

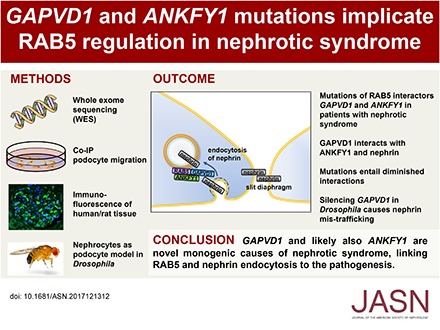
Abstract
Background
Steroid-resistant nephrotic syndrome (SRNS) is a frequent cause of CKD. The discovery of monogenic causes of SRNS has revealed specific pathogenetic pathways, but these monogenic causes do not explain all cases of SRNS.
Methods
To identify novel monogenic causes of SRNS, we screened 665 patients by whole-exome sequencing. We then evaluated the in vitro functional significance of two genes and the mutations therein that we discovered through this sequencing and conducted complementary studies in podocyte-like Drosophila nephrocytes.
Results
We identified conserved, homozygous missense mutations of GAPVD1 in two families with early-onset NS and a homozygous missense mutation of ANKFY1 in two siblings with SRNS. GAPVD1 and ANKFY1 interact with the endosomal regulator RAB5. Coimmunoprecipitation assays indicated interaction between GAPVD1 and ANKFY1 proteins, which also colocalized when expressed in HEK293T cells. Silencing either protein diminished the podocyte migration rate. Compared with wild-type GAPVD1 and ANKFY1, the mutated proteins produced upon ectopic expression of GAPVD1 or ANKFY1 bearing the patient-derived mutations exhibited altered binding affinity for active RAB5 and reduced ability to rescue the knockout-induced defect in podocyte migration. Coimmunoprecipitation assays further demonstrated a physical interaction between nephrin and GAPVD1, and immunofluorescence revealed partial colocalization of these proteins in rat glomeruli. The patient-derived GAPVD1 mutations reduced nephrin-GAPVD1 binding affinity. In Drosophila, silencing Gapvd1 impaired endocytosis and caused mistrafficking of the nephrin ortholog.
Conclusions
Mutations in GAPVD1 and probably in ANKFY1 are novel monogenic causes of NS. The discovery of these genes implicates RAB5 regulation in the pathogenesis of human NS.
Keywords: nephrotic syndrome, podocyte, nephrin, endocytosis, genetic renal disease, nephrocyte
Steroid-resistant nephrotic syndrome (SRNS) is characterized by edema, nephrotic-range proteinuria, and hyperlipidemia. Mutations in approximately 45 different genes have been discovered as monogenic causes of SRNS1–38 (Supplemental Figure 1A) and our understanding of the pathophysiology of SRNS and podocyte biology in general has been formed by the pathways delineated by discovery of these genes (Supplemental Figure 1A).1,3 The first gene to be found was nephrin (NPHS1), which encodes a major constituent of the slit diaphragm.4 Subsequently, mutations of several proteins associated with the slit diaphragm complex were identified. Linking the slit diaphragm to the actin cytoskeleton to maintain the complex podocyte morphology seems essential because actin-binding and -regulating proteins form the most numerous group among the monogenic causes of SRNS.1,3 More recently, mutations in CoQ10-biosynthesis genes,14–16 nucleoporins,39–41 and the KEOPS complex42 have been discovered as further monogenic causes of SRNS. This opened new avenues toward a better understanding of the complex pathogenesis of SRNS.
A role of endocytosis for podocyte biology has previously been proposed: The slit diaphragm protein nephrin is subject to endocytosis utilizing different branches of the endocytosis pathway.43–48 In mice, phosphoinositide 3-kinases49,50 and effectors of vesicular fission and clathrin uncoating are essential for the glomerular filtration barrier. Knockout of the murine isoforms of dynamin, synaptojanin, or endophilin each resulted in severe proteinuria.51,52 In humans, no direct endosomal regulator has previously been implicated in nephrotic syndrome.
Methods
Study Approval
Approval for human subject research was obtained from the University of Michigan and the Boston Children’s Hospital Institutional Review Boards. All participants or their guardians provided written informed consent.
Study Participants
After informed consent, clinical data and blood samples were obtained from individuals with nephrotic syndrome. Clinical data were obtained using an established questionnaire (http://www.renalgenes.org). The diagnosis of NS was made by (pediatric) nephrologists, on the basis of standardized clinical and renal histologic criteria. Renal biopsy samples were evaluated by renal pathologists.
Homozygosity Mapping, Whole-Exome Resequencing, and Mutation Calling
Homozygosity mapping, whole-exome resequencing, and mutation calling were performed as described previously.22
Plasmids, siRNAs, Cell Culture, and Transfection
Human full-length GAPVD1 cDNA was subcloned after PCR from human full-length cDNA (GenBank BC114937; GE Dharmacon). Mouse Gapvd1 was subcloned after PCR from murine full-length cDNA (RefSeq NM_025709) that was a gift from Dr. Alan Saltiel53 (University of California, San Diego). Human ANKFY1 isoform 1 cDNA (GenBank BC152991.1) and human NPHS1 cDNA (GenBank: BC156935.1) were obtained from the Harvard PlasmID Repository. Truncation constructs were generated by PCR. Primers are shown in Supplemental Table 1. ANKFY1 rescue constructs were obtained by introduction of two synonymous mutations within the shRNA target sequences.
The following expression vectors were used: pRK5-N-Myc, pCDNA6.2-N-GFP, pQCXIP mCherry, and pSirenRetroQ. Clones reflecting the mutations identified in individuals with nephrotic syndrome were introduced into the cDNA constructs using Quik change II XL site-directed mutagenesis kit (Agilent Technologies). The following constructs were obtained from addgene: mCherry-Rab5CAQ79L (#35138), mCherry-Rab5DNS34N (#35139), mCh-Rab5 (#49201), pRK5myc Rac1 wt (#37030), and pSpCas9(BB)-2A-GFP (PX458) (#48138).
The GAPVD1- and ANKFY1-specific and control scrambled siRNAs were purchased from GE Dharmacon.
Overexpression experiments were performed in HEK293T cells (ATCC biologic resource center). Immortalized human podocytes were a gift from Dr. Moin Saleem (University of Bristol, Bristol, UK).
HEK cells were maintained in DMEM, supplemented with 10% FBS, 50 IU/ml penicillin, and 50 μg/ml streptomycin. Podocytes were maintained in RPMI 1640 plus GlutaMAX-I (Gibco) supplemented with 10% FBS, 50 IU/ml penicillin/50 μg/ml streptomycin, and insulin-transferrin-selenium-X.
Plasmids and siRNAs were transfected into HEK293T cells at 37°C or podocytes grown at the permissive temperature of 33°C using Lipofectamine 2000 or Lipofectamine RNAiMax, respectively (Invitrogen). Knockdown in human podocyte cell lines employed pSirenRetroQ with 2–3 independent shRNAs directed against human GAPVD1 or ANKFY1 for retroviral transduction. Puromycin was used to select transduced cells. Knockdown efficiency was confirmed for all experiments, shRNA targets are shown in Supplemental Table 1. For rescue experiments, knockdown podocytes underwent a transient transfection using murine Gapvd1 or human ANKFY1 constructs that are resistant to shRNA.
