

Abstract
Background
Although estrogen receptor α (ERα) acts primarily as a transcription factor, it can also elicit membrane‐initiated steroid signaling. Pharmacological tools and transgenic mouse models previously highlighted the key role of ERα membrane‐initiated steroid signaling in 2 actions of estrogens in the endothelium: increase in NO production and acceleration of reendothelialization.
Methods and Results
Using mice with ERα mutated at cysteine 451 (ERaC451A), recognized as the key palmitoylation site required for ERα plasma membrane location, and mice with disruption of nuclear actions because of inactivation of activation function 2 (ERaAF20 = ERaAF2°), we sought to fully characterize the respective roles of nuclear versus membrane‐initiated steroid signaling in the arterial protection conferred by ERα. ERaC451A mice were fully responsive to estrogens to prevent atheroma and angiotensin II–induced hypertension as well as to allow flow‐mediated arteriolar remodeling. By contrast, ERαAF20 mice were unresponsive to estrogens for these beneficial vascular effects. Accordingly, selective activation of nuclear ERα with estetrol was able to prevent hypertension and to restore flow‐mediated arteriolar remodeling.
Conclusions
Altogether, these results reveal an unexpected prominent role of nuclear ERα in the vasculoprotective action of estrogens with major implications in medicine, particularly for selective nuclear ERα agonist, such as estetrol, which is currently under development as a new oral contraceptive and for hormone replacement therapy in menopausal women.
Keywords: arteriolar remodeling, atherosclerosis, estrogen, hypertension, nuclear receptor
Subject Categories: Vascular Disease, Atherosclerosis, Hypertension, Remodeling
Clinical Perspective
What Is New?
Although most vasculoprotective effects studied until now in mouse models were shown to be mediated by membrane estrogen receptor α actions, the present results reveal that experimental prevention against atheroma and hypertension required only 17β‐estradiol–mediated nuclear actions.
What Are the Clinical Implications?
Estetrol, as a selective nuclear estrogen receptor α agonist that does not alter circulating coagulation factors, could now be considered as an alternative estrogen for women's health treatment in contraception or menopause.
Introduction
Hypertension affects 1 of 4 women worldwide, and its prevalence is particularly high among women >60 years of age. Indeed, the first decade after menopause is accompanied by an increase in blood pressure and has been associated with a higher risk of cardiovascular diseases, such as myocardial infarction and stroke.1 In line with these epidemiological data, clinical and preclinical studies show considerable evidence that estrogen modulates cardiovascular physiological features and function.2 Even if the publication of the Women's Health Initiative in 20023, 4 did not initially confirm the expected protective action of estrogens against coronary heart disease and questioned their overall benefit, subgroup analysis suggested that women who initiated hormone therapy soon after the onset of menopause had reduced coronary artery events, in contrast to the increased risk in more aged women.5 All experimental data demonstrate major atheroprotective actions of estrogens. In particular, 17β‐estradiol strongly prevents lipid deposition in mouse models of atherosclerosis: apolipoprotein E–deficient6, 7 and low‐density lipoprotein receptor–deficient (LDLr−/−)8 mice. In addition, in several experimental models of hypertension, ovariectomy exacerbates, whereas estrogen replacement attenuates, the course of hypertension9, 10 and reduces arterial stiffening associated with hypertension or aging.11 17β‐estradiol also increases basal nitric oxide (NO) production,12 accelerates reendothelialization,8, 13 and prevents postinjury medial as well as neointimal hyperplasia after vessel injury.14 Finally, 17β‐estradiol plays a crucial role in the ability of resistance arteries to remodel in response to a long‐term increase in blood flow, which is necessary to optimize tissue perfusion.11, 15
Two nuclear receptors, estrogen receptors (ERs) α and β, have been reported to mediate cardiovascular protection conferred by estrogens.16 ERβ plays a key role in cardiac protection,17 and ERα, especially in the endothelium, is involved in the beneficial vascular actions of 17β‐estradiol.18 Indeed, most of the vascular actions of 17β‐estradiol are abrogated in mice lacking ERα in both endothelial and hematopoietic cells by breeding Tie2‐Cre transgenic mice (expressing the Cre recombinase under the control of the endothelial tyrosine‐protein kinase receptor (Tie2) promoter) with ERα‐floxed mice at exon 2 (ERαlox/lox).8, 15, 19 Thus, the endothelium, a guardian of arterial integrity, represents a promising cellular target for 17β‐estradiol via the activation of ERα.18 Beside the well‐recognized role of nuclear ERα, which regulates target gene transcription (genomic action) through activation functions (AFs) 1 and 2,16, 20 a subpopulation of ERα is present at or near the plasma membrane, where it can elicit rapid, nongenomic, membrane‐initiated steroid signaling (MISS) effects.21, 22, 23, 24, 25
Recently, we generated a mouse model in which membrane ERα localization was abrogated by a point mutation of the palmitoylation site of ERα (mice with ERα mutated at cysteine 451 [ERα‐C451A]), leading to an abrogation of endothelial NO synthase (eNOS) phosphorylation and acceleration of reendothelialization in response to 17β‐estradiol.22 The estrogen‐dendrimer conjugate (EDC) and “pathway preferential estrogens” (PaPEs) are highly effective in stimulating nonnuclear signaling, but they are inefficient in stimulating nuclear ER target gene expression.23, 26 More important, in vivo administration of EDC or PaPE‐1 accelerates repair of endothelial damage.23, 26 In addition, EDC and PaPE‐1 both favor the production of endothelial NO,22 a vasculoprotective agent of the arteries, initially viewed as an important mechanism of the protective action of 17β‐estradiol against the development of atheroma and hypertension.27 Our arsenal of pharmacological tools has recently been enriched with estetrol, a fetal estrogen produced exclusively by the liver of humans (and to a smaller extent in large apes). We demonstrated that although estetrol nicely induced ERα target gene transcription, it failed to promote eNOS activation and to accelerate endothelial healing, but also antagonized both of these 17β‐estradiol MISS‐dependent effects.28 Altogether, these powerful pharmacological tools allow us to uncouple nuclear and membrane actions of ERα with a selective activation of MISS using PaPE‐1 or EDC and of nuclear ERα using estetrol. The novel understanding of action of this new class of selective ER modulator–like molecules led to the idea that they would decrease the proliferative action of estrogens23, 29 and, thereby, lower the risk of breast and/or uterine cancer induced by sex hormones.30, 31
In the present work, using these pharmacological tools of gain of function (EDC, PaPE‐1, and estetrol) and genetically modified mouse models of ERα loss of function (Using mice with ERa mutated at cysteine 451, recognized as the key palmitoylation site required for ERa plasma membrane location (ERaC451A), and mice with disruption of nuclear actions because of inactivation of activation function 2 (ERaAF2°)), we sought to further explore the role of ERα MISS in the prevention of atherosclerotic lesions and hypertension and in the promotion of arteriolar remodeling, 3 crucial pathophysiological aspects of the vasculoprotective actions of estrogens.
Methods
The data, analytical methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Mice
All procedures involving experimental animals were performed in accordance with the principles and guidelines established by the National Institute of Medical Research and were approved by the local Animal Care and Use Committee. The investigation conforms to the directive 2010/63/EU of the European parliament. To generate the double‐deficient mice, LDLr−/− female mice, purchased from Charles River (L'Arbresle, France), were crossed with ERα‐C451A+/− mice.22 ERα‐AF20 mice were generated, as previously described,32 and were kindly provided by Professor P. Chambon (Strasbourg, France). Mice are on a C57Bl/6 background, and genetically manipulated mice are systematically compared with their wild‐type (WT) littermates controls (n=7–12 per group). For short‐term 17β‐estradiol treatment, bilateral ovariectomy was performed when the mice were 4 weeks old and then, 2 weeks after surgery, mice were injected SC with either vehicle or 17β‐estradiol (8 μg/kg). Mice were euthanized 6 hours after the injection to study the short‐term effect of 17β‐estradiol on gene expression. Before surgical and euthanasia procedures, mice were anesthetized with a combination of ketamine hydrochloride (100 mg/kg; Panpharma) and xylazine (5 mg/kg; Sigma‐Aldrich) by IP injection.
Real‐Time NO Production
Aortas from intact mice (10–12 weeks/n=6 per group) were quickly harvested and maintained in 200 μL Krebs‐Ringer oxygenated solution containing 2.5 mmol/L glucose at 37°C. A NO‐specific amperometric probe (ISO‐NOPF100; World Precision Instruments, Sarasota, FL) was implanted directly in the tissue, and NO release was monitored. The aortas were exposed to 17β‐estradiol (10−8 mol/L) or vehicle (dimethyl sulfoxide). The concentration of NO gas in the tissue was measured in real time with the data acquisition system LabTrax (World Precision Instruments) connected to the free radical analyzer Apollo1000 (World Precision Instruments). Data acquisition and analysis were performed with DataTrax2 software (World Precision Instruments). The NO‐specific amperometric probe was calibrated, as previously described.28
Analyses of Atherosclerosis Lesions
Bilateral ovariectomy was performed, as previously described. Two weeks after surgery, mice were switched to a hypercholesterolemic‐atherogenic diet (1.25% cholesterol, 6% fat, no cholate; TD96335; Harlan Teklad, Madison, WI) and concomitantly received long‐term estrogenic treatments. Mice were implanted with subcutaneous pellets releasing either placebo or 17β‐estradiol (60‐day release, 0.1 mg [ie, 80 μg/kg per day]) at weeks 6 and 12. To evaluate the atheroprotective role of selective activation of ERα MISS, mice received subcutaneous miniosmotic pumps (Alzet, model 2004; 0.25 μL/h, 28 days), releasing either empty dendrimer or EDC (240 μg/kg per day); or pellet, releasing either a placebo or equivalent dose of 17β‐estradiol (pellet, 0.25 mg [ie, 240 μg/kg per day]; Innovative Research of America, Sarasota, FL) or PaPE‐1 (8.5‐mg compound mixed with cholesterol to a total weight of 20 mg) at weeks 6 and 10. At the end of the protocol, overnight fasted mice were anesthetized with a combination of ketamine hydrochloride (100 mg/kg; Panpharma) and xylazine (5 mg/kg; Sigma‐Aldrich) via intraperitoneal injection, and blood was collected from retro‐orbital venous plexus. After euthanization, the heart, the thoracic aorta, the liver, and the uterus were carefully dissected.
