

Abstract
Background
Mineralocorticoid receptor (MR) has pathological roles in various cell types, including renal tubule cells, myocytes, and smooth muscle cells; however, the role of MR in intestinal epithelial cells (IECs) has not been sufficiently evaluated. The intestine is the sensing organ of ingested sodium; accordingly, intestinal MR is expected to have essential roles in blood pressure (BP) regulation.
Methods and Results
We generated IEC‐specific MR knockout (IEC‐MR‐KO) mice. With a standard diet, fecal sodium excretion was 1.5‐fold higher in IEC‐MR‐KO mice, with markedly decreased colonic expression of β‐ and γ‐epithelial sodium channel, than in control mice. Urinary sodium excretion in IEC‐MR‐KO mice decreased by 30%, maintaining sodium balance; however, a low‐salt diet caused significant reductions in body weight and BP in IEC‐MR‐KO mice, and plasma aldosterone exhibited a compensatory increase. With a high‐salt diet, intestinal sodium absorption markedly increased to similar levels in both genotypes, without an elevation in BP. Deoxycorticosterone/salt treatment elevated BP and increased intestinal sodium absorption in both genotypes. Notably, the increase in BP was significantly smaller in IEC‐MR‐KO mice than in control mice. The addition of the MR antagonist spironolactone to deoxycorticosterone/salt treatment eliminated the differences in BP and intestinal sodium absorption between genotypes.
Conclusions
Intestinal MR regulates intestinal sodium absorption in the colon and contributes to BP regulation. These regulatory effects are associated with variation in epithelial sodium channel expression. These findings suggest that intestinal MR is a new target for studying the molecular mechanism of hypertension and cardiovascular diseases.
Keywords: aldosterone, hypertension, intestine, mineralocorticoids, nuclear receptor, sodium channels
Subject Categories: High Blood Pressure
Clinical Perspective
What Is New?
We developed intestinal epithelial cell–specific mineralocorticoid receptor (MR) knockout mice to evaluate the physiological and pathological roles of intestinal MR.
We elucidated the significance of intestinal MR in sodium balance and blood pressure regulation by evaluating the response of knockout and control mice to a low‐ or high‐salt diet, with or without MR agonist/antagonist treatments.
What Are the Clinical Implications?
This study revealed that not only renal but also intestinal MR contributes to excessive sodium retention in mineralocorticoid hypertension.
Our findings suggest that intestinal MR has potential as a new target for studying MR‐associated cardiovascular diseases and will help further elucidate the underlying molecular mechanisms of these diseases.
Hypertension is a major cause of cardiovascular diseases and is estimated to contribute to 9.4 million deaths yearly worldwide.1 The global prevalence of uncontrolled hypertension in adults remains as high as ≈22%. Blood pressure (BP) is defined as cardiac output×systemic vascular resistance, and the sympathetic nervous system, the renin–angiotensin–aldosterone system, and plasma volume are primary determinants. Plasma volume is influenced by sodium intake, and sodium restriction effectively lowers BP.2 Multiple factors regulate sodium balance; in this study, we focused on mineralocorticoid receptor (MR). In recent studies, we identified novel coregulators of MR3, 4 and proposed a new pathological condition associated with aberrant MR activation, designated MR‐associated hypertension.5
MR is mainly expressed in epithelial cells of the renal tubules, intestine, and skin. In particular, renal MR regulates sodium balance by sodium reabsorption in the tubules. Renal tubule–specific MR knockout mice show significant sodium and body weight losses with a low‐salt diet.6, 7 In contrast, overactivated MR in the kidneys induces salt‐sensitive hypertension, characterized by an increase in BP in response to increased sodium intake.8 The elevated BP associated with overactivated MR is known as mineralocorticoid hypertension.9 These studies have shown that renal MR is essential to regulate the plasma sodium volume and BP and suggest that the antihypertensive effect of MR antagonists is mediated by renal MR inhibition.
Recent large clinical trials10, 11, 12 have demonstrated that the addition of an MR antagonist to conventional antihypertensive therapies reduces cardiovascular events and improves patient prognoses. These results have prompted further studies of the role of MR in cardiovascular tissues, including cardiomyocytes, smooth muscle cells, endothelia, and macrophages, to understand the cell‐specific contribution of MR to BP and cardiovascular diseases, using tissue‐specific MR knockout mouse models. The deletion of cardiomyocyte MR prevents cardiac remodeling after myocardial infarction13 and cardiac fibrosis with deoxycorticosterone acetate (DOCA)/salt treatment.14 Endothelial MR is involved in inflammatory and profibrotic responses15 and the regulation of vasomotor function.16 Although these studies showed that endothelial MR does not contribute to BP regulation, overexpression of MR in the endothelium was found to affect BP.17 MR in myeloid cells and macrophages has roles in cardiac fibrosis and DOCA/salt‐induced BP elevation.18, 19 Vascular smooth muscle cell MR functions in the stiffening of carotid arteries in hypertension induced by an aldosterone/salt challenge20 and in the age‐related increase in BP in mice.21 Although these previous studies have clarified MR functions in the kidneys and cardiovascular tissues, the roles of MR in other epithelial tissues, such as the intestinal epithelium, have not been elucidated. MR is reportedly expressed throughout the intestine,22 and aldosterone increases the expression of the epithelial sodium channel (ENaC) in rats23; however, there are no studies focusing on the functions of MR in intestinal epithelial cells (IECs) using MR deletion mutants, the implementation of which is required.24 Furthermore, it is not clear how intestinal MR contributes to sodium dynamics and BP regulation. Most studies of MR using hypertensive models apply aldosterone/salt treatment to induce hypertension, and either aldosterone or high salt loading alone is considered insufficient for significant BP elevation.8, 25, 26 These studies suggest the significance of excessive sodium retention in hypertension via MR activation. The kidneys regulate sodium excretion, whereas the intestine regulates its absorption; accordingly, intestinal MR is expected to be involved in sodium dynamics and, consequently, in BP regulation.
In this study, to evaluate the contribution of intestinal MR to sodium dynamics and BP regulation and to determine its significance in the development of hypertension, we generated IEC‐specific MR knockout (IEC‐MR‐KO) mice and investigated their response to a low‐salt diet, which stimulates aldosterone secretion; a high‐salt diet, which suppresses aldosterone secretion; and MR agonist (DOCA) treatment. MR antagonist (spironolactone) treatment of DOCA‐treated mice was used to assess the reversibility of DOCA effects and to confirm the relationship between intestinal sodium absorption and BP regulation.
Materials and Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. The sources of study materials can be specified in the following descriptions.
Generation of IEC‐MR‐KO Mice
IEC‐MR‐KO mice (MRflox/floxVillin‐Cre) were generated by crossing MRflox/flox mice27 with transgenic mice expressing Cre recombinase under the control of the Villin gene promoter (Villin Cre Tg: B6.SJL‐Tg[Vil‐Cre]997Gum/J; Jackson Laboratory, Bar Harbor, ME). The presence of the MRflox/flox and Villin Cre transgenes was determined by a polymerase chain reaction (PCR) analysis of genomic DNA from tail tips using primers listed in Table S1. MRflox/flox littermates were used as controls. Mice were group‐housed (4–5 mice/cage) in an air‐conditioned room under a 12‐hour dark/light cycle and allowed free access to water and a standard diet (including 0.5% NaCl). All animal procedures were approved by the Keio University institutional animal care and use committee.
