

Abstract
Background
Cholesterol oxides, the oxygenated derivatives of cholesterol, have been shown to cause programmed cell death in a variety of cell types. Using N9 microglia, this study was designed to investigate the molecular events induced by cholesterol oxides prior to the execution of programmed cell death.
Results
Microglia were very sensitive to 25-OH-cholesterol, such that a 2-day treatment of the cells with 5 μM 25-OH-cholesterol reduced cell viability to 5–10% of controls. There was a dose- and time-dependent increase in c-jun and phospho-c-jun levels in microglia prior to this 25-OH-cholesterol induced cell death. In contrast, 7-β-OH-cholesterol, which was relatively non-toxic to microglia, did not increase phospho-c-jun levels. Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptors that have important roles in atherogenesis. Results from this study indicate that PPAR agonists such as 15d-PGJ2, indomethacin and WY14643 can attenuate cholesterol oxide induced c-jun activation and cell death in microglia.
Conclusions
Peroxisome proliferator-activated receptor agonists may be useful in future development of pharmacological agents against cholesterol oxide induced cytotoxicity.
Background
High levels of serum cholesterol and low density lipoprotein (LDL) have been associated with the genesis of atherosclerosis, a leading cause of cardiovascular diseases that affect a large number of people all over the world. It has become clear that the pathological conditions associated with an excess level of LDL are actually caused by the oxidized products of LDL [1]. More specifically, cholesterol oxides (also termed oxysterols), the oxygenated derivatives of cholesterol, appear to be the major cytotoxic components in oxidized LDL [2,3]. These compounds have a hydroxyl- or a keto- group on the cholesterol molecule. Examples of cholesterol oxides include: 7-OH-, 7-keto-, 19-OH-, 22-OH- and 25-OH-cholesterol (see [4] for a recent review).
Tissue damage caused by cholesterol oxides has been the subject of many studies. Cholesterol oxides are cytotoxic to a variety of cell types [5]. These compounds can damage endothelial cells, smooth muscle cells and fibroblasts, all of which are major components of the arterial wall. Cholesterol oxides are also toxic to cells derived from the immune system, such as macrophages, thymocytes, lymphoma cells and leukemic T-cells. Pharmacological agents capable of reducing cholesterol oxide toxicity are yet to be discovered.
The mode of cell death caused by cholesterol oxides has generated much interest recently. There is evidence showing that programmed cell death (PCD, also known as apoptosis) occurs in some cells treated with cholesterol oxides [6-8]. PCD is a special type of cell death that can be induced by growth factor deprivation or toxins [9-11]. By using the PCD induced by nerve growth factor deprivation in sympathetic neurons as an example, the cell death process begins with the generation of reactive oxygen species followed by a significant decrease in glucose uptake, RNA and protein synthesis, activation of the immediate early protein, c-jun, and release of cytochrome c from mitochondria. Cell death is executed by the activation of a family of proteases termed caspases. Morphologically, cells dying of PCD appear atrophic and exhibit condensed nuclei, which can be stained with nuclear stains (bisbenzimide or propidium iodide), or by the TUNEL assay. DNA extracted from cells dying of PCD is often fragmented and shows a characteristic ladder-type pattern on the agarose gel upon electrophoresis.
MAP kinases (Mitogen-Activated Protein kinases) have very important roles in PCD. The MAP kinase pathway consists of 3 major parallel pathways designated as the ERK (p42/44, Extracellular signal-Regulated Kinase), SAPK/JNK (p46, Stress-Activated Protein Kinase/ c-Jun NH2-terminal Kinase) and p38 pathways [12-14]. Activation of each pathway above involves phosphorylation of a number of upstream and downstream family members of the pathway. During growth factor deprivation induced PCD in neuronal PC12 cells, there is a sustained activation of the JNK and p38 pathways while the activities of ERK pathway is inhibited. It was proposed that a dynamic balance of JNK-p38 and ERK can determine if the neuronal cell will live or die [15]. Similar interplay of the MAP kinases was shown in the PCD of non-neuronal cells [16]. It is known that oxidized LDL can cause activation of MAP kinases [17,18]. Whether cholesterol oxides, the main toxic components in oxidized LDL, can induce MAP kinase activation, and whether MAP kinase activation has a role in cholesterol oxide induced PCD remain to be investigated, and is the subject of this study.
Peroxisome proliferator-activated receptors (PPARs) belong to a group of nuclear receptors which includes steroid, retinoid, thyroid hormone receptors and others [19-21]. There are three types of PPARs: PPARα is found predominantly in the liver, heart, kidney, brown adipose and stomach mucosa, and is important for lipid catabolism. PPARγ is found in adipose tissues, and is important for adipogenesis. PPARβ is found in most tissues, but its role is less well-defined.
PPARγ is expressed in atherosclerotic lesions [22] and is important in the process of atherogenesis. Depending on the experimental system and the particular question asked, PPARγ can be involved with pro-atherogenesis or anti-atherogenesis. For example, PPARγ is involved in a positive feedback mechanism that induces the formation of foam cells [23]. On the other hand, activation of PPARγ was shown to inhibit monocyte [24] and macrophage [25] inflammatory responses by preventing the activation of nuclear transcription factors, such as NF-kB (nuclear factor-kappa B), AP-1 (activator protein-1) and STAT 1 (signal transducer and activator of transcription 1). Since inflammation plays an important role in atherogenesis, this anti-inflammatory effect of PPARγ should help to reduce the risk of atherogenesis. Development of PPAR agonists that retain the antiatherogenic activity but dismiss the proatherogenic activities will be of great value [26].
