice after hypertrophy was induced for 4 weeks (Figure 2B). Furthermore, cardiac functional assessment via echocardiogram showed that the CTMP‐CKO mouse heart exhibited a significantly increased LV end‐diastolic diameter and LV end‐systolic diameter but decreased fractional shortening (percentage) compared with the 2 control groups (Figure 2C). Moreover, picrosirius red staining revealed that the collagen content in both the perivascular and interstitial spaces of the CTMP‐CKO mice was significantly increased compared with that in the control litters after AB surgery (Figure 2D). Simultaneously, the expression levels of cardiac hypertrophy markers, including Anp, brain natriuretic peptide (Bnp), and myosin heavy chain 7 (Myh7), in the CTMP‐CKO hearts were markedly higher than those of the control hearts (Figure 2E) and were accompanied by higher mRNA levels of the fibrotic markers collagen Iα, collagen III, and connective tissue growth factor (Ctgf; Figure 2F). This evidence indicates that cardiac‐specific CTMP deficiency aggravates AB‐induced cardiac hypertrophy.
Figure 2.
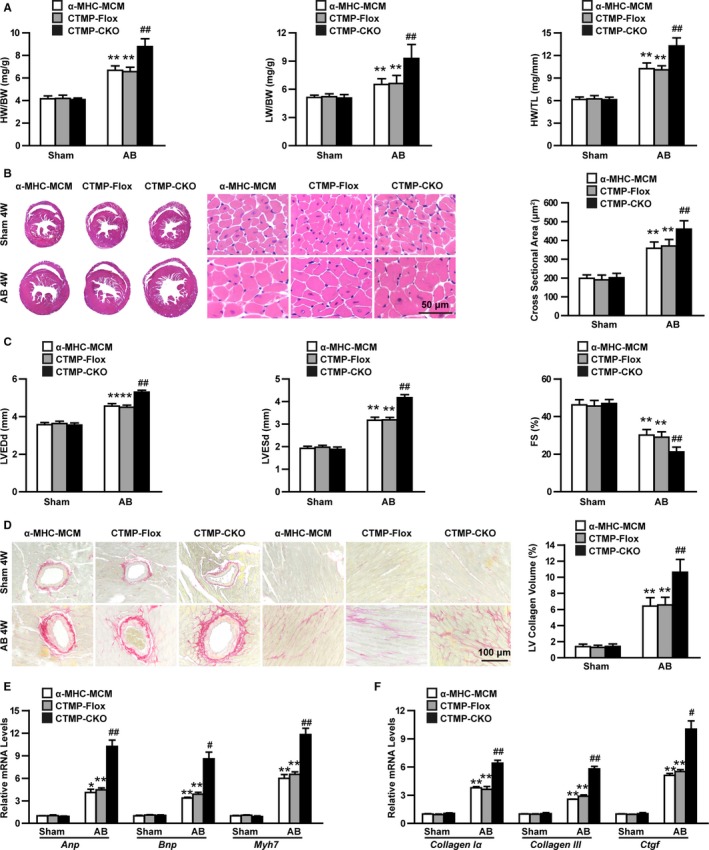
Cardiac‐specific carboxyl‐terminal modulator protein (CTMP) deficiency exacerbates pathological cardiac hypertrophy induced by aortic banding (AB). A, The ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) in different genotypic mice (ɑ‐major histocompatibility complex–MerCreMer [ɑ‐MHC‐MCM], CTMPloxP/loxP [CTMP‐Flox], and cardiac‐specific knockout of CTMP [CTMP‐CKO]) 4 weeks (4W) after sham or aortic banding (AB) surgery (n=11–13 mice per group). B, Representative images of hematoxylin and eosin staining of hearts from each group (n=5–6 mice per group; bar=50 μm), and statistical results for the cardiomyocyte cross‐sectional area (n>100 cells per group). C, Quantitative analysis of left ventricular (LV) end‐diastolic diameter (LVEDd), LV end‐systolic diameter (LVESd), and fractional shortening (FS) in each group 4W after AB surgery (n=11–13 mice per group). D, Representative images of picrosirius red staining of hearts from each group (n=6 mice per group; bar=100 μm), and statistical results for fibrotic areas in each group (n>30 fields per group). E and F, Real‐time polymerase chain reaction analyses of hypertrophic (E) and fibrotic (F) markers in each group (n=4 per group). *P<0.05 or **P<0.01 vs ɑ‐MHC‐MCM sham or CTMP‐Flox sham; # P<0.05 or ## P<0.01 vs ɑ‐MHC‐MCM AB or CTMP‐Flox AB (A through F) by Tamhane's T2 analysis (A, B, and D‐F [Collagen III and Ctgf]) or Bonferroni post hoc analysis (C and F [Collagen Iα]).
CTMP Overexpression Ameliorates Pressure Overload–Induced Cardiac Hypertrophy
To confirm the protective role of CTMP in the progression of cardiac hypertrophy, we generated 4 independent lines of cardiomyocyte‐specific CTMP‐overexpressing transgenic mice (Figure S3A and S3B). The second line, which expressed the highest level of CTMP, was used for further study. Similar to the CTMP‐CKO model, CTMP‐transgenic did not exhibit any effects on normal hearts (Figure 3A through 3C). When hypertrophy was induced through AB surgery after 4 weeks, in contrast to the results observed in CTMP‐CKO mice, the transgenic mice showed significantly ameliorated pathological cardiac hypertrophy, as indicated by a decrease in the HW/BW, lung weight/BW, and HW/tibia length ratios, the cardiac size, and the cross‐sectional area of cardiomyocytes, compared with the nontransgenic mice (Figure 3A and 3B). In accordance with these results, the cardiac structure and function, as measured by echocardiography analyses, were greatly improved in the transgenic mice (Figure 3C). In addition, the improving effect of CTMP overexpression on cardiac fibrosis was identified with histological staining and analysis (Figure 3D). Concomitant with mitigated cardiac hypertrophy and fibrosis, the mRNA expression of hypertrophic and fibrotic genes was lower in the transgenic mice than in the nontransgenic mice (Figure 3E and 3F). Collectively, these gain‐of‐function data demonstrate that cardiomyocyte‐specific CTMP overexpression alleviated the cardiac hypertrophy and fibrosis induced by pressure overload.