Animal Treatments
Male mice were grouped (n=6–8 per group) by standard diet, low‐salt, high‐salt, DOCA/salt, and DOCA/salt plus spironolactone (DOC‐SPL) treatment. For low‐ or high‐salt treatment, the standard diet (0.5% NaCl) was replaced with a low‐salt diet (0.025% NaCl) or a high‐salt diet (8% NaCl) at 5 weeks of age. At 10 and 13 weeks of age, the mice were placed in metabolic cages for 24 hours to collect urine and feces, and BP was measured. For the DOCA/salt treatment, 10‐week‐old mice on a standard diet were anesthetized and implanted subcutaneously with 21‐day–release 50‐mg DOCA pellets. These mice were administered a standard diet and 1% (wt/vol) NaCl solution to drink for 3 weeks. For DOC‐SPL treatment, DOCA/salt treatment was started at 10 weeks, as described, and spironolactone (100 mg/kg per day) dissolved in 0.5% (wt/vol) methylcellulose was orally administered daily from 11 to 13 weeks. After the start of each treatment, mice were placed in metabolic cages for 24 hours to collect urine and feces weekly. BP was measured once a week. At 13 weeks of age, blood samples and tissues were collected.
Metabolic Cage Studies: Urine and Fecal Analyses
Mice were fed in their home cage, and body weight was measured weekly. Ten‐week‐old control and IEC‐MR‐KO mice were individually placed in mouse metabolic cages for 24 hours to determine food consumption and to collect urine and feces. Samples of urine and feces were also obtained after the start of each treatment. Fecal samples were prepared for measurements, as reported previously.28, 29 In brief, fecal samples were weighed after drying at 80°C for 3 hours, resuspended in 0.75 mol/L nitric acid and left overnight at 4°C, and centrifuged. The supernatants were used for measurements. Urinary and fecal concentrations of sodium and potassium were measured using ion‐selective electrodes. Urinary creatinine and albumin concentrations were determined by an enzyme method at SRL Inc and by ELISA (AKRAL‐121; Shibayagi), respectively.
Systolic BP Measurement
At 10 and 13 weeks of age, systolic BP was measured noninvasively in the conscious state by tail‐cuff plethysmography (MK2000; Muromachi Kikai), according to the manufacturer's manual. Mice were trained for 2 days before the first measurement and gently handled to avoid stress. The mean of 10 measurements for each mouse was determined.
Tissue Collection
At the end of the experiment (at 13 weeks of age), blood samples were collected following euthanasia, and the intestines were dissected (duodenum, jejunum, ileum, proximal colon, and distal colon). In addition, the kidneys, heart, aorta, and liver were collected. Each organ was immediately divided into 2 parts. One part was immersion‐fixed in 4% paraformaldehyde for paraffin embedding, and the other was frozen in liquid nitrogen for DNA, RNA, and protein extraction. Paraffin‐embedded sections stained with hematoxylin and eosin were prepared for histologic examination.
Plasma Analyses
Plasma creatinine, sodium, and potassium concentrations were measured at SRL Inc. Plasma aldosterone and corticosterone levels were determined by radioimmunoassay and enzyme immunoassay, respectively.
RNA Extraction and Real‐Time Quantitative PCR
Total RNA and DNA were prepared from tissue samples using ISOGEN (Nippon Gene), according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using PrimeScript RT Master Mix (RR036A; TaKaRa), and quantitative PCR was carried out using SYBR Green Master Mix (Applied Biosystems) on an Mx3000 PCR cycler (Agilent Technologies). The target genes were as follows: MR, GR (glucocorticoid receptor); SCNN1A (sodium channel epithelial 1 α subunit; α‐ENaC), SCNN1B (β subunit; β‐ENaC), and SCNN1G (γ subunit; γ‐ENaC); SGK1 (serum/glucocorticoid regulated kinase 1); NHE3 (sodium–hydrogen exchanger 3); NKCC2 (sodium–potassium–chloride cotransporter); NCC (sodium–chloride cotransporter); and SGLT1 (sodium–glucose cotransporter 1) and SGLT2. Target gene expression was normalized to 18S rRNA expression. Primer sequences are given in Table S1. PCR conditions are available upon request.
Immunoblotting
Tissue samples from the intestine were lysed in radioimmunoprecipitation assay buffer with protease inhibitor cocktail. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Proteins were denatured with Laemmli sample buffer, separated on a 7.5% TGX‐gel (Bio‐Rad), and transferred to a nitrocellulose membrane. The membranes were incubated with primary antibodies (α‐ENaC: 1:1000 dilution; β‐ENaC: 1:1000 dilution; γ‐ENaC: 1:1000 dilution; SGK1: 1:500 dilution; β‐actin: 1:400 dilution), followed by incubation with a donkey antirabbit horseradish peroxidase–conjugated secondary antibody (1:2000 dilution). The blots were visualized by enhanced chemiluminescence using ECL Prime (Amersham). Band intensities were measured using ImageJ (National Institutes of Health). Protein amounts were normalized against levels of β‐actin.
Statistical Analysis
Data are presented as the mean±SEM. All data were tested for normality using the Shapiro–Wilk test. Differences in baseline characteristics and mRNA expression between genotypes were analyzed using unpaired Student t tests or Mann–Whitney U tests, as appropriate. Plasma hormone and mRNA and protein expression levels were compared between treatments and genotypes using 2‐way ANOVA followed by Bonferroni multiple comparison tests. Changes in BP and sodium excretion over time were assessed by 2‐way ANOVA with repeated measures. Statistical analysis was performed using SPSS version 24.0 (IBM Corp). P<0.05 was considered significant.
Results
IEC‐MR‐KO Mice Are Viable and Exhibit Normal Intestinal Histology
IEC‐MR‐KO (MRflox/floxVillin‐Cre) mice were born in the expected Mendelian frequencies and showed no postnatal mortality. IEC‐MR‐KO and control (MRflox/flox) mice were indistinguishable in appearance, growth, and body weight (Table 1). Macroscopically, the intestinal morphology from the duodenum to the distal colon did not differ between genotypes. A microscopic evaluation of intestinal epithelia from the duodenum to the colon revealed no obvious differences between genotypes, and colonic mucin‐secreting goblet cells appeared normal in IEC‐MR‐KO mice (Figure S1). DNA isolated from the duodenum, jejunum, ileum, proximal colon, and distal colon of IEC‐MR‐KO mice exhibited MR recombination (Figure S2A), which was not observed in control mice. DNA from the kidneys did not show MR recombination in either genotype. In IEC‐MR‐KO mice, the MR mRNA level in each portion of the intestine was ≈95% lower than that in control mice (Figure S2B). The mRNA levels in the kidneys, heart, aorta, and liver were similar in the 2 genotypes (Figure S2C). In addition, GR mRNA expression did not differ between genotypes (Figure S2D). These data confirmed that MR was effectively and specifically knocked out in the IECs of IEC‐MR‐KO mice.
Table 1.