Many naturally occurring or synthetic compounds are agonists for the PPARs [20,21]. For example, a number of prostaglandins are agonists for PPARγ . The prostaglandin, 15-deoxy-delta 12, 14-PGJ2 (15d-PGJ2), is the most potent endogenous PPARγ agonist known. On the other hand, many synthetic compounds such as the non-steroid anti-inflammatory drugs (NSAIDs) are PPARα and PPARγ agonists [27]. Since these are agents used clinically for other purposes, it is important to determine whether interaction of these agents with PPARs could modulate atherogenesis in a positive or negative manner.
While most research regarding cholesterol oxides are focused on the cardiovascular system, it should be noted that the nervous tissue has the ability to generate cholesterol oxides, which may exert toxic effect on cells and tissues of the nervous system. For example, it has been shown that synaptosomes and mitochondria prepared from CNS tissue can convert cholesterol to cholesterol oxides, which may play an important role in brain tissue damage during oxidative stress [28]. Consistent with this notion, we have shown cholesterol oxides are toxic to neuronal PC12 cells [29,30] and neurons derived from sympathetic ganglia [31], retina [32] and cerebellum [33].
Microglia in the central nervous system express scavenger receptors and are involved in the oxidized LDL uptake and metabolism [34]. These cells are, thus, the macrophage-equivalent in the central nervous system [35]. Results from this laboratory showed that cholesterol oxides can cause programmed cell death in microglia [7], which suggests that microglia, similar to macrophages, can be a potential target for oxidized LDL toxicity.
Based on the observations that: 1. cholesterol oxides cause PCD but the involvement of MAP kinases remains to be investigated; 2. PPARs are important in atherosclerosis but the effect of PPAR activation on cholesterol oxide induced PCD has not been determined, this study was designed to test if a dysregulation of MAP kinases was associated with the cholesterol oxide induced PCD, and to determine if PPAR activation could modulate the cholesterol oxide induced PCD.
Results
Cytotoxicity of 25-OH-cholesterol toward microglia
A set of dose- and time-response experiments indicated that 5, 10 and 20 μM 25-OH-cholesterol treatment for one day reduced the viability to ~97, 70 and 52% of controls, respectively (Fig. 1). This agent at 5 μM reduced the viability to ~9% of controls after a 2-day treatment. In contrast, 7-β-OH-cholesterol was much less toxic to microglia, such that this agent at 20 μM showed little toxicity to the cells after a 1-day treatment (Fig. 2). A 3-day treatment of the cells with 20 μM 7-β-OH-cholesterol reduced the viability to ~81% of controls. These results confirmed and extended those obtained from our previous study [7].
Figure 1.

Effect of 25-OH-cholesterol on cell viability. Microglia were treated with various concentrations of 25-OH-cholesterol for 1 or 2 days, then the viability of each treatment was determined.
Figure 2.

Effect of 7-β-OH-cholesterol on cell viability. Microglia were treated with various concentrations of 7-β-OH-cholesterol for 1, 2 or 3 days, then the viability of each treatment was determined. Results from Fig. 1 and Fig. 2 indicated that 25-OH-cholesterol was more toxic to these cells as compared with 7-β-OH-cholesterol.
Activation of c-jun as a result of 25-OH-cholesterol treatment
Because c-jun was shown to play important roles in programmed cell death in many cell types [20,21], and microglia were shown to undergo programmed cell death after cholesterol oxide treatment [7], experiments were performed to determine if c-jun activation was involved in cholesterol oxide induced microglial cell death. Cells were treated with 10 μM 25-OH-cholesterol or 7-β-OH-cholesterol for 12 hours, then processed for Western blot analyses. Antibodies against c-jun, phospho-c-jun (at position serine 63) or ERK 1/2 (an antibody which can recognize both ERK 1 and ERK 2) were used in this set of experiments. Results indicated that both cholesterol oxides caused an increase in c-jun in microglia, with 25-OH-cholesterol showing a more pronounced increase (Fig. 3). Furthermore, 25-OH-cholesterol caused the phosphorylation of c-jun, a phenomenon not observed in 7-β-OH-cholesterol treated microglia. The levels of ERK 1/2 were not altered by either treatment. These results suggested that c-jun activation was associated with the toxicity of cholesterol oxides.
Figure 3.

Activation of c-jun by 25-OH-cholesterol. Cells were treated with 10 μM 25-OH-cholesterol or 10 μM 7-β-OH-cholesterol for 12 hours, then processed for Western blot analyses. Antibodies against c-jun, phospho-c-jun (at serine 63) and ERK 1/2 were used in this set of experiments. Concurrent electrophoresis procedures were performed for each antibody. Results indicated that c-jun activation, i.e., phosphorylation of c-jun, was associated with the toxicity of 25-OH-cholesterol.
A set of experiments was performed to determine any dose-dependent change in c-jun activation by 25-OH-cholesterol. Microglia were treated with 0, 1.25, 2.5, 5, 10 or 20 μM 25-OH-cholesterol for 12 hours, then processed for Western blot analysis. Results indicated that 25-OH-cholesterol treatment led to an increase in c-jun and phospho-c-jun at serine 63. The levels of ERK 1/2 were not altered by this treatment (Fig. 4).
Figure 4.