Figure 3.
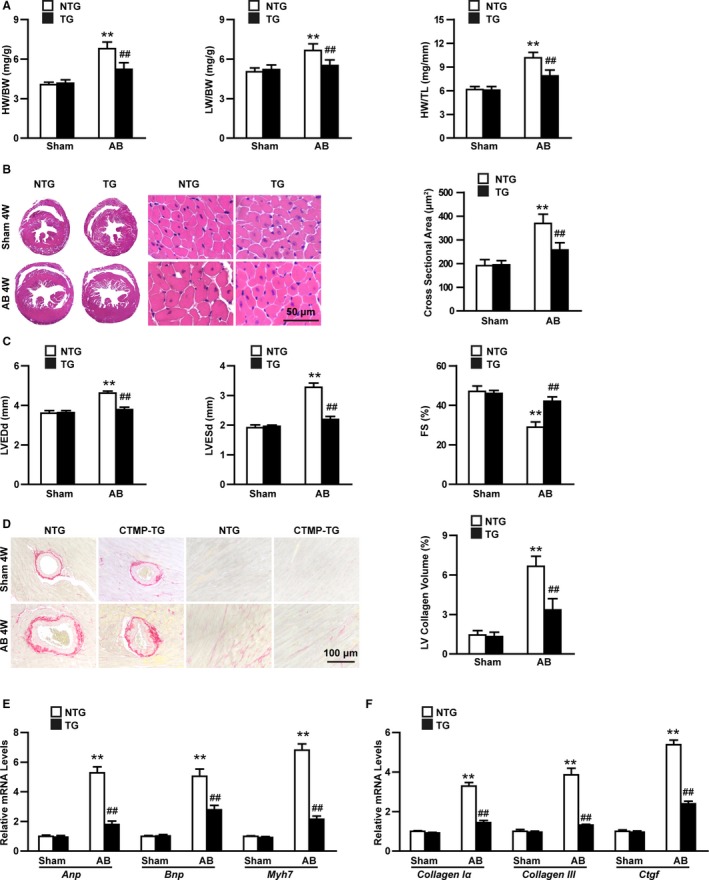
Cardiac‐specific carboxyl‐terminal modulator protein (CTMP) overexpression ameliorates pathological cardiac hypertrophy induced by pressure overload. A, The ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) in nontransgenic (NTG) and transgenic (TG) mice 4 weeks (4W) after sham or aortic banding (AB) surgery (n=11–13 mice per group). B, Representative images of hematoxylin and eosin–stained hearts from each group (n=6–7 mice per group; bar=50 μm), and statistical results for the cardiomyocyte cross‐sectional area (n>100 cells per group). C, Quantitative analysis of left ventricular (LV) end‐diastolic diameter (LVEDd), LV end‐systolic diameter (LVESd), and fractional shortening (FS) in each group 4W after AB surgery (n=10–13 mice per group). D, Representative images of picrosirius red–stained hearts from each group (n=6–7 mice per group; bar=100 μm), and statistical results for fibrotic areas in each group (n>30 fields per group). E and F, Real‐time polymerase chain reaction analyses of the hypertrophic (E) and fibrotic (F) markers in each group (n=4 per group). **P<0.01 vs NTG sham; ## P<0.01 vs NTG AB (A through F) by Tamhane's T2 analysis (A [HW/BW], B, C [LVESd], D, E [Bnp and Myh7], and F [Collagen III]) or Bonferroni post hoc analysis (A [LW/BW and HW/TL], C [LVEDd and FS], E [Anp], and F [Collagen Iα and Ctgf]).
CTMP Inhibits Hypertrophy of Cultured Cardiomyocytes Induced by AngII
To further investigate the effect of CTMP on cardiomyocytes, gain‐of‐function and loss‐of‐function studies were performed in vitro. NRCMs were infected with adenoviral short‐hairpin CTMP to knock down CTMP or with adenoviral CTMP to overexpress CTMP. The efficiency of the adenovirus‐mediated CTMP knockdown or overexpression was confirmed (Figure 4A). These infected cells were subsequently treated with 1 μmol/L AngII or PBS control for 48 hours. To visualize the area of the NRCMs after induction, α‐actinin staining was performed. We found that the cell size was remarkably enlarged by adenoviral short‐hairpin CTMP–medicated CTMP knockdown compared with the cells infected with adenoviral short‐hairpin RNA after AngII stimulation (Figure 4B and 4C). In contrast, CTMP overexpression significantly inhibited AngII‐induced expansion of NRCMs compared with the control group, although it did not exhibit this effect in PBS‐treated NRCMs (Figure 4B and 4D). Furthermore, although the expression of the established hypertrophic hallmarks Anp and Myh7 was drastically induced by AngII stimulation, it can be significantly promoted by CTMP knockdown but blunted by CTMP overexpression (Figure 4E and 4F). These in vitro results indicate that CTMP can protect against cardiomyocyte hypertrophy.
Figure 4.