Immunoblotting, Immunoprecipitation, Pull-Down Assay, and Immunofluorescence Staining
Immunoblotting, immunoprecipitation, and immunofluorescence staining were performed as described previously.39 Briefly, HEK293T cells were lysed and precleared using rec-Protein A-Sepharose 4B Conjugate (Life Technologies) overnight. Then, equal amounts of protein were incubated with EZview Red Anti-c-Myc Affinity Gel (Sigma-Aldrich) or GFP-nAb (Allele Biotechnology). Coimmunoprecipitation (co-IP) experiments were performed in three independent experiments. Immunoblotting was performed using rabbit anti-GAPVD1 (#A302–116; Bethyl Laboratories), mouse anti-ANKFY1 (sc-393353; Santa Cruz Biotechnology), mouse anti–c-Myc (sc-40; Santa Cruz Biotechnology), rabbit anti–c-Myc (sc-789; Santa Cruz Biotechnology), mouse anti-GFP (sc-9996; Santa Cruz Biotechnology), rabbit anti-GFP (sc-8334; Santa Cruz Biotechnology), and rabbit anti-mCherry (ab167452; Abcam).
Immunofluorescence of ANKFY1 was performed with mouse anti-ANKFY1 (sc-393353). Rabbit anti-murine GAPVD1 was a kind gift from Dr. Alan Saltiel.53 Other antibodies used were guinea pig anti-NPHS1 (GP-N2; Progen) and rabbit anti-RAB5 (#ab18211; Abcam). Paraffin-embedded human tissue sections (HP-901; amsbio) were deparaffinized in xylene for 10 minutes, rehydrated, and antigen retrieval was performed by incubating the slides for 40 minutes at 95°C in citrate buffer, pH 6.0.
Fluorescent images were obtained with a Leica SP5X or a Zeiss LSM 880 laser scanning microscope.
RAB5 Assay
RAB5 activity assay was performed using the Rab5 Activation Assay Kit (#83701; NewEast Biosciences) according to the manufacturer’s instructions. Protein concentrations were determined by Lowry’s method and equal amounts of protein were exposed to the RAB5-GTP–specific antibody.
Podocyte Dextran Internalization Assay
Human podocytes were exposed to culture medium containing 500 ng/ml Texas-Red-dextran 10 kD (ThermoFisher) for 30 minutes at 37°C. Afterward, cells were rinsed twice with ice-cold PBS, fixed in 4% paraformaldehyde for 15 minutes in the presence of Hoechst 33342 (1:1000), and mounted in Antifade Diamond (ThermoFisher). Cells were imaged using a Leica SP5X confocal microscope and uptake was quantified using ImageJ software in maximized intensity projections after thresholding.
Podocyte Migration Assay
Real-time migration assay was performed using the IncuCyte videomicroscopy system (Essen Bioscience) in 96-well plates according to the manufacturer’s instructions. Briefly, 24 hours after transfection scratch wounds were made using a 96-pin tool (WoundMaker) as per protocol. Cells were monitored automatically via live cell imaging and time-lapse images. Wound confluency was automatically acquired hourly and recorded by the IncuCyte software (ZOOM). Data processing and analysis for migration assay were performed using the IncuCyte 96-well Kinetic Cell Migration and Invasion Assay software module. Results are presented as time versus wound confluency.
Drosophila Studies
Drosophila melanogaster stable RNAi stocks Gapvd1-RNAi 1 (#108453) and Gapvd1-RNAi 2 (#19649) were obtained from the Vienna Drosophila Resource Center. Prospero-GAL454 was used to control expression in garland cell nephrocytes; actin-GAL4 (#3954; Bloomington stock) was used for ubiquitous expression.
Fluorescent tracer uptake in nephrocytes was performed as previously described.55 Briefly, nephrocytes were dissected in PBS and incubated with FITC-albumin (Sigma) for 30 seconds. After a fixation of 5 minutes in 8% paraformaldehyde, cells were rinsed in PBS and exposed to Hoechst 33342 (1:1000) for 20 seconds and mounted in Antifade Diamond. Cells were imaged using a Leica SP5X confocal microscope. Quantitation of fluorescent tracer uptake was performed with ImageJ software. The results are expressed as a ratio to a control experiment with EGFP-RNAi that was done in parallel.
For immunohistochemistry, nephrocytes were dissected, fixed for 15 minutes in PBS containing 4% paraformaldehyde, and stained according to the standard procedure. The following primary antibodies were used: rabbit anti-sns56 (1:500, gift from S. Abmayr) and guinea pig anti-Kirre57 (1:200, gift from S. Abmayr). For imaging, a Leica SP5X confocal microscope was used. Image processing was done by ImageJ and Adobe Photoshop CS4 software.
For transmission electron microscopy (TEM), nephrocytes were dissected and fixed in 4% formaldehyde and 0.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. TEM was carried out using standard techniques. One complete diameter of six cells from three different animals was analyzed for ectopic slit diaphragms. Areas where the labyrinthine channels are cut obliquely are recognizable by elongated stretches of higher electron density along the cell surface. These areas were excluded from the quantitation.
Quantitative Real-Time PCR
Total RNA was isolated using TRIzol (Invitrogen) and purified with the Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. Real-time PCR was performed using Taqman probes (ThermoFisher Scientific). The RPL39 gene was used to normalize expression data.
Statistical Analyses
Paired t test was used to determine the statistical significance between two interventions. ANOVA followed by Dunnett’s correction (unless otherwise indicated) was used for multiple comparisons (GraphPad Prism software). Asterisks indicate significance as follows: *P<0.05, **P<0.01, ***P<0.001. A statistically significant difference was defined as P<0.05. Error bars indicate SD.
Results
Mutations in GAPVD1 and ANKFY1, Two RAB5 Interactors, in Families with Nephrotic Syndrome
To discover novel monogenic causes of SRNS, we performed whole-exome sequencing (WES) in a cohort of 665 patients. We identified two homozygous missense mutations (p.Leu414Val and p.Arg937Gln) of the gene GTPase Activating Protein And VPS9 Domains 1 (GAPVD1) in two individuals (A4619 and B1391) with early-onset nephrotic syndrome (Figure 1A, Supplemental Figures 1, B–D, G–J, and 2, A and B, Table 1). The respective amino acid residues are conserved to C. intestinalis for both mutations (Figure 1, A, C, and D, Table 1) and causative mutations are located within a homozygosity peak (Supplemental Figure 2, A and B). The families were of Mexican and Arabic descent, respectively (Table 1). One patient (A4619) showed the unusual clinical course of congenital nephrotic syndrome with spontaneous remission after 18 months (Table 1), whereas nephrotic syndrome persisted in the other case after 2 years of observation. Spontaneous remission of congenital nephrotic syndrome has previously been described in certain alleles of nephrin (NPHS1).58–63 Neither patient exhibited extrarenal manifestations. The histology in both cases was characterized by mesangial hypercellularity with expansion of the extracellular matrix (Figure 1, F and G, Supplemental Figure 1, G–J). This uncommon histologic pattern is suggestive of glomerular injury and is frequently observed with mutations of nephrin.64,65 TEM revealed foot process effacement (Figure 1, I and J). Renal ultrasound of both cases showed increased echogenicity (Figure 1K, Table 1).
Figure 1.

WES identifies recessive disease-causing mutations in GAPVD1 and ANKFY1 in families with early-onset nephrotic syndrome. (A) Schematic of GAPVD1 cDNA with the corresponding protein including its functional domains. Arrows indicate the positions of two recessive mutations of GAPVD1 that were identified by WES in two families (B1391 and A4619) with nephrotic syndrome. (B) Schematic of ANKFY1 cDNA with the corresponding protein including its functional domains. Arrow indicates the position of one recessive missense mutation of ANKFY1 that was identified by WES in two individuals from family B1027 with nephrotic syndrome. (C) Alignment of GAPVD1 aa sequences for Homo sapiens, Mus musculus, Gallus gallus, Xenopus tropicalis, Danio rerio, and Ciona intestinalis shows conservation of the residue Leucine 414. (D) Alignment of aa sequence of GAPVD1 for H. sapiens, M. musculus, G. gallus, X. tropicalis, D. rerio, and C. intestinalis shows conservation of the residue Arginine 937. (E) Alignment of aa sequence of ANKFY1 for H. sapiens, M. musculus, G. gallus, X. tropicalis, D. rerio, C. intestinalis, C. elegans, and D. melanogaster shows conservation of the residue Arginine 95. (F) Renal histology (trichrome staining) of patient A4619 with the Leu414Val mutation of GAPVD1 shows mesangial hypercellularity and expansion of the extracellular matrix (asterisks). (G) Renal histology (Jones silver stain) of patient A4619 confirms extracellular matrix expansion (asterisk). (H) Renal histology of patient B1027 (HE staining) reveals FSGS. (I and J) Electron microscopy image shows podocyte foot process effacement (arrow heads) in (I) A4619 and (J) B1391. (K) Renal ultrasound image of B1391 shows increased echogenicity with renal echogenicity equal to liver echogenicity. aa, amino acid.
