

Abstract
Background
X-linked Alport syndrome (XLAS) is a progressive hereditary nephropathy caused by mutations in the COL4A5 gene. Genotype-phenotype correlation in male XLAS is relatively well established; relative to truncating mutations, nontruncating mutations exhibit milder phenotypes. However, transcript comparison between XLAS cases with splicing abnormalities that result in a premature stop codon and those with nontruncating splicing abnormalities has not been reported, mainly because transcript analysis is not routinely conducted in patients with XLAS.
Methods
We examined transcript expression for all patients with suspected splicing abnormalities who were treated at one hospital between January of 2006 and July of 2017. Additionally, we recruited 46 males from 29 families with splicing abnormalities to examine genotype-phenotype correlation in patients with truncating (n=21, from 14 families) and nontruncating (n=25, from 15 families) mutations at the transcript level.
Results
We detected 41 XLAS families with abnormal splicing patterns and described novel XLAS atypical splicing patterns (n=14) other than exon skipping caused by point mutations in the splice consensus sequence. The median age for developing ESRD was 20 years (95% confidence interval, 14 to 23 years) among patients with truncating mutations and 29 years (95% confidence interval, 25 to 40 years) among patients with nontruncating mutations (P=0.001).
Conclusions
We report unpredictable atypical splicing in the COL4A5 gene in male patients with XLAS and reveal that renal prognosis differs significantly for patients with truncating versus nontruncating splicing abnormalities. Our results suggest that splicing modulation should be explored as a therapy for XLAS with truncating mutations.
Keywords: X-linked Alport syndrome, splicing abnormalities, genotype-phenotype correlation, transcript analysis, renal prognosis
Alport syndrome is an inherited type IV collagen disease that causes kidney disorder and usually develops into ESRD, accompanied by sensorineural hearing loss and ocular abnormalities. X-linked Alport syndrome (XLAS) accounts for approximately 85% of cases of Alport syndrome; notably pathogenic variants can be detected in the COL4A5 (NM: 000495.4) gene, which encodes the α5 chain of type IV collagen [α5(IV)].1 The genotype-phenotype correlation in male XLAS is relatively well established; missense mutations (i.e., nontruncating mutations) exhibit milder phenotypes compared with truncating mutations.2–4 Jais et al.2 reported that, by the age of 30, the probability of developing ESRD was 90% in cases that included large rearrangements and small frameshift mutations, but 50% in cases with missense mutations. Bekheirnia et al.3 also reported that the average age at ESRD onset was 22 years for those with large or small deletions, 25 years for those with truncating mutations, and 37 years for those with missense mutations. Regarding splice site mutations, Jais et al.2 analyzed 29 families with mutations in consensus splice sites, revealing that 70% of those developed ESRD by 30 years of age. In addition, Bekheirnia et al.3 studied 24 families with consensus splice sites and reported that the average age of onset of ESRD was 28 years old. These data suggest that cases with splice site variants tend to show moderate severity (i.e., between the severities observed with missense and nonsense mutations). However, in these studies, transcript analysis was not conducted. Therefore, a comparison between truncating and nontruncating mutations (i.e., between cases where the number of deleted nucleotide numbers is or is not a multiple of 3) at the transcript level, to determine splicing abnormalities in the COL4A5 gene, has not yet been performed. In some other inherited diseases, in-frame splice site variants exhibit milder phenotypes relative to those caused by truncating mutations (e.g., Becker muscular dystrophy and Duchenne muscular dystrophy [DMD]).5 In XLAS, it is critical to know the difference between truncating transcripts, which result in a premature stop codon, and nontruncating transcripts, in order to estimate renal prognosis, perform genetic counseling, and develop further treatment strategies (e.g., exon skipping therapy, which is already approved by the US Food and Drug Administration [FDA] for DMD).
However, it is very difficult to make a reliable prediction of splicing patterns without transcriptional analysis. Thus far, we have detected 41 families with splicing abnormalities, including typical (n=16) and atypical (n=25) splicing patterns. Among them, we have previously reported on 11 families with atypical splicing patterns6; since then, we have analyzed an additional 14 families.