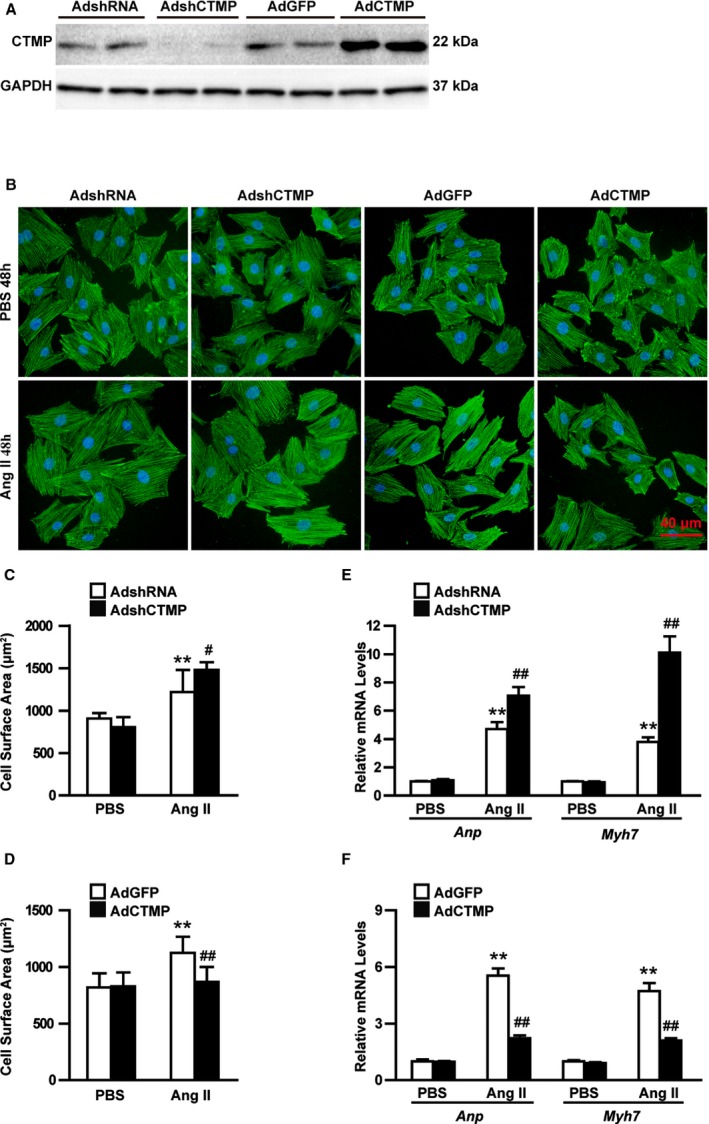
Carboxyl‐terminal modulator protein (CTMP) alleviates angiotensin II (AngII)–induced cardiomyocyte hypertrophy. A, Western blot analysis of CTMP expression in neonatal rat cardiomyocytes (NRCMs) infected with adenoviral short‐hairpin CTMP (AdshCTMP) and adenoviral CTMP (AdCTMP) and their controls (n=3 independent experiments). B, Representative images of NRCMs infected with adenoviral short‐hairpin RNA (AdshRNA), AdshCTMP, adenoviral green fluorescent protein (AdGFP), and AdCTMP treated with PBS or AngII for 48 hours (48h; green, α‐actinin; blue, nuclear; bar=40 μm). C and D, Quantitative analysis of the cell surface area of NRCMs infected with AdshRNA or AdshCTMP (C) and AdGFP or AdCTMP (D) treated with PBS or AngII for 48h (n>50 cells per group; **P<0.01 vs AdshRNA+PBS or AdGFP+PBS; # P<0.05 vs AdshRNA+AngII; ## P<0.01 vs AdGFP+AngII). E and F, Real‐time polymerase chain reaction analyses of the hypertrophic biomarkers atrial natriuretic peptide (Anp) and myosin heavy chain 7 (Myh7) in NRCMs infected with AdshRNA or AdshCTMP (E) and AdGFP or AdCTMP (F) treated with PBS or AngII (n=3 independent experiments; **P<0.01 vs AdshRNA+PBS or AdGFP+PBS; ## P<0.01 vs AdshRNA+AngII or AdGFP+AngII) by Tamhane's T2 analysis (C and E) or Bonferroni post hoc analysis (D and F).
CTMP Protects Against Pathological Cardiac Hypertrophy Mainly by Blocking the AKT Signaling Pathway
The aforementioned results suggest that CTMP plays a protective role in pressure overload–induced cardiac hypertrophy. However, the molecular mechanisms by which CTMP regulates the hypertrophic response remain unknown. To delineate these underlying mechanisms, we first investigated whether CTMP influenced the AB‐induced MAPK signaling response because the MAPK signaling pathway has been confirmed to play a vital role in pathological cardiac hypertrophy. Compared with the sham operation, AB was associated with increased phosphorylation levels of MAPK members, including MAPK/ERK kinase 1/2, ERK 1/2, c‐Jun N‐terminal kinase 1/2, and P38, in response to hypertrophic stimuli. Unexpectedly, neither the deficiency nor the overexpression of CTMP altered the phosphorylation of MAPKs compared with controls in response to pressure overload (Figure S4A and S4B). These data indicate that the MAPK signaling pathway might not be involved in the CTMP‐regulated hypertrophic response. Therefore, we further explored whether the effect of CTMP on pathological cardiac hypertrophy is associated with the AKT signaling pathway, which also plays a crucial role in the development of cardiac hypertrophy. We observed that the phosphorylation of AKT, mTOR, GSK3β, and P70S6K was markedly elevated in CTMP‐deficient mice, whereas the activation of the AKT signaling cascade was inhibited in transgenic mice compared with their control groups in response to the induction of hypertrophy (Figure 5A and 5B).
Figure 5.