Baseline Characteristics of Control and IEC‐MR‐KO Mice at 13 Weeks of Age
| Control (n=7) | IEC‐MR‐KO (n=7) | P Value | |
|---|---|---|---|
| Body weight, g | 28.9±1.0 | 28.7±1.1 | 0.858 |
| Food intake, g/24 h | 3.93±0.14 | 3.98±0.15 | 0.825 |
| Feces output, g/24 h | 1.31±0.05 | 1.25±0.08 | 0.553 |
| Urine output, μL/24 h | 1490±143 | 1360±120 | 0.649 |
| Serum Cr, mg/dL | 0.10±0.00 | 0.10±0.01 | 0.161 |
| Serum sodium, mEq/L | 145.3±2.2 | 146.0±1.3 | 0.803 |
| Fecal sodium, μmol/24 h | 127.5±7.7 | 183.6±9.2 | 0.001 |
| Fecal potassium, μmol/24 h | 110.5±6.6 | 86.3±4.2 | 0.005 |
| Urinary sodium concentration, mmol/L | 144.3±11.1 | 104.2±8.9 | 0.017 |
| Urinary sodium, μmol/24 h | 197.5±14.9 | 138.2±9.5 | 0.006 |
| Urinary potassium, μmol/24 h | 387.0±36.7 | 442.7±32.4 | 0.253 |
| Urinary albumin, μg/mg Cr | 45.0±3.9 | 43.0±3.0 | 0.798 |
| Blood pressure, mm Hg | 105.4±1.1 | 104.4±1.1 | 0.560 |
Data are presented as the mean±SEM. Cr indicates creatinine; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout.
IEC‐MR‐KO Increases Fecal and Decreases Urinary Sodium Excretion With a Standard Diet
IEC‐MR‐KO mice on a standard diet showed higher sodium and lower potassium excretion in feces than control mice, whereas no differences were observed in 24‐hour food intake or feces and urine volumes between genotypes (Table 1). Urinary sodium excretion was significantly lower, whereas potassium excretion tended to be higher in IEC‐MR‐KO than control mice. Plasma aldosterone was higher, albeit not significantly, in IEC‐MR‐KO than control mice (Table 2). Consequently, sodium balance was maintained in IEC‐MR‐KO mice, and systolic BP was similar in both genotypes. To examine the cause of increased fecal sodium excretion in IEC‐MR‐KO mice, we measured the mRNA expression of ENaC, an MR target, in each portion of the intestine. Although the transcript level of α‐ENaC was similar in both genotypes throughout the intestine, mRNA levels of β‐ and γ‐ENaC were markedly lower in IEC‐MR‐KO than in control mice in the proximal and distal colon (Figure 1A). Notably, these genes were hardly expressed in the small intestine, even in control mice. The mRNA expression of the other MR target, SGK1, was also decreased in the proximal and distal colon of IEC‐MR‐KO mice (Figure 1B). Furthermore, the transcript level of NHE3, the other intestinal sodium channel, was significantly lower in the colons of IEC‐MR‐KO mice than in those of control mice (Figure 1C). In the kidneys, the mRNA levels of these genes were similar in both genotypes (Figure 1D). In addition, we measured mRNA expression of other sodium channels, including NKCC2, NCCT, and SGLT, in the kidneys. The results confirmed similar expression levels for each of these in both genotypes (Figure S3).
Table 2.
Plasma Renin Activity and Aldosterone and Corticosterone Levels in Control and IEC‐MR‐KO Mice With Each Treatment
| Standard Diet | High‐Salt Diet | DOCA/Salt | ||||
|---|---|---|---|---|---|---|
| Control (n=7) | IEC‐MR‐KO (n=7) | Control (n=7) | IEC‐MR‐KO (n=6) | Control (n=8) | IEC‐MR‐KO (n=8) | |
| Plasma renin activity, ng/mL/h | 53.7±5.8 | 48.7±3.0 | 30.7±3.7* | 36.2±5.2* | 3.0±0.7*† | 2.8±1.1*† |
| Plasma aldosterone, pg/mL | 485.6±50.0 | 537.4±48.3 | 91.0±20.3* | 87.8±20.8* | 285.4±56.9‡, † | 360.3±58.2‡, † |
| Plasma corticosterone, ng/mL | 130.6±19.7 | 224.8±29.2 | 168.9±34.1 | 163.2±48.4 | 46.9±6.4*† | 47.3±4.8*† |
Data are presented as the mean±SEM. DOCA indicates deoxycorticosterone acetate; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout.
P<0.001 vs *standard diet, †high‐salt diet.
P<0.005 vs ‡standard diet.
Figure 1.
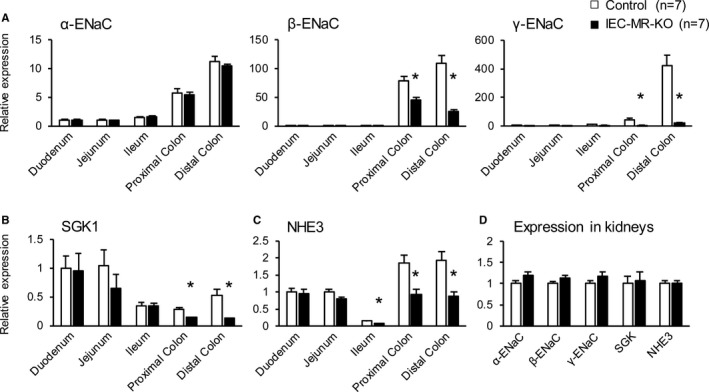
The mRNA expression levels of the ENaC family, SGK1, and NHE3 in the kidneys and various intestinal portions. The mRNA expression of α‐, β‐, and γ‐ENaC (A), SGK1 (B), and NHE3 (C) in various intestinal parts and in the kidneys (D), as measured by quantitative polymerase chain reaction. Gene expression in each intestinal portion is normalized to that of control mouse duodenum. The number of animals per group is indicated in parentheses. Data are presented as mean±SEM. *P<0.05 vs control. ENaC indicates epithelial sodium channel; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; NHE3, sodium–hydrogen exchanger 3; SGK1, serum/glucocorticoid regulated kinase 1.
Salt Restriction Reduces Body Weight and BP and Markedly Increases Plasma Aldosterone in IEC‐MR‐KO Mice
Unlike the standard diet, with the low‐salt diet, body weight and BP significantly decreased in IEC‐MR‐KO mice compared with control mice (Table 3). Plasma aldosterone in IEC‐MR‐KO mice significantly increased. Although fecal sodium in IEC‐MR‐KO mice was higher than that in control mice, similar to the observations with a standard diet, urinary output of IEC‐MR‐KO mice was also higher owing to increased water intake. Consequently, despite the lower sodium concentration in the urine of IEC‐MR‐KO mice, urinary sodium excretion was higher than that of control mice. Next, we evaluated the mRNA expression of the ENaC family and SGK1 in the distal colon and kidneys in this condition. Consistent with the higher aldosterone level with a low‐salt diet than with a standard diet, the expression levels of β‐ and γ‐ENaC and SGK1 in the distal colon of control mice significantly increased with a low‐salt diet (Figure 2A). Although these increases were also observed in IEC‐MR‐KO mice, the elevation was milder than in control mice. As for the kidneys, significantly increased aldosterone elevated the expression of α‐ENaC and SGK1 in IEC‐MR‐KO mice with a low‐salt diet (Figure 2B).
Table 3.
Effects of Low‐Salt Diet
| Control (n=7) | IEC‐MR‐KO (n=7) | P Value | |
|---|---|---|---|
| Body weight, g | 27.4±0.8 | 17.4±0.3 | 0.002 |
| Water intake, μL/24 h | 2023±144 | 7215±148 | <0.001 |
| Urine output, μL/24 h | 920±114 | 5391±191 | <0.001 |
| Urinary sodium concentration, mmol/L | 33.6±3.4 | 12.8±2.0 | 0.001 |
| Urinary sodium, μmol/24 h | 31.2±4.9 | 70.3±11.2 | 0.013 |
| Fecal sodium, μmol/24 h | 8.5±0.6 | 30.8±2.3 | <0.001 |
| Blood pressure, mm Hg | 98.4±3.5 | 82.6±2.4 | 0.014 |
| Plasma aldosterone, pg/mL | 982.0±157.7 | 17 587.5±3477.0 | <0.001 |
Data are presented as the mean±SEM. IEC‐MR‐KO indicates intestinal epithelial cell–specific mineralocorticoid receptor knockout.