In this study, we report on patients with atypical splicing patterns (n=14) and examine the genotype-phenotype correlation in male patients with XLAS with variants in the COL4A5 gene that cause aberrant splicing, including those typical and atypical splicing patterns that have been proven by transcript analysis (46 male patients from 29 families).
Methods
Ethical Consideration
All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine. Informed consent was obtained from patients or their parents.
Participants and Inclusion Criteria
A total of 279 families were genetically defined as having XLAS between January of 2006 and July of 2017 at Kobe University. Among these, 71 (25%) exhibited truncating variants, 159 (57%) exhibited nontruncating variants, and 49 (18%) exhibited splicing variants. Among the 49 families with splicing variants, eight families were excluded for the following reasons: possession of COL4A5 variants with somatic mosaic in a male patient, one case; the 47,XXY karyotype, two cases; merging with COL4A4 variant, one case; merging with missense COL4A5 variant, one case; merging with membranoproliferative GN, one case; and impossible to analyze mRNA, two cases. We included the remaining 41 families and described 14 patients with atypical splicing patterns; other than exon skipping caused by point mutations in the splice consensus sequence, these patterns had never been reported. In addition, to examine the genotype-phenotype correlation of male patients with XLAS, we excluded 11 families with only female patients and one family with exon 49 skipping because exons 49–51 are known as a noncollagenous domain and the skipping of exon 49 may result in a severe phenotype. Ultimately, we included 29 families with 46 male patients; we divided these patients into two groups with truncating or nontruncating variants at the transcript level, then compared their clinical severity of disease. In our analysis, we included family members who had not undergone their own DNA analysis but exhibited obvious urine abnormalities or renal dysfunction. Most patients were followed in a variety of local hospitals throughout Japan. Blood samples and data were sent to our laboratory after acceptance of the request for mutational analysis. When we have detected variants that may possibly affect RNA splice processing, we have routinely analyzed the transcripts to confirm the mutation-induced splicing abnormalities. In this study, we selected only patients with proven splicing variants.
Mutational Analyses
Mutational analysis of COL4A5 was performed by several methods: (1) targeted next-generation sequencing using a custom disease panel, including COL4A3, COL4A4, and COL4A5 genes; (2) conventional direct sequencing using the Sanger method for all exons and exon-intron boundaries; (3) multiplex ligation-dependent probe amplification to detect copy-number variations; and (4) RT-PCR of mRNA and direct sequencing to detect abnormal splicing. We initially performed methods (1) or (2); if no mutations were detected, we then performed methods (3) and/or (4). In addition, when we detected a suspected splicing site variant with methods (1) or (2), we also performed method (4). Genomic DNA was isolated from patient peripheral blood leukocytes using the Quick Gene Mini 80 System (Kurabo Industries Ltd., Tokyo, Japan), according to the manufacturer’s instructions. Amplification of all 51 specific exons of COL4A5, next-generation sequencing, and multiplex ligation-dependent probe amplification were conducted as described previously.6–8 RNA from leukocytes was isolated using RNAlater RNA Stabilization Reagent (Qiagen Inc., Chatsworth, CA), then reverse transcribed into cDNA using the EcoDry Kit (Clontech Laboratories, Inc., Palo Alto, CA). In all patients in this study, mRNA was extracted from peripheral blood leukocytes.