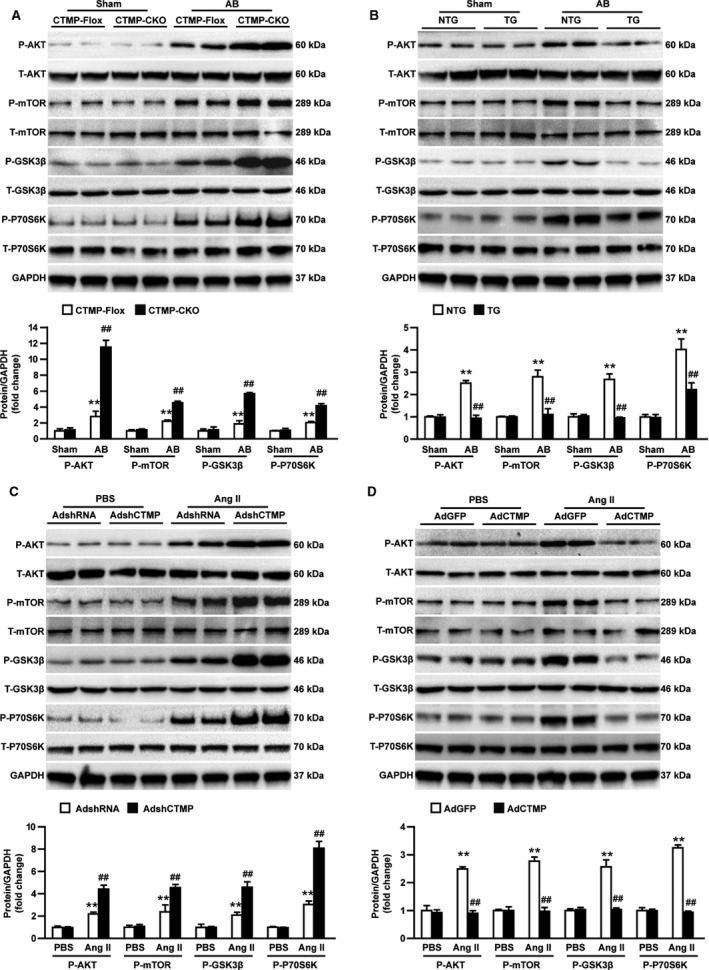
Carboxyl‐terminal modulator protein (CTMP) inhibits the activation of protein kinase B (AKT) signaling in hypertrophic hearts and myocytes. A, Western blot analysis and quantitative results of total (T) and phosphorylated (P) levels of AKT, mammalian target of rapamycin (mTOR), glycogen synthase kinase 3β (GSK3β), and ribosomal protein S6 kinase β‐1 (P70S6K) in the left ventricular tissues of CTMPloxP/loxP (CTMP‐Flox) and cardiac‐specific knockout of CTMP (CTMP‐CKO) mice 4 weeks after sham or aortic banding (AB) surgery (n=4 mice per group; **P<0.01 vs CTMP‐Flox sham; ## P<0.01 vs CTMP‐Flox AB). B, Western blot analysis and quantitative results of T and P levels of AKT, mTOR, GSK3β, and P70S6K in left ventricular tissues from transgenic (TG) and nontransgenic (NTG) mice 4 weeks after sham or AB surgery (n=4 mice per group; **P<0.01 vs NTG sham; ## P<0.01 vs NTG AB). C, Western blot analysis and quantitative results of the T and P levels of AKT, mTOR, GSK3β, and P70S6K in neonatal rat cardiomyocytes (NRCMs) infected with adenoviral short‐hairpin RNA (AdshRNA) and adenoviral short‐hairpin CTMP (AdshCTMP) and treated with PBS or angiotensin II (AngII; n=3 independent experiments; **P<0.01 vs AdshRNA+PBS; ## P<0.01 vs AdshRNA+AngII). D, Western blot analysis and quantitative results of the T and P levels of AKT, mTOR, GSK3β, and P70S6K in NRCMs infected with adenoviral green fluorescent protein (AdGFP) and adenoviral CTMP (AdCTMP) and treated with PBS or AngII (n=3 independent experiments; **P<0.01 vs AdGFP+PBS; ## P<0.01 vs AdGFP+AngII) by Bonferroni post hoc analysis (A [P‐AKT and P‐mTOR], C [P‐mTOR], and D [P‐mTOR and P‐P70S6K]) or Tamhane's T2 analysis (A [P‐GSK3β and P‐P70S6K], B, C [P‐AKT, P‐GSK3β, and P‐P70S6K], and D [P‐AKT and P‐GSK3β]).
To further verify the regulatory effect of CTMP on the AKT signaling pathway, NRCMs were subjected to hypertrophic stimuli after infection with adenoviral short‐hairpin CTMP, adenoviral short‐hairpin RNA, adenoviral CTMP, and adenoviral green fluorescent protein. Consistent with the in vivo data, the in vitro results indicated that CTMP knockdown strengthened the phosphorylation levels of AKT and its downstream kinases in response to AngII, whereas CTMP overexpression weakened the activities of AKT‐related pathways in AngII‐treated NRCMs (Figure 5C and 5D). These results indicate that CTMP ameliorates pathological cardiac hypertrophy by suppressing the overactive AKT signaling pathway but not the MAPK signaling pathway.
Inhibition of AKT Signaling Blunted the Cardiac Hypertrophy–Promoting Effect of CTMP‐CKO
The previous results showed that CTMP is an inhibitor of AKT signaling in response to hypertrophic stress. To further identify the causal association between AKT signaling and CTMP‐regulated cardiac hypertrophy, CTMP‐CKO mice and CTMP‐Flox mice were injected with the AKT inhibitor LY294002 for 4 weeks after the AB operation. Western blotting revealed that the phosphorylation of AKT signaling induced by AB was significantly inhibited by the AKT inhibitor LY294002 in both CTMP‐CKO and CTMP‐Flox mice (Figure 6A). More important, administration of the AKT inhibitor completely blunted the effect of CTMP ablation on the activation of AKT signaling (Figure 6A). Furthermore, the cardiac abnormalities, including cardiac hypertrophy (Figure 6B and 6C), cardiac dysfunction (Figure 6D), and cardiac fibrosis (Figure 6E), were significantly ameliorated by the AKT inhibitor LY294002 in both CTMP‐CKO and CTMP‐Flox mice subjected to AB; LY294002 abolished the difference between the CTMP‐CKO and CTMP‐Flox mice. Taken together, the results demonstrate that the effect of CTMP on cardiac hypertrophy is mediated by its inhibition of AKT activity.
Figure 6.