Lipid deposition size was estimated at the aortic sinus, as previously described.8, 32 Briefly, each heart was frozen on a cryostat mount with optimal cutting temperature compound. One hundred 10‐μm‐thick sections were prepared from the top of the left ventricle, where the aortic valves were first visible, up to a position in the aorta where the valve cusps were just disappearing from the field. After drying for 2 hours, the sections were stained with oil red O and counterstained with Mayer's hematoxylin. Ten sections of the 100, each separated by 90 μm, were used for specific morphometric evaluation of intimal lesions using a computerized Biocom morphometry system. The first and most proximal section to the heart was taken 90 μm distal to the point where the aorta first becomes rounded. The mean lesion size (expressed in μm2) in these 10 sections was used to evaluate the lesion size of each animal.
En face aorta analyses were analyzed at 8 months of age. The heart was perfusion fixed in situ with 10% phosphate‐buffered formalin for 10 minutes. After the perfusion, the abdominal cavity was opened and the internal organs were removed. The aorta from the arch to bifurcation was carefully cleaned from periadventitial connective tissue. Then, the aorta was opened longitudinally by an incision along its ventral aspect. The thoracic and abdominal parts of the aorta were pinned out flat (using 10‐mm tips of dental root‐canal needles).
Determination of Plasma Lipids
Overnight fasted mice were anesthetized, and blood samples were collected from the retro‐orbital venous plexus. Total plasma cholesterol was assayed using the CHOD‐PAD kit (Horiba ABX, Montpellier, France). The high‐density lipoprotein fraction was isolated from 10 μL of serum and assayed using the “C‐HDL+Third generation” kit (Roche, Lyon, France).
Analysis of mRNA Levels by Real‐Time Quantitative Reverse Transcription–Polymerase Chain Reaction
Tissues were dissected and frozen. They were homogenized using a Precellys tissue homogenizer (Bertin Technology, Cedex, France), and total RNA from tissues was prepared using Trizol reagent (Invitrogen, Carlsbad, CA). A total of 500 ng (aorta) or 1 μg (liver) was reverse transcribed for 10 minutes at 25°C and 2 hours at 37°C in a 20‐μL final volume using the High Capacity cDNA reverse transcriptase kit (Applied Biosystems). Real‐time quantitative polymerase chain reactions were performed on the StepOne instrument (Applied Biosystems). Primers were validated by testing polymerase chain reaction efficiency using standard curves (95% ≤ efficiency ≤ 105%). Gene expression was quantified using the comparative threshold cycle method; tumor protein, translationally‐controlled 1 (TPT1) (aorta) or Peptidyl‐prolyl cis‐trans isomerase A (PPIA) (liver) was used as reference.
Blood Pressure Measurement
As previously shown,33 systolic blood pressure (SBP) was measured every day using photoplethysmography (BP‐2000 Blood Pressure Analysis System; Visitech Systems). Female mice (aged 12–16 weeks) were divided into the following groups: ERα−/−, ERα‐C451A, and ERαAF20 mice associated with their corresponding littermates. In all groups, mice were acclimated to the technique for 2 weeks (week “−2” to “−1”) before measuring basal SBP for 1 week (week “0”). Mice were then subjected to sham operation or were implanted SC with osmotic minipumps (Alzet, model 2004; 0.25 μL/h, 28 days) releasing angiotensin II (AngII; 0.5 mg/kg per day; Bachem, no. 4006473; solubilized in NaCl 0.9%) for 4 weeks to induce hypertension.34 After surgery, mice were allowed to recover for 3 to 5 days, and SBP was measured daily during 4 weeks. The weekly average of SBP evolution pre‐ and post‐AngII treatment was represented as mean of 5 days of SBP measurements (from week 0 to week 4). To assess the effect of estetrol in AngII‐treated mice, ovariectomy was performed 2 weeks before implantation of osmotic minipump containing AngII and estetrol (AngII, 0.5 mg/kg per day, and estetrol, 6 mg/kg per day; solubilized in 50% PBS+50% dimethyl sulfoxide). Surgery was performed under isoflurane (2%) anesthesia. Analgesia was obtained with buprenorphine (Temgesic; 0.1 mg/kg, SC) before and after surgery.
Histomorphometry
AngII arterial remodeling was evaluated on thoracic aorta. Transverse aortic segments were dissected, embedded in Tissue‐tek (Sakura Finetek), and cut in 7‐μm sections (Cryostat CM3050 S; Leica). Orceine was used to stain elastic fibers in the thoracic aorta media to measure media thickness/surface and lumen surface. Image acquisition was performed using Histolab, and measurements were performed using ImageJ 1.47v.
Blood Flow–Mediated Arterial Remodeling
Increase in blood flow, applied to the mesenteric artery, was performed, as previously described, on 4‐ to 5‐month‐old female mice.15 Briefly, 3 consecutive first‐order mesenteric arteries were used, and surgery consisted of ligatures of second‐order branches. The artery located between the 2 ligated arteries was designated as the high‐flow artery. Arteries located at a distance of the ligated arteries were used as controls (normal flow). After 14 days, mice were euthanized and mesenteric arteries were collected. In each protocol, animals were anesthetized with isoflurane (2.5%). They were treated with buprenorphine (Temgesic; 0.1 mg/kg, SC) before and after surgery. The protocol was approved by the ethical committee (Protocol CEEA PdL 2012‐105).
Flow‐Mediated Dilation
First‐ or second‐order mesenteric arterial segments were isolated and mounted onto an arteriograph system (Living System; LSI, Burlington, VT) in physiological saline solution (37°C, pH 7.4; partial pressure of O2, 160 mm Hg; and partial pressure of CO2, 37 mm Hg). This video‐monitored perfusion system allowed us to follow changes in arterial internal diameter of pressurized arteries (Pression: 75 mm Hg; diameter, 270 μm) after they were cannulated on glass microcannulas connected to 2 peristaltic pumps, 1 controlling flow rate and 1 under the control of a pressure‐servo control system.35 Pressure at both ends of the artery segment was monitored using pressure transducers. After the arteries were pressurized at 75 mm Hg, a 20‐minute equilibration period was provided and endothelial function was assessed through vasodilatory response to acetylcholine (1 μmol/L) after precontraction with phenylephrine (10−6 mol/L). Arteries were then washed and allowed to recover for 20 minutes. After an equilibration period at low flow (3 μL/min) for 10 minutes, arteries were precontracted (phenylephrine, 10−6 mol/L) and step increases in intraluminal flow (2‐minute step from 3–50 μL/min) were generated through the distal pipette with a peristaltic pump. The effect of estetrol on flow‐mediated dilation was evaluated ex vivo on arteries incubated with estetrol (10−6 mol/L) for 30 minutes before assessing arterial responses to flow. At the end of the experiment, arteries were bathed in a Ca2+‐free PSS containing ethylene‐bis‐(oxyethylenenitrolo) tetra‐acetic acid (2 mol/L) and sodium nitroprusside (10 μmol/L) to measure maximal passive diameter.36
Statistical Analyses
To test the respective roles of 17β‐estradiol treatment and genotype, a 2‐way ANOVA was performed for atherosclerosis experiment. When an interaction was observed between the 2 factors, the effect of 17β‐estradiol treatment was studied in each genotype using a Bonferroni post test. A 2‐way ANOVA for repeated measures was used for hypertension, flow‐mediated remodeling (FMR), and flow‐mediated dilation experiments. To test the respective roles of 17β‐estradiol and EDC treatment, a 1‐way ANOVA was performed, followed by a Bonferroni post test. P<0.05 was considered statistically significant.
Results
Short‐Term 17β‐Estradiol Treatment Induces Transcriptional Response But Not NO Production in Aorta From ERα‐C451A Mice
To evaluate the potential cross talk between MISS and nuclear ERα in blood vessels, we first compared gene expression profiles in the aorta from ERα−/−, ERα‐C451A, and ERαAF20 mice, including their littermate controls, after short‐term 17β‐estradiol administration (6 hours). Two genes among the most induced by 17β‐estradiol in the aorta32, 37 (ie, Gremlin 2 and UDP‐N‐acetyl‐α‐d‐galactosamine:polypeptide N‐acetylgalactosaminyltransferase‐like 2) were analyzed. Their transcriptional regulation is entirely ERα dependent because no gene regulation was observed in aortas from ERα−/− mice (Figure 1A). Similarly, induction of these ERα target genes was totally abrogated in ERαAF20 mice, whereas targeting membrane ERα, in ERα‐C451A mice, has a minor impact, affecting only modestly the most regulated gene by 17β‐estradiol, Gremlin 2 (Figure 1A). By contrast, although 17β‐estradiol (10−8 mol/L) rapidly induced NO production by aorta in WT mice, it failed to induce NO production in aorta from ERα‐C451A mice (Figure 1B). These results further demonstrate that, in ERα‐C451A mice, eNOS activation in response to 17β‐estradiol is abrogated, indicating the abolition of ERα MISS, whereas nuclear ERα activity in the arterial wall is preserved.
Figure 1.