Table 1.
Mutations in GAPVD1 and ANKFY1, in four individuals from three families with SRNS
| Family_Individual | Nucleotide Change | Amino Acid Change | Zygosity | Exon | PPH2 | SIFT | MT | Amino Acid Conserved to Species | ExAC | gnomAD | Sex | Ethnic Origin | Parental Consanguinity | Response to Steroids | Age of Onset (Proteinuria) | Renal Function | Renal Biopsy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GAPVD1 | |||||||||||||||||
| A4619 | c.1240 C>G | p.Leu414Val | Hom | 5 | 0, 99 | Tol | DC | C.i. | 0/27/120,882 | 0/71/272,940 | F | Mexican | yes | not done | 2 moa | normal | Mesangial hypercellularity |
| B1391 | c.2810 G>A | p.Arg937Gln | Hom | 16 | 0, 99 | Tol | DC | C.i. | 0/13/121,408 | 0/25/277,226 | F | Arabic | yes | SRNS | 18 mo | normalb | Mesangial hypercellularity |
| ANKFY1 | |||||||||||||||||
| B1027_21 | c.284 G>T | p.Arg95Leu | Hom | 2 | 1 | Del | DC | D.m. | 0 | 0 | M | Indian | not known | SRNS | 11 yr | normal | FSGS (11 yr) |
| B1027_22 | c.284 G>T | p.Arg95Leu | Hom | 2 | 1 | Del | DC | D.m. | 0 | 0 | F | Indian | not known | not done | 8 yr | normal | ND |
PPH2, PolyPhen-2 prediction score (http://genetics.bwh.harvard.edu/pph2/); SIFT, Sorting Tolerant From Intolerant prediction score (http://sift.jcvi.org/); MT, Mutation Taster (http://www.mutationtaster.org/); ExAC, Exome Aggregation Consortium database (http://exac.broadinstitute.org); gnomAD, Genome Aggregation Database (http://gnomad.broadinstitute.org); Hom, homozygous; Tol, tolerated; DC, disease causing; C.i., Ciona intestinalis; F, female; Del, deleteriousness; D.m., D. melanogaster; M, male; ND, no data.
Proteinuria spontaneously resolved age 18 mo.
Kidneys enlarged on renal ultrasound.
GAPVD1 has been described as an endosomal regulator that interacts with RAB5.53,66–71 GAPVD1 harbors both a GTPase activating and an inactivating domain, the RasGAP and VPS9 domains, respectively (Figure 1A). An activating role as a guanine nucleotide exchange factor (GEF) for RAB5 has been suggested.66,70
In an unbiased approach by WES, we further identified a homozygous mutation of the gene Ankyrin Repeat And FYVE Domain Containing 1 (ANKFY1) in two siblings of a family with FSGS and proteinuria (Figure 1B, Table 1). Similar to GAPVD1, ANKFY1 interacts with RAB5, serving as a versatile RAB5 effector.72–75 The identified mutation (p.Arg95Leu) is conserved to D. melanogaster and locates within a homozygosity peak that is shared between both siblings (Supplemental Figure 2, C and D). Renal biopsy revealed FSGS (Figure 1H). Renal ultrasound was without pathologic findings (Supplemental Figure 1F).
Sanger sequencing of parental DNA confirmed segregation of the mutations according to the status of affection for the families B1391 and B1027, whereas parental DNA could not be obtained for A4619 (Supplemental Figure 1). WES detected no other likely causative mutations in our patients.
We thus identify mutations of GAPVD1 as a novel monogenic cause of early-onset nephrotic syndrome, and ANKFY1 most likely shares this role. Interestingly, both encoded proteins play a regulatory role for RAB5.
GAPVD1 and ANKFY1 Are Interaction Partners and Colocalize in HEK293T Cells
All genes that may cause monogenic SRNS in humans are expressed in podocytes.3 Immunoblotting revealed expression of GAPVD1 and ANKFY1 in immortalized podocytes (Figure 2, A and B) and also in HEK293T cells (Figure 2, C and D).
Figure 2.

GAPVD1 and ANKFY1 proteins are expressed in a podocyte cell line and are interaction partners. (A) Transient, siRNAi-mediated silencing of GAPVD1 (arrow head) in human podocytes is demonstrated by immunoblot. This indicates endogenous expression of GAPVD1 in immortalized podocytes. (B) Transient, siRNAi-mediated silencing of ANKFY1 (arrow head) in human podocytes is demonstrated by immunoblot. This indicates endogenous expression of ANKFY1 in immortalized podocytes. (C) Western blot shows CRISPR/Cas9-mediated knockdown of GAPVD1 in HEK293T cells for two independent gRNAs (arrow head). This indicates endogenous expression of GAPVD1 in HEK293T cells and confirms the efficiency of the gRNAs. (D) Western blot shows CRISPR/Cas9-mediated silencing of ANKFY1 in HEK293T cells for two independent gRNAs (arrow head). This indicates endogenous expression of ANKFY1 in HEK293T cells and confirms the efficiency of the gRNAs. (E) Overexpressed N-GFP-GAPVD1 localizes to the cytosol in cultured podocytes. (F and F’) CRISPR/Cas9 mediated silencing of ANKFY1 in human podocytes is marked by coexpression of GFP (shown in [F]) and abrogates the signal from small, cytosolic vesicles using an anti-ANKFY1 antibody (gRNA-expressing cells are outlined by dashed line and marked by arrow heads in [F’]). (G and H) Staining human renal tissue sections with anti-ANKFY1 results in (G and G’’) a diffuse signal that was more intense than (H and H’’) the control signal (secondary antibody alone). Enhanced resolution microscopy by Airyscan technology revealed (insets G–G’’) fine vesicles in all glomerular cells including podocytes, whereas (H–H’’) no vesicular pattern was detected in the control. Nephrin staining is shown in (G’ and G’’) and (H’ and H’’). Scale bars represent 10 µm. (I and J) Overexpressed GFP-ANKFY1 and mCherry-GAPVD1 colocalize in HEK293T cells (I–I’’). (J) Colocalization of GFP-ANKFY1 and mCherry-GAPVD1 is confirmed by a scatter plot and a high Pearson’s coefficient (0.98). (K and L) Colocalization of GFP-ANKFY1 and an mCherry control appears incomplete (K–K’’). (L) Pearson coefficient is 0.35. (M) Upon overexpression and co-IP N-Myc–tagged GAPVD1 precipitates GFP-ANKFY1, but not Mock-GFP, in HEK293T cells. (N) Upon overexpression and co-IP with anti-GFP antibody, GFP-tagged GAPVD1 precipitates Myc-ANKFY1, but not Mock-GFP, in HEK293T cells. GAPVD1 cDNA constructs that reflect the mutations from patients B1391 (asterisk) and A4619 (dagger) show reduced binding affinity to Myc-ANKFY1. (O) Quantitation of density from (N) shows a significantly reduced affinity of mutant GAPVD1 (R937Q and L414V) to Myc-ANKFY1. Densitometry results from (N) were expressed as ANKFY1/GAPVD1wild type/mutant (n=3, P<0.05 for R937Q and P<0.01 for L414V). contr, control; IP, immunoprecipitation; PC, Pearson's coefficient; scra, scrambled; wt/mut, wild type/mutant.