In Vitro Splicing Assay
For patient A422, we conducted a “minigene” in vitro splicing assay because we detected a remarkably abnormal splicing pattern of exon skipping in an exonic variant. To create hybrid minigene constructs, we used the H492 vector, on the basis of the pcDNA 3.0 mammalian expression vector (Invitrogen, Carlsbad, CA) (Supplemental Figure 1A) that we developed previously.9 We amplified genomic DNA from the patient and from a sample of control peripheral leukocytes, using primers for COL4A5 intron 8 to intron 13 that contained additional restriction sites. This enabled cloning of the PCR products into the multiple cloning site of the vector, located within an intron between exons A and B. The forward primer contained an NheI site (GCTAGC): (5′-GCAGCTAGCCAGTGTACTCTGGCCACTTCC-3′), and the reverse primer contained a BamHI site (GGATCC): (5′-CGTGGATCCTGTTTGCAAGATAAAATAAGACAGTG-3′). PCR products and the H492 vector were digested by NheI and BamHI, then ligated together to create both wild-type and variant hybrid (NM_000495.4: c.548dupG) minigenes. The hybrid minigenes were checked by sequencing, then transfected into HEK293T and HeLa cells via Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA). Twenty-four hours later, total RNA was extracted from the cells using the RNeasy Plus Mini Kit (QIAGEN GmbH, Hilden, Germany). Two micrograms of total RNA was subjected to reverse transcription using the EcoDry Kit, then PCR was performed with a forward primer corresponding to a segment upstream of exon A and a reverse primer complementary to a segment downstream of exon B, as previously described (Supplemental Figure 1A).9 PCR products were analyzed by electrophoresis on a 1.2% agarose gel, followed by direct sequencing.
In Silico Splicing Assay
We analyzed the strength of the splicing domain of all 29 families by in silico analysis, using the Human Splicing Finder (http://www.umd.be/HSF3/).
Immunohistochemical Analyses
Immunohistochemical analyses were performed with frozen sections of kidney or skin tissue. The immunohistochemical procedure has been described previously.10–12 A mixture of FITC-conjugated rat mAb for the human α5(IV) chain (H53) and Texas red–conjugated rat mAb for the human α2(IV) chain (H25) was purchased from Shigei Medical Research Institute (Okayama, Japan). The epitopes were EAIQP at positions 675–679 of the α2(IV) chain and IDVEF at positions 251–255 of the α5(IV) chain.
Statistical Analyses
All calculations were performed using standard statistical software (SAS version 9.4 for Windows; SAS, Cary, NC). The occurrence of events (age of developing ESRD) was analyzed via the Kaplan–Meier method. To calculate P value, we used a shared frailty model.13 Under the model, the correlation within a frailty was modeled by shared frailty, following a log normal distribution. We considered an association to be significant when the P value was <0.05.
Results
Abnormal Splicing Patterns Detected in Our Study
In total, 41 families were included in the study. Among these 41 families, 11 exhibit splicing variants that we have already reported, along with corresponding transcript analysis.6,7,14 Furthermore, for two families (A43, A302), transcript analysis results have been reported by another group.15 These cases were included in this study for purposes of genotype-phenotype correlation analysis.
Here, we reveal the transcript analysis results of 14 families whose transcripts had never been reported and who showed atypical splicing patterns, not the mere exon skipping by variation in the consensus AG-GT sequence (Figure 1, for larger view see Supplemental Figure 2).
Figure 1.