Figure 2.
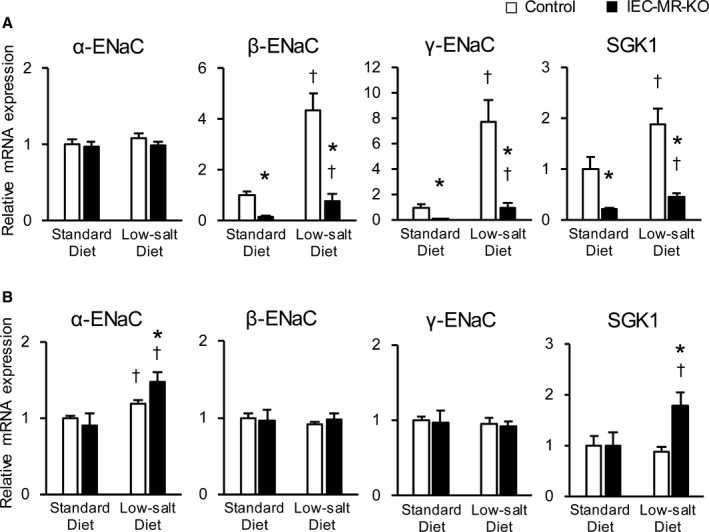
The mRNA expression levels of ENaC family members and SGK1 in the distal colon and kidneys with a low‐salt diet. The mRNA expression of ENaC and SGK1 in the distal colon (A) and in the kidneys (B) with a standard diet and a low‐salt diet, as measured by quantitative polymerase chain reaction. Gene expression is normalized to that of 18S rRNA and is expressed relative to that in control mice on a standard diet. Data are presented as mean±SEM. n=7 per genotype and group. P<0.05 vs *control, †standard diet. ENaC indicates epithelial sodium channel; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; SGK1, serum/glucocorticoid regulated kinase 1.
High‐Salt Diet Increases Fecal and Urinary Sodium Excretion to Similar Levels in Control and IEC‐MR‐KO Mice
With a standard diet, urinary and fecal sodium were consistently decreased and increased, respectively, in IEC‐MR‐KO mice aged 10 to 13 weeks (Figure 3A and 3D), resulting in the maintenance of BP, which was similar in both genotypes (Figure 3G). A high‐salt diet slightly increased fecal sodium and significantly (P<0.001) increased urinary sodium excretion compared with the standard diet, with no difference between genotypes (Figure 3B and 3E). Plasma aldosterone was lower with a high‐salt diet (Table 2). Consequently, systolic BP was nearly equal to that with the standard diet (Figure 3H).
Figure 3.
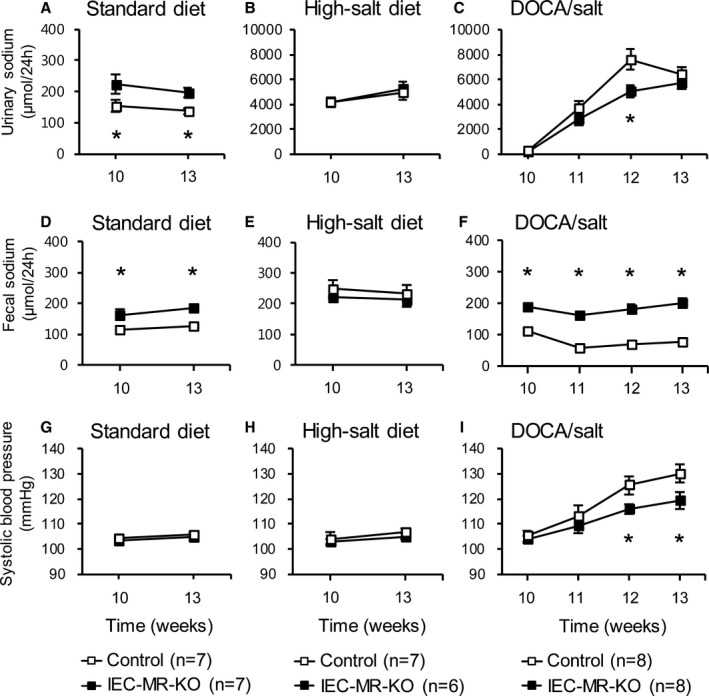
Changes in fecal and urinary sodium excretion and systolic blood pressure under each treatment. Urinary (A–C) and fecal (D–F) sodium excretion and systolic blood pressure (G–I) with a standard diet, a high‐salt diet, and DOCA/salt treatment. Data are presented as mean±SEM. *P<0.05 vs control. DOCA indicates deoxycorticosterone acetate; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout.
IEC‐MR‐KO Suppresses BP Elevation in Response to DOCA/Salt
DOCA/salt treatment from 10 to 13 weeks of age markedly increased urinary volume and sodium excretion compared with the standard diet (Figure 3C). Urinary sodium tended to be higher in control than IEC‐MR‐KO mice at all time points, with a significant difference at 2 weeks of treatment. In contrast to a high‐salt diet, DOCA/salt induced a significant difference in fecal sodium between genotypes at each time point (Figure 3F). Concomitant with the increase in urinary sodium, systolic BP increased in both genotypes. Notably, 2 and 3 weeks after DOCA/salt treatment, the increase in BP was significantly higher in control than in IEC‐MR‐KO mice (Figure 3I; control: 25.0±2.5 mm Hg, IEC‐MR‐KO: 15.3±2.5 mm Hg, P<0.05). Plasma renin activity was markedly suppressed by DOCA/salt in both genotypes and was significantly lower than that with a high‐salt diet (Table 2). Plasma aldosterone was also decreased but was higher than that with a high‐salt diet because DOCA showed cross‐reactivity in the aldosterone assay.
Spironolactone Suppresses the Differential Response of IEC‐MR‐KO Mice to DOCA/Salt
In the DOC‐SPL group, DOCA/salt treatment was started at 10 weeks of age, and spironolactone was administered daily between 11 and 13 weeks. At 11 weeks, markedly increased urinary sodium excretion and slightly elevated BP were observed in both genotypes, as with the DOCA/salt treatment (Figure 4A, 4B, 4D, and 4E). The addition of spironolactone prevented further elevations of BP in both genotypes and of urinary sodium excretion at 12 and 13 weeks, mainly in control mice. Thus, spironolactone suppressed the differences in urinary sodium excretion and BP observed between control and IEC‐MR‐KO mice in response to DOCA/salt treatment (Figure 4C and 4F).
Figure 4.