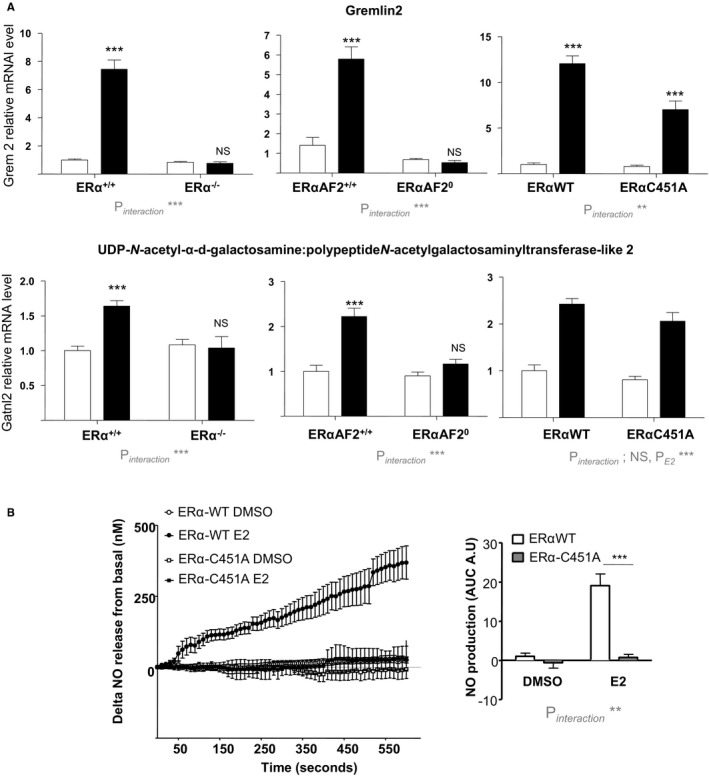
17β‐Estradiol (E2) induces transcriptional response but not NO production in aorta from estrogen receptor α mutated at cysteine 451 (ERα‐C451A) mice. A, Four‐week‐old ovariectomized ERα−/−, deficient for activation function‐2 ERα‐AF20, and ERα‐C451A mice or their respective littermates control mice (n=7–9 per group) received short‐term E2 subcutaneous injection. Six hours after, mRNA levels of the indicated gene from aorta were quantified by real‐time quantitative polymerase chain reaction and normalized to TPT1 mRNA levels. Results are expressed according to the level in aorta from vehicle‐treated ERα–wild type (WT) set as 1. B, Representative trace of ex vivo amperometric measurements of NO release of aorta from 10‐ to 12‐week‐old ERα‐C451A or their littermate control mice exposed to E2 (10−8 mol/L) and area under the curve (AUC) quantification. Results are expressed as means±SEM. To test the respective roles of E2 treatment and genotype, a 2‐way ANOVA was performed. When an interaction was observed between the 2 factors, effect of E2 treatment was studied in each genotype using a Bonferroni post test. AU indicates arbitrary unit; DMSO, dimethyl sulfoxide; Gatnl2, UDP‐N‐acetyl‐α‐d‐galactosamine:polypeptide N‐acetylgalactosaminyltransferase‐like 2; Grem2, Gremlin 2; NS, not significant. ***P<0.001.
By Contrast With Nuclear ERα‐AF2, ERα MISS Is Neither Necessary Nor Sufficient for Estrogen‐Mediated Atheroprotection
We then investigated the role of ERα MISS actions on arteries submitted to an atherogenic stress using ERα‐C451A mice bred with LDLr−/− mice (Figure 2A). As expected, ovariectomy led to uterine atrophy, whereas exogenous 17β‐estradiol replacement induced uterine hypertrophy in both ERα‐WT/LDLr−/− and ERα‐C451A/LDLr−/− mice (Figure 2B and 2C). In addition, 17β‐estradiol treatment similarly decreased fatty streak deposits at the aortic sinus of both ERα‐WT/LDLr−/− and ERα‐C451A/LDLr−/− mice (Figure 2D and 2E). Accordingly, gene expression of atherosclerosis markers, such as vascular cell adhesion molecule 1 or Monocyte Chemoattractant Protein‐1 (MCP‐1), was decreased in 17β‐estradiol–treated mice aorta, irrespective of the genotype (Figure 2F). As expected, 17β‐estradiol significantly decreased cholesterolemia in ovariectomized ERα‐WT/LDLr−/− mice and in ERα‐C451A/LDLr−/− mice (Figure 2G).
Figure 2.
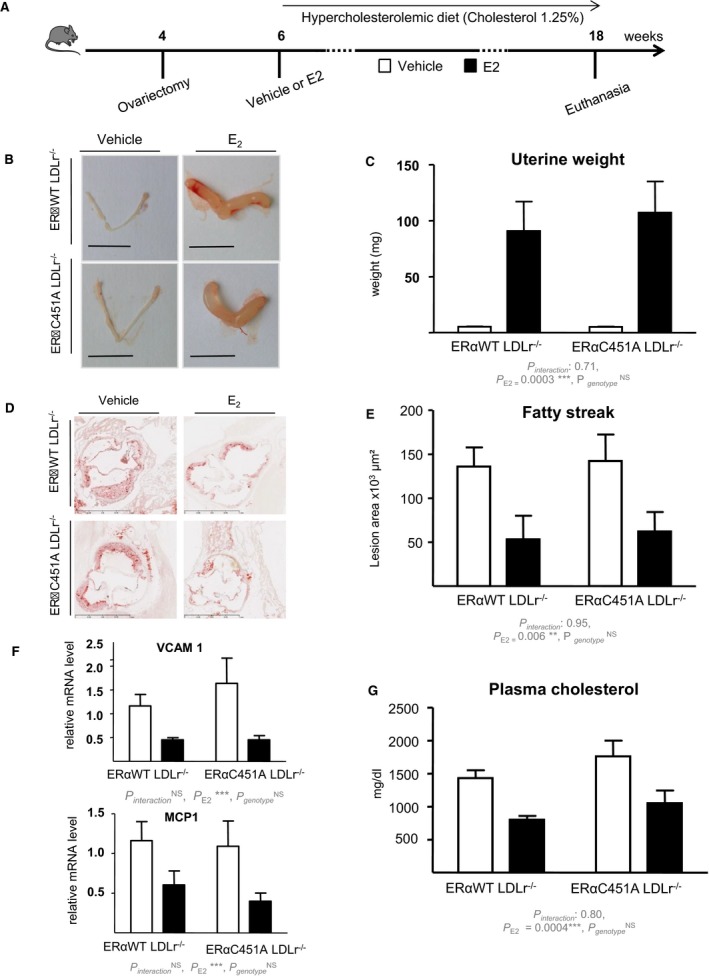
17β‐Estradiol (E2) decreases fatty streak deposits at the aortic sinus in estrogen receptor α mutated at cysteine 451 (ERα‐C451A)/low‐density lipoprotein receptor–deficient (LDLr−/−) mice. A, Four‐week‐old ovariectomized ERα–wild‐type (WT)/LDLr−/− and ERα‐C451A/LDLr−/− mice were given either placebo or E2 during 12 weeks and switched to an atherogenic diet from the age of 6 to 18 weeks (n=7–10). Representative uteri (B), uterine weights (C), representative micrographs of oil red O lipid‐stained cryosections of the aortic sinus (D), and quantification of lipid deposition (E) and vascular cell adhesion molecule (VCAM‐1) and MCP1 mRNA levels from aorta, quantified by real‐time quantitative polymerase chain reaction and normalized to TPT1 mRNA levels (F) and cholesterol content (G) from these mice.
In addition, endogenous estrogens decreased the lesion size on later stages of atheroma, as demonstrated by analysis of “en face” preparations of the aortic tree from 8‐month‐old ovariectomized versus sham‐operated female WT mice (Figure 3A). This protective effect was abolished in ERαAF20/LDLr−/− mice (Figure 3B), confirming that atheroprotection required nuclear ERα, as previously shown in aortic sinus at earlier stages.32 However, compared with ERα‐WT/LDLr−/− mice, endogenous 17β‐estradiol similarly prevented atheroma in intact ERα‐C451A/LDLr−/− mice, demonstrating that ERα MISS is fully dispensable for the atheroprotective effect of endogenous estrogens (Figure 3C).
Figure 3.
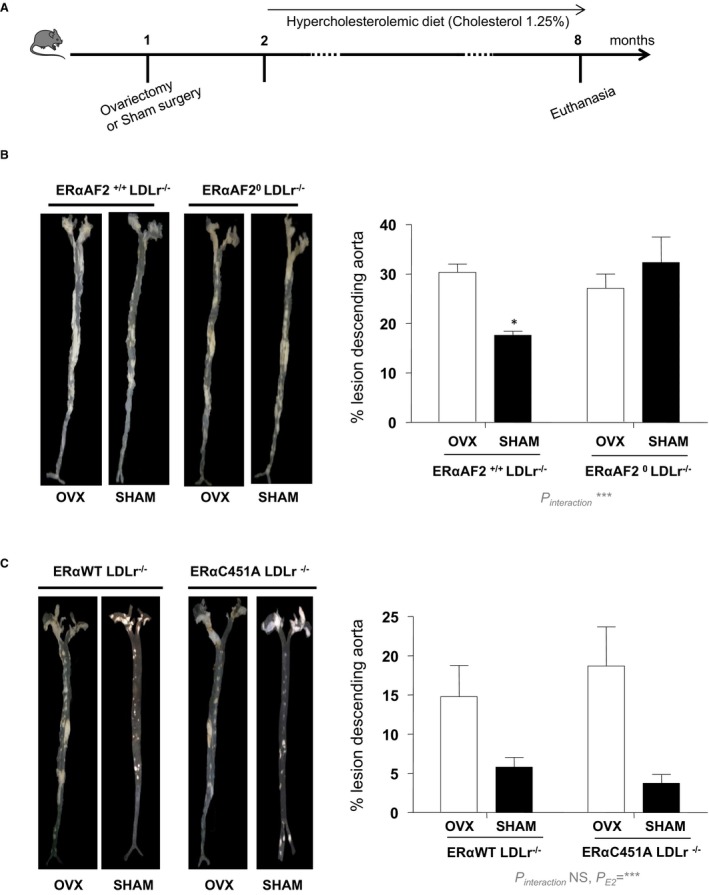
The atheroprotective effect of endogenous estrogens persists in 8‐month‐old estrogen receptor α mutated at cysteine 451 (ERα‐C451A)/low‐density lipoprotein receptor–deficient (LDLr−/−) mice, whereas it is abolished in ERα‐AF20/LDLr−/− mice. A, Four‐week‐old mice (n=5–7) were ovariectomized (OVX) or not (SHAM) and switched to atherogenic diet from the age of 6 weeks until euthanasia. Representative en face aorta preparations are shown with respective quantification of lesions from descending aorta of ERα‐AF20/LDLr−/− (B) or ERα‐C451A/LDLr−/− (C) mice and their respective littermates. Data are mean ± SEM. WT indicates wild type.