Mutations and their consequences (the same figures are shown in Supplemental Figure 2 with larger scales). Upper panels show schemas of aberrant splicing (red lines). Normal splicing is indicated by black lines. The original and new splice sites and flanking sequences are shown below. Patients’ flanking genomic DNA and cDNA sequences are shown in the lower panels. (A) Patient ID A196. IVS23–1 G>A eliminated the splice acceptor site of intron 23 to activate a new splice site, one nucleotide downstream. (B) Patient ID A333. IVS49+1 G>A disrupted the splicing donor site of intron 49, resulting in an intron 49 insertion, which creates a transcript with a 345-bp insertion. (C) Patient ID A424. IVS 6–1 G>A altered the splice acceptor site of intron 6 one nucleotide downstream, which creates a transcript with a 1-bp deletion. (D) Patient ID A231, A258, A298. IVS35–4 A>G altered the splice acceptor site of intron 35 three nucleotides upstream, which creates a transcript with a 3-bp insertion. (E) Patient ID A247. IVS 12+5 G>A disrupted the splice donor site of intron 12, resulting in exon 12 skipping, which creates a transcript with a 42-bp deletion. (F) Patient ID A299. IVS29+3 A>G disrupted the splice donor site of intron 29, resulting in exon 29 skipping, which creates a transcript with a 151-bp deletion. (G) Patient ID A323. IVS40–9 C>G altered the splice acceptor site of intron 40 nine nucleotides upstream, which creates a transcript with a 9-bp insertion. (H) Patient ID A371. IVS 18+3_6 del AAGT disrupted the splice donor site of intron 18, resulting in exon 18 skipping, which creates a transcript with a 42-bp deletion. (I) Patient ID A384. IVS48–11A>G altered the splice acceptor site of intron 48 ten nucleotides upstream, which creates a transcript with a 10-bp insertion. (J) Patient ID A402. IVS27+4 del T disrupted the splice donor site of intron 27, resulting in exon 27 skipping, which creates a transcript with a 105-bp deletion. (K) Patients ID A452. IVS29+5 G>A disrupted the splice donor site of intron 29, resulting in exon 29 skipping, which creates a transcript with a 151-bp deletion. (L) Patient ID A329. IVS21–367 C>T produced a new splice donor site, resulting in a cryptic exon activation between exons 21 and 22 and creating a transcript with a 93-bp insertion. (M) Patient ID A375. Mutation in last nucleotide of exon 25, C1948 G>T, disrupted the splice donor site of intron 25, resulting in exon 25 skipping, which creates a transcript with a 169-bp deletion. (N) Patient ID A422. Mutation of the second nucleotide of exon 10, C548 dup G, disrupted the splicing acceptor site of intron 9, resulting in exon 10 skipping, which creates a transcript with a 63-bp deletion. gDNA, genomic DNA.
Of these 41 families, 32 families (78%) exhibited splice site variants (19 families possess variants at the consensus sequence of either the AG or GT site; the remaining 13 possess variants out of these sites) (Table1) and five families (12%) exhibited deep intronic variants creating novel exons; the remaining four families (10%) exhibited exonic variants, one of which created an aberrant splice site, whereas the other three induced exon skipping. Twenty-two families (54%) exhibited exon skipping, 13 families (32%) revealed production of a new splice site, five families (12%) revealed production of a cryptic exon, and one family (2%) revealed production of a large exon (Figure 1, Supplemental Figure 2, Table 1).
Table 1.
Clinical and genetic findings in this study
| Family Number | Sex | Effect | ESRD Age (yr) | Mutation | Intron/Exon | Transcription | Splicing Variant | Previously Reported cDNA Analysis | Figure |
|---|---|---|---|---|---|---|---|---|---|
| Splice consensus sequence mutations | |||||||||
| A7 | Male | Truncating | 9 | c.610–2A>T | Intron 10 | 19-bp deletion | Creating a novel splice site | Nozu et al.6a | |
