

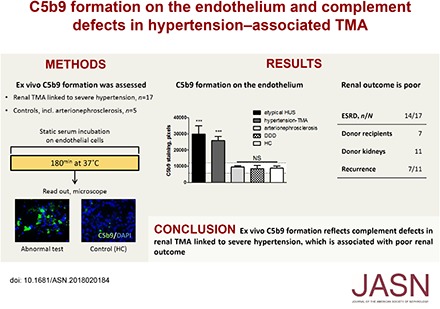
Abstract
Background Severe hypertension can induce thrombotic microangiopathy (TMA) in the renal vasculature, the occurrence of which has been linked to mechanical stress to the endothelium. Complement defects may be the culprit of disease in patients who present with severe renal disease and often progress to ESRD, despite BP control.
Methods We studied a well defined cohort of 17 patients with hypertension-associated TMA to define the prevalence of complement defects by a specific ex vivo serum-based microvascular endothelial cell assay.
Results Compared with normal human serum and samples from patients with hypertensive arterionephrosclerosis, 14 of 16 (87.5%) serum samples collected at presentation from 16 patients with hypertension-associated TMA induced abnormal C5b9 formation on microvascular endothelial cells. We detected rare variants in complement genes in eight of 17 (47%) patients. ESRD occurred in 14 of 17 (82%) patients, and recurrent TMA after transplant occurred in seven of 11 (64%) donor kidneys. Eculizumab improved the renal function in three patients and prevented TMA recurrence in an allograft recipient.
Conclusions These observations point to complement defects as the key causative factor of ESRD and recurrent TMA after transplant in patients presenting with severe hypertension. Complement defects can be identified by measurements of complement activation on microvascular endothelial cells, which should substantially influence treatment and prognosis.
Keywords: hemolytic uremic syndrome, complement, hypertension, renal biopsy
Long-standing uncontrolled mild to moderate hypertension can induce renal failure due to hypertensive arterionephrosclerosis with a slowly progressive disease course, whereas a more acute and potentially life-threatening thrombotic microangiopathy (TMA) can occur in the setting of severe hypertension. The transition to TMA among patients with severe hypertension remains poorly understood, but it can arise from mechanical stress to the endothelium,1 assuming that the kidneys are the victim rather than culprit of disease.2 Recently, we identified a clinically distinct phenotype in patients with severe hypertension and renal TMA who present with complement defects as the driving factor of disease.3 Most patients with severe hypertension achieve stable disease and do not require RRT after BP control,4,5 whereas the prognosis of those with complement defects is dismal.3
The complement cascade is an ancient and conserved effector system involved in the defense against pathogens and host homeostasis, which can be initiated via the classic pathway, the lectin pathway, and the alternative pathway (AP).6 The AP is a continuously active surveillance system operating in the circulation and on cell surfaces, which is tightly regulated to prevent damage to the self.7 Defective AP regulation, however, has been linked to another syndrome of TMA: that is, the atypical hemolytic uremic syndrome (HUS). AP defects can be due to mutations in genes encoding proteins that either regulate or activate the AP and/or autoantibodies that inhibit regulatory proteins.8,9 TMA arises from an inciting trigger to the endothelium that exceeds the complement regulatory capacity, leading to unrestrained complement activation, consumptive thrombocytopenia, microangiopathic hemolytic anemia, and ischemic organ damage.10 The pivotal role of complement defects has been confirmed by prospective trials, showing the efficacy of eculizumab, an anti-C5 mAb that blocks C5, in atypical HUS.11,12
In contrast to atypical HUS, most patients with “so-called” hypertension-associated TMA do not present with systemic hemolysis (that is, thrombocytopenia and hemolysis), and thus, renal biopsies are often needed to detect the TMA. In clinical practice, it is of utmost importance, although challenging, to identify patients with complement defects as the causative factor of disease.13 If complement defects remain unrecognized, patients may progress to ESRD without receiving optimal treatment.3 At present, however, no standard tests to detect the relevant complement defects are available. Noris et al.14 recently developed a highly specific serum-based test to detect endothelium-restricted complement dysregulation and activation in patients with atypical HUS. Here, we questioned whether the test also can be used to detect complement defects in patients presenting with severe hypertension and TMA on renal biopsy. We hypothesized that abnormal test results can differentiate patients who develop TMA on the background of complement defects from those with mechanical stress as the cause of disease. Furthermore, we extended our previous data regarding the presentation, long-term outcome, and prevalence of genetic complement defects in patients presenting with renal TMA in the setting of severe hypertension.
Methods
Patient Population
From 1980 onward, consecutive patients with hypertension-associated TMA were recruited from the Limburg Renal Registry.15 Hypertension-associated TMA was presumed in patients with TMA and typical pathologic features of severe hypertension (i.e., myxoid intimal changes, hypertrophy of the arterial vessel walls, and/or fibrinoid necrosis of arterioles) on renal biopsy,2 severe hypertension (i.e., BP levels of ≥180 mm Hg systolic and/or ≥120 mm Hg diastolic), and evidence of impending or progressive target organ dysfunction secondary to hypertension3,16; other causes of TMA were excluded according to recent guidelines.13,17 Systemic hemolysis was defined as microangiopathic hemolytic anemia (hematocrit <30%, hemoglobin <10 g/L, lactate dehydrogenase >500 U/L, and schistocytes on peripheral blood smear) and thrombocytopenia (<150,000/μl).