Next, we explored the subcellular localization of GAPVD1. Overexpression constructs of GAPVD1, including constructs representing the patient mutations, predominantly localized to the cytosol (Figure 2E, Supplemental Figure 3, A–C). Truncations of GAPVD1 lacking the RasGAP domain localized to (RAB5-positive) vesicles (Supplemental Figure 3, D–F), although deleting the VPS9 domain had no overt effect (Supplemental Figure 3G). Eight different antibodies detected overexpressed GAPVD1 in podocytes using immunofluorescence but never the endogenous protein (data not shown).
We analyzed the subcellular localization of ANKFY1 in podocytes using a conditional CRISPR/Cas approach.76 ANKFY1 was observed in intracellular vesicles that were absent from the gRNA/GFP-expressing cells (Figure 2, F and F’). Applying Airyscan technology for enhanced resolution in human tissue sections, we confirmed a vesicular staining pattern of ANKFY1 in podocytes that was absent in the control (Figure 2, G–H’’). GFP-tagged overexpression constructs of ANKFY1 localized to vesicles in cultured human podocytes that in turn fully colocalized with mCherry-RAB5 (Supplemental Figure 4, A–B’’), whereas endogenous RAB5 and ANKFY1 showed partial colocalization (Supplemental Figure 4, C–C’’). The deletion of the FYVE domain of ANKFY1 abrogated vesicular localization of overexpressed ANKFY1 (Supplemental Figure 4, D and E), whereas the patient mutation ANKFY1-R95L showed no overt mislocalization (Supplemental Figure 4F). We conclude that the SRNS-causing mutation does not alter endosomal localization of ANKFY1.
Studying HEK293T cells, we noted that overexpressed GFP-ANKFY1 and mCherry-GAPVD1 colocalize completely (Figure 2, I and J), whereas Mock-mCherry (Figure 2, K and L) showed partial colocalization. In cultured podocytes, tagged overexpression constructs of both proteins partially colocalized (Supplemental Figure 4, G–G’’). The extent of colocalization of GAPVD1 and ANKFY1 thus appears cell-type dependent.
We hypothesized that GAPVD1 and ANKFY1 proteins interact. Using co-IP, we demonstrated interaction between GFP-GAPVD1 and Myc-ANKFY1 (Figure 2M). Reciprocally, GFP-ANKFY1 precipitates Myc-GAPVD1 (Supplemental Figure 5A). Introducing the mutations of GAPVD1 that caused nephrotic syndrome reduced binding of GAPVD1 to ANKFY1 (Figure 2N, quantitation Figure 2O). These mutations thus weaken the interaction of GAPVD1 with ANKFY1.
GAPVD1 Interacts with Nephrin
Nephrin is subject to endocytosis51 and IQGAP1, another RasGAP domain protein, interacts with nephrin.77 We therefore hypothesized that GAPVD1 might interact with nephrin. Performing co-IP experiments in HEK293T cells, we found that Myc-nephrin precipitated with GFP-GAPVD1 (Figure 3A). The reverse experiment confirmed this finding (Supplemental Figure 5B).
Figure 3.

The N-terminal cytosolic domain of nephrin interacts with the RasGAP and VPS9 domains of GAPVD1 and both proteins partially colocalize in neonatal rat kidney. (A) Upon overexpression in HEK293T cells and co-IP, GFP-tagged GAPVD1, but not Mock-GFP, precipitates Myc-tagged nephrin, yielding only the lower of two bands (arrow head). (B) GAPVD1 cDNA constructs that reflect the mutations from patients A4619 (p.Leu414Val) and B1391 (p.Arg937Gln) exhibit reduced binding affinity to nephrin. (C) Quantitation of density from (B) shows a significantly reduced affinity of GAPVD1 mutants to nephrin. Densitometry results from (B) were expressed as nephrin/GAPVD1wild type/mutant (n=3, P<0.05). (D) Schematic of truncation constructs of GAPVD1 and their ability to interact with nephrin (indicated by “+” versus “–,” also see below). (E) Schematic of truncation constructs of nephrin and their ability to interact with GAPVD1 (indicated by “+” versus “–,” also see below). (F) Upon overexpression and co-IP, full-length nephrin does not precipitate GFP-tagged GAPVD1 that lacks both functional domains (ΔRasGAP, ΔVPS9), whereas full-length nephrin precipitates the respective GAPVD1 constructs that contain the RasGAP or the VPS9 domain alone. This suggests that both functional domains of GAPVD1 show affinity for nephrin independently. (G) Mock-GFP and a truncated cDNA construct of nephrin (aa 1160–1241) that reflects the C-terminal half of the ICD of nephrin do not interact with GFP-GAPVD1. A GFP-tagged truncation construct (aa 1084–1160) that represents the N-terminal half of the ICD of nephrin interacts with Myc-tagged GAPVD1 (asterisk). This suggests that this subdomain of 76 aa mediates binding to GAPVD1. (H–I’’) Frozen sections of neonatal rat kidney were stained with anti-nephrin (red) and anti-GAPVD1 (green) or control. Nuclei are marked by Hoechst 33342 in blue. (I–I’’) Nephrin and GAPVD1 colocalize partially in newborn rat kidney. GAPVD1 is not restricted to podocytes but localizes to mesangial cells as well. (H–H’’) The third elution fraction obtained during the antibody purification of the GAPVD1 antibody (that contains only traces of antibody) was used as control and shows virtually no GAPVD1 signal. All images were recorded with identical confocal settings. Scale bars represent 10 µm. aa, amino acid; IP, immunoprecipitation; term., terminal.
Analyzing the GAPVD1 patient mutations R937Q and L414V, we observed a significantly reduced binding to Myc-nephrin for cDNA constructs reflecting these mutations (Figure 3B, quantitation Figure 3C). The mutations of GAPVD1 that cause nephrotic syndrome thus affect the interaction with nephrin.
To map the interacting domains on GAPVD1, we performed truncation mapping by co-IP and found that the RasGAP domain of GAPVD1 (aa 1–458) and the VPS9 domain of GAPVD1 (aa 1355–1460) were precipitated by full-length nephrin, whereas the large interjacent domain of GAPVD1 (aa 458–1355) was not (Figure 3, D and F). Each of the functional domains of GAPVD1 seem to be sufficient for interaction with nephrin (Figure 3, D and F).
We similarly employed truncation mapping for nephrin. The N-terminal half of the cytosolic domain (intracellular domain, ICD) of nephrin (nephrin aa 1084–1160) was precipitated by Myc-GAPVD1, whereas Mock-GFP or the C-terminal half of the ICD (aa 1160–1241) were not (Figure 3, E and G). Consequently, we found that a nephrin lacking the last 82 aa (R1160Stop) shows robust interaction with GAPVD1 (Supplemental Figure 5C). We conclude that a domain of 76 residues located at the N-terminal half of the ICD of nephrin (aa 1084–1160) is sufficient for GAPVD1 interaction.
GAPVD1 and Nephrin Partially Colocalize in Newborn Rat Glomeruli
To analyze the localization of GAPVD1 in vivo together with nephrin, we stained newborn rat kidney sections using antibodies directed against nephrin and murine Gapvd1.53 The control staining (Figure 3, H–I’’) resulted in virtual absence of a fluorescent signal, whereas the antibody recorded at identical confocal settings revealed expression of GAPVD1 in podocytes and mesangial cells. Interestingly, GAPVD1 and nephrin partially colocalize in podocytes, further supporting a functional connection between these proteins.