| A8 | Male | Truncating | 16 | c.3247–2A>C | Intron 36 | 127-bp deletion | Exon 37 skipping | ||
| A17 | Female | Truncating | no (11 yo) | c.3455–1G>A | Intron 38 | 1-bp deletion | Creating a novel splice site | Nozu et al.6a | |
| A27 | Male | Truncating | no (5 yo) | c.991–1G>A | Intron 17 | 1-bp deletion | Creating a novel splice site | Nozu et al.6a | |
| A28 | Female | Nontruncating | no (6 yo) | c.2147–2A>G | Intron 27 | 18-bp deletion | Creating a novel splice site | Nozu et al.6a | |
| A30 | Female | Nontruncating | no (8 yo) | c.990+1G>T | Intron 17 | 54-bp deletion | Exon 17 skipping | ||
| A43 | Male | Truncating | no (2 yo) | c.1948+1G>A | Intron 25 | 169-bp deletion | Exon 25 skipping | Wang et al.15 | |
| A91 | Female | Nontruncating | no (3 yo) | c.646–2A>G | Intron 11 | 42-bp deletion | Exon 12 skipping | ||
| A97 | Female | Nontruncating | no (18 yo) | c.1166–1G>A | Intron 19 | 174-bp deletion | Exon 20 skipping | ||
| A116 | Female | Nontruncating | no (10 yo) | c.646–1G>C | Intron 11 | 42-bp deletion | Exon 12 skipping | ||
| A141 | Female | Truncating | no (25 yo) | c.3247–2A>G | Intron 36 | 127-bp deletion | Exon 37 skipping | ||
| A196 | Male | Truncating | no (7 yo) | c.1588–1G>A | Intron 23 | 1-bp deletion | Creating a novel splice site | 1A | |
| A244 | Female | Nontruncating | no (18 yo) | c.4069+1G>A | Intron 44 | 72-bp deletion | Exon 44 skipping | ||
| A276 | Male | Nontruncating | 29 | c.892–2A>G | Intron 15 | 45-bp deletion | Exon 16 skipping | ||
| A302 | Female | Truncating | no (10 yo) | c.1948+1G>A | Intron 25 | 169-bp deletion | Exon 25 skipping | Wang et al.15 | |
| A326 | Male | Nontruncating | no (12 yo) | c.2678–1G>A | Intron 31 | 90-bp deletion | Exon 32 skipping | ||
| A333 | Male | Truncating | 23 | c.4803+1G>A | Intron 49 | 345-bp insertion | Creating a large exon | 1B | |
| A365 | Male | Nontruncating | no (3 yo) | c.3604+1G>A | Intron 40 | 51-bp deletion | Exon 40 skipping | ||
| A424 | Male | Truncating | no (11 yo) | c.385–1G>A | Intron 6 | 1-bp deletion | Creating a novel splice site | 1C | |
| Suspected splice site mutations | |||||||||
| A121 | Male | Nontruncating | no (25 yo) | c.546+2_3insT | Intron 9 | 81-bp deletion | Exon 9 skipping | Hashimura et al.7a | |
| A128 | Male | Nontruncating | no (46 yo) | c.2042–18A>G | Intron 26 | 105-bp deletion | Exon 27 skipping | Nozu et al.6a | |
| A158 | Female | Nontruncating | no (14 yo) | c.2245–8T>A | Intron 28 | 6-bp insertion | Creating a novel splice site | Nozu et al.6a | |
| A231 | Female | Nontruncating | no (3 yo) | c.3107–4A>G | Intron 35 | 3-bp insertion | Creating a novel splice site | 1D | |
| A247 | Female | Nontruncating | no (19 yo) | c.687+5G>A | Intron 12 | 42-bp deletion | Exon 12 skipping | 1E | |
| A258 | Male | Nontruncating | no (3 yo) | c.3107–4A>G | Intron 35 | 3-bp insertion | Creating a novel splice site | 1D | |
| A298 | Male | Nontruncating | 25 | c.3107–4A>G | Intron 35 | 3-bp insertion | Creating a novel splice site | 1D | |
| A299 | Female | Truncating | no (2 yo) | c.2395+3A>G | Intron 29 | 151-bp deletion | Exon 29 skipping | 1F | |
| A323 | Female | Non-truncating | no (18 yo) | c.3605–9C>G | Intron 40 | 9-bp insertion | Creating a novel splice site | 1G | |
| A371 | Male | Nontruncating | no (12 yo) | c.1032+3_6 delAAGT | Intron 18 | 42-bp deletion | Exon 18 skipping | 1H | |
| A384 | Female | Truncating | no (6 yo) | c.4511–11A>G | Intron 47 | 10-bp insertion | Creating a novel splice site | 1I | |
| A402 | Male | Nontruncating | no (7 yo) | c.2146+4delT | Intron 27 | 105-bp deletion | Exon 27 skipping | 1J | |
| A452 | Female | Truncating | no (14 yo) | c.2395+5G>A | Intron 29 | 151-bp deletion | Exon 29 skipping | 1K | |
| Deep intronic mutations creating cryptic exon | |||||||||