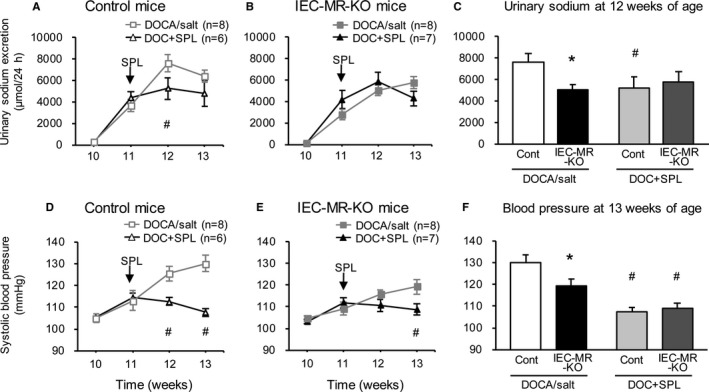
Changes in urinary sodium excretion and blood pressure under DOC+SPL treatment. Gray lines indicate the changes under DOCA/salt treatment, as shown in Figure 3C and 3I. Black lines indicate the changes under DOC+SPL treatment. Urinary sodium excretion (A and B) and systolic blood pressure (D and E) in control and IEC‐MR‐KO mice. C, Urinary sodium excretion in all groups at 12 weeks of age. F, Blood pressure in all groups at 13 weeks of age. Arrows indicate the start of spironolactone treatment. Data are presented as mean±SEM. P<0.05 vs *control (Cont), # DOCA/salt. DOC+SPL indicates deoxycorticosterone acetate/salt treatment plus spironolactone; DOCA, deoxycorticosterone acetate; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout.
Gene Expression in Response to Treatments
Next, we evaluated mRNA expression of sodium channels in the intestine and kidneys with standard diet, high‐salt diet, and DOCA/salt treatment. A high‐salt diet decreased the mRNA levels of β‐ and γ‐ENaC in the distal colon of control mice by 57% and 72%, respectively, compared with a standard diet (Figure 5B and 5C). In contrast, these mRNA levels were increased by DOCA/salt in the distal colon of control mice by 3.5‐ and 3.9‐fold compared with the levels observed on a standard diet, although excessive sodium was noted in this treatment group. In IEC‐MR‐KO mice, β‐ and γ‐ENaC expression remained at low levels even under DOCA/salt treatment. SGK1 expression showed the same trend as β‐ and γ‐ENaC expression, but the reduction in SGK1 expression in IEC‐MR‐KO mice was not as pronounced as the reductions in β‐ and γ‐ENaC expression (Figure 5D). We also measured the protein expression of the ENaC family and SGK1 in the distal colon under each treatment. As for γ‐ENaC, consistent with the results of mRNA expression, the density of the bands centered around 70 kDa, representing the active γ‐ENaC form, was significantly decreased under a high‐salt diet and increased under DOCA/salt treatment in control mice. In IEC‐MR‐KO mice, the protein expression level of γ‐ENaC was low under each treatment (Figure 6). Although the protein expression of α‐ and β‐ENaC also showed the same trends as their mRNA expression, SGK1 protein expression did not reveal significant differences between genotypes and treatments. In the kidneys, the mRNA levels of α‐ENaC were slightly lower with a high‐salt diet than a standard diet (Figure 7A). As expected, DOCA/salt treatment increased kidney α‐, β‐, and γ‐ENaC expression ≈2.0‐, 2.1‐, and 2.0‐fold, respectively (Figure 7A through 7C). Kidney ENaC expression did not differ between control and IEC‐MR‐KO mice under any treatment. In contrast, a high‐salt diet significantly increased NHE3 expression in the colon and kidneys, although the difference in the colon between genotypes was maintained (Figures 5E and 7D). However, with DOCA/salt treatment, NHE3 expression did not differ from the control levels in both tissues and genotypes. Thus, NHE3 might not contribute to BP regulation, and β‐ and γ‐ENaC expression levels are critical determinants of BP change in this context. Under DOC‐SPL treatment, spironolactone suppressed the expression of each ENaC, especially γ‐ENaC, in the colon compared with expression levels for DOCA/salt treatment (Figure 5A through 5C). This decreased expression is consistent with the BP decline under DOC‐SPL treatment. These results indicated that BP changes in mice lacking intestinal MR are associated with parallel changes in colonic β‐ and γ‐ENaC expression levels.
Figure 5.
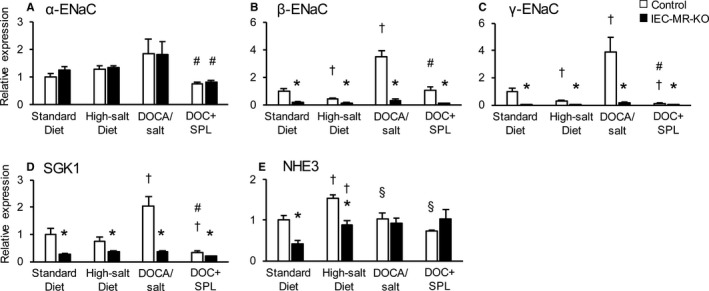
The mRNA expression levels of ENaC family members and NHE3 in the distal colon under each treatment. The mRNA expression in the distal colon of α‐ENaC (A), β‐ENaC (B), γ‐ENaC (C), SGK1 (D), and NHE3 (E) with a standard diet, a high‐salt diet, DOCA/salt treatment, and DOC+SPL treatment, as measured by quantitative polymerase chain reaction. Gene expression is normalized to that of 18S rRNA and is expressed relative to that in control mice on a standard diet. Data are presented as mean±SEM. n=6 to 8 per genotype and group. P<0.05 vs *control, †standard diet, §high‐salt diet, # DOCA/salt. DOC+SPL indicates deoxycorticosterone acetate/salt treatment plus spironolactone; DOCA, deoxycorticosterone acetate; ENaC, epithelial sodium channel; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; NHE3, sodium–hydrogen exchanger 3; SGK1, serum/glucocorticoid regulated kinase 1.
Figure 6.

Protein expression levels of ENaC family and SGK1 in the distal colon. A, Representative immunoblot showing the protein expression of each gene. The arrowhead indicates the β‐ENaC band and the arrow indicates the broad bands centered around 70 kDa, which represent the active γ‐ENaC form. B, Quantification of β‐ and γ‐ENaC protein expression levels after analysis using ImageJ. Levels are normalized to those of β‐actin and expressed relative to that in control mice on a standard diet. Data are presented as mean±SEM. n=5 per genotype and group. P<0.05 vs *control, †standard diet, §high‐salt diet. DOCA indicates deoxycorticosterone acetate; ENaC, epithelial sodium channel; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; SGK1, serum/glucocorticoid regulated kinase 1.
Figure 7.
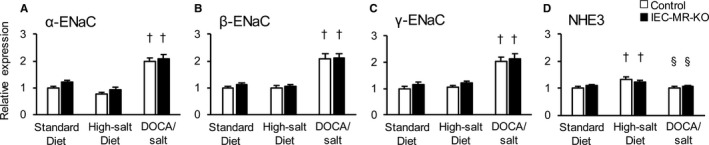
The mRNA expression levels of ENaC family members and NHE3 in the kidneys with each treatment. The mRNA expression in the kidneys of α‐ENaC (A), β‐ENaC (B), γ‐ENaC (C), and NHE3 (D) with a standard diet, a high‐salt diet, and DOCA/salt treatment, as measured by quantitative polymerase chain reaction. Gene expression was normalized to that of 18S rRNA and is expressed relative to that in control mice on a standard diet. Data are expressed as mean±SEM. n=6 to 8 per genotype and group. P<0.05 vs †standard diet, §high‐salt diet. DOCA indicates deoxycorticosterone acetate; ENaC, epithelial sodium channel; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; NHE3, sodium–hydrogen exchanger 3.