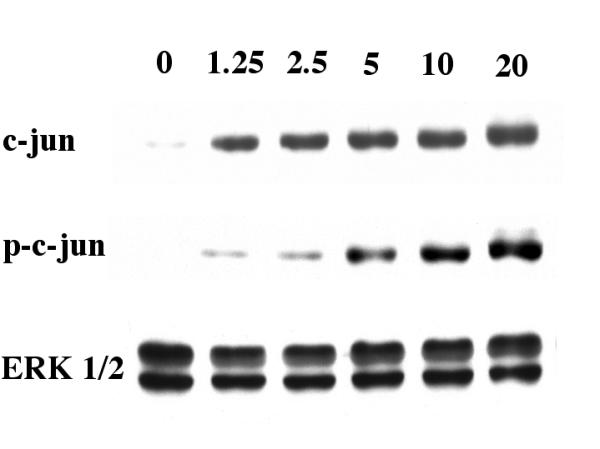
Dose-dependent effect of 25-OH-cholesterol on c-jun activation. Cells were treated with 0. 1.25, 2.5, 5, 10 or 20 μM 25-OH-cholesterol for 12 hours, then processed for Western blot analyses. Antibodies against c-jun, phospho-c-jun (at serine 63) and ERK 1/2 were used in this set of experiments. Concurrent electrophoresis procedures were performed for each antibody. Results indicated that 25-OH-cholesterol caused a dose-dependent increase in c-jun activation.
Time-dependent change of MAP kinase activities
Further studies were performed to determine the effect of 25-OH-cholesterol on different members of MAP kinases. The time-dependent change in c-jun, ERK and p38 levels was determined. Cells were treated with 10 μM 25-OH-cholesterol for 1, 3, 6, 12 or 24 hours, then processed for Western blot analyses. Results indicated that there was an increase in c-jun after 25-OH-cholesterol treatment between 1–12 hours of treatment. This protein dropped to a non-detectable level at 24 hours after treatment. In addition, there was an increase in the levels of phospho-c-jun on the serine residues (at both position 63 and position 73). The levels of JNK (JNK 1 and JNK 2), however, were not altered (Fig. 5).
Figure 5.
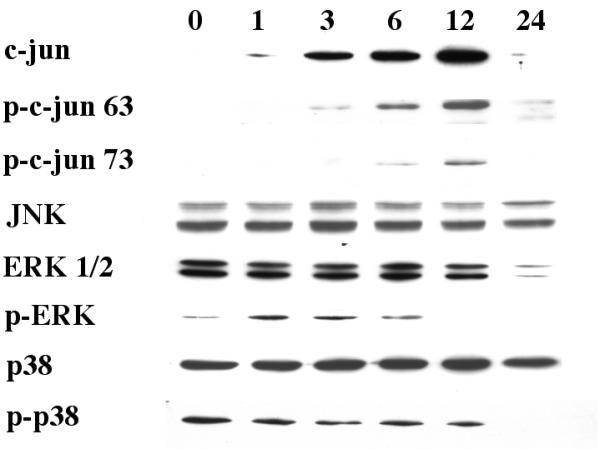
Time-dependent alterations of MAP kinases activity. Cells were treated with 10 μM 25-OH-cholesterol for 0, 1, 3, 6, 12 or 24 hours, then processed for Western blot analysis. Antibodies against c-jun, phospho-c-jun (serine 63 or serine 73), JNK, ERK 1/2, phospho-ERK, p38 and phospho-p38 were used in this series of experiments. See text for an analysis of the results.
The levels of ERK1/2 were similar during the first 6 hours, then dropped between 12–24 hours after treatment. The levels of phospho-ERK increased between 1–6 hours after treatment, then decreased to a non-detectable level afterwards (Fig. 5). This decrease in phospho-ERK expression occurred at a time coincided with the appearance of phospho-c-jun. The changes in p38 or phospho-p38 levels were minimal (Fig. 5).
Modulation of c-jun activation by PPAR agonists
It was reported that PPAR activation could down-regulate the activity of AP-1 by interfering with the functions of c-jun [36,37]. Therefore, the following set of experiments was designed to test if PPAR activation could reduce c-jun activation in 25-OH-cholesterol treated microglia. PPAR agonists including the prostaglandin, 15d-PGJ2, and two non-steroid anti-inflammatory drugs, indomethacin and ibuprofen, were tested in this series of experiments. Cells were treated with 5 μM 25-OH-cholesterol together with these PPAR agonists for 12 hours, then processed for Western blot analyses. Antibodies against c-jun, phospho-c-jun (at serine 63) and ERK 1/2 were used in this set of experiments. Results indicated that while 25-OH-cholesterol induced the expression of c-jun and phospho-c-jun, these PPAR agonists reduced the expression to various degrees. The apparent potency to reduce phospho-c-jun among the three agents tested was: 15d-PGJ2 > indomethacin > ibuprofen (Fig. 6). These results indicated that while 15d-PGJ2 was very effective in inhibiting c-jun activation, ibuprofen appeared to be ineffective in this regard.
Figure 6.
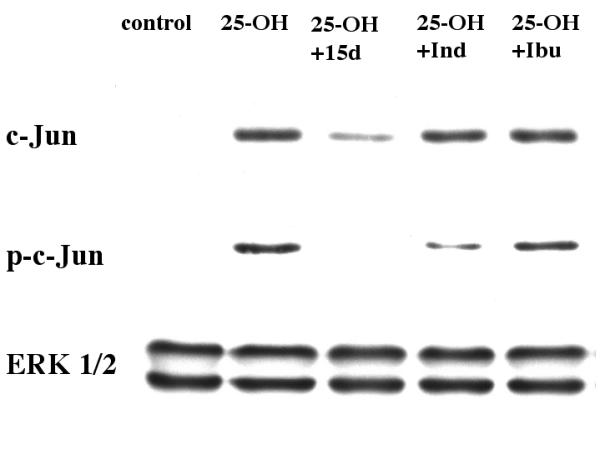
PPAR agonists reduced c-jun activation in 25-OH-cholesterol treated microglia. Cells were treated with 5 μM 25-OH-cholesterol together with 10 μM 15d-PGJ2, 100 μM indomethacin or 100 μM ibuprofen for 12 hours, then processed for Western blot analyses. Antibodies against c-jun, phospho-c-jun (at serine 63) and ERK 1/2 were used in this set of experiments. Results indicated that these agents reduced 25-OH-cholesterol induced c-jun activation to various degrees.