Patients with severe hypertension and arterionephrosclerosis without features of TMA on renal biopsy also were included. Arterionephrosclerosis was defined as obsolescence of glomeruli, intimal fibrosis, and medial thickening of arteries without morphologic evidence of other renal disease.
Eight healthy individuals without a relevant medical history who were not using any drugs were enrolled; serum samples were pooled and used as normal human serum (NHS).
At the time of renal biopsy and during follow-up, serum and heparin plasma samples were obtained, processed, and immediately stored at −80°C to prevent in vitro complement activation.18 Clinical data were obtained from the Limburg Renal Registry and the patients’ medical records. The study was approved by the local ethics committee of the Maastricht University Medical Center and is in accordance with the Declaration of Helsinki.
Routine Complement Assays
C4 and C3 plasma levels were determined using nephelometry. Classic pathway functional assays (Eurodiagnostica, Malmo, Sweden) were completed according to the manufacturer’s instructions.19 Furthermore, plasma sC5b9 levels were determined using a capture ELISA (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions.
Complement Activation on Human Microvascular Endothelial Cells of Dermal Origin
Ex vivo complement activation on human microvascular endothelial cells of dermal origin (HMEC-1; ATCC, Manassas, VA) was assessed as described with modifications.14 Briefly, HMEC-1 were plated on glass culture slides and used when >80% confluent. Before serum incubation, HMEC-1 were activated with 10 μM ADP for 10 minutes or were not activated. HMEC-1 were incubated with serum diluted in test medium (1:2, final volume 300 μl) for 3 hours at 37°C, fixed in 3% formaldehyde, and blocked with 2% BSA for 1 hour.
To analyze complement activation, HMEC-1 were stained with FITC-labeled anti-human C3c (1:20; Dako, Heverlee, Belgium) or rabbit polyclonal anti-human C5b9 (1:100; Calbiochem, San Diego, CA) followed by Alexa488-labeled anti-rabbit IgG antibody (1:100; Life Technologies, Carlsbad, CA). In selected experiments, HMEC-1 were stained with FITC-labeled goat anti-human IgG (1:100; ICN Biomedicals, Irvine, CA) or mouse anti-human C4d (1:200; Quidel, Alkmaar, The Netherlands) followed by FITC-labeled rabbit anti-mouse IgG (1:60; Dako). Nuclei were stained with 4′,6-diamidino-2-phenylindole. Fluorescent staining was analyzed by using ImageJ (National Institutes of Health, Bethesda, MD) on 15 systematically acquired fields and expressed as mean pixels per field; complement activation was compared with NHS run in parallel. Samples from patients with atypical HUS and dense deposit disease were used as positive and negative disease controls, respectively. The experimental design can be found in Supplemental Material.
Renal Immunofluorescence
Sections from snap-frozen renal tissue specimens were stained with FITC-labeled rabbit anti-human C3c (1:20) or rabbit polyclonal anti-human C5b9 (1:100) followed by Alexa488-labeled goat anti-rabbit antibody (1:100).
Genetic Testing
Coding regions of CFH, CFI, CD46, CFB, and C3 were amplified and screened for mutations and polymorphisms using DNA sequencing.20,21 Rearrangements in the CFH-CFHR1-5 genomic region were analyzed by multiplex ligation probe amplification.22 In selected cases, the presence of circulating factor H autoantibodies was assessed by ELISA.23
Statistical Analyses
Continuous variables were presented as mean (±SD) or median (interquartile range) as appropriate. Comparisons were made for each patient comparing serum-induced complement deposits for the patient and NHS run in parallel by using the paired sample t test or Wilcoxon signed rank test as appropriate. Between-group differences were analyzed by ANOVA. P<0.05 was considered statistically significant.
Results
Patient Population
Seventeen consecutive white patients who fulfilled the inclusion criteria of hypertension-associated TMA were recruited from January 1980 onward. The baseline characteristics are depicted in Table 1. Patients 5 and 7–14 have been previously reported.3 Women-to-men ratio was 0.8, and the median age at diagnosis was 38 (interquartile range, 34–45) years old. Patients invariably presented with severe hypertension, AKI, and proteinuria; therefore, a diagnosis of hypertensive kidney disease was assumed. Renal biopsies revealed characteristic lesions of TMA accompanied by prominent intimal fibrosis, myxoid intimal alterations, and/or fibrinoid necrosis of the renal arterioles, reflecting severe hypertension. Indeed, 12 (71%) of 17 patients had a known medical history of hypertension, including three patients with documented episodes of preeclampsia and/or hemolysis, elevated liver enzymes, and low platelets. No precipitating events were identified in the others. In 12 (71%) of 17 patients, no systemic hemolysis was observed. The enzymatic activity of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 appeared normal in eight patients; the other patients presented with platelet counts of over 95,000×109/L, making thrombotic thrombocytopenic purpura unlikely.24 Drug use, infection, autoimmune disease, and pregnancy as causes of TMA were ruled out.17 Familial disease was not noted. Extrarenal manifestations included severe hypertensive retinopathy (n/n=12/15), cardiac disease (n/n=11/14), and/or neurologic disease (n/n=4/17). Thus, a diagnosis of hypertension-associated TMA was made.