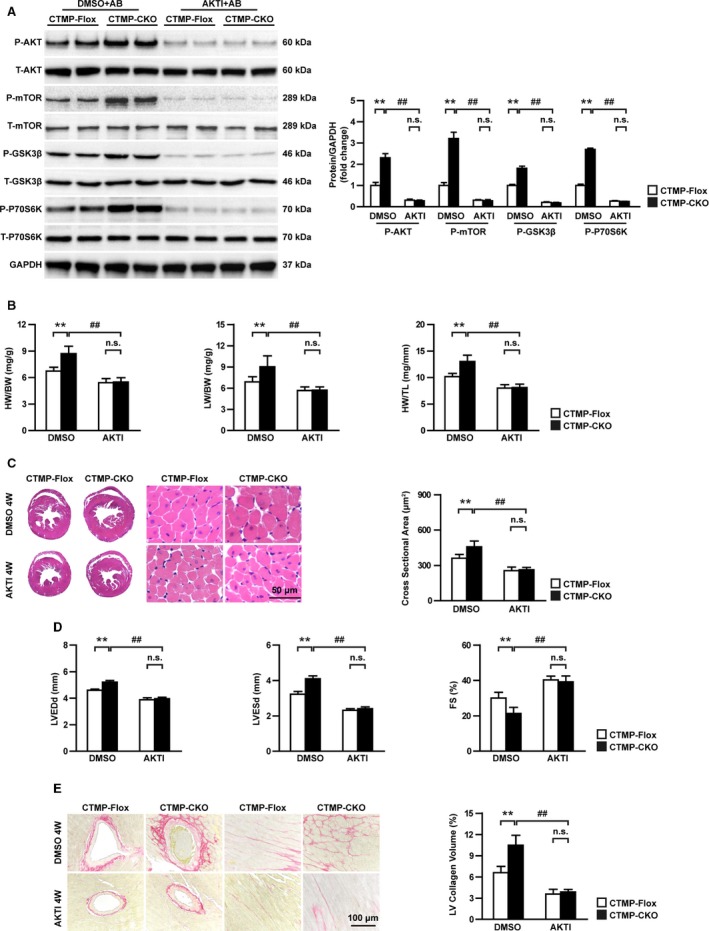
Inhibition of the protein kinase B (AKT) signaling pathway rescues cardiac injury induced by pressure overload in cardiac‐specific carboxyl‐terminal modulator protein (CTMP) deficiency mice. A, Western blot analysis and quantitative results of an AKT inhibitor (AKTI), LY294002, and dimethyl sulfoxide (DMSO) on CTMPloxP/loxP (CTMP‐Flox) and cardiac‐specific knockout of CTMP (CTMP‐CKO) mice 4 weeks (4W) after aortic banding (AB) surgery (n=4 mice per group). B, The ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) in different genotypic mice (CTMP‐Flox and CTMP‐CKO) mice treated with AKTI or DMSO 4W after AB surgery (n=12–14 mice per group). C, Representative images of hematoxylin and eosin–stained hearts from each group (n=6 mice per group; bar=50 μm), and statistical results for the cardiomyocyte cross‐sectional area (n>100 cells per group). D, Quantitative analysis of left ventricular (LV) end‐diastolic diameter (LVEDd), LV end‐systolic diameter (LVESd), and fractional shortening (FS) in each group (n=11–13 mice per group). E, Representative images of picrosirius red–stained hearts from each group (n=6 mice per group; bar=100 μm), and statistical results for fibrotic areas in each group (n>30 fields per group). n.s. indicates no significant difference; and T, total. **P<0.01 vs CTMP‐Flox DMSO+AB; ## P<0.01 vs CTMP‐CKO DMSO+AB) by Bonferroni post hoc analysis (A [phosphorylated {P}‐AKT and P‐ribosomal protein S6 kinase β‐1 {P70S6K}] and D) or Tamhane's T2 analysis (A [P‐mammalian target of rapamycin {mTOR} and P‐glycogen synthase kinase 3β {GSK3β}], B, C, and E).
Discussion
Although considerable progress has been made in the treatment of heart failure in recent decades, its prognosis remains poor. Pathological cardiac hypertrophy is a crucial independent risk factor for heart failure, but few methods have been able to reverse pathological cardiac hypertrophy to date. Thus, it is indispensable to explore possible targets for improving the outcome of pathological cardiac hypertrophy. The present study characterized CTMP as a negative regulator of pathological cardiac hypertrophy and fibrosis by using gain‐of‐function and loss‐of‐function approaches in vivo and in vitro. Mechanistically, we discovered that the beneficial effect of CTMP on cardiac hypertrophy was mainly dependent on the blockade of AKT signaling. Clinically, decreased expression was observed in the hearts of patients with dilated cardiomyopathy or hypertrophic cardiomyopathy. Taken together, these results demonstrate that CTMP may be a therapeutic target for pathological cardiac hypertrophy in basic and clinical studies.
In the present study, we found that CTMP exerts a protective effect on pathological cardiac hypertrophy by suppressing the activation and phosphorylation of AKT‐mTOR‐GSK3β‐P70S6K signaling. Previously, multiple studies strongly suggested that the AKT signaling pathway is associated with cardiac hypertrophy, which involves the transition from adaptive hypertrophy to maladaptive hypertrophy, with significant changes in structure and function.23, 24, 25 In addition, recent studies concluded that transient activation of AKT contributes to physiological cardiac hypertrophy, whereas persistent activation causes pathological cardiac hypertrophy.26 Therefore, AKT is considered an essential regulatory switch that triggers the development of cardiac hypertrophy in a time‐dependent manner. The underlying mechanisms of the effects of AKT on cardiac hypertrophy may involve angiogenesis, cell death, and metabolism in the myocardium.26 Moreover, the phosphorylation of serine 473 in the carboxyl terminal regulatory domain of AKT is necessary for the full activation of AKT.25 When activated, AKT phosphorylates its downstream molecules mTOR and GSK3β via the phosphorylation of tuberous sclerosis complex and the inactivation of Ras homolog enriched in brain; mTOR phosphorylates P70S6K.27, 28 Furthermore, the expression of mTOR is increased in the cardiac tissue of patients with heart failure compared with healthy controls.29 The inhibition of mTOR with rapamycin could ameliorate pathological cardiac hypertrophy in humans and in animal models induced by pressure overload.30, 31 In addition, a previous study suggested that mTOR might restrain fibrosis via the inhibition of CTGF.32 Thus, the AKT signaling cascade is of vital importance in pathological cardiac hypertrophy. CTMP, as a pivotal target of AKT, also plays an important role in pathological cardiac hypertrophy. Although PTEN33 and CTMP are negative regulators of AKT, their roles are unexpectedly different in pathological cardiac hypertrophy induced by AB, which is associated with basal myocardial contractility33, 34 and molecular mechanisms in pathological cardiac hypertrophy. Intriguingly, PTEN33 and CTMP exert the same effect in pathological cardiac hypertrophy induced by AngII. Except for CTMP, many molecules have been demonstrated to play crucial roles on cardiac remodeling induced by pressure overload by regulating AKT signaling.4, 35, 36, 37, 38, 39