Discussion
With a standard diet, IEC‐MR‐KO mice showed higher fecal sodium excretion and lower β‐ and γ‐ENaC and NHE3 expression levels in the colon than did control mice. In particular, γ‐ENaC expression was markedly (≈90%) lower. ENaC is a heterotrimer of α, β, and γ subunits, all of which are essential for channel functionality.30 The main target of MR in the kidneys is α‐ENaC, whereas in the intestine, β‐ and γ‐ENaC are the main targets.31, 32, 33 The reduction in β‐ and γ‐ENaC but not α‐ENaC expression in the colons of IEC‐MR‐KO mice was consistent with these previous findings. ENaC and NHE3 are thought to have major roles in sodium absorption in the intestine.34 NHE3 expression was ≈50% lower in IEC‐MR‐KO mice than in control mice. Because NHE3 expression is regulated by several factors, including aldosterone, glucocorticoid, acidosis, high salt, and hyperosmolarity,35, 36, 37 the residual expression of NHE3 in IEC‐MR‐KO mice might be related to regulatory factors other than MR. These results revealed that intestinal MR regulates intestinal sodium absorption mainly via the expression of its target proteins β‐ and γ‐ENaC. Previous findings in colon‐specific α‐ENaC KO mice28 support the interpretation that increased fecal sodium excretion is caused by decreased colonic ENaC expression. Conversely, urinary sodium excretion was lower in IEC‐MR‐KO mice than in control mice. In this condition, plasma aldosterone and ENaC expression in the kidneys of IEC‐MR‐KO mice did not significantly increase. In addition, there were no significant differences in the expression of other sodium channels, including NHE3, NKCC2, NCCT, and SGLT, in the kidneys between genotypes. These findings suggest that IEC‐MR‐KO mice did not lack sodium under a standard diet, and urinary sodium excretion decreased merely because of decreased intestinal sodium absorption. Consequently, sodium balance was maintained, and reduced body weight and BP were not observed in IEC‐MR‐KO mice.
Conversely, a low‐salt diet induced significant differences in not only fecal sodium excretion but also body weight, BP, and plasma aldosterone levels between IEC‐MR‐KO and control mice. These changes are consistent with previous reports of colon‐specific α‐ENaC knockout mice28 and reveal that a compensatory mechanism, via increased plasma aldosterone, is active with a low‐salt diet. Notably, urine output and water intake were significantly increased in IEC‐MR‐KO compared with control mice. This result suggests that a drastic lack of sodium in IEC‐MR‐KO mice with a low‐salt diet caused remarkably increased water intake. The maximum dilution ability of the kidneys in mice is known to be ≈50 mOsmol/L, similar to that in humans.38 The urinary sodium concentration in IEC‐MR‐KO mice was significantly suppressed to be 12.8 mmol/L, which was significantly lower than that of control mice. Therefore, the remarkably increased water intake of IEC‐MR‐KO mice with salt deficiency was thought to have caused the significant increase in urine volume and the consequent increase in daily urinary sodium excretion compared with control mice. Notably, however, these drastic changes in water intake and urine output were not observed in α‐ENaC–deficient mice. MR is known to regulate the expression of several genes other than ENaC, including SGK1 and NHE3, as described in our study. The expression of SGK1, which activates ENaC function, was suppressed in the distal colon of IEC‐MR‐KO mice compared with control mice with a low‐salt diet, as with a standard diet. This reduction would not have occurred in α‐ENaC–deficient mice because ENaC is not an upstream regulator of SGK1. In addition, other unknown target genes of MR might have affected urinary volume with a low‐salt diet. This means that the lack of MR upstream of ENaC can induce more drastic phenotypes, such as massive diuresis and body weight loss, than ENaC deficiency. Further evaluation is warranted to elucidate the causes of this discrepancy.
A high‐salt diet considerably increased urinary sodium excretion in both control and IEC‐MR‐KO mice. Because the amount of sodium excreted in the urine reflects the amount absorbed in the intestine, intestinal sodium absorption markedly increased under a high‐salt diet. Of the 2 main sodium channels in the intestine, NHE3 expression significantly increased under a high‐salt diet in both genotypes; as mentioned, a high‐salt diet and hyperosmolarity might increase NHE3 expression. Alternatively, plasma aldosterone was suppressed to about one‐fifth, and colonic β‐ and γ‐ENaC expression levels were markedly decreased with a high‐salt diet. Together, these findings suggested that a high‐salt diet enhanced NHE3 expression but did not elevate BP because of suppressed aldosterone and decreased intestinal ENaC expression. Although the tail‐cuff method that we used to measure BP in this study might not be able to detect slight differences, this lack of BP elevation under salt loading alone is consistent with the results of previous studies that used radiotelemetry to measure BP after a high‐salt diet and reported no increase in BP.26, 39 Although NHE3 expression in the distal colon was significantly lower in IEC‐MR‐KO than control mice, intestinal sodium absorption was similar in the 2 genotypes. The physiological significance of the change in NHE3 expression requires further study.
Whereas BP did not increase with a high‐salt diet for either genotype, treatment with DOCA in addition to high salt loading elevated BP in both genotypes, indicating that MR agonist activity is pivotal in BP elevation. Notably, this elevation was significantly suppressed in IEC‐MR‐KO mice. Although MR in vascular smooth muscle cells and myeloid cells is involved in BP regulation, as mentioned earlier, MR in nonepithelial tissues does not affect sodium balance in the body. These findings suggest that intestinal and renal MR are more essential in DOCA/salt‐induced BP elevation than MR in nonepithelial tissues.
Comparing both genotypes, fecal and urinary sodium excretion and intestinal β‐ and γ‐ENaC expression were significantly different with this treatment, as with the standard diet, but to a greater degree. The difference in sodium dynamics between genotypes likely caused the difference in BP. Lower urinary sodium excretion in IEC‐MR‐KO than in control mice indicates lower intestinal sodium absorption in IEC‐MR‐KO mice. Because the expression levels of the 2 main intestinal sodium channels ENaC and NHE3 on the standard diet were lower in IEC‐MR‐KO than in control mice, the lower intestinal sodium absorption in IEC‐MR‐KO mice with DOCA/salt treatment was expected to be related to the lower expression of these channels; however, whereas ENaC expression differed markedly between genotypes, NHE3 expression showed similar levels in both genotypes with DOCA/salt treatment. Although NHE3 expression was significantly lower in IEC‐MR‐KO mice on a standard diet and MR regulates NHE3 expression, NHE3 is also regulated by several factors other than MR, such as acidosis, high salt, hyperosmolarity, and proinflammatory cytokines.35, 36, 37 With DOCA/salt treatment, several conditions, such as salt loading, osmolarity, and perhaps proinflammatory cytokines, would have been changed. These factors likely affected NHE3 expression, thereby eliminating the difference between genotypes. This finding suggests that the differences in intestinal sodium absorption and urinary sodium excretion could be entirely attributed to differential colonic ENaC expression. Together, our findings suggest that this difference in ENaC expression explains the difference in BP elevation between genotypes with DOCA/salt treatment. Further studies should focus on establishing the causal relationship among ENaC expression, BP, and sodium excretion.
IEC‐MR‐KO mice showed elevated BP in response to DOCA/salt, whereas colonic ENaC expression remained low with this treatment. Myeloid MR might contribute to this residual effect of DOCA/salt on BP in IEC‐MR‐KO mice because previous studies have reported its involvement in aldosterone/salt‐induced hypertension. In addition, because DOCA/salt increases the renal expression of all ENaC subunits, renal MR may have essential roles in BP elevation in this state. The activation of renal MR is considered to have accelerated excessive sodium reabsorption and, consequently, to have contributed to BP elevation in IEC‐MR‐KO mice.