Table 1.
Baseline clinical features and laboratory evaluation
| No. | Age, yr | Sex | Relevant History | BP, mm Hg | Hypertensive Retinopathy, Grade | Cardiac Disease | Neurologic Disease | SCr, μmol/L | Hb, mmol/L | LDH, U/L | Platelets, ×109/L | Hemolysis, Systemic | ADAMTS13 Activity, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 37.2 | M | HTN | 220/130 | 3 | Y | N | 275 | 5.7 | 597 | 244 | N | ND |
| 2 | 47.1 | M | HTN | 280/160 | 4 | Y | Y | 1980 | 6.1 | 700 | 272 | N | 42 |
| 3 | 38.7 | M | N/a | 239/162 | 4 | Y | Y | 726 | 4.9 | 1375 | 62 | Y | 78 |
| 4 | 47.4 | M | HTN | 185/140 | 4 | Y | Y | 835 | 10.1 | 2949 | 285 | N | 61 |
| 5 | 44.0 | W | HTN | 220/120 | 2 | N | N | 649 | 8.2 | 339 | 339 | N | 76 |
| 6 | 72.3 | W | N/a | 225/125 | 3 | N | N | 356 | 4.7 | 1068 | 75 | Y | >10 |
| 7 | 27.9 | M | N/a | 240/150 | 4 | ND | N | 673 | 7.9 | 165 | 133 | N | ND |
| 8 | 28.5 | W | HELLP, HTN | 224/112 | 3 | N | N | 1065 | 5.1 | 298 | 228 | N | 82 |
| 9 | 41.1 | W | HTN | 180/120 | 3 | Y | N | 334 | 6.1 | 219 | 291 | N | ND |
| 10 | 65.0 | M | HTN | 195/105 | ND | Y | N | 162 | 7.9 | 271 | 98 | N | ND |
| 11 | 32.0 | W | PE, HTN | 180/120 | ND | Y | N | 1138 | 4.7 | 1486 | 142 | Y | 96 |
| 12 | 37.7 | M | HTN | 200/120 | 3 | Y | N | 586 | 5.3 | 2125 | 100 | Y | >10 |
| 13 | 40.3 | M | HTN, CKD | 205/114 | 2 | Y | N | 1195 | 5.7 | 1104 | 158 | N | ND |
| 14 | 38.4 | W | PE, HTN | 184/140 | 2 | ND | N | 1730 | 5.1 | 1800 | 228 | N | ND |
| 15 | 37.2 | W | HTN | 300/140 | 4 | Y | N | 1030 | 6.0 | 1128 | 204 | N | ND |
| 16 | 39.1 | M | N/a | 190/120 | 3 | ND | Y | 1089 | 6.1 | 680 | 101 | Y | ND |
| 17 | 23.4 | W | N/a | 185/120 | 3 | Y | N | 645 | 6.3 | 1053 | 179 | N | ND |
SCr, serum creatinine; Hb, hemoglobin; LDH, lactate dehydrogenase; ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; M, man; HTN, hypertension; Y, yes; N, no; ND, not determined; N/a, not applicable; W, woman; HELLP, hemolysis, elevated liver enzymes, and low platelets; PE, preeclampsia.
Five patients with hypertensive arterionephrosclerosis were included as disease controls. They presented at older age with lower diastolic BP and less severe renal failure compared with those with TMA (Supplemental Material).
Routine Complement Assays
Lower than normal C3 levels were found in five (31%) of 16 patients with hypertension-associated TMA, whereas C4 levels and functional studies of the classic pathway of complement activation were normal in all but one patient. Plasma levels of sC5b9, however, were higher than normal in patients with hypertension-associated TMA (n/n=14/14), but they did not differ from those with hypertensive arterionephrosclerosis (Supplemental Material).
Ex Vivo Complement Activation
At the time of presentation, sera from 14 (88%) of 16 patients with hypertension-associated TMA induced extensive C5b9 formation on resting HMEC-1 (25,926±9533 versus 9025±3144 pixels; P<0.01) compared with NHS (n=8) run in parallel (Figure 1A, Table 2); of note, treatment-naive serum from patient 2 was not available. In additional experiments, HMEC-1 were preincubated with ADP to mimic a perturbed endothelium,14 after which comparable results were found (Figure 1B, Table 2). The intensities of C5b9 deposits on ADP-activated HMEC-1 exposed to sera from patients with abnormal test results (n=12) were comparable with those exposed to atypical HUS sera (26,588±8229 versus 31,206±2866 pixels, respectively) (Figure 1B). Also, C3c was found (n=5; 31,098±12,690 versus 8421±3093 pixels [NHS]; P<0.05) but neither C4d nor IgG were found on ADP-activated HMEC-1, indicating selective activation of the AP. HMEC-1 exposed to ADP alone without serum or with heat-inactivated patient serum (n=3; 30 minutes at 56°C) showed neither C3c nor C5b9 staining.
Figure 1.