AKT represents a vital regulator of a wide variety of biological functions, including proliferation, survival, angiogenesis, and migration.40 AKT signaling plays important roles in various physiological and pathological processes.40 Supporting our findings, CTMP was originally identified via a yeast 2‐hybrid assay as an endogenous inhibitor of AKT that binds to the carboxyl terminal regulatory domain of AKT, reduces AKT activities by inhibiting phosphorylation on serine 473 and threonine 308, and reverts the phenotype of viral homolog‐AKT (v‐AKT) transformed cells.10 Thus, CTMP could negatively regulate AKT signaling. Although dysregulation of AKT signaling is implicated in a broad range of diseases, including cancer, brain disorders, and diabetes mellitus, the CTMP‐AKT axis has been reported to possess different functions in certain contexts because of different pathological processes. Inhibiting AKT signaling with CTMP may have an ameliorative effect in the treatment of liver cancer,12 lung cancer,13, 41 and pancreatic adenocarcinoma.8 In contrast, CTMP had the opposite effect on neuroprotection after brain injury9, 14, 17, 42 and insulin resistance,15 via restraining AKT signaling. Notably, a few reports have suggested that CTMP directly binds to AKT through the N‐terminal domain 1 to 64 amino acids at the plasma membrane,16 enhances AKT phosphorylation,43 and functions as an oncogenic molecule in head and neck squamous cell carcinoma44 and breast cancer.16, 45 These findings challenge the association of CTMP with AKT's phosphorylation condition and functional effects. Clinically, CTMP contributes to poor outcome and prognosis in head and neck squamous cell carcinoma44 and breast cancer,45 with low disease‐free survival and overall survival. In the present study, we clearly showed that CTMP predominantly extinguishes AKT phosphorylation and activity in pathological cardiac hypertrophy in vivo and in vitro. The reason CTMP has opposite effects on the function of AKT under different stresses remains unclear. Hence, this question places great importance on targeting AKT with CTMP in the treatment of major clinical diseases.
Several limitations of this study need to be mentioned. First, this study does not elucidate the mechanism of the reduction of CTMP in transcriptional levels, which requires further research. Second, this study demonstrated CTMP plays a key role in the early stage of cardiac dysfunction, but whether CTMP still has protective effects on intermediate and advanced heart failure may be worth further study. Third, because of differences in species, the ameliorative effect of CTMP in pathological cardiac hypertrophy needs further validation in nonhuman primates and clinic.
In summary, this study first identified CTMP as a novel negative regulator of pathological cardiac hypertrophy and fibrosis via its inhibition of the AKT signaling pathway. These findings suggest that CTMP may be a new therapeutic target for pathological cardiac hypertrophy.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81270184, 81670044, and 81770053), the National Science and Technology Support Project (2015BAI08B01), the National Key Research and Development Program (2016YFF0101504), and the Science and Technology Support Program of Hubei Province (2017BEC015).
Disclosures
None.
Supporting information
Table S1. Detailed Information of Human Heart Samples
Table S2. PCR Primers CTMP‐P1 to CTMP‐P6 Used for Identification
Table S3. Primers for Real‐Time Quantitative PCR
Figure S1. Construction and phenotypes of cardiac‐specific CTMP knockout mice. A, Schematic workflow showing the establishment of the cardiac‐specific CTMP knockout (CTMP‐CKO) mouse strain. B, In vitro Cre/loxP‐mediated recombination of CTMP conditional allele. The genomic DNA of founder #15 and #26 was incubated with Cre recombinase and used as PCR template. Upon Cre‐dependent excision, primers CTMP‐P1 and CTMP‐P2 flanking the floxed allele produce shorter products. CTMP‐P3 and CTMP‐P4 detect the circular molecule, which only form upon Cre‐loxP recombination. The position of each primer is shown in (A). C, Representative genotyping PCR amplification of wild type (CTMP +/+), CTMP loxp/loxp, and CTMP loxp/+. Allele with loxP 393 bp, wild‐type allele 320 bp. D, Cardiac‐specific CTMP deletion was confirmed by Western blot analysis.
Figure S2. Real‐time polymerase chain reaction (PCR) analyses of CTMP in the left ventricular tissues of mice subjected to sham operation, 4 weeks after aortic banding (AB) and 8 weeks after AB. **P<0.01 vs sham, # P<0.05 vs AB 4 weeks; by Bonferroni post hoc analysis.
Figure S3. Generation of cardiac‐specific CTMP transgenic mice. A, Schematic diagram for the construction of cardiac‐specific CTMP transgenic (TG) mouse lines. B, Western blot analysis for cardiac‐specific CTMP expression in TG mice and their non‐transgenic (NTG) controls.
Figure S4. Mitogen‐activated protein kinase (MAPK) signaling is not involved in CTMP‐regulated pathological cardiac hypertrophy. A and B, Western blot analysis and quantitative results of the total and phosphorylated levels of MAPK/ERK kinase 1/2 (MEK1/2), extracellular‐signal‐regulated kinase 1/2 (ERK1/2), c‐Jun N‐terminal kinase 1/2 (JNK1/2) and P38 of MAPK signaling in left ventricular tissues from CTMP‐Flox and CTMP‐CKO mice (A) and transgenic (TG) and non‐transgenic (NTG) mice (B) 4 weeks after sham or aortic banding (AB) surgery. **P<0.01 vs CTMP‐Flox sham or NTG sham; n.s. means no significant difference vs CTMP‐Flox AB or NTG AB; by Tamhane's T2 analysis (A [P‐MEK, P‐ERK and P‐JNK] and B [P‐MEK and P‐P38]) or Bonferroni post hoc analysis (A [P‐P38] and B [P‐ERK and P‐JNK]).
Acknowledgments
We thank Song Tian, Li Zhang, Hongjie Shi, Yan Zhang, and Ling Yang for providing technical support.
(J Am Heart Assoc. 2018;7:e008654 DOI: 10.1161/JAHA.118.008654.)
Contributor Information
Hao Xia, Email: xiahao1966@163.com.
Zhi‐Gang She, Email: zgshe@whu.edu.cn.