Spironolactone suppressed intestinal MR activation and prevented further increases in urinary sodium excretion and BP induced by DOCA/salt in control mice; therefore, no difference in BP between genotypes was observed under this treatment. These results confirmed that BP elevation was mediated by the activation of intestinal MR. Moreover, because the reduction in BP with additional spironolactone treatment was also observed in IEC‐MR‐KO mice, this reduction is considered to be caused by the inhibition of MR activation in other tissues, such as the kidneys, and further supports the proposed mechanism. Taken together, these findings suggest that intestinal MR contributes to sodium dynamics and BP regulation, comparable to renal MR.
A limitation of this study was that we used only male mice to evaluate intestinal MR function. The evaluation of sex‐related differences in the role of intestinal MR deserves further study.
From a clinical perspective, a direct association between intestinal MR and BP has not been established to date; however, several interesting studies have assessed the efficiency and safety of MR antagonists in dialysis patients.40, 41 In some, but not all, of these studies, BP improvement was observed using MR antagonists. The effects of MR antagonists on BP in anuric dialysis patients are controversial owing to the complexity of evaluating antihypertensive effects in these patients. However, the only randomized study with a primary outcome of reduced BP demonstrated a significant BP reduction in dialysis patients using MR antagonists.42 Although the mechanism underlying the antihypertensive effect was not clarified, this clinical finding is noteworthy because it suggests the potential involvement of intestinal MR in BP regulation.
In summary, this study is the first to evaluate the physiological and pathological roles of intestinal MR by generating IEC‐MR‐KO mice. We clarified the significance of intestinal MR in sodium balance and BP regulation. Our findings revealed that not only renal but also intestinal MR contributes to excessive sodium retention in mineralocorticoid hypertension and indicates the significant role of intestinal MR in the pathophysiology of hypertension. The model and data generated in this study will help further elucidate the molecular mechanism underlying MR‐associated cardiovascular diseases.
Sources of Funding
This work was supported by JSPS KAKENHI grant number 17K16095 (to Nakamura) and 16K09656, Keio Gijuku Academic Development Funds (to Kurihara).
Disclosures
None.
Supporting information
Table S1. Primers Used for Genotyping and Gene Expression Analyses
Figure S1. Histologic evaluation of intestinal mucosal morphology by hematoxylin and eosin staining. Intestines, including the duodenum to the distal colon, of control (upper panels) and IEC‐MR‐KO mice (lower panels) are shown. Magnification ×200. Scale bar=100 μm. IEC‐MR‐KO indicates intestinal epithelial cell–specific mineralocorticoid receptor knockout.
Figure S2. Specific deletion of MR from intestinal epithelial cells in IEC‐MR‐KO mice. A, Genomic DNA was amplified with primers specific for LoxP MR or recombined MR. Recombination in MR occurs throughout the intestine of IEC‐MR‐KO mice (+), including the duodenum, jejunum, ileum, proximal colon, and distal colon. Recombination does not occur in the intestine of control mice (−) or the kidneys of either mouse line. B and C, The mRNA expression level of MR throughout the intestine (B) and in the kidney, heart, aorta, or liver (C) of IEC‐MR‐KO and control mice. D, The mRNA expression of GR in each intestinal portion between genotypes. The expression level in each intestinal portion was normalized to that in the duodenum of control mice. Data are presented as mean±SEM. *P<0.05 vs control. GR indicates glucocorticoid receptor; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; MR, mineralocorticoid receptor.
Figure S3.The mRNA expression levels of NKCC2, NCC, SGLT1, and SGLT2 in the kidneys. The mRNA expression in the kidneys of NKCC2, NCC, SGLT1, and SGLT2 with a standard diet, as measured by quantitative polymerase chain reaction. Gene expression was normalized to that of 18S rRNA and is expressed relative to that in control mice. Data are expressed as mean±SEM. n=7 per genotype. NKCC2, sodium–potassium–chloride cotransporter; NCC, sodium–chloride cotransporter; SGLT1, sodium–glucose cotransporter 1; SGLT2, sodium–glucose cotransporter 2.
Acknowledgments
We are grateful to Stefan Berger (German Cancer Research Center) for kindly providing us with the MRflox/flox mouse line.
(J Am Heart Assoc. 2018;7:e008259 DOI: 10.1161/JAHA.117.008259.)
References
- 1. Mendis S. Global status report on noncommunicable diseases 2014. World Health Organization; Available at: http://www.who.int/nmh/publications/ncd-status-report-2014/en/. Accessed December 18, 2017. [DOI] [PubMed] [Google Scholar]
- 2. Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM; American Heart Association . Dietary approaches to prevent and treat hypertension: a scientific statement from the American Heart Association. Hypertension. 2006;47:296–308. [DOI] [PubMed] [Google Scholar]
- 3. Yokota K, Shibata H, Kurihara I, Kobayashi S, Suda N, Murai‐Takeda A, Saito I, Kitagawa H, Kato S, Saruta T, Itoh H. Coactivation of the N‐terminal transactivation of mineralocorticoid receptor by Ubc9. J Biol Chem. 2007;282:1998–2010. [DOI] [PubMed] [Google Scholar]
- 4. Murai‐Takeda A, Shibata H, Kurihara I, Kobayashi S, Yokota K, Suda N, Mitsuishi Y, Jo R, Kitagawa H, Kato S, Saruta T, Itoh H. NF‐YC functions as a corepressor of agonist‐bound mineralocorticoid receptor. J Biol Chem. 2010;285:8084–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shibata H, Itoh H. Mineralocorticoid receptor‐associated hypertension and its organ damage: clinical relevance for resistant hypertension. Am J Hypertens. 2012;25:514–523. [DOI] [PubMed] [Google Scholar]
- 6. Ronzaud C, Loffing J, Bleich M, Gretz N, Gröne H‐J, Schütz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol. 2007;18:1679–1687. [DOI] [PubMed] [Google Scholar]
- 7. Ronzaud C, Loffing J, Gretz N, Schutz G, Berger S. Inducible renal principal cell‐specific mineralocorticoid receptor gene inactivation in mice. Am J Physiol Renal Physiol. 2011;300:F756–F760. [DOI] [PubMed] [Google Scholar]
- 8. Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, Kawarazaki W, Takeuchi M, Ayuzawa N, Miyoshi J, Takai Y, Ishikawa A, Shimosawa T, Ando K, Nagase M, Fujita T. Rac1 GTPase in rodent kidneys is essential for salt‐sensitive hypertension via a mineralocorticoid receptor‐dependent pathway. J Clin Invest. 2011;121:3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stewart PM. Mineralocorticoid hypertension. Lancet. 1999;353:1341–1347. [DOI] [PubMed] [Google Scholar]
- 10. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 11. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. [DOI] [PubMed] [Google Scholar]
- 12. Zannad F, McMurray JJV, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21. [DOI] [PubMed] [Google Scholar]
- 13. Fraccarollo D, Berger S, Galuppo P, Kneitz S, Hein L, Schütz G, Frantz S, Ertl G, Bauersachs J. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation. 2011;123:400–408. [DOI] [PubMed] [Google Scholar]
- 14. Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston G a, Shen JZ, Reichelt ME, Delbridge LM, Young MJ. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt‐mediated inflammation and cardiac fibrosis. Hypertension. 2012;60:1443–1450. [DOI] [PubMed] [Google Scholar]