Hypertension-associated thrombotic microangiopathy (TMA) sera induced C5b9 formation on resting and ADP-activated human microvascular endothelial cells of dermal origin (HMEC-1) at the time of presentation. (A) Resting HMEC-1 were incubated for 3 hours with serum from patients with atypical hemolytic uremic syndrome (HUS; C3 and/or CFB, n=2; deficiency of CFHR plasma proteins and factor H autoantibody positive [DEAP] HUS, n=1; no mutations, n=1), hypertension-associated TMA (n=14), hypertensive arterionephrosclerosis (n=4), or dense deposit disease (DDD; n=5) and healthy controls (HCs; n=8). ***P<0.001 versus normal human serum (NHS) run in parallel. (B) ADP-activated HMEC-1 were incubated for 3 hours with serum from patients with atypical HUS (C3 and/or CFB, n=3), hypertension-associated TMA (n=12), hypertensive nephrosclerosis (n=4), or DDD (n=4) and HCs (n=7). Dotted horizontal areas: control, mean±SD. ***P<0.001 versus NHS run in parallel. (C) Representative immunofluorescence microscopy images of C5b9 (green) staining of ADP-activated HMEC-1 exposed to sera from patients with hypertension-associated TMA, atypical HUS, or hypertensive arterionephrosclerosis and NHS serum. The data of patients 3 and 15 have not been included. Original magnification, ×400.
Table 2.
Complement defects at the time of renal biopsy
| No. | Mutation(s) | CFH-H3 | ΔCFHR1-3 | FHAA | C4, g/L (0.11–0.35) | C3, g/L (0.75–1.35) | CPFA, % (>75) | sC5b9, ng/ml (<337) | C5b9 Formation on HMEC-1, % of the Control | |
|---|---|---|---|---|---|---|---|---|---|---|
| Resting | ADP Activated | |||||||||
| TMA and severe hypertension | ||||||||||
| 1 | No mutations | N | Y | ND | 0.35 | 1.45 | 119 | 1252 | 223a | 190a |
| 2 | CFI-P50A,37 THBD-T478I | Y | Y | ND | ND | ND | ND | ND | ND | ND |
| 3 | No mutations | Y | Y | None | 0.36 | 1.40 | ND | 4200 | 160 | 117 |
| 4 | No mutations | N | Y | None | 0.24 | 0.81 | 113 | 740 | 353a | 205a |
| 5 | No mutations | N | N | None | 0.35 | 1.56 | 113 | 3800 | 198a | 204a |
| 6 | No mutations | N | N | None | 0.23 | 1.17 | ND | 480 | 245a | 340a |
| 7 | No mutations | Y | N | None | 0.26 | 0.82 | 90 | 440 | 224a | ND |
| 8 | C3-R161W38 | N | Yb | None | 0.14 | 0.72 | 110 | 1840 | 373a | 246a |
| 9 | CFI-N151S37 | N | N | ND | 0.27 | 1.20 | 97 | 4200 | 395a | 252a |
| 10 | No mutations | N | N | ND | 0.19 | 0.87 | 99 | 1800 | 253a | ND |
| 11 | CFH-C853R | Y | N | ND | 0.21 | 0.63 | 104 | 1840 | 339a | 253a |
| 12 | C3-R161W38 | Y | N | ND | 0.30 | 0.88 | 94 | 640 | 463a | 404a |
| 13 | CD46-ΔD237/S238,39 CFH-Q950H27 | Y | Y | None | 0.28 | 0.89 | 97 | 1000 | 284a | 272a |
| 14 | C3-R161W38 | N | N | None | 0.20 | 0.69 | 95 | 2800 | 310a | 325a |
| 15 | No mutations | N | N | None | 0.25 | 1.10 | 105 | 1800 | 140 | 92 |
| 16 | C3-R161W38 | Y | N | ND | 0.47 | 0.74 | ND | ND | 336a | 283a |
| 17 | No mutations | Y | N | ND | ND | 0.64 | 50 | ND | 306a | 255a |
| Hypertensive arterionephrosclerosis | ||||||||||
| 1 | ND | ND | ND | ND | 0.33 | 1.37 | 122 | 2360 | 70 | ND |
| 2 | ND | ND | ND | ND | 0.37 | 1.36 | 122 | 888 | 89 | 71 |
| 3 | ND | ND | ND | ND | 0.26 | 0.95 | 67 | ND | 76 | 87 |
| 4 | ND | ND | ND | ND | 0.27 | 2.00 | 110 | 780 | ND | 81 |
| 5 | ND | ND | ND | ND | 0.38 | 1.22 | 103 | 1168 | 67 | 77 |
ΔCFHR1-3, deletion of complement factor H–related genes CFHR1 and CFHR3; FHAA, factor H autoantibodies; CPFA, classic pathway functional activity; HMEC-1, human microvascular endothelial cells of dermal origin; TMA, thrombotic microangiopathy; N, no; Y, yes, ND, not determined.
P value <0.05.
Genetic abnormality was found in heterozygosity.
Follow-up samples from nine patients with hypertension-associated TMA were available (Table 3). C5b9 formation normalized on resting but did not normalize on ADP-activated HMEC-1 (n=4 untreated patients; 21,972±7373 versus 7894±4136 pixels [NHS]; P<0.05) at the time of quiescent disease (Figure 2), ruling out the possibility that complement activation was a secondary phenomenon triggered by acute TMA (that is, ischemic tissue injury and/or platelet activation). As expected, C5b9 formation on resting (n=5) and ADP-activated HMEC-1 (n=4) was attenuated when incubated with samples from patients on eculizumab treatment (Figure 2), underlining the specificity of C5b9 staining.