PPAR agonists attenuate cholesterol oxide induced cytotoxicity in microglia
Since PPAR agonists appeared to reduce the c-jun activation caused by 25-OH-cholesterol treatment, the following experiment was designed to test if these agents could reduce 25-OH-cholesterol induced cytotoxicity. Cells were treated with 5 μM 25-OH-cholesterol together with PPAR agonists to be tested for 2 days, then the viability of each treatment was determined. Results indicated that 15d-PGJ2 could prevent 25-OH-cholesterol induced cell death in a dose-dependent manner. While treatment of the cells with 5 μM 25-OH-cholesterol reduced the cell viability to ~7% of controls, addition of 0.62, 1.25, 2.5, 5 or 10 μM 15d-PGJ2 raised the viabilities to ~10, 14, 18, 34 or 48%, respectively (Fig. 7). The rescue by 5 μM and 10 μM 15d-PGJ2 was statistically significant (p < 0.001). This agent at concentrations above 10 μM was toxic to microglia. The solvent for 15d-PGJ2, methylacetate, was without any rescue effect.
Figure 7.
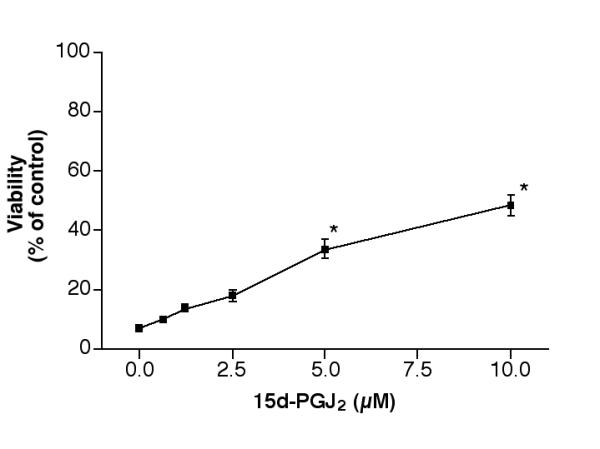
Effect of 15d-PGJ2 on 25-OH-cholesterol induced cell death. Cells were treated with 5 μM 25-OH-cholesterol plus various concentrations of 15d-PGJ2 for 2 days, then the viability of each treatment was determined. *P< 0.001
Indomethacin also prevented 25-OH-cholesterol induced cell death in a dose-dependent manner (Fig. 8). This agent at 12.5, 25, 50, or 100 μM raised the viability from ~8% to ~11, 13, 19 or 31% of controls, respectively. A further increase in indomethacin to 200 μM did not increase cell viability. The rescue by 50–200 μM indomethacin was statistically significant (p < 0.001). Ibuprofen, on the other hand, had little rescue effect even at 200 μM (Fig. 9). It is important to note that this agent also had no inhibitory effect on c-jun activation (Fig. 6).
Figure 8.

Effect of indomethacin on 25-OH-cholesterol induced cell death. Cells were treated with 5 μM 25-OH-cholesterol plus various concentrations of indomethacin for 2 days, then the viability of each treatment was determined. *P < 0.001
Figure 9.
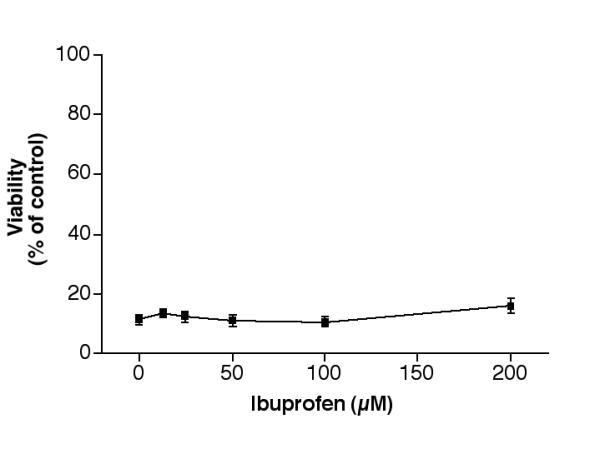
Effect of ibuprofen on 25-OH-cholesterol induced cell death. Cells were treated with 5 μM 25-OH-cholesterol plus various concentrations of ibuprofen for 2 days, then the viability of each treatment was determined.
While 15d-PGJ2 is considered to be a PPARγ agonist, indomethacin and ibuprofen can activate both PPARγ and PPARα [27]. The following experiment was conducted to determine if a pure PPARα agonist, WY14643, could modulate the 25-OH-cholesterol induced cytotoxicity. Results indicated that this agent was without rescue effect at 25, 50 or 100 μM. At 200 μM, WY14643 raised the viability from ~10% to ~28% of controls. The difference was statistically significant (p < 0.001). No further increase in viability could be achieved by this agent (Fig. 10).
Figure 10.