Mutations of GAPVD1 that Cause Nephrotic Syndrome Increase Binding to Active RAB5
RAB5 exists in a GTP-bound active state and a GDP-bound inactive state (Figure 4A). As is characteristic for a GEF, GAPVD1 was shown to specifically bind to inactive RAB5.53,66,70 To test if the GAPVD1 mutations alter the RAB5 interaction, we performed co-IP using constitutively active and dominant negative cDNA constructs of RAB5. These RAB5 derivatives with substitution of a single amino acid are locked in the active and inactive state, respectively (Figure 4B). We quantified the relative density of the respective RAB5 constructs that precipitated with GAPVD1. The low active/inactive RAB5 ratio of wild-type GAPVD1 confirms the published findings (Figure 4C, quantitation in Figure 4D). Evaluating the mutants R937Q and L414V in this fashion, we observed a significantly higher ratio indicating an increased binding of mutant GAPVD1 to active RAB5. The mutations that cause nephrotic syndrome thus impair the specificity of GAPVD1 for inactive RAB5. Further, we observed strongly reduced binding of the mutant ANKFY1-R95L to active RAB5 (Figure 4E, quantitation Figure 4F). The patient mutations of both genes thus alter RAB5-binding.
Figure 4.

Mutations of GAPVD1 that cause nephrotic syndrome increase the affinity to active RAB5, and GAPVD1 promotes dextran endocytosis. (A and B) Schematic showing that (A) RAB5 shuttles between active and inactive states dependent on binding to GTP/GDP, whereas (B) dominant negative and constitutively active RAB5 constructs are clamped to active and inactive states, respectively. (C) Overexpression and co-IP of mCherry-tagged RAB5 dominant negative (RAB5 dom. neg.) and constitutively active (RAB5 const. act.) constructs together with Myc-GAPVD1 reflecting the wild-type (WT) sequence or mutations causing nephrotic syndrome (R937Q and L414V). WT and mutant GAPVD1 interact with active and inactive RAB5. The mutant constructs of GAPVD1 show a stronger affinity to constitutively active RAB5 (asterisks) compared with WT GAPVD1 († sign). (D) Quantitation of density from precipitates analogous to (C) normalized to respectively precipitated RAB5 construct for co-IPs and shown as a ratio of RAB5 const. act. divided by RAB5 dom. neg. (n=4, P<0.05 or 0.01, respectively). (E) Overexpression and co-IP of mCherry-tagged constitutively active RAB5 together with Myc-ANFKY1 reflecting the WT sequence or the R95L mutation shows strongly reduced amounts of RAB5 precipitating with the mutant ANKFY1 (asterisk), indicating a reduced binding affinity. (F) Quantitation of density from precipitates of RAB5 protein analogous to (E) normalized to respectively precipitated Myc-ANKFY1 WT or mutant protein (n=3, P<0.05). (G–H’) Human podocytes transfected with plasmids expressing gRNA, Cas9, and GFP are exposed to Texas-Red-dextran (10 kD) for 30 minutes. Nuclei are marked by Hoechst 33342 in blue. (G and G’) Podocytes expressing control gRNA exhibit comparable Texas-Red-dextran endocytosis to the neighboring cells, whereas (H and H’) podocytes expressing a gRNA targeting GAPVD1 exhibit reduced tracer endocytosis. Scale bars represent 10 µm. (I) Quantitation of results from (G–H’). For both control gRNA or GAPVD1 targeting gRNA fluorescence intensity ratio is shown between gRNA-expressing cells and their nontransfected neighboring cells (n=2, approximately 25 cells each, P<0.001).
To analyze the functional consequence of the altered RAB5 interaction of the GAPVD1 mutations, we used a RAB5 activity assay that employs a RAB5-GTP–specific antibody. We observed an increase of active RAB5 compared with wild-type GAPVD1 for mutation R937Q but not L414V (Supplemental Figure 6A, quantitation Supplemental Figure 6B). In this assay, we observed a relative increase of mutant GAPVD1 precipitating with active RAB5 (Supplemental Figure 6A). This suggests increased binding to endogenous RAB5-GTP.
RAB5 is an essential regulator of endocytosis. To evaluate endocytic activity in podocytes, we applied Texas-Red-dextran (10 kD). The extent of tracer endocytosis reflects fluid-phase endocytosis. We observed reduced tracer uptake upon CRISPR/Cas-mediated loss of function of GAPVD1 (Figure 4, G–I). GAPVD1 thus appears to promote endocytic activity. Surprisingly, overexpression of GAPVD1 did not result in increased RAB5 activity or increased uptake of dextran (Supplemental Figure 6, A–E).
Analysis of Podocyte Migration Rate Indicates Functional Significance of the Patient Mutations of GAPVD1 and ANKFY1
The evaluation of podocyte migration rate is an established functional assay in podocyte cell lines and has been extensively used to characterize the deleteriousness of monogenic mutations in SRNS genes.2,7,14,22,24,32,38 To analyze the role of GAPVD1, we generated stable shRNA-expressing human podocyte lines and measured the podocyte migration rate using the IncuCyte videomicroscopy system. Two independent shRNAs caused a reduced podocyte migration rate (Supplemental Figure 7, A–G). Transient expression of a murine wild-type Gapvd1 cDNA construct rescued the reduced migration rate, whereas constructs corresponding to the patient mutations resulted in a partial rescue. This indicates a hypomorphic function of these alleles regarding podocyte migration rate (Figure 5, A–E, quantitation in Figure 5F). Knockdown and expression of the rescue constructs was confirmed by immunoblotting (Supplemental Figure 7, A and B).
Figure 5.

GAPVD1 and ANKFY1 regulate podocyte migration rate. (A–E) Podocyte migration rate is analyzed by IncuCyte videomicroscopy. Representative images show human podocytes after induction of scratch wound (light blue) and 22 hours thereafter. Scratch wound area (light blue) and podocytes that have migrated (dark blue) are shown at 22 hours. Serum addition strongly increases podocyte migration rate. Podocytes stably expressing scrambled shRNA (negative control) show (A) complete wound closure, whereas (B–F) silencing GAPVD1 results in reduced migration. The decrease in podocyte migration was (C) strongly reversed by transfection of mouse Gapvd1 but (D and E) only partially rescued by murine Gapvd1 constructs reflecting mutations R937Q and L414V detected in patients with nephrotic syndrome. (F) Graph shows wound confluence versus time for conditions described in (A–E). Error bars indicate SD of 12 wells with identical conditions (n=3). (G–J) Podocyte migration is observed, indicating (G) complete wound closure upon stable expression of scrambled shRNA (negative control), whereas (H) silencing ANKFY1 in the presence of a mock-rescue results in reduced migration. The decrease in podocyte migration was (I) strongly reversed by transfection of shRNA-resistant ANKFY1, whereas (J) introduction of the mutation from family B1027 (R95L) into the rescue construct abrogates this ability. (K) Graph shows wound confluence versus time for the conditions described in (G–J). Error bars indicate SD of 12 wells with identical conditions (n=3).
To analyze a role of ANKFY1 for podocyte migration, we generated shRNA directed against human ANKFY1 and confirmed efficient silencing (Supplemental Figure 8A). Upon knockdown of ANKFY1, we observed a significantly reduced podocyte migration rate for three independent shRNAs (Supplemental Figure 8, C–G). ANKFY1, similar to GAPVD1, is thus required for podocyte migration. Transient expression of shRNA-resistant ANFKY1 completely rescued the reduced migration, whereas introducing the mutation R95L into the rescue construct abrogated the ability to rescue. This indicates a pronounced functional effect of this mutation (Figure 5, G–J, quantitation Figure 5K). The expression of the rescue constructs was verified by immunoblotting (Supplemental Figure 8B).