Table 3.
C5b9 formation on resting and ADP-activated human microvascular endothelial cells of dermal origin when incubated with follow-up serum samples from patients with hypertension-associated thrombotic microangiopathy
| No. | Hb, mmol/L | LDH, U/L | Platelets, ×109/L | Creatinine, μmol/L | C5b9 formation on HMEC-1, % of the control | |
|---|---|---|---|---|---|---|
| Resting | Activated | |||||
| Eculizumab treatment | ||||||
| 2 | 7.2 | 135 | 199 | 297 | 21a | ND |
| 5 | 5.5 | 143 | 492 | ESRD | 0a | 0a |
| 6 | 6.3 | 154 | 298 | 243 | 17a | 14a |
| 9 | 5.5 | 179 | 291 | 315 | 0a | 24a |
| 11 | 6.8 | 138 | 216 | 132 | 8a | 29a |
| No treatment | ||||||
| 8 | 7.0 | 239 | 333 | ND | 160 | 231a |
| 11 | 6.8 | 197 | 174 | 205 | 163 | 293a |
| 12 | 6.1 | 181 | 232 | 220 | 87 | 171a |
| 14 | 7.5 | ND | 198 | 98 | 116 | 248a |
Hb, hemoglobin; LDH, lactate dehydrogenase; HMEC-1, human microvascular endothelial cells of dermal origin; ND, not determined.
P value <0.05 versus control serum run in parallel.
Figure 2.

C5b9 formation normalized on resting but not activated human microvascular endothelial cells of dermal origin (HMEC-1) at the time of quiescent disease, whereas eculizumab blocked C5b9 to form. (A) C5b9 formation on resting and ADP-activated HMEC-1 after incubation with serum from patients with hypertension-associated thrombotic microangiopathy (TMA) at the time of acute disease (n=7), quiescent disease (n=4), and/or during eculizumab treatment (Ecu; n=5 or n=4, respectively); individual data are shown in Table 3. (B) Representative immunofluorescence microscopy images of either resting or ADP-activated HMEC-1 exposed to sera from patients with hypertension-associated TMA at the time of quiescent disease and healthy control (HC; i.e., normal human serum[NHS]). *P<0.01 versus NHS run in parallel; ***P<0.001 versus NHS run in parallel; ^^P<0.01 versus quiescent disease samples. DAPI, 4',6-diamidino-2-phenylindole.
In contrast, sera from four patients with hypertensive arterionephrosclerosis induced normal C5b9 formation on resting HMEC-1 (n=4; 9753±1214 versus 9025±3144 pixels [NHS]); preincubation with ADP did not affect C5b9 formation (Figure 1B). Dense deposit disease samples also induced normal C5b9 formation on ADP-activated HMEC-1 (n=4; 9011±941 versus 8989±3184 pixels [NHS]) (Figure 1B). Dense deposit disease, however, has been associated with AP dysregulation in the circulation but not on the cell surface,25,26 indicating that C5b9 formation on HMEC-1 reflects endothelium-restricted complement activation. The data of the disease controls can be found in Supplemental Material.
Renal Immunofluorescence
Renal tissue specimens of ten of 14 patients with hypertension-associated TMA and abnormal C5b9 formation on resting HMEC-1 were sufficient for analysis and revealed C3c as well as C5b9 deposits along the vasculature and/or glomerular capillary wall, confirming in vivo complement activation (Figure 3, A and B). Eight (80%) of ten tissue specimens revealed unspecific entrapment of IgM, whereas staining for IgG, IgA, and light chains was negative. Electron dense deposits were not found on electron microscopy, supporting the paucity of immune complex deposits (Figure 3D). In contrast, C3c was not found in the setting of arterionephrosclerosis (tissue specimens, n=5), excluding complement activation.
Figure 3.

C3c and C5b9 staining reflect in vivo complement activation in hypertension–associated thrombotic microangiopathy (TMA) but not hypertensive arterionephrosclerosis. Representative immunofluorescence microscopy images of (A) C3c and (B) C5b9 deposits along the renal vasculature in severely hypertensive patients with TMA, whereas (D) electron dense deposits were not found; widening of the subendothelial space (asterisk) was often acknowledged. (C) C3c was not linked to hypertensive arterionephrosclerosis. Original magnification, ×400 in A and B; ×200 in C; ×1900 in D.
Rare Variants in Complement Genes
Genetic AP abnormalities were found in eight (47%) of 17 patients with hypertension-associated TMA (Supplemental Material, Table 2). The mutated genes included C3 (n=4), CFH (n=2), CFI (n=2), and/or CD46 (n=1). The CD46 mutation was found in association with CFH-Q950H, the latter of which is a rare variant of unknown significance27 also present in the normal population. Furthermore, a novel variant of unknown significance was found in THBD. Eight patients carried the −322C>T and c.2808G>T single-nucleotide polymorphisms that tag the CFH-H3 haplotype.28,29 The homozygous genomic deletion of CFHR1 and CFHR3 was identified in one patient, whereas circulating factor H autoantibodies were not found.