We then assessed whether selective activation of ERα MISS was sufficient to prevent early atheroma in LDLr−/− mice under an atherogenic diet using 2 unique pharmacological tools: PaPE‐1 and EDC. As previously described, these 2 molecules did not induce uterine hypertrophy (Figure 4A and 4B) but were able to induce transcriptional responses, such as increase in adiponectin mRNA level in the liver (Figure 4C), as previously reported.26 However, although 17β‐estradiol decreases plasma cholesterol, PaPE‐1 or EDC do not (Figure 4D). In addition, doses of EDC or PaPE‐1, previously shown to accelerate reendothelialization,23, 26 did not prevent lipid deposition at the aortic sinus from LDLR−/− mice fed a hypercholesterolemic diet (Figure 4E and 4F). By contrast, equivalent dose of 17β‐estradiol prevented atheroma deposition by up to 70%. Altogether, the use of both genetically deficient mice and pharmacological tools consistently demonstrated that ERα MISS is neither necessary nor sufficient for the atheroprotective effect mediated by ERα activation, by contrast with nuclear ERαAF2.
Figure 4.
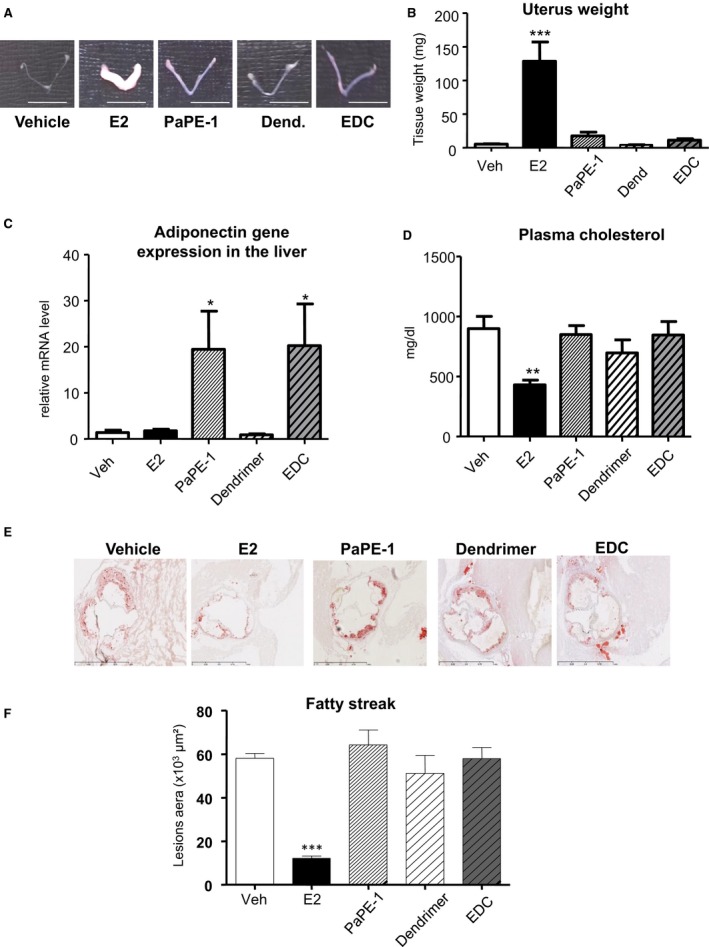
Selective activation of estrogen receptor α membrane‐initiated steroid signaling does not prevent early plaque formation. Four‐week‐old ovariectomized low‐density lipoprotein receptor–deficient mice were given 17β‐estradiol (E2), pathway preferential estrogen (PaPE)‐1 pellet or vehicle (Veh), or empty dendrimer (Dend) or estrogen‐Dend conjugate (EDC) (n=6–9) and switched to an atherogenic diet from the age of 6 to 14 weeks. Representative uteri (A), uterine weight (B), adiponectin mRNA levels from liver quantified by real‐time quantitative polymerase chain reaction and normalized to PPIA mRNA levels (C), plasma cholesterol content (D), representative micrographs of oil red O lipid‐stained cryosections of the aortic sinus (E), and quantification of lipid deposition from these mice (F). Data are mean ± SEM. To test the impact of the different treatments, a 1‐way ANOVA was performed. *P <0.05 , ***P <0.001.
Endogenous Estrogens Prevent Hypertension in ERα‐C451A Mice But Not in ERα‐AF20 Mice
We then explored the role of ERα MISS actions in animals submitted to hypertensive stress. Endogenous estrogens protected WT female mice from AngII‐induced hypertension, as demonstrated by a greater increase in SBP in ovariectomized mice than in intact mice (Figure S1A). We then evaluated the respective protective role of ERα, ERα MISS, and nuclear ERα in AngII‐induced hypertension using ERα−/−, ERα‐C451A, and ERα‐AF20 intact mice and their respective littermate controls (Figure 5A). In the different WT littermate control mice, 4 weeks of AngII infusion increased SBP. The hypertensive effect of AngII was significantly exacerbated in ERα−/− mice (Figure 5B) but not in ERβ−/− mice (Figure S2), showing that the beneficial effect of endogenous estrogens is ERα dependent. Moreover, the blood pressure increase induced by AngII infusion was significantly greater in ERα‐AF20 mice than in WT mice (Figure 5C), demonstrating that the nuclear ERαAF2 is necessary for the protective effect of ERα in hypertension. By contrast, the increase in blood pressure was similar in ERα‐C451A and WT mice, demonstrating the dispensable role of membrane ERα in this process (Figure 5D). Histomorphometric analysis showed the absence of aortic smooth muscle hypertrophy in intact WT mice treated with AngII, contrasting with the media remodeling in ovariectomized WT mice given AngII (Figure S1B). Interestingly, AngII treatment led to arterial remodeling in ERα−/− and ERα‐AF20, but not in ERα‐C451A, intact mice (Figure 5E through 5G), confirming that aortic structural changes paralleled the level of hypertension. Because hypertension was induced by AngII, we measured the gene expression level of the angiotensin‐converting enzyme (Ace) and angiotensin‐converting enzyme 2 (Ace2) and of the AngII type 1 receptors (AT1Ra and AT1Rb) in arteries isolated, and we found no difference in expression level in ERα−/−, ERα‐C451A, and ERα‐AF20 compared with WT mice (Figure S3).
Figure 5.
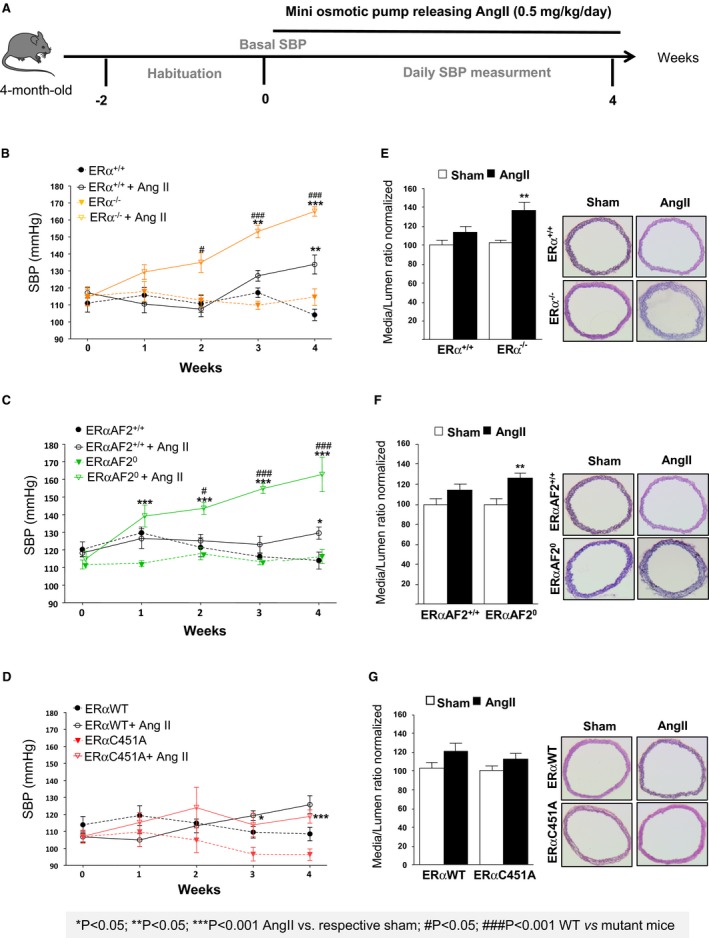
Endogenous estrogens prevent hypertension and medial remodeling in estrogen receptor α mutated at cysteine 451 (ERα‐C451A) mice but not in ERα‐AF20 mice. A, Intact female mice were implanted with osmotic minipumps delivering angiotensin II (AngII; 0.5 mg/kg per day, 1 month). Mean of systolic blood pressure (SBP) measurements for 5 days was represented as weekly evolution of blood pressure pre‐AngII treatment (week 0) and post‐AngII treatment (weeks 1–4). SBP measurements (B through D) and and medial remodeling (E through G) from thoracic aorta were evaluated in ERα−/− (n=8; B and E), ERα‐AF20 (n=7; C and F), ERα‐C451A (n=7; D and G) intact mice and their respective littermate controls (n=7). WT indicates wild type.
Endogenous Estrogens Allow Flow‐Mediated Arteriolar Remodeling in ERα‐C451A Mice But Not in ERα‐AF20 Mice
Besides pressure‐associated medial hypertrophic remodeling that characterizes hypertension, at the level of arterioles, outward remodeling occurs in response to increased blood flow and shear stress at the surface of endothelial cells. This remodeling is essential in postischemic revascularization or collateral artery growth.38 To better characterize the respective role of nuclear versus ERα MISS in this remodeling, we investigated in vivo FMR of resistance arteries from ERα‐AF20 and ERα‐C451A mice. Two weeks after arterial ligation, arterial diameter was determined in vitro in response to stepwise increases in intraluminal pressure (Figure 6A). As expected, passive arterial diameter was significantly higher in high‐flow than in normal‐flow arteries in WT (Figure 6B), but not in ERα−/−, mice (Figure 6C). Interestingly, the effect of endogenous estrogens on FMR was normal in ERα‐C451A (Figure 6D), whereas it was completely abrogated in ERαAF20 (Figure 6E), highlighting the crucial role of nuclear ERα in FMR.