Silencing the Drosophila Ortholog of GAPVD1 Impairs Nephrocytes and Results in Mislocalization of Fly Nephrin
To further validate a causative role of GAPVD1 for hereditary nephrotic syndrome, we employed the Drosophila model. Garland cell nephrocytes are podocyte-like cells that are an established model for glomerular disease,54,78 including monogenic SRNS.55 In nephrocytes, the ortholog of nephrin forms autocellular slit diaphragms across membrane invaginations called labyrinthine channels.54,55,78
By sequence analysis, we identified the uncharacterized Drosophila gene CG1657 as the ortholog of human GAPVD1 (Figure 6A). This gene encodes a protein of 1712 aa that contains both a RasGAP and a VPS9 functional domain. We introduce the term Gapvd1 for the Drosophila ortholog CG1657.
Figure 6.

Silencing the Drosophila ortholog of GAPVD1 in nephrocytes affects slit diaphragm restriction and nephrocyte function. (A) Amino acid sequence of human GAPVD1 is 29% identical to the Drosophila ortholog CG1657 (Gapvd1), which shares the RasGAP and VPS9 domain as functional domains. (B) Silencing the GAPVD1 ortholog by two independent RNAi lines in nephrocytes using prospero-GAL4 significantly reduces uptake of FITC-albumin as an established assay of nephrocyte function. (C) Quantitation of data in (B) (n=3 per genotype, P<0.001). (D–D’’) Equatorial cross-section of a negative control garland cell nephrocyte costained for the nephrin ortholog Sns (green) and the KIRREL/NEPH1 ortholog Kirre (red). Slit diaphragm proteins localize at the cell periphery in a fine line. Inset shows a subcortical section; Sns and Kirre are restricted to the plasma membrane. Nuclei are marked by Hoechst 33342 in blue. Scale bar represents 5 µm. (E–E’’) Equatorial cross-section of garland cell nephrocytes expressing Gapvd1-RNAi shows appearance of vesicles (arrow heads) and broadening of the line of slit diaphragm proteins. Inset shows a subcortical section; Sns and Kirre are observed in puncta and are not restricted to the membrane. Nuclei are marked by Hoechst 33342 in blue. Scale bar represents 5 µm. (F) Nephrocyte expressing control-RNAi shows regular formation of slit diaphragms (black arrow heads). Labyrinthine channels are slender and restricted to the cortical area (white asterisks). (G and H) EM image from a section through the surface of a nephrocyte expressing Gapvd1-RNAi shows cross-section of slit diaphragms (black arrow heads) on the surface but also in an ectopic localization deeper in the labyrinthine channels (red arrow heads, see also inset). Labyrinthine channels are dilated, fused, and protrude deeply into the cell (white asterisks). (I) Quantitation of the number of ectopic intracellular slit diaphragms underneath the cell surface formed per micrometer of the cell surface length. Note that ectopic slit diaphragms are absent under negative control conditions, whereas more than one ectopic slit diaphragm per micrometer is formed upon expression of Gapvd1-RNAi 1 (quantitation of six cells from three different animals per genotype, P<0.001). (J and K) Compared with (J) control nephrocytes, (K) cells expressing Gapvd1-RNAi show cortical areas of lower electro-density (black asterisks). These areas correspond to the enlarged labyrinthine channels (see [G and H]). Pros, prospero-GAL4.
To analyze conservation of a functional role of the ortholog of GAPVD1 in this Drosophila model, we expressed two independent RNAis directed against Gapvd1 in nephrocytes. Using the established read-out of FITC-albumin endocytosis,55 we observed significantly reduced nephrocyte function for both Gapvd1-RNAi lines (Figure 6, B and C). Knockdown efficiency was confirmed by quantitative PCR (Supplemental Figure 9D). Staining the slit diaphragm proteins Sns (ortholog of nephrin) and Kirre (ortholog of NEPH1), we found both slit diaphragm proteins to adhere to the nephrocyte surface in a fine, well defined line under control conditions (Figure 6, D–D’’). Silencing of Gapvd1 resulted in the appearance of subcortical vesicles/puncta and the line of slit diaphragm proteins appeared distorted, including protrusions from the surface toward the interior of the cell (Figure 6, E–E’’). To visualize the slit diaphragms directly, we performed TEM. In control nephrocytes, slit diaphragms were formed at regular intervals along the surface but never ectopically. Labyrinthine channels were narrow and limited to the cortical area of the cell (Figure 6F). Upon knockdown of Gapvd1, the slit diaphragms on the surface were formed properly but ectopic slit diaphragms appeared in addition (Figure 6, G and H). On average, about one ectopic slit diaphragm per micrometer surface area was formed upon Gapvd1-silencing, whereas ectopic slit diaphragms were absent in control nephrocytes (Figure 6I). The mislocalized slit diaphragms correspond to the protrusions of slit diaphragm proteins that were observed by immunofluorescence. We did not detect a transcriptional upregulation of the ortholog of nephrin upon Gapvd1 silencing (Supplemental Figure 9D), suggesting that the excess of nephrin reflects reduced protein degradation.
TEM further revealed that the labyrinthine channels became strongly dilated and protruded deeper into the cell (Figure 6, G and H). These enlarged channels impressed as cortical areas of reduced electro-density in lower magnifications that were absent from control cells (Figure 6, J and K). Because the channel formation is dependent on the slit diaphragm proteins, the enlarged channels are potentially caused by the ectopic formation of slit diaphragms.
We performed immunostaining of Sns/Kirre and TEM upon expression of a second, independent RNAi line and observed a similar phenotype to the first Gapvd1-RNAi (Supplemental Figure 9, A–C). Taken together, these findings imply that loss of Gapvd1 impairs the function of the podocyte-like nephrocytes and results in altered trafficking of the ortholog of nephrin.
Discussion
Here, we describe the discovery of recessive mutations in GAPVD1 and likely also ANKFY1 as novel monogenic causes of hereditary nephrotic syndrome in humans. Interestingly, we found that both proteins are interaction partners that are expressed in podocytes. We demonstrate that GAPVD1 interacts with nephrin and both proteins partially colocalize in rat glomeruli. Overexpressing GAPVD1 that reflects mutations from patients with nephrotic syndrome resulted in reduced ANKFY1 and nephrin affinity, whereas the mutations conversely increased binding to GTP-bound RAB5. GAPVD1 and ANKFY1 were required for podocyte migration and this assay indicated functional significance for the mutations of both genes. Finally, silencing the ortholog of GAPVD1 in Drosophila nephrocytes impaired tracer uptake in these podocyte-like cells and resulted in mislocalization of fly nephrin.
Our findings implicate RAB5 regulation in human hereditary nephrotic syndrome. The interaction of GAPVD1 and nephrin suggests that endocytic trafficking of nephrin may play a role in an RAB5-mediated pathogenesis. Consistent with such a role, the mutations that we discovered in our patients affected RAB5 binding and the interaction with nephrin. This is supported by our findings in Drosophila nephrocytes, where slit diaphragms are mislocalized. This may be caused by a lack of endocytic removal of ectopic fly nephrin and/or impaired degradation. Ectopic slit diaphragms have been reported in pericardial nephrocytes upon silencing of RAB7, which blocks endo-lysosomal function further downstream.79 From these findings we derived a hypothetic working model (shown in Supplemental Figure 10). In line with previous reports,43–48,51,80,81 our data thus underscore the importance of the endocytic pathway for nephrin trafficking. This pathway may further be involved in the delivery of nephrin to the slit diaphragm and/or slit diaphragm maintenance by endocytic turnover. Similar to dynamin, endophilin, and synaptojanin,52 GAPVD1 has been implicated in clathrin uncoating71 but a connection with these proteins currently remains unclear. Actin regulation and endocytosis are intertwined, and RAB5 also promotes activation of RAC1.82 As suggested by the effect on podocyte migration rate, actin dysregulation thus may contribute to the roles of GAPVD1 and ANKFY1 in nephrotic syndrome.