Clinical Outcome
Follow-up ranged from 4 months to 37 years. In all patients, intravenous administration of antihypertensive agents was started, and although BP normalized, 14 (82%) of 17 patients initially needed dialysis (Figure 4). Since 2015, eculizumab was started in five patients (patients 2, 3, 5, 6, and 9) not responding to conventional treatment. Patients were started on four weekly doses of 900 mg eculizumab followed by a single dose of 1200 mg every 2 weeks (Supplemental Material). Renal function improved in three patients, whereas two patients remained dialysis dependent.
Figure 4.

Clinical outcome of patients with hypertension-associated thrombotic microangiopathy who were treated with eculizumab (Ecu). Green bars, renal survival. Red bars, dialysis. TX, transplant recipient.
At the time of quiescent disease, treatment was stopped (patients 5 and 6) or tapered to 900 mg (patient 9) every 4 weeks; during tapering, C5b9 formation on ADP-activated HMEC-1 remained suppressed (not shown). Patient 2’s renal function is still improving, and the dosing regimen has not been tapered yet. No renal response, however, was observed in patient 3, whose serum induced normal C5b9 formation; moreover, no genetic variants were found. He died from duodenal perforation probably due to pancreatic cancer, suggesting that TMA did not occur on the background of complement defects.
Eleven allografts (cadaveric, n=5 and living [un-]related, n=6) were transplanted in seven patients (Figure 5), all of whom received calcineurin inhibitors (tacrolimus, n=10 or cyclosporin, n=1) and steroids. TMA recurrence manifested in seven grafts (recipients, n=5) either with (n=4) or without (n=3) systemic hemolysis. BP was tightly regulated, and the presence of donor-specific alloantibodies as well as infections as endothelium-damaging events was ruled out, linking TMA recurrence to complement defects. Recipients with recurrent disease showed abnormal C5b9 formation on HMEC-1 at the time of presentation. Four patients carried rare variants in complement genes (C3, n=3 and CD46, n=1). Graft loss occurred in five (71%) of seven disease episodes, although plasma exchange was initiated in five patients. At the time of transplantation, upfront eculizumab was started in a high-risk recipient (patient 11), preventing disease recurrence. The recipients with normal test results at presentation did not develop TMA recurrence.
Figure 5.

Allograft outcome. Black arrows indicated thrombotic microangiopathy recurrence. White arrows indicated retransplantation. Patient no. 13 was preemptively retransplanted. Blue bars, allograft survival. Red bars, dialysis. T, transplant recipient.
Discussion
Complement defects have been linked to various syndromes of TMA, and they have recently been acknowledged as the key causative factor of ESRD in a subset of patients with severe hypertension.3 Many of these patients, however, do not present with systemic hemolysis, and complement defects may, therefore, remain unrecognized. This study showed that abnormal serum-induced C5b9 formation on microvascular endothelial cells can identify these particular patients, reflecting complement-mediated TMA. The high prevalence of rare variants in complement genes as well as the favorable response to eculizumab treatment support this premise.
This study supports our previous hypothesis that complement defects are the key causative factor of ESRD in patients presenting with renal TMA in the setting of severe hypertension.3 Serum samples from most patients with hypertension-associated TMA but not from those with hypertensive arterionephrosclerosis induced abnormal C5b9 formation on resting and ADP-activated HMEC-1 at the time of acute TMA, reflecting massive complement activation via the AP. C5b9 deposits along the renal vasculature and/or glomerular capillary wall provided the in vivo counterparts of complement activation. At variance, C5b9 formation normalized on resting but not on ADP-activated HMEC-1 at the time of quiescent disease, underlining that TMA developed on the background of complement defects was triggered by a concomitant condition. Rare variants in complement genes associated with AP dysregulation confirmed complement defects in approximately of one half the patients. The complement gene mutations were considered pathogenic on the basis of proven functional abnormalities of the respective proteins. The presence of the CFH-H3 haplotype also might have affected disease penetrance. In our cohort, most patients had a history of hypertension, suggesting that hypertension triggered complement activation,30 leading to unrestricted AP activation and the occurrence of TMA.
In line with previous studies focusing on severe hypertension, including our patient series, systemic hemolysis seemed uncommon,3,4,31 leading to a low suspicion of TMA. In these patients with difficult cases, renal biopsies are needed to detect the TMA, the presence of which should prompt screening for complement defects.3 Routine complement assays appeared normal in most of our patients and therefore, cannot be used to detect complement defects. Elevated levels of sC5b9, however, can be found during the acute phase but lack specificity, because sC5b9 concentrations overlap with conditions not linked to systemic complement activation,32 including arterionephrosclerosis. At present, genetic studies can be considered the reference method for the determination of complement defects,13 although they are suboptimal for diagnostic purposes, because genetics are time consuming and lack sensitivity. Noris et al.14 recently showed that abnormal C5b9 formation on microvascular endothelial cells is highly specific for atypical HUS. In this study, we validate their data and reproducibility in patients who present with renal TMA in the setting of severe hypertension, differentiating patients with complement defects from those with mechanical stress as the cause of disease.