Figure 6.
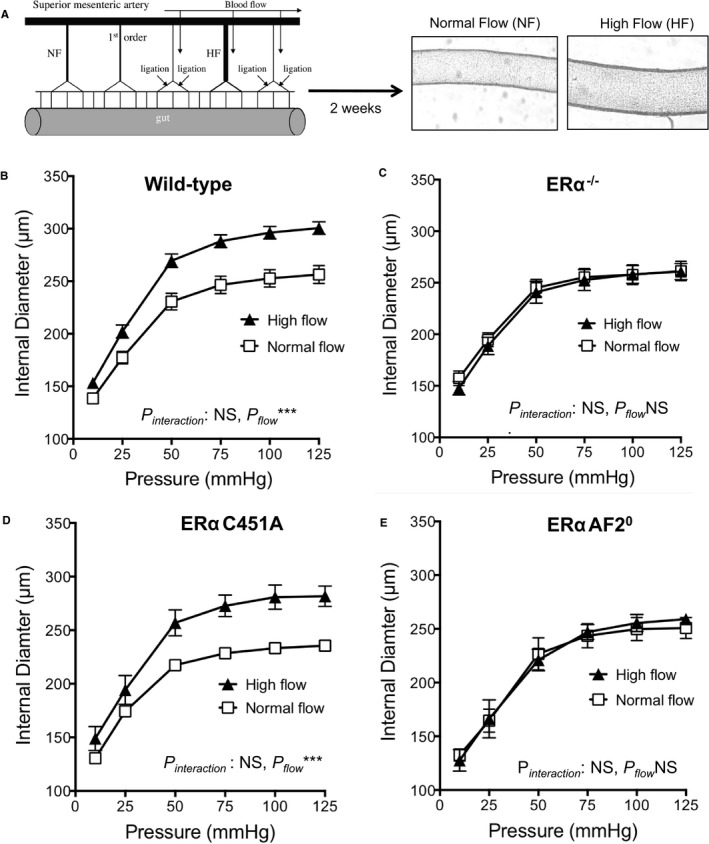
Endogenous estrogens allow flow‐mediated outward remodeling of resistance arteries in estrogen receptor α mutated at cysteine 451 (ERα‐C451A) mice but not in ERα‐AF20 mice. A, Arterial diameter was measured in response to stepwise increases in pressure in mesenteric arteries submitted chronically to high flow (HF) or to normal flow (NF). Arteries were isolated from wild‐type (n=12; B), ERα−/− (n=8; C), ERα‐C451A (n=5; D), or ERα‐AF20 (n=7; E) mice. NS indicates not significant.
Selective Activation of Nuclear ERα Using Estetrol Prevents AngII‐Induced Hypertension and Favors Flow‐Mediating Remodeling
Because nuclear ERα appears to be crucial to the prevention of atheroprotection and hypertension as well as to allow FMR, we decided to evaluate the impact of estetrol (Figure 7A). This natural selective ER modulator–like molecule was recently recognized to activate selectively nuclear ERα and to be devoid of vascular MISS action.28 We also previously demonstrated that estetrol conferred atheroprotection.28 In addition, although short incubation with estetrol was not able to modulate the acute vasodilating response to flow (Figure 7B), a long‐term treatment with estetrol allowed flow arteriolar remodeling to occur in ovariectomized mice (Figure 7C). Estetrol also prevented the increase in SBP induced by AngII treatment in ovariectomized mice (Figure 7D). These data clearly show that nuclear ERα activation is sufficient to attenuate AngII‐induced hypertension and to promote FMR.
Figure 7.
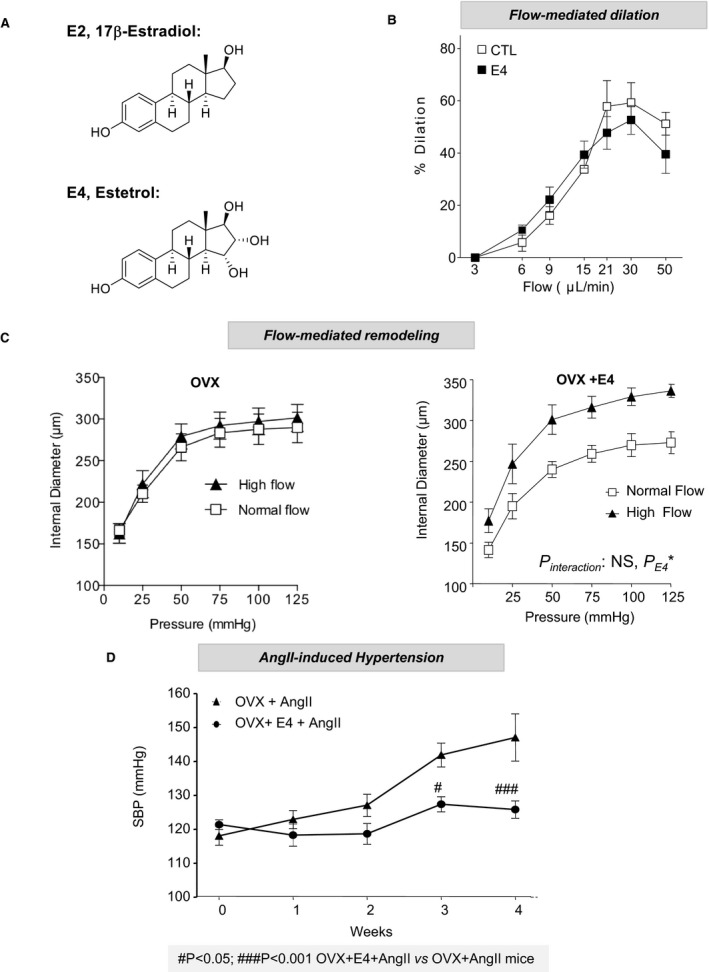
Selective activation of nuclear estrogen receptor α using estetrol (E4) prevents angiotensin II (AngII)–induced hypertension and favors flow‐mediating remodeling. A, Chemical structures of 17β‐estradiol (E2) and E4. B, Short‐term flow‐mediated dilation was assessed in pressurized precontracted mesenteric arteries. Vasodilating response to stepwise increase in intraluminal flow was evaluated in the presence of E4 (30 minutes) or vehicle (n=8). C, Flow‐mediated remodeling was evaluated in mesenteric arteries isolated from ovariectomized (OVX) mice treated chronically with vehicle or E4 over 2 weeks (n=6). Arterial diameter was measured in response to stepwise increases in pressure in mesenteric arteries submitted chronically to high flow or to normal flow. D, Effect of E4 on AngII treatment was evaluated in wild‐type female mice OVX 2 weeks before sham surgery or simultaneous implantation with osmotic minipumps delivering AngII (1 month) and E4 (n=7). Mean of systolic blood pressure (SBP) measurements for 5 days were represented as weekly evolution of blood pressure pre‐AngII (week 0) and post‐AngII treatment. CTL indicates control.
Discussion
Estrogen effects are mainly mediated by ERα, which acts primarily as a nuclear receptor/transcriptional factor, but which can also elicit rapid MISS. Because ERα MISS is necessary and sufficient to increase NO production and to accelerate postinjury reendothelialization without impact on sex targets, it was initially proposed that selective activation of membrane ERα could confer arterial protection with minimal risk of uterine or breast cancer.23, 26 In the present study, we investigated the role of ERα eliciting membrane actions in the prevention of atheroma and hypertension as well as in the promotion of arteriolar outward remodeling.
First, we addressed the question of the necessary role of ERα MISS on 3 major vasculoprotective effects of estrogens. To this aim, on the basis of in vitro work demonstrating that the palmitoylation site at Cys447 of human ERα (the mouse counterpart of ERα Cys451) is crucial for the membrane localization of ERα,39 we generated a mouse model with a point mutation of this amino acid (ERα‐C451A).22 The decrease of membrane localization of ERα was confirmed in primary hepatocytes and resulted in female infertility, an increase in luteinizing hormone levels, and hemorrhagic ovaries lacking corpora lutea.22 By contrast, the transcriptional action of 17β‐estradiol on the uterus was preserved: gene expression was similar in ERα‐C451A and WT mice.22 Herein, we show that the impact of MISS on short‐term 17β‐estradiol–dependent gene regulation in the aorta is also minimal, extending the previous observation to an arterial tissue. We also demonstrate that palmitoylation of ERα‐C451, and thereby the activation of ERα MISS, is dispensable for the prevention by 17β‐estradiol of early stages of atheroma development at the aortic sinus, but also of more advanced lesions on the thoracic and abdominal aorta.
In addition, long‐term treatment with AngII induced a significantly higher increase in SBP and media aortic hypertrophy in the absence of endogenous estrogens (ovariectomized mice) or of ERα (ERα−/− mice). Our finding is in agreement with previous works showing that hypertension induced by AngII is lower in female than in male mice.40, 41 The protective effect of 17β‐estradiol is, at least in part, mediated by the AngII type 2 receptor.42 This receptor is counteracting the effect of the type 1 receptor with vasodilator effect not subjected to desensitization,43 although in hypertension, this balance is not necessarily in favor of a vasodilator tone.44, 45 The measurement of blood pressure in mice using plethysmography (tail cuff) can be considered as a limitation, because this technique is less accurate than telemetry. However, this limitation has to be taken with caution as regard to baseline or central blood pressure with the tail cuff method and some concern about the impact of the surgery for telemetry.46 Nevertheless, the increase in pressure induced by AngII is the same whether it is measured using plethysmography or telemetry.46 Our work is in agreement with previous works showing that ERα was involved in the protective effect of 17β‐estradiol against AngII‐induced hypertension.47 Interestingly, a protective role for ERα has been also suggested in pulmonary hypertension.48 The present study definitively demonstrates the role of ERα in this beneficial effect of endogenous estrogens independently on ERα MISS.