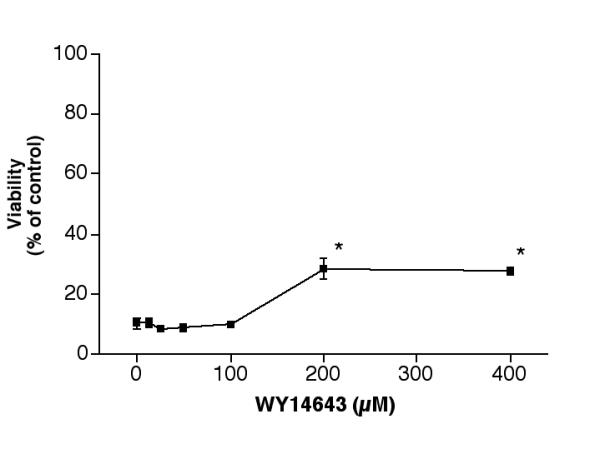
Effect of WY14643 on 25-OH-cholesterol induced cell death. Cells were treated with 5 μM 25-OH-cholesterol plus various concentrations of WY14643 for 2 days, then the viability of each treatment was determined. *P < 0.001
Since 25-OH-cholesterol induced an alteration of phospho-ERK 1/2 during the first 6 hours of treatment (Fig. 5), an experiment was performed to test if inhibition of ERK pathways could affect 25-OH-cholesterol induced cell death. Microglia were treated with 5 μM 25-OH-cholesterol together with 10 μM AG126 (ERK inhibitor), PD98059 (MEK inhibitor, an up-stream member in the ERK pathway) for 2 days, then the viability of each treatment was determined. Results indicated that neither of the pharmacological agents tested had any rescue effect toward 25-OH-cholesterol induced cytotoxicity. Similarly, the p38 pathway inhibitor, SB203580, had no rescue effect under the same experimental conditions (results not shown).
Discussion
Cholesterol oxides are formed in cholesterol-containing food (powdered milk, cheese, egg products) during storage [38,39] or during cholesterol catabolism in animals [40]. The level of cholesterol oxide in meat product increases significantly during the cooking process [39]. Animals can take up cholesterol oxides in a way similar to the absorption of cholesterol. Cholesterol oxides given exogenously can be cleared from plasma rapidly and be widely redistributed in different parts of the body [41]. Cholesterol oxides and authentic cholesterol are transported within the cell by a similar mechanism, but the transport is more efficient for cholesterol oxides [42]. High levels of cholesterol oxides have been observed in hypercholesterolemic animals. For example, the plasma level of 7-OH-cholesterol increases from 15 μM to approximately 200 μM in rabbits fed with high cholesterol diet for 6 weeks [43].
Cholesterol oxide treatment can lead to alterations of cellular functions. These compounds can activate some nuclear receptors including steroidogenic factor 1 [44] and LXR, which may be partially responsible for their biological activities [45,46]. Cholesterol oxides are inhibitors of HMG-CoA reductase, the key enzyme of cholesterol biosynthesis pathway [47]. The ability to bind to an intracellular receptor, oxysterol binding protein, may be responsible for their inhibition of cholesterol biosynthesis [48,49]. It should be noted that addition of cholesterol does not prevent cholesterol oxide induced cell death, which leads to the suggestion that inhibition of cholesterol synthesis is not the cause of cholesterol oxide induced PCD [50,51]. The critical events that result in cholesterol oxide induced cytotoxicity remain to be determined.
Results from this study indicated that 25-OH-cholesterol was very cytotoxic to microglia, such that a 2-day treatment with 5 μM 25-OH-cholesterol reduced cell viability to 5–10% of controls. In contrast, 7-β-OH-cholesterol was much less toxic to microglia (Fig. 1,2). Selective toxicity of cholesterol oxides toward a particular cell type was also observed in our previous studies [29,31-33]. Further experiments with Western blot analyses indicated that the cytotoxicity of cholesterol oxides was associated with their abilities to cause c-jun activation, as evidenced by the increase in activated c-jun, i.e., phospho-c-jun, in 25-OH-cholesterol treated cells but not in 7-β-OH-cholesterol treated cells (Fig. 3). There was a dose-dependent increase in c-jun activation in cells treated with 25-OH-cholesterol (Fig. 4).
It has been suggested that a dysregulation of MAP kinases is responsible for the growth factor deprivation [15] or chemotherapeutic agent [16] induced PCD. Consistent with this idea, results from this study suggest that an unfavorable balance of the JNK and ERK causes the cell death observed in cholesterol oxide treated microglia. Cells treated with 25-OH-cholesterol showed a time-dependent increase in c-jun protein as well as phospho-c-jun (Fig. 5). This c-jun activation was apparent at 6 hours after treatment. In contrast, 25-OH-cholesterol caused a slight, initial increase in phospho-ERK, which decreased to a non-detectable level 6 hours after treatment. The change in p38 and phospho-p38, on the other hand, was not as apparent. It was reported that rabbits fed with high-cholesterol diet exhibited high JNK protein levels and JNK activity in their atherosclerotic lesions, and oxidized LDL caused JNK activation in cultured smooth muscle cells [18]. Based on this study, there is a possibility that cholesterol oxides in oxidized LDL were responsible for those previous observations.
Activation of the JNK pathway can lead to the phosphorylation of downstream members including c-jun that belongs to the AP-1 transcription factor family [12,52]. Because results from this study indicated that JNK pathway was activated by 25-OH-cholesterol, one could expect that AP-1 would be activated in cells treated with cholesterol oxides. Consistent with this idea, it was shown that LDL could induce AP-1 activity in endothelial cells [53] and cholesterol oxide treatment could lead to increased AP-1 DNA binding activity and AP-1 dependent transcription in other cell types [54,55]. Results from this study, thus, complemented and extended the findings of those reports.