Endocytosis is a fundamental cellular process. Although it may seem surprising that recessive mutations in a RAB5 regulator such as GAPVD1 manifest exclusively as nephrotic syndrome, the functional redundancy with RAB5-GEFs like RIN1, ALS2, and RABGEF1 may compensate for most cellular functions. Similarly, a complete GAPVD1 knock out was found to be viable in Caenorhabditis elegans.70
GAPVD1 and ANKFY1 appear to be rare causes of nephrotic syndrome. This is consistent with a number of recently discovered novel monogenic causes1,19,22,24,32 that are similarly uncommon. Nevertheless, the identification of monogenic causes, including the less frequent ones, has shaped our understanding of podocyte biology.3 In conclusion, our findings implicate two RAB5-interacting proteins in hereditary nephrotic syndrome and suggest RAB5 regulation as a novel pathogenetic mechanism.
Accession Numbers
Human GAPVD1 full-length protein is GenBank accession number NM_015635.3.
Human ANKFY1 full-length is GenBank accession number NM_001257999.2.
Disclosures
F.H. is a cofounder of Goldfinch-Bio and receives royalties from Claritas. The remaining authors declare that they have no competing financial interests.
Supplementary Material
Acknowledgments
We are grateful to the families and study individuals for their contribution. We thank the Yale Center for Mendelian Genomics and the Broad Center for Mendelian Genomics for whole-exome sequencing analysis. F.H. is the William E. Harmon Professor. We thank R. Nitschke, Life Imaging Centre, University of Freiburg, for help with confocal microscopy.
This research was supported by grants from the National Institutes of Health to F.H. (DK076683), H.L.R., and D.M. (UM1 HG008900), and to the Yale Center for Mendelian Genomics (U54HG006504); by the Deutsche Forschungsgemeinschaft (DFG) to T.H. (HE 7456/1-1), A.T.v.d.V. (VE916/1-1), and T.J.-S. (Jo 1324/1-1); by the Iinuma-Tsuchiya Foundation for Overseas Research to M.N.; and by the German National Academy of Sciences Leopoldina to E.W. (LPDS-2015-07). The Broad Center for Mendelian Genomics was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung, and Blood Institute.
T.H., R.S., S.S., D.S., D.A.B., J.K.W., A.D., A.T.v.d.V., E.W., M.N., T.J.-S., A.J.M., J.R., S.A., R.P.L., S.M., K.L., M.L., D.M., and H.L.R. generated total genome linkage analysis, performed exome capture and massively parallel sequencing, and performed whole-exome evaluation and mutation analysis. R.S. and T.H. performed cDNA cloning, protein purification, tracer endocytosis and immunofluorescence, and subcellular localization studies in cell lines by confocal microscopy. R.S., E.W., and A.T.v.d.V. performed migration assays in immortalized human podocytes. T.H. and R.S. performed coimmunoprecipitation and the experiments in Drosophila. V.T., J.D.H., A.B., S.E.D., J.A.K., V.T., and F.H. recruited patients and gathered detailed clinical information for the study. L.S.F. and S.M.J. analyzed renal histology and electron microscopy and provided images. F.H. conceived of and directed the study. T.H. wrote the manuscript with help from F.H. The manuscript was critically reviewed by all of the authors.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017121312/-/DCSupplemental.
References
- 1.Bierzynska A, Soderquest K, Dean P, Colby E, Rollason R, Jones C, et al.; NephroS; UK study of Nephrotic Syndrome : MAGI2 mutations cause congenital nephrotic syndrome. J Am Soc Nephrol 28: 1614–1621, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi WI, et al.: Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest 127: 912–928, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovric S, Ashraf S, Tan W, Hildebrandt F: Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol Dial Transplant 31: 1802–1813, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al.: Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Löwik MM, Groenen PJ, Pronk I, Lilien MR, Goldschmeding R, Dijkman HB, et al.: Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int 72: 1198–1203, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Ebarasi L, Ashraf S, Bierzynska A, Gee HY, McCarthy HJ, Lovric S, et al.: Defects of CRB2 cause steroid-resistant nephrotic syndrome. Am J Hum Genet 96: 153–161, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gee HY, Sadowski CE, Aggarwal PK, Porath JD, Yakulov TA, Schueler M, et al.: FAT1 mutations cause a glomerulotubular nephropathy. Nat Commun 7: 10822, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al.: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Ozaltin F, Li B, Rauhauser A, An SW, Soylemezoglu O, Gonul II, et al.: DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol 24: 377–384, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozaltin F, Ibsirlioglu T, Taskiran EZ, Baydar DE, Kaymaz F, Buyukcelik M, et al.; PodoNet Consortium : Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet 89: 139–147, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Has C, Spartà G, Kiritsi D, Weibel L, Moeller A, Vega-Warner V, et al.: Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med 366: 1508–1514, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kambham N, Tanji N, Seigle RL, Markowitz GS, Pulkkinen L, Uitto J, et al.: Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis 36: 190–196, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Zenker M, Aigner T, Wendler O, Tralau T, Müntefering H, Fenski R, et al.: Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13: 2625–2632, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al.: ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 123: 5179–5189, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al.: COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 18: 2773–2780, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, et al.: COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121: 2013–2024, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, et al.: Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 79: 1125–1129, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al.: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, et al.: Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 25: 1991–2002, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al.: Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, et al.: ARHGDIA: A novel gene implicated in nephrotic syndrome. J Med Genet 50: 330–338, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, et al.: ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123: 3243–3253, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al.: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42: 72–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, et al.: KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 125: 2375–2384, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al.; PodoNet Consortium : MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365: 295–306, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, et al.: Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet 69: 1033–1045, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ovunc B, Otto EA, Vega-Warner V, Saisawat P, Ashraf S, Ramaswami G, et al.: Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J Am Soc Nephrol 22: 1815–1820, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, et al.: Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 82: 673–684, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, et al.: Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30: 215–220, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Vollrath D, Jaramillo-Babb VL, Clough MV, McIntosh I, Scott KM, Lichter PR, et al.: Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet 7: 1091–1098, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, et al.: Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38: 1397–1405, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Gee HY, Ashraf S, Wan X, Vega-Warner V, Esteve-Rudd J, Lovric S, et al.: Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 94: 884–890, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. : Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway–Mowat syndrome. Am J Hum Genet 95: 637–648, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sethi S, Fervenza FC, Zhang Y, Smith RJ: Secondary focal and segmental glomerulosclerosis associated with single-nucleotide polymorphisms in the genes encoding complement factor H and C3. Am J Kidney Dis 60: 316–321, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeanpierre C, Denamur E, Henry I, Cabanis MO, Luce S, Cécille A, et al.: Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 62: 824–833, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K: Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J Biol Chem 275: 4251–4257, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al.: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Rao J, Ashraf S, Tan W, van der Ven AT, Gee HY, Braun DA, et al.: Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J Clin Invest 127: 4257–4269, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, et al.: Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48: 457–465, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyake N, Tsukaguchi H, Koshimizu E, Shono A, Matsunaga S, Shiina M, et al.: Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am J Hum Genet 97: 555–566, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park E, Ahn YH, Kang HG, Miyake N, Tsukaguchi H, Cheong HI: NUP107 mutations in children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 32: 1013–1017, 2017 [DOI] [PubMed] [Google Scholar]