| A19 | Male | Truncating | no (22 yo) | c.1948+894C>G | Intron 25 | 106-bp insertion with stop codon | Creating a novel exon | Nozu et al.6a | |
| A48 | Male | Truncating | no (6 yo) | c.4511–345A>G | Intron 47 | 74-bp insertion with stop codon | Creating a novel exon | Nozu et al.6a | |
| A126 | Male | Truncating | no (12 yo) | c.609+875G>T | Intron 10 | 123-bp insertion with stop codon | Creating a novel exon | Nozu et al.6a | |
| A217 | Male | Truncating | no (3 yo) | c.4510+1754T>G | Intron 47 | 84-bp insertion with stop codon | Creating a novel exon | Nozu et al.6a | |
| A329 | Male | Nontruncating | 22 | c.1424–367C>T | Intron 21 | 93-bp insertion | Creating a novel exon | 1L | |
| Exonic mutations | |||||||||
| A21 | Male | Nontruncating | 24 | c.3790G>A | Exon 41 | 186-bp deletion | Exon 41 skipping | Nozu et al.6a | |
| A178 | Male | Truncating | 18 | c.876A>T | Exon 15 | 35-bp deletion | Creating a novel splice site | Fu et al.14a | |
| A375 | Male | Truncating | 11 | c.1948G>T | Exon 25 | 169-bp deletion | Exon 25 skipping | 1M | |
| A422 | Male | Nontruncating | 29 | c.548dupG | Exon 10 | 63-bp deletion | Exon 10 skipping | 1N | |
yo, years old.
Shows the exact reported case.
In Vitro Splicing Assay
One patient (A422) showed an atypical splicing abnormality of exon 10 skipping by exonic variation (c.548dupG, Figure 1N, Supplemental Figure 2N). To clearly assess the probability, we performed an in vitro splicing assay. Electrophoresis of cDNA from the patient and from a wild-type fragment showed double and triple bands, respectively (Supplemental Figure 1B). Although some bands revealed an absence of exons 9 and 10 even in the wild-type fragments, a direct connection between exon 9 and exon 11 was observed only within the sequence of the patient (Supplemental Figure 1C). This indicates that c.548dupG led to the skipping of exon 10, which was supported by both in vivo and in vitro data.
In Silico Splicing Assay
Among 41 families, 19 were completely compatible with the analytic result of the Human Splicing Finder (http://www.umd.be/HSF3/), including complete exon skipping and deep intronic variants; 17 of the remaining families yielded data that enabled prediction of splicing defects, but could not predict a precise novel splice site; and the final five families yielded data that were not sufficient for the prediction of splicing abnormalities (Supplemental Table 1).
Immunohistochemical Analyses
The results of immunohistochemical analysis for α5(IV) collagen are shown in Supplemental Table 3. Of 41 total patients, α5(IV) collagen staining was performed in 31 patients. Among them, 17 were male patients and these data are shown in Supplemental Table 4. In the nontruncating group (n=7), three of seven patients were positive for α5(IV) staining (43%). In contrast, none showed α5(IV) positivity in the truncating group (n=10). All three patients who were α5(IV) positive belonged to the nontruncating group (100%). These results indicate that α5(IV) positivity in patients with splicing variants at the genomic DNA level can predict nontruncating transcript variants and later onset of ESRD.
Patients Included in the Genotype-Phenotype Correlation Analysis
During genotype-phenotype correlation analysis, we excluded one patient (A333) with an insertion in intron 49 that resulted in the creation of a stop codon; this was because exons 49–51 are known as a noncollagenous domain, the lack of which may cause a severe phenotype. Thus, we specifically limited the analysis to families with variants in the collagenous domain.
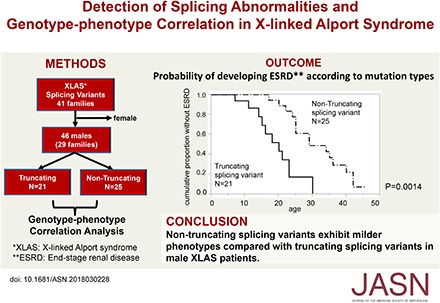
Therefore, we included 46 male patients, from 29 families who exhibited 27 variants, in our genotype-phenotype correlation analysis (Figure 2, Supplemental Table 2).