Nevertheless, a role for ERβ has been also shown in basal arterial blood pressure. Indeed, basal arterial blood pressure increases more in ERβ−/− mice during aging than in age‐matched control mice.49 In addition, the pharmacological activation of ERβ leads to blood pressure lowering in spontaneously hypertensive rats with reduced myocardial hypertrophy.50 Arias‐Loza et al also show that both ERα and ERβ activation attenuates cardiovascular remodeling in aldosterone salt‐treated rats.51 In the present study, we observed no protective role of ERβ against AngII‐mediated hypertension in adult female mice (Figure S2). Thus, both ERα and ERβ could contribute to the control of arterial blood pressure, according to the context (aging versus AngII‐induced hypertension), and their precise role and how they interact remain to be further investigated. In addition, beside ERs, G protein‐coupled estrogen receptor 1 (GPER), has emerged as a third ER. Despite conflicting results obtained with the different mice models of GPER inactivation,52 GPER activation has been reported to exert several beneficial effects in the cardiovascular system, including protection against atherosclerosis and hypertension.53, 54 Treatment with the selective agonist G‐1 reduces atherosclerosis in ovariectomized mice, and the beneficial effects of GPER in this model are associated with a reduction in macrophage and T‐cell recruitment, indicating an anti‐inflammatory mechanism.55 In addition, activation of GPER has been reported to protect female mice from salt‐ and pressure‐induced vascular damage.56, 57 Altogether, although activation of nuclear, but not membrane, ERα is necessary to induce a protection against atherosclerosis and hypertension, we cannot exclude the participation of other membrane‐initiated steroid signal elicited through GPER‐mediated pathway.
Because hypertension is a major risk factor for cardiovascular diseases and especially for ischemic disorders, we also investigated FMR, which has a major role in the homeostasis of tissue perfusion. Indeed, FMR consists of vascular enlargement or outward remodeling induced by flow (shear stress) in small collateral arteries surrounding ischemic areas.38 It contributes to the prevention of further tissue injury (eg, in limb or myocardial ischemia). FMR has been shown to be reduced by hypertension,58, 59 diabetes mellitus,60, 61 and aging.11, 62 More important and rather unexpectedly, we previously reported that FMR depends on the presence of ERα15, 63 and could contribute to the better resistance to ischemia/necrosis of female compared with male mice. Herein, we report that ERα MISS is also dispensable to mediate this beneficial action of estrogens.
We previously demonstrated22 and confirmed herein, using another experimental approach, that ERα MISS is critical for the production of NO. Because NO is a well‐recognized vasculoprotective mediator and guardian of arterial integrity,27 and because 17β‐estradiol stimulates endothelial NO production through ERα MISS activation,22 one of the main surprises and unexpected findings in the present study is the absence of a role of ERα MISS on vascular protection. Indeed, it is largely believed that the increase in endothelium‐derived NO plays an important role in the vasculoprotective actions of estrogens, in particular in their atheroprotective action.18, 64 Interestingly, present finding is strikingly consistent with our previous work published 20 years ago, reporting that the atheroprotective effect of exogenous 17β‐estradiol was not altered by NOS inhibition.65 Maeda's group subsequently refined this conclusion using hypercholesterolemic eNOS−/− mice, showing that eNOS gene inactivation favored atheroma through hypertension, whereas the lack of local endothelial NO after blood pressure normalization did not contribute to the atheroprotective effect of 17β‐estradiol.66 In addition, the absence of caveolin‐1, which is involved in ERα‐associated eNOS activation at the level of the plasma membrane, had no impact on FMR.67
Then, in addition to genetic model of loss of function, pharmacological tools, such as PaPE and EDC, represent a complementary approach to evaluate ERα MISS action. Contrary to their stimulatory action on reendothelialization and on NO production,22, 23 none of these molecules was able to prevent atheroma LDLr−/− mice. Interestingly, a similar finding was previously reported in EDC‐treated apolipoprotein E–deficient mice.68 Accordingly, we previously showed that 17β‐estradiol failed to induce any atheroprotective action using mouse model in which nuclear effects are abrogated (ERα‐AF20/LDLr−/− mice), highlighting the requirement of nuclear/transcriptional actions of ERα for early atheroprotection at the level of the aortic sinus.32 Herein, we further extend the crucial role of ERαAF2 in atheroma prevention by endogenous estrogens through en face analysis of the thoracic and abdominal aorta (ie, in arterial sites where the shear stress is less dramatically altered compared with that at the aortic sinus). Moreover, the present study allows us to identify the crucial role of ERαAF2 in the protective effect of estrogens against high blood pressure and to promote FMR. In line with the genetic approach showing the necessary role of nuclear ERα, the response of estetrol, which has an unusual profile of ERα activation, and uncoupling nuclear and membrane activation28 demonstrate the vasculoprotective potential of nuclear ERα activation alone. Indeed, we previously reported the potent atheroprotective effect of estetrol28 as well as its capacity to prevent neointimal hyperplasia after endovascular femoral artery injury.14 Herein, we show that estetrol prevents hypertension and vascular hypertrophy induced by AngII and allows FMR. These experimental data complement previous clinical studies reporting that estetrol has less effect than ethinyl‐estradiol on hepatic‐derived hemostatic biomarkers69 and, therefore, could be the only oral estrogen that does not increase the risk of thromboembolic events. Because estetrol, in combination with a progestin, is able to block ovulation, this fetal estrogen is being evaluated as both a new contraceptive and a new hormonal treatment for menopause.69
To conclude, the combination of genetically modified mouse models (membrane and nuclear loss of function) and pharmacological approaches (membrane and nuclear gain of function) is unique in the area of nuclear receptors. This combination has led to the following conclusions, summarized in Figure 8: (1) ERα MISS is necessary and sufficient to promote 2 short‐term actions of 17β‐estradiol on the endothelium: the acceleration of endothelial wound healing, which involves mainly endothelial cell migration,70 and the increase in endothelial NO production. (2) By contrast, ERα MISS is neither necessary nor sufficient for eliciting the long‐term effect of estrogens on arteries (ie, protection against neointimal hyperplasia,14 atheroma,28 hypertension, and induction of FMR). (3) Accordingly, the nuclear ERα activation elicited by estetrol (that does not activate ERα MISS) is sufficient to confer these 4 major vasculoprotective actions. These results shed a new light on the mechanisms of the vasculoprotective effects of estrogens and will help to understand the tissue‐specific actions of selective ER modulators, one of the major mysteries of ER biological features.
Figure 8.
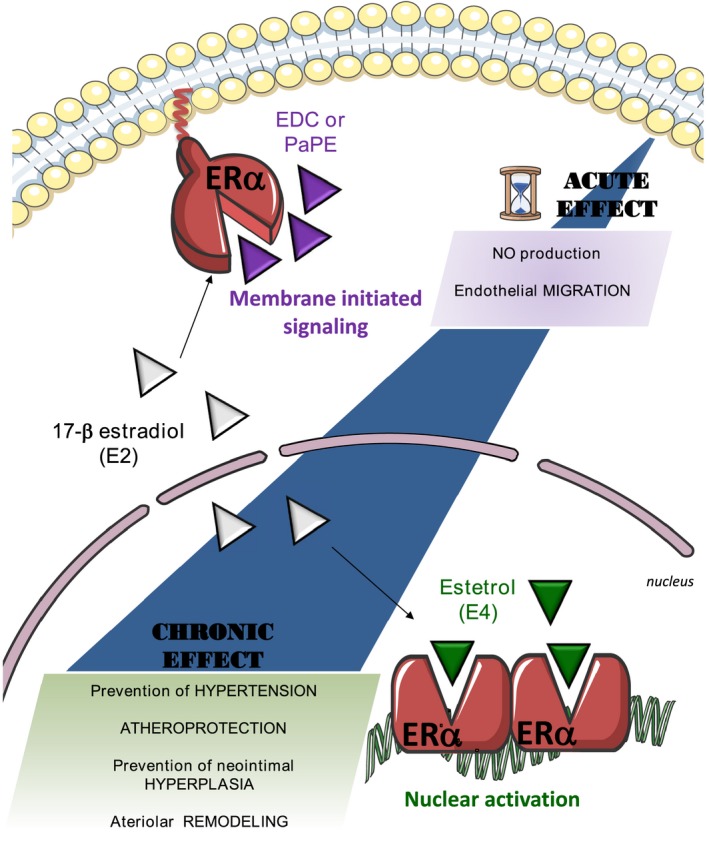
The beneficial actions of estrogen receptor α (ERα) activation on atheroma, hypertension, neointimal hyperplasia, and arterial remodeling all rely on nuclear ERα, whereas ERα membrane‐initiated signaling is restricted to the rapid endothelial actions. E2 indicates 17β‐estradiol; E4, estetrol; EDC, estrogen‐dendrimer conjugate; and PaPE, pathway preferential estrogen.
Sources of Funding
The work at INSERM U1048 was supported by INSERM, centre hospitalo universitaire (CHU) de Toulouse, Université de Toulouse III, Faculté de Médecine Toulouse‐Rangueil, Institut Universitaire de France, Fondation de France, Fondation pour la Recherche Médicale (DEQ20160334924), Agence Nationale de la Recherche (ANR‐14‐CE12‐0021‐01), Conseil Régional Midi‐Pyrénées, and the Eurostar project Septime. Support was also provided by the National Institutes of Health (PHS 5R01 DK015556 to J.A. Katzenellenbogen and P50 AT006268 to B.S. Katzenellenbogen). The work at Mitovasc was supported by INSERM, Centre national de la recherche scientifique (CNRS) and the centre hospitalo universitaire (CHU) d'Angers, Université of Angers, Fondation de France, and the Region des Pays de Loire (Mitovasc grant). Favre was financially supported by the Société d'Hypertension Artérielle and the Foundation de Recherche sur l'Hypertension Artérielle.
Disclosures
Foidart is a consultant at Mithra, the company that develops estetrol‐based women's healthcare products and has received a research grant. This relationship is significant. The remaining authors have no disclosures to report.
Supporting information
Figure S1. Effect of estrogen depletion on systolic blood pressure and aortic remodeling. A, Systolic blood pressure measurements in intact wild type (WT) and in ovariectomized (OVX) mice implanted with osmotic minipumps delivering angiotensin II (Ang II, 500 ng/kg per day, 1 month) to induce hypertension or sham operated (Sham). B, Media/Lumen Ratio and (C) Representative images of aortic remodeling in WT and WT‐OVX mice. Values are presented as mean±SEM (n=6), and statistically compared to respective control group. *P<0.05;** P<0.01; *** P<0.001 AngII vs respective sham; ## P<0.01 WT vs WT‐OVX.