The prognosis of our patients with abnormal C5b9 formation on HMEC-1 and/or rare variants in complement genes seemed dire, with 80% of patients requiring dialysis at presentation, resembling historical atypical HUS cohorts.8,9 These clinical observations recapitulate our previous data linking complement defects to ESRD in an extended cohort of patients considered to have hypertension-associated TMA,3 pointing to a new target for treatment. Indeed, eculizumab blocked C5b9 to form on HMEC-1, and moreover, renal function recovered and/or improved in all but one patient with complement defects. Dialysis, however, could be tapered in the latter, and further improvement in renal function might have been expected on extended treatment.12 To date, two additional patient studies concerning the use of eculizumab for the treatment of TMA in the setting of severe hypertension have been published with conflicting results.33,34
Thus, the questions are how to diagnose complement-mediated TMA and when to consider treatment in patients presenting with severe hypertension. In our experience, complement defects should be considered in patients who do not respond to BP control (that is, no decrease in serum creatinine of over 25%),35 particularly when abnormal C5b9 formation is apparent. Furthermore, we feel that it is important to stress that most of our patients presented with fundoscopic lesions consistent with severe hypertension, and thus, retinal lesions should not be used to exclude underlying complement defects. Patients with complement defects appeared to benefit from treatment11; eculizumab should, therefore, be considered in these patients. Dosing schedule and treatment duration, however, remain controversial, and prospective studies are needed to establish the optimal use of eculizumab for the treatment of renal TMA in patients with severe hypertension.36 Also, it has to be proven whether longitudinal measurements of ex vivo C5b9 formation can guide treatment.
After transplantation, recurrent disease appeared to be common and linked to abnormal C5b9 formation at baseline. TMA onset after transplantation was associated with poor allograft survival, particularly among carriers of rare variants in complement genes. Thus, TMA can reoccur on the background of complement defects after transplantation. Patients who present with TMA and severe hypertension should, therefore, be screened for complement defects before renal transplantation so that prophylactic measures can be adopted,13 which is supported by the favorable outcome in a high-risk recipient who received preemptive eculizumab treatment.
This study is limited by the number of included patients. Future validation in other larger cohorts is, therefore, warranted. However, a strong aspect of our study is the fact that we studied a well defined cohort of patients who had been classified according to clinical and pathologic data.
Taken together, our data show that complement defects are the key causative factor of severe renal sequelae in patients with renal TMA and severe hypertension at presentation, indicating that these patients should be classified as complement-mediated TMA, although systemic hemolysis seemed to be uncommon. Patients with complement defects can be identified by abnormal C5b9 formation on microvascular endothelial cells, which is associated with a dismal prognosis. Finally, targeting the complement defect should be evaluated as a novel approach to treatment of renal TMA in patients with severe hypertension.
Disclosures
P.v.P. declares that he has received an unrestricted educational grant from Alexion Pharmaceuticals Inc., which has not had any influence on the results or interpretation in this article. All of the other authors declare no competing interests.
Supplementary Material
Acknowledgments
We gratefully thank the nephrologists affiliated with the Limburg Renal Registry for providing data for this study: M. Christiaans, T. Fung, M. Gelens, J. Kooman, K. Leunissen, E. Litjens, J. van der Net, F. van der Sande, E. Duijnhoven (Maastricht University Medical Center, Maastricht, The Netherlands); S. Boorsma, J. Huitema, J. Wirtz (St. Laurentius Hospital, Roermond, The Netherlands); F. de Heer, M. Krekels, F. Stifft, G. Verseput (Zuyderland Medical Center, Geleen, The Netherlands); N. ter Braak, L. Frenken, and S. Gaertner (Zuyderland Medical Center, Heerlen, The Netherlands). Furthermore, we appreciate the technical assistance of N. Bijnens, E. Geelkens, and H. van Rie (Laboratory of Clinical Immunology, Maastricht University Medical Center, Maastricht, The Netherlands).
This work was supported in part by funding from The Netherlands Organisation for Scientific Research and The Netherlands Organisation for Health Research and Development grant MKMD 40-42600-98-13007.
This study was presented in part at the 2017 Kidney Week (New Orleans, LA) on November 3, 2017.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018020184/-/DCSupplemental.