Figure 2.

Probability of developing ESRD according to mutation types. Solid line indicates patients with truncating splicing mutations (n=21). The median age for developing ESRD was 20 years. Dash-dots indicate nontruncating splicing mutations (n=25). The median age for developing ESRD was 29 years.
Genotype-Phenotype Correlation Analysis
Twenty-one participants from 14 families revealed truncating transcriptional variants and 25 participants from 15 families revealed nontruncating transcriptional variants (Supplemental Table 2). Age at onset of ESRD differed significantly between these two groups. The median time (95% confidence interval) from birth to ESRD was as follows: truncating, 20 years (14 to 23 years); nontruncating, 29 years (25 to 40 years; P=0.001; Figure 2); overall, 25 years (21 to 34 years) (Table 2).
Table 2.
Distribution of the participants
| Variable | Total (Censored Case) | ESRD Age, Median (95% CI) |
|---|---|---|
| Nontruncating | 25 (10) | 29 (25 to 40) |
| Truncating | 21 (11) | 20 (14 to 23) |
| Total | 46 (21) | 25 (21 to 34) |
95% CI, 95% confidence interval.
Discussion
This is our second report of a case series for male patients with XLAS with aberrant splicing; concurrently, it is the first report of a comparative analysis of male patients with XLAS with aberrant splicing, thereby enabling distinction between truncating and nontruncating transcriptional variants.6
Genotype-phenotype shows a strong correlation in male XLAS; missense mutations result in milder phenotypes, relative to truncating mutations such as nonsense mutations and deletions/insertions.2–4 However, all of the prior studies have grouped splice site variants into a single group without characterizing the specific patterns of their transcriptional variations. Moreover, the effects of splicing mutations that result in a premature stop codon, designated “truncating transcript,” or nontruncating mutations caused by abnormal splicing, designated “nontruncating transcript,” have yet to be elucidated. Jais et al.2 reported that splice site variants confer a 70% probability for development of ESRD at age 30 years. Bekheirnia et al.3 reported that the average age of onset of ESRD was 28 years for patients with splice site variants. However, our study clearly showed that patients with disease caused by aberrant splicing exhibit different prognoses, depending on whether their mutations are truncating or nontruncating transcript. To most accurately predict kidney prognosis, it is important to conduct transcriptional analysis when detecting either splice site mutations or aberrant-splicing mutations.
Transcriptional analysis is frequently difficult to conduct because transcripts are not stable within tissues. An in silico analysis tool to predict the effect of transcriptional variants, such as the Human Splicing Finder (http://www.umd.be/HSF3/), may aid in prediction of the disruption of the original splice site. However, our study revealed that in silico analysis may not precisely identify the aberrant splice site and abnormal transcript. Indeed, 19 families were compatible with the in silico analysis in our study; however, for 22 families, the analysis tool could not predict the exact splicing sites (Supplemental Table 1). Furthermore, the presence of a consensus splicing variant at the AG-GT site is usually suspected to result in exon skipping; however, in our study results, seven of 19 cases with variation in the AG-GT site did not show mere exon skipping. Thus, in silico analysis is frequently unpredictable.
Previously, it was reported that male patients with XLAS with positive staining for α5(IV) in kidney tissue tended to show milder clinical phenotypes and carried nontruncating mutations (mostly missense mutations).7,16 In our cohort, three of seven patients carrying nontruncating transcriptional variants were α5(IV) positive. In contrast, no patients carrying nontruncating transcriptional variants were α5(IV) positive (Supplemental Table 4). Although the sample number was not large, our study revealed the possibility that, for patients with splice site variants at the genome level, α5(IV) positivity in patient tissues can predict nontruncating transcript variants. Therefore, immunohistochemical analysis might aid in predicting the prognosis of those patients, potentially replacing mRNA analysis that is labor-intensive, expensive, and not readily available in most countries.