Figure S2. Effect of the absence of estrogen receptor beta (ERbeta) on systolic blood pressure in mice. Systolic blood pressure measurements in intact wild type (ERbeta+/+) and in mice lacking ERbeta (ERbeta−/−) mice implanted with osmotic minipumps delivering angiotensin II (AngII, 500 ng/kg per day, 1 month) to induce hypertension or sham operated. Values are presented as mean±SEM (n=6 per group), and statistically compared to respective control group.*P<0.05; AngII vs respective control.
Figure S3. Gene expression level of the angiotensin converting enzymes Ace andAce2 and of the angiotensin II type 1 receptor (AT1Ra and AT1Rb) measured in intact wild type (WT) and in ERa−/−, ERaC451A and ERaAF20 mice. Values are presented as mean±SEM (n=5 per group), and statistically compared to the WT group. No siginficant difference was observed between groups.
Acknowledgments
The animal facilities and the “Plateforme d'Experimentation Fonctionnelle” staff are acknowledged for skillful technical assistance. Estrogen receptor α–deficient and estrogen receptor α activation function‐2 deficient mice (ERaAF2°) mice were kindly provided by Professor P. Chambon and Dr A. Krust.
(J Am Heart Assoc. 2018;7:e008950 DOI: 10.1161/JAHA.118.008950.)
References
- 1. Barton M, Meyer MR. Postmenopausal hypertension: mechanisms and therapy. Hypertension. 2009;54:11–18. [DOI] [PubMed] [Google Scholar]
- 2. Regitz‐Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. 2017;97:1–37. [DOI] [PubMed] [Google Scholar]
- 3. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA. 2002;288:321–333. [DOI] [PubMed] [Google Scholar]
- 4. Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski‐Wende J, Wallace R, Wassertheil‐Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the women's health initiative randomized controlled trial. JAMA. 2004;291:1701–1712. [DOI] [PubMed] [Google Scholar]
- 5. Vitale C, Mercuro G, Cerquetani E, Marazzi G, Patrizi R, Pelliccia F, Volterrani M, Fini M, Collins P, Rosano GM. Time since menopause influences the acute and chronic effect of estrogens on endothelial function. Arterioscler Thromb Vasc Biol. 2008;28:348–352. [DOI] [PubMed] [Google Scholar]
- 6. Elhage R, Arnal JF, Pierragi M‐T, Duverger N, Fiévet C, Faye JC, Bayard F. Estradiol‐17β prevents fatty streak formation in apolipoprotein e‐deficient mice. Arterioscler Thromb Vasc Biol. 1997;17:2679–2684. [DOI] [PubMed] [Google Scholar]
- 7. Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta‐estradiol's atheroprotective effects on lesion size in apoe‐/‐ mice. J Clin Invest. 2001;107:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Billon‐Gales A, Fontaine C, Douin‐Echinard V, Delpy L, Berges H, Calippe B, Lenfant F, Laurell H, Guery JC, Gourdy P, Arnal JF. Endothelial estrogen receptor‐alpha plays a crucial role in the atheroprotective action of 17beta‐estradiol in low‐density lipoprotein receptor‐deficient mice. Circulation. 2009;120:2567–2576. [DOI] [PubMed] [Google Scholar]
- 9. Pollow DP Jr, Romero‐Aleshire MJ, Sanchez JN, Konhilas JP, Brooks HL. Ang II‐induced hypertension in the VCD mouse model of menopause is prevented by estrogen replacement during perimenopause. Am J Physiol Regul Integr Comp Physiol. 2015;309:R1546–R1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chappell MC, Gallagher PE, Averill DB, Ferrario CM, Brosnihan KB. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2: Lewis rat. Hypertension. 2003;42:781–786. [DOI] [PubMed] [Google Scholar]
- 11. Tarhouni K, Guihot AL, Vessieres E, Toutain B, Procaccio V, Grimaud L, Loufrani L, Lenfant F, Arnal JF, Henrion D. Determinants of flow‐mediated outward remodeling in female rodents: respective roles of age, estrogens, and timing. Arterioscler Thromb Vasc Biol. 2014;34:1281–1289. [DOI] [PubMed] [Google Scholar]
- 12. Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J, Chambon P, Bayard F, Arnal JF. Estradiol alters nitric oxide production in the mouse aorta through the alpha‐, but not beta‐, estrogen receptor. Circ Res. 2002;90:413–419. [DOI] [PubMed] [Google Scholar]
- 13. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor‐alpha but not estrogen receptor‐beta. Circulation. 2001;103:423–428. [DOI] [PubMed] [Google Scholar]
- 14. Smirnova NF, Fontaine C, Buscato M, Lupieri A, Vinel A, Valera MC, Guillaume M, Malet N, Foidart JM, Raymond‐Letron I, Lenfant F, Gourdy P, Katzenellenbogen BS, Katzenellenbogen J, Laffargue M, Arnal JF. The activation function‐1 of estrogen receptor alpha prevents arterial neointima development through a direct effect on smooth muscle cells. Circ Res. 2015;117:770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tarhouni K, Guihot AL, Freidja ML, Toutain B, Henrion B, Baufreton C, Pinaud F, Procaccio V, Grimaud L, Ayer A, Loufrani L, Lenfant F, Arnal JF, Henrion D. Key role of estrogens and endothelial estrogen receptor alpha in blood flow‐mediated remodeling of resistance arteries. Arterioscler Thromb Vasc Biol. 2013;33:605–611. [DOI] [PubMed] [Google Scholar]
- 16. Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, Fontaine C, Gourdy P, Chambon P, Katzenellenbogen B, Katzenellenbogen J. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol Rev. 2017;97:1045–1087. [DOI] [PubMed] [Google Scholar]
- 17. Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. Estrogen receptor‐beta prevents cardiac fibrosis. Mol Endocrinol. 2010;24:2152–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arnal JF, Fontaine C, Billon‐Gales A, Favre J, Laurell H, Lenfant F, Gourdy P. Estrogen receptors and endothelium. Arterioscler Thromb Vasc Biol. 2010;30:1506–1512. [DOI] [PubMed] [Google Scholar]
- 19. Toutain CE, Filipe C, Billon A, Fontaine C, Brouchet L, Guery JC, Gourdy P, Arnal JF, Lenfant F. Estrogen receptor alpha expression in both endothelium and hematopoietic cells is required for the accelerative effect of estradiol on reendothelialization. Arterioscler Thromb Vasc Biol. 2009;29:1543–1550. [DOI] [PubMed] [Google Scholar]
- 20. Arnal JF, Fontaine C, Abot A, Valera MC, Laurell H, Gourdy P, Lenfant F. Lessons from the dissection of the activation functions (Af‐1 and AF‐2) of the estrogen receptor alpha in vivo. Steroids. 2013;78:576–582. [DOI] [PubMed] [Google Scholar]
- 21. Fu XD, Simoncini T. Extra‐nuclear signaling of estrogen receptors. IUBMB Life. 2008;60:502–510. [DOI] [PubMed] [Google Scholar]
- 22. Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond‐Letron I, Guihot AL, Boudou F, Sautier L, Vessieres E, Kim SH, Liere P, Fontaine C, Krust A, Chambon P, Katzenellenbogen JA, Gourdy P, Shaul PW, Henrion D, Arnal JF, Lenfant F. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue‐specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111:E283–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak‐Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non‐nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrington WR, Kim SH, Funk CC, Madak‐Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. [DOI] [PubMed] [Google Scholar]
- 25. Keung W, Chan ML, Ho EY, Vanhoutte PM, Man RY. Non‐genomic activation of adenylyl cyclase and protein kinase G by 17beta‐estradiol in vascular smooth muscle of the rat superior mesenteric artery. Pharmacol Res. 2011;64:509–516. [DOI] [PubMed] [Google Scholar]
- 26. Madak‐Erdogan Z, Kim SH, Gong P, Zhao YC, Zhang H, Chambliss KL, Carlson KE, Mayne CG, Shaul PW, Korach KS, Katzenellenbogen JA, Katzenellenbogen BS. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal. 2016;9:ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713–735. [DOI] [PubMed] [Google Scholar]
- 28. Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, Drougard A, Rajan S, Laine M, Milon A, Muller I, Henrion D, Adlanmerini M, Valera MC, Gompel A, Gerard C, Pequeux C, Mestdagt M, Raymond‐Letron I, Knauf C, Ferriere F, Valet P, Gourdy P, Katzenellenbogen BS, Katzenellenbogen JA, Lenfant F, Greene GL, Foidart JM, Arnal JF. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor alpha modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. 2014;6:1328–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerard C, Blacher S, Communal L, Courtin A, Tskitishvili E, Mestdagt M, Munaut C, Noel A, Gompel A, Pequeux C, Foidart JM. Estetrol is a weak estrogen antagonizing estradiol‐dependent mammary gland proliferation. J Endocrinol. 2015;224:85–95. [DOI] [PubMed] [Google Scholar]
- 30. Acconcia F, Marino M. The effects of 17beta‐estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front Physiol. 2011;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2008;108:351–361. [DOI] [PubMed] [Google Scholar]
- 32. Billon‐Gales A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, Berges H, Gadeau AP, Lenfant F, Gourdy P, Chambon P, Arnal JF. Activation function 2 (AF2) of estrogen receptor‐{alpha} is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci USA. 2011;108:13311–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roy C, Tabiasco J, Caillon A, Delneste Y, Merot J, Favre J, Guihot AL, Martin L, Nascimento DC, Ryffel B, Robson SC, Sevigny J, Henrion D, Kauffenstein G. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase‐1/CD39 in hypertension. Purinergic Signal. 2018;14:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dowell FJ, Henrion D, Benessiano J, Poitevin P, Levy B. Chronic infusion of low‐dose angiotensin ii potentiates the adrenergic response in vivo. J Hypertens. 1996;14:177–182. [DOI] [PubMed] [Google Scholar]