- 15. Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt‐mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension. 2014;63:1033–1040. [DOI] [PubMed] [Google Scholar]
- 16. Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension. 2015;66:988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cat AND, Griol‐Charhbili V, Loufrani L, Labat C, Benjamin L, Farman N, Lacolley P, Henrion D, Jaisser F. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. 2010;24:2454–2463. [DOI] [PubMed] [Google Scholar]
- 18. Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt‐induced cardiac fibrosis and increased blood pressure. Hypertension. 2009;54:537–543. [DOI] [PubMed] [Google Scholar]
- 19. Shen JZ, Morgan J, Tesch GH, Rickard AJ, Chrissobolis S, Drummond GR, Fuller PJ, Young MJ. Cardiac tissue injury and remodeling is dependent upon MR regulation of activation pathways in cardiac tissue macrophages. Endocrinology. 2016;157:3213–3223. [DOI] [PubMed] [Google Scholar]
- 20. Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard‐Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone‐salt to induce vascular stiffness. Hypertension. 2014;63:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheppard KE, Li KX, Autelitano DJ. Corticosteroid receptors and 11beta‐hydroxysteroid dehydrogenase isoforms in rat intestinal epithelia. Am J Physiol. 1999;277:G541–G547. [DOI] [PubMed] [Google Scholar]
- 23. Bertog M, Cuffe JE, Pradervand S, Hummler E, Hartner A, Porst M, Hilgers KF, Rossier BC, Korbmacher C. Aldosterone responsiveness of the epithelial sodium channel (ENaC) in colon is increased in a mouse model for Liddle's syndrome. J Physiol. 2008;586:459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cole TJ, Young MJ. 30 Years of the mineralocorticoid receptor: mineralocorticoid receptor null mice: informing cell‐type‐specific roles. J Endocrinol. 2017;234:T83–T92. [DOI] [PubMed] [Google Scholar]
- 25. Kennedy CRJ, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt‐sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. [DOI] [PubMed] [Google Scholar]
- 26. Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GTJ, Okamura H. Salt‐sensitive hypertension in circadian clock‐deficient Cry‐null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16:67–74. [DOI] [PubMed] [Google Scholar]
- 27. Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, Chepkova AN, Welzl H, Haas HL, Lipp H‐P, Schütz G. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci USA. 2006;103:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malsure S, Wang Q, Charles R‐P, Sergi C, Perrier R, Christensen BM, Maillard M, Rossier BC, Hummler E. Colon‐specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol. 2014;25:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meneton P, Schultheis PJ, Greeb J, Nieman ML, Liu LH, Clarke LL, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Increased sensitivity to K+ deprivation in colonic H,K‐ATPase‐deficient mice. J Clin Invest. 1998;101:536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure‐function, tissue distribution, and associated inherited diseases. Gene. 2016;579:95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asher C, Wald H, Rossier BC, Garty H. Aldosterone‐induced increase in the abundance of Na+ channel subunits. Am J Physiol. 1996;271:C605–C611. [DOI] [PubMed] [Google Scholar]
- 32. Greig ER, Baker EH, Mathialahan T, Boot‐Handford RP, Sandle GI. Segmental variability of ENaC subunit expression in rat colon during dietary sodium depletion. Pflugers Arch. 2002;444:476–483. [DOI] [PubMed] [Google Scholar]
- 33. Epple HJ, Amasheh S, Mankertz J, Goltz M, Schulzke JD, Fromm M. Early aldosterone effect in distal colon by transcriptional regulation of ENaC subunits. Am J Physiol Gastrointest Liver Physiol. 2000;278:G718–G724. [DOI] [PubMed] [Google Scholar]
- 34. Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82:245–289. [DOI] [PubMed] [Google Scholar]
- 35. He P, Yun CC. Mechanisms of the regulation of the intestinal exchanger NHE3. J Biomed Biotechnol. 2010;2010:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pasham V, Rotte A, Gu S, Yang W, Bhandaru M, Rexhepaj R, Pathare G, Lang F. Upregulation of intestinal NHE3 following saline ingestion. Kidney Blood Press Res. 2013;37:48–57. [DOI] [PubMed] [Google Scholar]
- 37. Ambühl P, Amemiya M, Preisig PA, Moe OW, Alpern RJ. Chronic hyperosmolality increases NHE3 activity in OKP cells. J Clin Invest. 1998;101:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jeff MS, Harold EL. The urine concentrating mechanism and urea transporters function In: Alpern RJ, Caplan MJ, Moe OW, ed. Seldin and Giebisch's The Kidney: Physiology & Pathophysiology. 5th ed New York, NY: Academic Press; 2013:1463–1510. [Google Scholar]
- 39. Milia AF, Gross V, Plehm R, De Silva JA Jr, Bader M, Luft FC. Normal blood pressure and renal function in mice lacking the bradykinin B(2) receptor. Hypertension. 2001;37:1473–1479. [DOI] [PubMed] [Google Scholar]
- 40. Quach K, Lvtvyn L, Baigent C, Bueti J, Garg AX, Hawley C, Haynes R, Manns B, Perkovic V, Rabbat CG, Wald R, Walsh M. The safety and efficacy of mineralocorticoid receptor antagonists in patients who require dialysis: a systematic review and meta‐analysis. Am J Kidney Dis. 2016;68:591–598. [DOI] [PubMed] [Google Scholar]
- 41. Zhao Y, Yan B, Zhao Z, Wang S, Weng X. Safety and cardiovascular effects of mineralocorticoid receptor antagonists for patients receiving hemodialysis: a systematic review and meta‐analysis. Ren Fail. 2016;38:589–599. [DOI] [PubMed] [Google Scholar]
- 42. Ni X, Zhang J, Zhang P, Wu F, Xia M, Ying G, Chen J. Effects of spironolactone on dialysis patients with refractory hypertension: a randomized controlled study. J Clin Hypertens. 2014;16:658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Used for Genotyping and Gene Expression Analyses
Figure S1. Histologic evaluation of intestinal mucosal morphology by hematoxylin and eosin staining. Intestines, including the duodenum to the distal colon, of control (upper panels) and IEC‐MR‐KO mice (lower panels) are shown. Magnification ×200. Scale bar=100 μm. IEC‐MR‐KO indicates intestinal epithelial cell–specific mineralocorticoid receptor knockout.
Figure S2. Specific deletion of MR from intestinal epithelial cells in IEC‐MR‐KO mice. A, Genomic DNA was amplified with primers specific for LoxP MR or recombined MR. Recombination in MR occurs throughout the intestine of IEC‐MR‐KO mice (+), including the duodenum, jejunum, ileum, proximal colon, and distal colon. Recombination does not occur in the intestine of control mice (−) or the kidneys of either mouse line. B and C, The mRNA expression level of MR throughout the intestine (B) and in the kidney, heart, aorta, or liver (C) of IEC‐MR‐KO and control mice. D, The mRNA expression of GR in each intestinal portion between genotypes. The expression level in each intestinal portion was normalized to that in the duodenum of control mice. Data are presented as mean±SEM. *P<0.05 vs control. GR indicates glucocorticoid receptor; IEC‐MR‐KO, intestinal epithelial cell–specific mineralocorticoid receptor knockout; MR, mineralocorticoid receptor.
Figure S3.The mRNA expression levels of NKCC2, NCC, SGLT1, and SGLT2 in the kidneys. The mRNA expression in the kidneys of NKCC2, NCC, SGLT1, and SGLT2 with a standard diet, as measured by quantitative polymerase chain reaction. Gene expression was normalized to that of 18S rRNA and is expressed relative to that in control mice. Data are expressed as mean±SEM. n=7 per genotype. NKCC2, sodium–potassium–chloride cotransporter; NCC, sodium–chloride cotransporter; SGLT1, sodium–glucose cotransporter 1; SGLT2, sodium–glucose cotransporter 2.