- 42.Braun DA, Rao J, Mollet G, Schapiro D, Daugeron MC, Tan W, et al.: Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat Genet 49: 1529–1538, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, et al.: beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci U S A 103: 14110–14115, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teng B, Schroder P, Müller-Deile J, Schenk H, Staggs L, Tossidou I, et al.: CIN85 deficiency prevents nephrin endocytosis and proteinuria in diabetes. Diabetes 65: 3667–3679, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin XS, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T: Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol 20: 2534–2545, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babayeva S, Rocque B, Aoudjit L, Zilber Y, Li J, Baldwin C, et al.: Planar cell polarity pathway regulates nephrin endocytosis in developing podocytes. J Biol Chem 288: 24035–24048, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Königshausen E, Zierhut UM, Ruetze M, Potthoff SA, Stegbauer J, Woznowski M, et al.: Angiotensin II increases glomerular permeability by β-arrestin mediated nephrin endocytosis. Sci Rep 6: 39513, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quack I, Woznowski M, Potthoff SA, Palmer R, Königshausen E, Sivritas S, et al.: PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem 286: 12959–12970, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris DP, Vogel P, Wims M, Moberg K, Humphries J, Jhaver KG, et al.: Requirement for class II phosphoinositide 3-kinase C2alpha in maintenance of glomerular structure and function. Mol Cell Biol 31: 63–80, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bechtel W, Helmstädter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, et al.: Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol 24: 727–743, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inoue K, Ishibe S: Podocyte endocytosis in the regulation of the glomerular filtration barrier. Am J Physiol Renal Physiol 309: F398–F405, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al.: Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest 122: 4401–4411, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lodhi IJ, Chiang SH, Chang L, Vollenweider D, Watson RT, Inoue M, et al.: Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab 5: 59–72, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weavers H, Prieto-Sánchez S, Grawe F, Garcia-López A, Artero R, Wilsch-Bräuninger M, et al.: The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature 457: 322–326, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hermle T, Braun DA, Helmstädter M, Huber TB, Hildebrandt F: Modeling monogenic human nephrotic syndrome in the Drosophila garland cell nephrocyte. J Am Soc Nephrol 28: 1521–1533, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bour BA, Chakravarti M, West JM, Abmayr SM: Drosophila SNS, a member of the immunoglobulin superfamily that is essential for myoblast fusion. Genes Dev 14: 1498–1511, 2000 [PMC free article] [PubMed] [Google Scholar]
- 57.Galletta BJ, Chakravarti M, Banerjee R, Abmayr SM: SNS: Adhesive properties, localization requirements and ectodomain dependence in S2 cells and embryonic myoblasts. Mech Dev 121: 1455–1468, 2004 [DOI] [PubMed] [Google Scholar]
- 58.Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, et al.: Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet 11: 379–388, 2002 [DOI] [PubMed] [Google Scholar]
- 59.Machuca E, Benoit G, Nevo F, Tête MJ, Gribouval O, Pawtowski A, et al.: Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol 21: 1209–1217, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abid A, Khaliq S, Shahid S, Lanewala A, Mubarak M, Hashmi S, et al.: A spectrum of novel NPHS1 and NPHS2 gene mutations in pediatric nephrotic syndrome patients from Pakistan. Gene 502: 133–137, 2012 [DOI] [PubMed] [Google Scholar]
- 61.Santín S, García-Maset R, Ruíz P, Giménez I, Zamora I, Peña A, et al.; FSGS Spanish Study Group : Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int 76: 1268–1276, 2009 [DOI] [PubMed] [Google Scholar]
- 62.Philippe A, Nevo F, Esquivel EL, Reklaityte D, Gribouval O, Tête MJ, et al.: Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J Am Soc Nephrol 19: 1871–1878, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Godefroid N, Dahan K: Expanding the clinical spectrum of congenital nephrotic syndrome caused by NPHS1 mutations. Nephrol Dial Transplant 25: 2837–2839, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Kaukinen A, Kuusniemi AM, Helin H, Jalanko H: Changes in glomerular mesangium in kidneys with congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol 25: 867–875, 2010 [DOI] [PubMed] [Google Scholar]
- 65.Patrakka J, Kestilä M, Wartiovaara J, Ruotsalainen V, Tissari P, Lenkkeri U, et al.: Congenital nephrotic syndrome (NPHS1): Features resulting from different mutations in Finnish patients. Kidney Int 58: 972–980, 2000 [DOI] [PubMed] [Google Scholar]
- 66.Hunker CM, Galvis A, Kruk I, Giambini H, Veisaga ML, Barbieri MA: Rab5-activating protein 6, a novel endosomal protein with a role in endocytosis. Biochem Biophys Res Commun 340: 967–975, 2006 [DOI] [PubMed] [Google Scholar]
- 67.Su X, Kong C, Stahl PD: GAPex-5 mediates ubiquitination, trafficking, and degradation of epidermal growth factor receptor. J Biol Chem 282: 21278–21284, 2007 [DOI] [PubMed] [Google Scholar]
- 68.Lodhi IJ, Bridges D, Chiang SH, Zhang Y, Cheng A, Geletka LM, et al.: Insulin stimulates phosphatidylinositol 3-phosphate production via the activation of Rab5. Mol Biol Cell 19: 2718–2728, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kitano M, Nakaya M, Nakamura T, Nagata S, Matsuda M: Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature 453: 241–245, 2008 [DOI] [PubMed] [Google Scholar]
- 70.Sato M, Sato K, Fonarev P, Huang CJ, Liou W, Grant BD: Caenorhabditis elegans RME-6 is a novel regulator of RAB-5 at the clathrin-coated pit. Nat Cell Biol 7: 559–569, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Semerdjieva S, Shortt B, Maxwell E, Singh S, Fonarev P, Hansen J, et al.: Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J Cell Biol 183: 499–511, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schnatwinkel C, Christoforidis S, Lindsay MR, Uttenweiler-Joseph S, Wilm M, Parton RG, et al.: The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol 2: E261, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang J, Reiling C, Reinecke JB, Prislan I, Marky LA, Sorgen PL, et al.: Rabankyrin-5 interacts with EHD1 and Vps26 to regulate endocytic trafficking and retromer function. Traffic 13: 745–757, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nehru V, Voytyuk O, Lennartsson J, Aspenström P: RhoD binds the Rab5 effector Rabankyrin-5 and has a role in trafficking of the platelet-derived growth factor receptor. Traffic 14: 1242–1254, 2013 [DOI] [PubMed] [Google Scholar]
- 75.Ito K, Ishii N, Miyashita A, Tominaga K, Kuriyama H, Maruyama H, et al.: Molecular cloning of a novel 130-kDa cytoplasmic protein, Ankhzn, containing Ankyrin repeats hooked to a zinc finger motif. Biochem Biophys Res Commun 257: 206–213, 1999 [DOI] [PubMed] [Google Scholar]
- 76.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F: Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Liang W, Yang Y, Pan Y, Yang Q, Chen X, et al.: IQGAP1 regulates actin cytoskeleton organization in podocytes through interaction with nephrin. Cell Signal 27: 867–877, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhuang S, Shao H, Guo F, Trimble R, Pearce E, Abmayr SM: Sns and Kirre, the Drosophila orthologs of Nephrin and Neph1, direct adhesion, fusion and formation of a slit diaphragm-like structure in insect nephrocytes. Development 136: 2335–2344, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu Y, Zhu JY, Zhang F, Richman A, Zhao Z, Han Z: Comprehensive functional analysis of Rab GTPases in Drosophila nephrocytes. Cell Tissue Res 368: 615–627, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeon J, Leibiger I, Moede T, Walter B, Faul C, Maiguel D, et al.: Dynamin-mediated Nephrin phosphorylation regulates glucose-stimulated insulin release in pancreatic beta cells. J Biol Chem 287: 28932–28942, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, et al.: Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One 5: e10185, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diaz J, Mendoza P, Ortiz R, Diaz N, Leyton L, Stupack D, et al. : Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J Cell Sci 127: 2401–2406, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.