References
- 1. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. [DOI] [PubMed] [Google Scholar]
- 2. Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1‐dependent signalling. Nat Commun. 2016;7:11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bisping E, Wakula P, Poteser M, Heinzel FR. Targeting cardiac hypertrophy: toward a causal heart failure therapy. J Cardiovasc Pharmacol. 2014;64:293–305. [DOI] [PubMed] [Google Scholar]
- 4. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, Zhao Y, Gao L, Yang Q, Zhu XH, Wei X, Pu J, Li H. Suppressor of IKKɛ is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–1438. [DOI] [PubMed] [Google Scholar]
- 6. Zhang XJ, Zhang P, Li H. Interferon regulatory factor signalings in cardiometabolic diseases. Hypertension. 2015;66:222–247. [DOI] [PubMed] [Google Scholar]
- 7. Ghigo A, Laffargue M, Li M, Hirsch E. PI3K and calcium signaling in cardiovascular disease. Circ Res. 2017;121:282–292. [DOI] [PubMed] [Google Scholar]
- 8. Simon PO Jr, McDunn JE, Kashiwagi H, Chang K, Goedegebuure PS, Hotchkiss RS, Hawkins WG. Targeting AKT with the proapoptotic peptide, TAT‐CTMP: a novel strategy for the treatment of human pancreatic adenocarcinoma. Int J Cancer. 2009;125:942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao S, Fu J, Liu F, Rastogi R, Zhang J, Zhao Y. Small interfering RNA directed against CTMP reduces acute traumatic brain injury in a mouse model by activating Akt. Neurol Res. 2014;36:483–490. [DOI] [PubMed] [Google Scholar]
- 10. Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, Hemmings BA. Carboxyl‐terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v‐Akt at the plasma membrane. Science. 2001;294:374–380. [DOI] [PubMed] [Google Scholar]
- 11. Parcellier A, Tintignac LA, Zhuravleva E, Cron P, Schenk S, Bozulic L, Hemmings BA. Carboxy‐terminal modulator protein (CTMP) is a mitochondrial protein that sensitizes cells to apoptosis. Cell Signal. 2009;21:639–650. [DOI] [PubMed] [Google Scholar]
- 12. Shin JY, Chung YS, Kang B, Jiang HL, Yu DY, Han K, Chae C, Moon JH, Jang G, Cho MH. Co‐delivery of LETM1 and CTMP synergistically inhibits tumor growth in H‐ras12V liver cancer model mice. Cancer Gene Ther. 2013;20:186–194. [DOI] [PubMed] [Google Scholar]
- 13. Hwang SK, Lim HT, Minai‐Tehrani A, Lee ES, Park J, Park SB, Beck GR Jr, Cho MH. Repeated aerosol delivery of carboxyl‐terminal modulator protein suppresses tumor in the lungs of K‐rasLA1 mice. Am J Respir Crit Care Med. 2009;179:1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyawaki T, Ofengeim D, Noh KM, Latuszek‐Barrantes A, Hemmings BA, Follenzi A, Zukin RS. The endogenous inhibitor of Akt, CTMP, is critical to ischemia‐induced neuronal death. Nat Neurosci. 2009;12:618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park J, Li Y, Kim SH, Yang KJ, Kong G, Shrestha R, Tran Q, Park KA, Jeon J, Hur GM, Lee CH, Kim DH, Park J. New players in high fat diet‐induced obesity: LETM1 and CTMP. Metabolism. 2014;63:318–327. [DOI] [PubMed] [Google Scholar]
- 16. Liu YP, Liao WC, Ger LP, Chen JC, Hsu TI, Lee YC, Chang HT, Chen YC, Jan YH, Lee KH, Zeng YH, Hsiao M, Lu PJ. Carboxyl‐terminal modulator protein positively regulates Akt phosphorylation and acts as an oncogenic driver in breast cancer. Cancer Res. 2013;73:6194–6205. [DOI] [PubMed] [Google Scholar]
- 17. Chen Y, Cai M, Deng J, Tian L, Wang S, Tong L, Dong H, Xiong L. Elevated expression of carboxy‐terminal modulator protein (CTMP) aggravates brain ischemic injury in diabetic db/db mice. Neurochem Res. 2016;41:2179–2189. [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y, Li H. Reprogramming interferon regulatory factor signaling in cardiometabolic diseases. Physiology (Bethesda). 2017;32:210–223. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y, Zhang XJ, Wang PX, Zhang P, Li H. Reprogramming innate immune signaling in cardiometabolic disease. Hypertension. 2017;69:747–760. [DOI] [PubMed] [Google Scholar]
- 20. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng KQ, Zhao GN, Wang Z, Fang J, Jiang Z, Gong J, Yan FJ, Zhu XY, Zhang P, She ZG, Li H. Targeting TMBIM1 alleviates pathological cardiac hypertrophy. Circulation. 2018;137:1486–1504. [DOI] [PubMed] [Google Scholar]
- 22. Fang J, Li T, Zhu X, Deng KQ, Ji YX, Fang C, Zhang XJ, Guo JH, Zhang P, Li H, Wei X. Control of pathological cardiac hypertrophy by transcriptional corepressor IRF2BP2 (interferon regulatory factor‐2 binding protein 2). Hypertension. 2017;70:515–523. [DOI] [PubMed] [Google Scholar]
- 23. Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. [DOI] [PubMed] [Google Scholar]
- 24. Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aoyagi T, Matsui T. Phosphoinositide‐3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. 2011;17:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chaanine AH, Hajjar RJ. Akt signalling in the failing heart. Eur J Heart Fail. 2011;13:825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E‐BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. [DOI] [PubMed] [Google Scholar]
- 28. Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz‐Vega S, Yonezawa K. Insulin and amino‐acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–6372. [DOI] [PubMed] [Google Scholar]
- 29. Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer‐Crosbie M, Hara K, Rosenzweig A, Matsui T. Mtor attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–C1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. [DOI] [PubMed] [Google Scholar]