The PPARγ agonist, 15d-PGJ2, functions as a general inhibitor of microglial functions [56-58]. For example, this agent inhibits the expression of inducible nitric oxide synthase, tumor necrosis factor-α, interleukin-1 β and major histocompatibility complex class II in activated microglia. Inhibition of the signal transducer and activator of transcription 1 (STAT-1) and the nuclear factor kB (NF-kB) may be partially responsible for the inhibitory effects. Results from this study further indicated that PPAR agonists could reduce c-jun activation (Fig. 6). Given the suggestion that an up-regulation of c-jun is a cause of PCD [15,16], a reduction in c-jun activation by PPAR agonists should attenuate 25-OH-cholesterol induced cytotoxicity. Consistent with this idea, results from this study indicated that 15d-PGJ2, indomethacin and WY14643 could attenuate the cytotoxicity caused by 25-OH-cholesterol to various degrees (Fig. 7,8,10). Since the protective effect of WY14643 could be observed only at concentrations higher than 200 μM, there is a possibility that this agent acted on the PPARγ receptor to achieve the protection. Similar protective roles of PPAR agonists have been shown in other nervous tissues. For example, PPARγ agonists were shown to protect cytokine-induced PCD in cerebellar granule cells [59]. These agents could also block the neurotoxic effects caused by β-amyloid activated microglia, and thus might be beneficial in the treatment of Alzheimer's disease [60]. In non-neuronal tissues, PPARα activation was shown to suppress both spontaneous rat hepatocyte PCD and the PCD induced by transforming growth factor-β1 [61].
Conclusions
Treatment of microglia with 25-OH-cholesterol caused an induction of c-jun, phospho-c-jun and a reduction in cellular viability. PPAR agonists reduced the 25-OH-cholesterol induced c-jun activation and attenuated the cytotoxicity. This group of compounds may be useful in future development of pharmacological agents against cholesterol oxide induced cytotoxicity.
Materials and methods
Cell culture
The N9 murine microglia cell line was a gift kindly provided by Dr. P. Ricciardi-Castagnoli [62]. This cell line has been used extensively as a model for microglia [63-65]. These cells were derived by immortalization of day 13 embryonic brain cultures with a retrovirus carrying an activated v-myc oncogene. The cultures were maintained in Minimum Essential Medium (MEM, GibcoBRL, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Sigma, St. Louis, MO), 1.4 mM glutamine (GibcoBRL, Grand Island, NY) and 20 μM 2-mercaptoethanol (Sigma, St. Louis, MO). WY14643 and 15d-PGJ2 were purchased from Cayman (Ann Arbor, MI). Indomethacin, ibuprofen and all other pharmacological and general biochemical reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated.
Cell viability
Cells grown in 96-well plates (20,000 cells/well, ~90% confluency) were treated with testing agents for a period of time as indicated in the figure legends; then, the viability from each treatment was determined by the MTT (3-(4,5-dimethylthiazole-2yl)-2,5-diphenyltetrazolium bromide) assay [66]. Culture medium was removed after treatment, then 100 μl MTT solution (100 μg/ml prepared in MEM) was added to each well. The cultures were incubated at 37°C for one hour in a tissue culture incubator. The MTT solution was then removed, and the cells in each well were lysed by the addition of 100 μl dimethly sulfoxide. The plate was placed on a shaker for one hour at room temperature to complete the lysing process, then the optical density of each well was measured by a 96-well plate reader with a filter setting at 570 nm (reference filter setting was 630 nm).
Western blot analysis
Protein isolated from cytoplasm was separated by SDS-PAGE on 10% polyacrylamide gels and transferred to nitrocellulose membranes (NitroBind, MSI, Westborough, MA), then incubated with primary antibody overnight at 4°C. Specific bands were detected by incubating the membrane with horseradish peroxidase conjugated secondary antibody for 45 minutes at room temperature, then with ECL Western Blotting Detection Reagents (Amersham Life Science, Arlington Heights, IL) according to the manufacturer's protocol. The primary antibodies used in this set of experiments were from New England Biological (anti-phospho-c-jun at serine 73, used at 1:1000) or Santa Cruz (anti-c-jun, used at 1:3000; anti-phospho-c-jun at serine 63, used at 1:1000, anti-JNK, used at 1:3000; anti-ERK 1/2, used at 1:3000; anti-phospho-ERK, used at 1:2000; anti-p38, used at 1:3000; anti-phospho-p38, used at 1:1000).
Statistical analysis
Unless otherwise stated, results of cell viability experiments were pooled from 12 replicate samples derived from 3 independent experiments, and expressed as mean ± SEM. Statistical analysis was performed by analysis of variance (one-way ANOVA) followed by the Bonferroni test to determine the significance of difference.
List of abbreviations
15d-PGJ2, 15-deoxy-delta 12, 14-PGJ2; AP-1, activator protein-1; ERK, p42/44, Extracellular signal-Regulated Kinase; LDL, low density lipoprotein; MAP kinases, Mitogen-Activated Protein kinases; MTT (3-(4,5-dimethylthiazole-2yl)-2,5-diphenyltetrazolium bromide); PCD, programmed cell death; PPARs, peroxisome proliferator-activated receptors; SAPK/JNK, Stress-Activated Protein Kinase/ c-Jun NH2-terminal Kinase.
Acknowledgments
Acknowledgments
This work was supported by the Arkansas Science & Technology Authority, Fight For Sight, and Research to Prevent Blindness.
Contributor Information
Jason Y Chang, Email: changjasony@uams.edu.
Ling-Zhi Liu, Email: lingzhil@bcm.tmc.edu.