Contributor Information
Collaborators: M. Christiaans, T. Fung, M. Gelens, J. Kooman, K. Leunissen, E. Litjens, J. van der Net, F. van der Sande, E. Duijnhoven, S. Boorsma, J. Huitema, J. Wirtz, F. de Heer, M. Krekels, F. Stifft, G. Verseput, N. ter Braak, L. Frenken, and S. Gaertner
References
- 1.George JN, Nester CM: Syndromes of thrombotic microangiopathy. N Engl J Med 371: 654–666, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Chang A: Thrombotic microangiopathy and the kidney: A nephropathologist’s perspective. Diagn Histopathol 19: 158–165, 2013 [Google Scholar]
- 3.Timmermans SAMEG, Abdul-Hamid MA, Vanderlocht J, Damoiseaux JGMC, Reutelingsperger CP, van Paassen P; Limburg Renal Registry : Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int 91: 1420–1425, 2017 [DOI] [PubMed] [Google Scholar]
- 4.Zhang B, Xing C, Yu X, Sun B, Zhao X, Qian J: Renal thrombotic microangiopathies induced by severe hypertension. Hypertens Res 31: 479–483, 2008 [DOI] [PubMed] [Google Scholar]
- 5.González R, Morales E, Segura J, Ruilope LM, Praga M: Long-term renal survival in malignant hypertension. Nephrol Dial Transplant 25: 3266–3272, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT: Complement system part I - molecular mechanisms of activation and regulation. Front Immunol 6: 262, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V: Endothelial cells: Source, barrier, and target of defensive mediators. Immunol Rev 274: 307–329, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al.: Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5: 1844–1859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al.: Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8: 554–562, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jorge EG, Macor P, Paixão-Cavalcante D, Rose KL, Tedesco F, Cook HT, et al.: The development of atypical hemolytic uremic syndrome depends on complement C5. J Am Soc Nephrol 22: 137–145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al.: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169–2181, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al.: Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87: 1061–1073, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al.; Conference Participants : Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int 91: 539–551, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al.: Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood 124: 1715–1726, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Paassen P, van Breda Vriesman PJ, van Rie H, Tervaert JW: Signs and symptoms of thin basement membrane nephropathy: A prospective regional study on primary glomerular disease-The Limburg Renal Registry. Kidney Int 66: 909–913, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Cremer A, Amraoui F, Lip GY, Morales E, Rubin S, Segura J, et al.: From malignant hypertension to hypertension-MOD: A modern definition for an old but still dangerous emergency. J Hum Hypertens 30: 463–466, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al.; HUS International : An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31: 15–39, 2016 [DOI] [PubMed] [Google Scholar]
- 18.Yang S, McGookey M, Wang Y, Cataland SR, Wu HM: Effect of blood sampling, processing, and storage on the measurement of complement activation biomarkers. Am J Clin Pathol 143: 558–565, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Seelen MA, Roos A, Wieslander J, Mollnes TE, Sjöholm AG, Wurzner R, et al.: Functional analysis of the classical, alternative, and MBL pathways of the complement system: Standardization and validation of a simple ELISA. J Immunol Methods 296: 187–198, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Westra D, Volokhina E, van der Heijden E, Vos A, Huigen M, Jansen J, et al.: Genetic disorders in complement (regulating) genes in patients with atypical haemolytic uraemic syndrome (aHUS). Nephrol Dial Transplant 25: 2195–2202, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Volokhina E, Westra D, Xue X, Gros P, van de Kar N, van den Heuvel L: Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol 27: 1519–1524, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ: A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant 26: 739–741, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al.: Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 16: 555–563, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Coppo P, Schwarzinger M, Buffet M, Wynckel A, Clabault K, Presne C, et al.; French Reference Center for Thrombotic Microangiopathies : Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: The French TMA reference center experience. PLoS One 5: e10208, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, et al.: Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest 118: 608–618, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sethi S, Vrana JA, Fervenza FC, Theis JD, Sethi A, Kurtin PJ, et al.: Characterization of C3 in C3 glomerulopathy. Nephrol Dial Transplant 32: 459–465, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Mohlin FC, Nilsson SC, Levart TK, Golubovic E, Rusai K, Müller-Sacherer T, et al.: Functional characterization of two novel non-synonymous alterations in CD46 and a Q950H change in factor H found in atypical hemolytic uremic syndrome patients. Mol Immunol 65: 367–376, 2015 [DOI] [PubMed] [Google Scholar]
- 28.Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, et al.; International Registry of Recurrent and Familial HUS/TTP : Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: The C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet 12: 3385–3395, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al.; European Working Party on Complement Genetics in Renal Diseases : Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24: 475–486, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin W, Ghebrehiwet B, Weksler B, Peerschke EI: Regulated complement deposition on the surface of human endothelial cells: Effect of tobacco smoke and shear stress. Thromb Res 122: 221–228, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van den Born BJ, Honnebier UP, Koopmans RP, van Montfrans GA: Microangiopathic hemolysis and renal failure in malignant hypertension. Hypertension 45: 246–251, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Cataland SR, Holers VM, Geyer S, Yang S, Wu HM: Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood 123: 3733–3738, 2014 [DOI] [PubMed] [Google Scholar]
- 33.Asif A, Nayer A, Haas CS: Atypical hemolytic uremic syndrome in the setting of complement-amplifying conditions: Case reports and a review of the evidence for treatment with eculizumab. J Nephrol 30: 347–362, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruderman I, Finlay M, Barbour T: The perfect storm. Kidney Int 92: 267, 2017 [DOI] [PubMed] [Google Scholar]
- 35.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V; French Study Group for aHUS/C3G : Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol 8: 643–657, 2012 [DOI] [PubMed] [Google Scholar]
- 36.Timmermans SAMEG, van Paassen P; Limburg Renal Registry : The authors reply. Kidney Int 92: 267–268, 2017 [DOI] [PubMed] [Google Scholar]
- 37.Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al.: Mutations in components of complement influence the outcome of factor I-associated atypical hemolytic uremic syndrome. Kidney Int 77: 339–349, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Roumenina LT, Frimat M, Miller EC, Provot F, Dragon-Durey MA, Bordereau P, et al.: A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood 119: 4182–4191, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al.: Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A 100: 12966–12971, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.