- 31. Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, Abel ED, Symons JD, Jalili T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finckenberg P, Inkinen K, Ahonen J, Merasto S, Louhelainen M, Vapaatalo H, Muller D, Ganten D, Luft F, Mervaala E. Angiotensin II induces connective tissue growth factor gene expression via calcineurin‐dependent pathways. Am J Pathol. 2003;163:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oudit GY, Kassiri Z, Zhou J, Liu QC, Liu PP, Backx PH, Dawood F, Crackower MA, Scholey JW, Penninger JM. Loss of PTEN attenuates the development of pathological hypertrophy and heart failure in response to biomechanical stress. Cardiovasc Res. 2008;78:505–514. [DOI] [PubMed] [Google Scholar]
- 34. Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie‐Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira‐dos‐Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K‐PTEN signaling pathways. Cell. 2002;110:737–749. [DOI] [PubMed] [Google Scholar]
- 35. Deng KQ, Li J, She ZG, Gong J, Cheng WL, Gong FH, Zhu XY, Zhang Y, Wang Z, Li H. Restoration of circulating MFGE8 (milk fat globule‐EGF factor 8) attenuates cardiac hypertrophy through inhibition of Akt pathway. Hypertension. 2017;70:770–779. [DOI] [PubMed] [Google Scholar]
- 36. Zhang ZZ, Wang W, Jin HY, Chen X, Cheng YW, Xu YL, Song B, Penninger JM, Oudit GY, Zhong JC. Apelin is a negative regulator of angiotensin II‐mediated adverse myocardial remodeling and dysfunction. Hypertension. 2017;70:1165–1175. [DOI] [PubMed] [Google Scholar]
- 37. Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation. 2015;131:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin SIRT6 blocks IGF‐Akt signaling and development of cardiac hypertrophy by targeting c‐Jun. Nat Med. 2012;18:1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46. [DOI] [PubMed] [Google Scholar]
- 40. Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hwang SK, Minai‐Tehrani A, Yu KN, Chang SH, Kim JE, Lee KH, Park J, Beck GR Jr, Cho MH. Carboxyl‐terminal modulator protein induces apoptosis by regulating mitochondrial function in lung cancer cells. Int J Oncol. 2012;40:1515–1524. [DOI] [PubMed] [Google Scholar]
- 42. Huang CY, Chen JJ, Wu JS, Tsai HD, Lin H, Yan YT, Hsu CY, Ho YS, Lin TN. Novel link of anti‐apoptotic ATF3 with pro‐apoptotic CTMP in the ischemic brain. Mol Neurobiol. 2015;51:543–557. [DOI] [PubMed] [Google Scholar]
- 43. Ono H, Sakoda H, Fujishiro M, Anai M, Kushiyama A, Fukushima Y, Katagiri H , Ogihara T, Oka Y, Kamata H, Horike N, Uchijima Y, Kurihara H, Asano T. Carboxy‐terminal modulator protein induces Akt phosphorylation and activation, thereby enhancing antiapoptotic, glycogen synthetic, and glucose uptake pathways. Am J Physiol Cell Physiol. 2007;293:C1576–C1585. [DOI] [PubMed] [Google Scholar]
- 44. Chang JW, Jung SN, Kim JH, Shim GA, Park HS, Liu L, Kim JM, Park J, Koo BS. Carboxyl‐terminal modulator protein positively acts as an oncogenic driver in head and neck squamous cell carcinoma via regulating Akt phosphorylation. Sci Rep. 2016;6:28503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen YC, Li HY, Liang JL, Ger LP, Chang HT, Hsiao M, Calkins MJ, Cheng HC, Chuang JH, Lu PJ. CTMP, a predictive biomarker for trastuzumab resistance in HER2‐enriched breast cancer patient. Oncotarget. 2017;8:29699–29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed Information of Human Heart Samples
Table S2. PCR Primers CTMP‐P1 to CTMP‐P6 Used for Identification
Table S3. Primers for Real‐Time Quantitative PCR
Figure S1. Construction and phenotypes of cardiac‐specific CTMP knockout mice. A, Schematic workflow showing the establishment of the cardiac‐specific CTMP knockout (CTMP‐CKO) mouse strain. B, In vitro Cre/loxP‐mediated recombination of CTMP conditional allele. The genomic DNA of founder #15 and #26 was incubated with Cre recombinase and used as PCR template. Upon Cre‐dependent excision, primers CTMP‐P1 and CTMP‐P2 flanking the floxed allele produce shorter products. CTMP‐P3 and CTMP‐P4 detect the circular molecule, which only form upon Cre‐loxP recombination. The position of each primer is shown in (A). C, Representative genotyping PCR amplification of wild type (CTMP +/+), CTMP loxp/loxp, and CTMP loxp/+. Allele with loxP 393 bp, wild‐type allele 320 bp. D, Cardiac‐specific CTMP deletion was confirmed by Western blot analysis.
Figure S2. Real‐time polymerase chain reaction (PCR) analyses of CTMP in the left ventricular tissues of mice subjected to sham operation, 4 weeks after aortic banding (AB) and 8 weeks after AB. **P<0.01 vs sham, # P<0.05 vs AB 4 weeks; by Bonferroni post hoc analysis.
Figure S3. Generation of cardiac‐specific CTMP transgenic mice. A, Schematic diagram for the construction of cardiac‐specific CTMP transgenic (TG) mouse lines. B, Western blot analysis for cardiac‐specific CTMP expression in TG mice and their non‐transgenic (NTG) controls.
Figure S4. Mitogen‐activated protein kinase (MAPK) signaling is not involved in CTMP‐regulated pathological cardiac hypertrophy. A and B, Western blot analysis and quantitative results of the total and phosphorylated levels of MAPK/ERK kinase 1/2 (MEK1/2), extracellular‐signal‐regulated kinase 1/2 (ERK1/2), c‐Jun N‐terminal kinase 1/2 (JNK1/2) and P38 of MAPK signaling in left ventricular tissues from CTMP‐Flox and CTMP‐CKO mice (A) and transgenic (TG) and non‐transgenic (NTG) mice (B) 4 weeks after sham or aortic banding (AB) surgery. **P<0.01 vs CTMP‐Flox sham or NTG sham; n.s. means no significant difference vs CTMP‐Flox AB or NTG AB; by Tamhane's T2 analysis (A [P‐MEK, P‐ERK and P‐JNK] and B [P‐MEK and P‐P38]) or Bonferroni post hoc analysis (A [P‐P38] and B [P‐ERK and P‐JNK]).