References
- Steinberg D. Lewis A. Conner Memorial Lecture: Oxidative modification of LDL and atherogenesis. Circulation. 1997;95:1062–1071. doi: 10.1161/01.cir.95.4.1062. [DOI] [PubMed] [Google Scholar]
- Hubbard RW, Ono Y, Sanchez A. Atherogenic effect of oxidized products of cholesterol. Prog Fd Nutr Sci. 1989;13:17–44. [PubMed] [Google Scholar]
- Imai H, Werthessen NT, LeQuesne PW, Soloway AH, Kanisawa M. Angiotoxicity of oxygenated sterols and possible precursors. Science. 1980;207:651–653. doi: 10.1126/science.7352277. [DOI] [PubMed] [Google Scholar]
- Schroepfer GJ., Jr Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80:361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- Smith LL, Johnson BH. Biological activities of oxysterols. Free Rad Biol & Med. 1989;7:285–332. doi: 10.1016/0891-5849(89)90136-6. [DOI] [PubMed] [Google Scholar]
- AyalaTorres S, Moller PC, Johnson BH, Thompson EB. Characteristics of 25-hydroxycholesterol-induced apoptosis in the human leukemic cell line CEM. Exp Cell Res. 1997;235:35–47. doi: 10.1006/excr.1997.3630. [DOI] [PubMed] [Google Scholar]
- Chang JY, Chavis JA, Liu L, Drew PD. Cholesterol oxides induce programmed cell death in microglial cells. Biochem Biophys Res Comm. 1998;249:817–821. doi: 10.1006/bbrc.1998.9237. [DOI] [PubMed] [Google Scholar]
- Nishio E, Watanabe Y. Oxysterols induced apoptosis in cultured smooth muscle cells through CPP32 protease activation and bcl-2 protein downregulation. Biochem Biophys Res Commun. 1996;226:928–934. doi: 10.1006/bbrc.1996.1452. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM. Programmed cell death in neurons: Focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.neuro.14.1.453. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival – Lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Force T, Bonventre JV. Growth factors and mitogen-activated protein kinases. Hypertension. 1998;31:152–161. doi: 10.1161/01.hyp.31.1.152. [DOI] [PubMed] [Google Scholar]
- Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann N Y Acad Sci. 1998;851:139–146. doi: 10.1111/j.1749-6632.1998.tb08987.x. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Stone AA, Chambers TC. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp Cell Res. 2000;254:110–119. doi: 10.1006/excr.1999.4731. [DOI] [PubMed] [Google Scholar]
- Kusuhara M, Chait A, Cader A, Berk BC. Oxidized LDL stimulates mitogen-activated protein kinases in smooth muscle cells and macrophages. Arterioscler Thromb Vasc Biol. 1997;17:141–148. doi: 10.1161/01.atv.17.1.141. [DOI] [PubMed] [Google Scholar]
- Metzler B, Hu Y, Dietrich H, Xu Q. Increased expression and activation of stress-activated protein kinases/c-Jun NH(2)-terminal protein kinases in atherosclerotic lesions coincide with p53. Am J Pathol. 2000;156:1875–1886. doi: 10.1016/S0002-9440(10)65061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Corton JC, Anderson SP, Stauber A. Central role of peroxisome proliferator-activated receptors in the actions of peroxisome proliferators. Annu Rev Pharmacol Toxicol. 2000;40:491–518. doi: 10.1146/annurev.pharmtox.40.1.491. [DOI] [PubMed] [Google Scholar]
- Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol. 2000;129:823–834. doi: 10.1038/sj.bjp.0703149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- Vatassery GT, Quach HT, Smith WE, Ungar F. Oxidation of cholesterol in synaptosomes and mitochondria isolated from rat brains. Lipids. 1997;32:879–886. doi: 10.1007/s11745-997-0113-1. [DOI] [PubMed] [Google Scholar]
- Chang JY, Liu LZ. 25-Hydroxycholesterol causes death but does not prevent NGF-induced neurite outgrowth in PC12 cells. Neurochem Int. 1997;31:517–523. doi: 10.1016/S0197-0186(97)00020-X. [DOI] [PubMed] [Google Scholar]
- Chang JY, Phelan KD, Liu LZ. Neurotoxicity of 25-OH-cholesterol on NGF-differentiated PC12 cells. Neurochem Res. 1998;23:7–16. doi: 10.1023/A:1022437000893. [DOI] [PubMed] [Google Scholar]
- Chang JY, Phelan KD, Chavis JA. Neurotoxicity of 25-OH-cholesterol on sympathetic neurons. Brain Res Bull. 1998;45:615–622. doi: 10.1016/S0361-9230(97)00461-9. [DOI] [PubMed] [Google Scholar]
- Chang JY, Liu LZ. Toxicity of cholesterol oxides on cultured neuroretinal cells. Curr Eye Res. 1998;17:95–103. doi: 10.1076/ceyr.17.1.95.5252. [DOI] [PubMed] [Google Scholar]
- Chang JY, Liu LZ. Neurotoxicity of cholesterol oxides on cultured cerebellar granule cells. Neurochem Int. 1998;32:317–323. doi: 10.1016/S0197-0186(97)00103-4. [DOI] [PubMed] [Google Scholar]
- Mato M, Ookawara S, Sakamoto A, Aikawa E, Ogawa T, Mitsuhashi U, Masuzawa T, Suzuki H, Honda M, Yazaki Y, et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc Natl Acad Sci USA. 1996;93:3269–3274. doi: 10.1073/pnas.93.8.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron KD. The microglial cell. A historical review. J Neurol Sci. 1995;134:57–68. doi: 10.1016/0022-510X(95)00209-K. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative crosstalk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- Addis PB. Occurrence of lipid oxidation products in foods. Fd Chem Toxic. 1986;24:1021–1030. doi: 10.1016/0278-6915(86)90283-8. [DOI] [PubMed] [Google Scholar]