- 35. Henrion D, Terzi F, Matrougui K, Duriez M, Boulanger CM, Colucci‐Guyon E, Babinet C, Briand P, Friedlander G, Poitevin P, Levy BI. Impaired flow‐induced dilation in mesenteric resistance arteries from mice lacking vimentin. J Clin Invest. 1997;100:2909–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubroca C, Loyer X, Retailleau K, Loirand G, Pacaud P, Feron O, Balligand JL, Levy BI, Heymes C, Henrion D. Rhoa activation and interaction with caveolin‐1 are critical for pressure‐induced myogenic tone in rat mesenteric resistance arteries. Cardiovasc Res. 2007;73:190–197. [DOI] [PubMed] [Google Scholar]
- 37. Lu Q, Schnitzler GR, Vallaster CS, Ueda K, Erdkamp S, Briggs CE, Iyer LK, Jaffe IZ, Karas RH. Unliganded estrogen receptor alpha regulates vascular cell function and gene expression. Mol Cell Endocrinol. 2017;442:12–23. [DOI] [PubMed] [Google Scholar]
- 38. Silvestre JS, Smadja DM, Levy BI. Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol Rev. 2013;93:1743–1802. [DOI] [PubMed] [Google Scholar]
- 39. Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, Visca P , Marino M. Palmitoylation‐dependent estrogen receptor alpha membrane localization: regulation by 17beta‐estradiol. Mol Biol Cell. 2005;16:231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low‐dose angiotensin II in female rats. Hypertension. 2008;52:666–671. [DOI] [PubMed] [Google Scholar]
- 41. Barsha G, Denton KM, Mirabito Colafella KM. Sex‐ and age‐related differences in arterial pressure and albuminuria in mice. Biol Sex Differ. 2016;7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mirabito KM, Hilliard LM, Head GA, Widdop RE, Denton KM. Pressor responsiveness to angiotensin II in female mice is enhanced with age: role of the angiotensin type 2 receptor. Biol Sex Differ. 2014;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Widdop RE, Matrougui K, Levy BI, Henrion D. AT2 receptor‐mediated relaxation is preserved after long‐term AT1 receptor blockade. Hypertension. 2002;40:516–520. [DOI] [PubMed] [Google Scholar]
- 44. Matrougui K, Levy BI, Henrion D. Tissue angiotensin II and endothelin‐1 modulate differently the response to flow in mesenteric resistance arteries of normotensive and spontaneously hypertensive rats. Br J Pharmacol. 2000;130:521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. You D, Loufrani L, Baron C, Levy BI, Widdop RE, Henrion D. High blood pressure reduction reverses angiotensin II type 2 receptor‐mediated vasoconstriction into vasodilation in spontaneously hypertensive rats. Circulation. 2005;111:1006–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilde E, Aubdool AA, Thakore P, Baldissera L Jr, Alawi KM, Keeble J, Nandi M, Brain SD. Tail‐cuff technique and its influence on central blood pressure in the mouse. J Am Heart Assoc. 2017;6:e005204 DOI: 10.1161/jaha.116.005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor‐alpha mediates estrogen protection from angiotensin II‐induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol. 2007;292:H1770–H1776. [DOI] [PubMed] [Google Scholar]
- 48. Wright AF, Ewart MA, Mair K, Nilsen M, Dempsie Y, Loughlin L, Maclean MR. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res. 2015;106:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–508. [DOI] [PubMed] [Google Scholar]
- 50. Jazbutyte V, Arias‐Loza PA, Hu K, Widder J, Govindaraj V, von Poser‐Klein C, Bauersachs J, Fritzemeier KH, Hegele‐Hartung C, Neyses L, Ertl G, Pelzer T. Ligand‐dependent activation of ER{beta} lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc Res. 2008;77:774–781. [DOI] [PubMed] [Google Scholar]
- 51. Arias‐Loza PA, Hu K, Dienesch C, Mehlich AM, Konig S, Jazbutyte V, Neyses L, Hegele‐Hartung C, Heinrich Fritzemeier K, Pelzer T. Both estrogen receptor subtypes, alpha and beta, attenuate cardiovascular remodeling in aldosterone salt‐treated rats. Hypertension. 2007;50:432–438. [DOI] [PubMed] [Google Scholar]
- 52. Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C. A critical review of fundamental controversies in the field of GPR30 research. Steroids. 2010;75:603–610. [DOI] [PubMed] [Google Scholar]
- 53. Meyer MR, Barton M. Estrogens and coronary artery disease: new clinical perspectives. Adv Pharmacol. 2016;77:307–360. [DOI] [PubMed] [Google Scholar]
- 54. Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. Gper‐novel membrane oestrogen receptor. Clin Sci. 2016;130:1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, Amann K, Arterburn JB, Barton M, Prossnitz ER. G protein‐coupled estrogen receptor protects from atherosclerosis. Sci Rep. 2014;4:7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein‐coupled receptor 30 agonist G‐1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu L, Kashyap S, Murphy B, Hutson DD, Budish RA, Trimmer EH, Zimmerman MA, Trask AJ, Miller KS, Chappell MC, Lindsey SH. GPER activation ameliorates aortic remodeling induced by salt‐sensitive hypertension. Am J Physiol Heart Circ Physiol. 2016;310:H953–H961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tuttle JL, Sanders BM, Burkhart HM, Fath SW, Kerr KA, Watson WC, Herring BP, Dalsing MC, Unthank JL. Impaired collateral artery development in spontaneously hypertensive rats. Microcirculation. 2002;9:343–351. [DOI] [PubMed] [Google Scholar]
- 59. Dumont O, Kauffenstein G, Guihot AL, Guerineau NC, Abraham P, Loufrani L, Henrion D. Time‐related alteration in flow‐ (shear stress‐) mediated remodeling in resistance arteries from spontaneously hypertensive rats. Int J Hypertens. 2014;2014:859793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. deBelin Chantemele EJ , Vessieres E, Guihot AL, Toutain B, Maquignau M, Loufrani L, Henrion D. Type 2 diabetes severely impairs structural and functional adaptation of rat resistance arteries to chronic changes in blood flow. Cardiovasc Res. 2009;81:788–796. [DOI] [PubMed] [Google Scholar]
- 61. Freidja ML, Tarhouni K, Toutain B, Fassot C, Loufrani L, Henrion D. The AGE‐breaker ALT‐711 restores high blood flow‐dependent remodeling in mesenteric resistance arteries in a rat model of type 2 diabetes. Diabetes. 2012;61:1562–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dumont O, Pinaud F, Guihot AL, Baufreton C, Loufrani L, Henrion D. Alteration in flow (shear stress)‐induced remodelling in rat resistance arteries with aging: improvement by a treatment with hydralazine. Cardiovasc Res. 2008;77:600–608. [DOI] [PubMed] [Google Scholar]
- 63. Tarhouni K, Freidja ML, Guihot AL, Vessieres E, Grimaud L, Toutain B, Lenfant F, Arnal JF, Loufrani L, Henrion D. Role of estrogens and age in flow‐mediated outward remodeling of rat mesenteric resistance arteries. Am J Physiol Heart Circ Physiol. 2014;307:H504–H514. [DOI] [PubMed] [Google Scholar]
- 64. Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;23:665–686. [DOI] [PubMed] [Google Scholar]
- 65. Elhage R, Bayard F, Richard V, Holvoet P, Duverger N, Fievet C, Arnal JF. Prevention of fatty streak formation of 17beta‐estradiol is not mediated by the production of nitric oxide in apolipoprotein E‐deficient mice. Circulation. 1997;96:3048–3052. [DOI] [PubMed] [Google Scholar]
- 66. Hodgin JB, Knowles JW, Kim HS, Smithies O, Maeda N. Interactions between endothelial nitric oxide synthase and sex hormones in vascular protection in mice. J Clin Invest. 2002;109:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERS associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100–115. [DOI] [PubMed] [Google Scholar]
- 68. Chambliss KL, Barrera J, Umetani M, Umetani J, Kim SH, Madak‐Erdogan Z, Huang L, Katzenellenbogen BS, Katzenellenbogen JA, Mineo C, Shaul PW. Nonnuclear estrogen receptor activation improves hepatic steatosis in female mice. Endocrinology. 2016;157:3731–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kluft C, Zimmerman Y, Mawet M, Klipping C, Duijkers IJ, Neuteboom J, Foidart JM, Bennink HC. Reduced hemostatic effects with drospirenone‐based oral contraceptives containing estetrol vs. ethinyl estradiol. Contraception. 2017;95:140–147. [DOI] [PubMed] [Google Scholar]
- 70. Filipe C, Lam Shang Leen L, Brouchet L, Billon A, Benouaich V, Fontaine V, Gourdy P, Lenfant F, Arnal JF, Gadeau AP, Laurell H. Estradiol accelerates endothelial healing through the retrograde commitment of uninjured endothelium. Am J Physiol. 2008;294:H2822–H2830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of estrogen depletion on systolic blood pressure and aortic remodeling. A, Systolic blood pressure measurements in intact wild type (WT) and in ovariectomized (OVX) mice implanted with osmotic minipumps delivering angiotensin II (Ang II, 500 ng/kg per day, 1 month) to induce hypertension or sham operated (Sham). B, Media/Lumen Ratio and (C) Representative images of aortic remodeling in WT and WT‐OVX mice. Values are presented as mean±SEM (n=6), and statistically compared to respective control group. *P<0.05;** P<0.01; *** P<0.001 AngII vs respective sham; ## P<0.01 WT vs WT‐OVX.
Figure S2. Effect of the absence of estrogen receptor beta (ERbeta) on systolic blood pressure in mice. Systolic blood pressure measurements in intact wild type (ERbeta+/+) and in mice lacking ERbeta (ERbeta−/−) mice implanted with osmotic minipumps delivering angiotensin II (AngII, 500 ng/kg per day, 1 month) to induce hypertension or sham operated. Values are presented as mean±SEM (n=6 per group), and statistically compared to respective control group.*P<0.05; AngII vs respective control.
Figure S3. Gene expression level of the angiotensin converting enzymes Ace andAce2 and of the angiotensin II type 1 receptor (AT1Ra and AT1Rb) measured in intact wild type (WT) and in ERa−/−, ERaC451A and ERaAF20 mice. Values are presented as mean±SEM (n=5 per group), and statistically compared to the WT group. No siginficant difference was observed between groups.