- Maraschiello C, Esteve E, Garcia Regueiro JA. Cholesterol oxidation in meat from chickens fed alpha-tocopherol- and beta-carotene-supplemented diets with different unsaturation grades. Lipids. 1998;33:705–713. doi: 10.1007/s11745-998-0260-4. [DOI] [PubMed] [Google Scholar]
- Smith LL, Teng JI, Lin YY, Seitz PK. Sterol metabolism-XLVII. Oxidized cholesterol esters in human tissues. J Steroid Biochem. 1981;14:889–900. doi: 10.1016/0022-4731(81)90238-7. [DOI] [PubMed] [Google Scholar]
- Krut LH, Yang JW, Schonfeld G, Ostlund RE. The effect of oxidizing cholesterol on gastrointestinal absorption, plasma clearance, tissue distribution, and processing by endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:778–785. doi: 10.1161/01.atv.17.4.778. [DOI] [PubMed] [Google Scholar]
- Lange Y, Ye J, Strebel F. Movement of 25-hydroxycholesterol from the plasma membrane to the rough endoplastic reticulum in cultured hepatoma cells. J Lipid Res. 1995;36:1092–1097. [PubMed] [Google Scholar]
- Hodis HN, Crawford DW, Sevanian A. Cholesterol feeding increases plasma and aortic tissue cholesterol oxides levels in parallel: Further evidence for the role of cholesterol oxidation in atherosclerosis. Atherosclerosis. 1991;89:117–126. doi: 10.1016/0021-9150(91)90051-4. [DOI] [PubMed] [Google Scholar]
- Lala DS, Syka PM, Lazarchik SB, Mangelsdorf DJ, Parker KL, Heyman RA. Activation of the orphan nuclear receptor steroidogenic factor 1 by oxysterols. Proc Natl Acad Sci USA. 1997;94:4895–4900. doi: 10.1073/pnas.94.10.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. 1977;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J Biol Chem. 1974;249:7306–7314. [PubMed] [Google Scholar]
- Taylor FR, Saucier SE, Shown EP, Parish EJ, Kandutsch AA. Correlation between oxysterol binding to a cytosolic binding protein and potency in the repression of hydroxymethylglutaryl coenzyme A reductase. J Biol Chem. 1984;259:12382–12387. [PubMed] [Google Scholar]
- Taylor F. Correlation among oxysterol potencies in the regulation of the degradation of 3-hydroxy-3-methylglutaryl CoA reductase, the depression of 3-hydroxy-3-methylglutaryl CoA synthase and affinities for the oxysterol receptor. Biochem Biophys Res Comm. 1992;186:182–189. doi: 10.1016/s0006-291x(05)80791-0. [DOI] [PubMed] [Google Scholar]
- Rusinol AE, Yang L, Thewke D, Panini SR, Kramer MF, Sinensky MS. Isolation of a somatic cell mutant resistant to the induction of apoptosis by oxidized low density lipoprotein. J Biol Chem. 2000;275:7296–7303. doi: 10.1074/jbc.275.10.7296. [DOI] [PubMed] [Google Scholar]
- Thompson EB, Ayala-Torres S. Oxysterols and apoptosis: evidence for gene regulation outside the cholesterol pathway. Crit Rev Biochem Mol Biol. 1999;34:25–32. doi: 10.1080/10409239991209174. [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Lin JH, Liao HL, Friedli O, Jr, Verna L, Marten NW, Straus DS, Stemerman MB. LDL induces transcription factor activator protein-1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:473–480. doi: 10.1161/01.atv.18.3.473. [DOI] [PubMed] [Google Scholar]
- Hanley K, Ng DC, He SS, Lau P, Min K, Elias PM, Bikle DD, Mangelsdorf DJ, Williams ML, Feingold KR. Oxysterols induce differentiation in human keratinocytes and increase Ap-1-dependent involucrin transcription. J Invest Dermatol. 2000;114:545–353. doi: 10.1046/j.1523-1747.2000.00895.x. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Vogel R, Holloway MK, Rutledge SJ, Friedman O, Yang Z, Rodan GA, Friedman E. Transcription control and neuronal differentiation by agents that activate the LXR nuclear receptor family. Mol Cell Endocrinol. 1999;155:51–60. doi: 10.1016/S0303-7207(99)00115-X. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-Delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur J Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Koppal T, Petrova TV, Van Eldik LJ. Cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J(2) acts as a general inhibitor of inflammatory responses in activated BV-2 microglial cells. Brain Res. 2000;867:115–121. doi: 10.1016/S0006-8993(00)02270-8. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol. 1999;100:156–168. doi: 10.1016/S0165-5728(99)00192-7. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, James NH, Woodyatt NJ, Macdonald N, Tugwood JD. Evidence for the suppression of apoptosis by the peroxisome proliferator activated receptor alpha (PPAR alpha). Carcinogenesis. 1998;19:43–48. doi: 10.1093/carcin/19.1.43. [DOI] [PubMed] [Google Scholar]
- Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli P. Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia. 1993;7:255–262. doi: 10.1002/glia.440070309. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner J, Vorpahl J, Preston K. Interferon-gamma induces apoptosis and augments the expression of Fas and Fas ligand by microglia in vitro. Exp Neurol. 2000;162:290–296. doi: 10.1006/exnr.1999.7345. [DOI] [PubMed] [Google Scholar]
- Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Balazs R, Jorgensen OS, Hack N. N-Methyl-D-Aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience. 1988;27:437–451. doi: 10.1016/0306-4522(88)90279-5. [DOI] [PubMed] [Google Scholar]