Recently, our group has reported that a splicing minigene assay is valuable in determining the variant’s effect on splicing in transfected cells, in cases of inherited kidney diseases.9,17 In addition, Malone et al.18 have recently reported that the minigene assay was effective for detecting splicing abnormality in a patient with XLAS. We also conducted the minigene assay in our patient A422, which revealed an exonic variant that caused the exon skipping. Although some unexpected variants were observed both in the patient sequence and the wild-type fragment, it may be appropriate to assume that exon 10 skipping was caused by the exonic variant detected in the patient.
Recently, splicing modulation of exon skipping by antisense oligonucleotide has been revealed as particularly valuable in cases with truncating mutations; hence, the splicing modulation induces a nontruncating transcription and can produce a protein that might partially compensate for the loss of full-length protein, thereby ameliorating the symptoms of the disease.19 Antisense oligonucleotide was approved by the FDA as a treatment for DMD, with the aim of changing truncating mutations into exon skipping that may lead to nontruncating mutations.20 Similar therapeutic strategies may be available in the future for patients with genetically diagnosed XLAS; importantly, we have shown that transcriptional in-frame variants can lead to the milder phenotype in our study, which may serve as factual evidence of the efficiency of exon skipping therapy for XLAS with truncating mutations.
In most cases, mRNA extracted from the kidney itself is hard to obtain. Fortunately, peripheral blood leukocytes express a sufficient quantity of COL4A5 transcripts and have been used as an alternative source for transcript analysis, in order to confirm the pathogenicity of suspected splice site variants.6 However, alternative splicing could occur in a tissue-specific manner.21,22 Regarding COL4A5, multiple organ-specific splicing patterns have been reported6,23; notably, these differences were NS, and these reports largely served to confirm the existence of additional splicing variants. One limitation of our study is that we solely confirmed the splicing variants by mRNA extracted from leukocytes; however, the issues associated with this method may be limited.
Our study has additional limitations. First, clinical information that might influence the prognosis, such as continuous angiotensin-converting enzyme inhibitor or angiotensin receptor blocker treatment, hypertension, nonsteroidal anti-inflammatory drug use, and/or incidental glomerular disease, was not fully analyzed. Most of the patients that developed ESRD were not treated by angiotensin-converting enzyme inhibitor or angiotensin receptor blocker; however, younger patients have begun treatment with those drugs, and we could not include these in our analysis. Another limitation is that our sample size was relatively small; thus, our study contains some familial cases or cases with the same nucleotide variant.
In conclusion, this is the first study to provide a comparison of the kidney prognosis between truncating and nontruncating splicing variants in male patients with XLAS. We showed that nontruncating splicing variants exhibit milder phenotypes compared with truncating splicing variants. This suggests that transcriptional analysis is necessary to accurately estimate renal prognosis and provide better information during genetic counseling. Our results also present factual evidence supporting the efficiency of therapeutic approaches that use splicing modulation that change truncating transcription into nontruncating transcription within patients with XLAS.
Disclosures
K.I. has received grant support from Daiichi Sankyo Co., Ltd. K.I. has received consulting fees from Takeda Pharmaceutical Company and Kyowa Hakko Kirin Co., Ltd. K. Nozu has received lecture fees from Novartis Pharmaceuticals Corporation. K.I. and K. Nozu have filed a patent application on the development of antisense nucleotides for exon skipping therapy in Alport syndrome.
Supplementary Material
Acknowledgments
We thank Ryan Chastain-Gross from the Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
This study was supported by a grant from the Ministry of Health, Labour and Welfare of Japan for Research on Rare Intractable Diseases in the Kidney and Urinary Tract (H24-nanchitou [nan]-ippan-041 to K.I.) in the “Research on Measures for Intractable Diseases” Project; by Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (subject ID: 15K09691 to K. Nozu and 17H04189 to K.I.); and by Japan Agency for Medical Research and Development (AMED) under grant number 7930006 to K. Nozu and K.I.
T.H. and K.I. designed the study concept and wrote the manuscript. K. Nozu interpreted the data and wrote the manuscript. T.Y., S.M., Keita Nakanishi, J.F., A.A., M. Kitamura, M. Kawano, W.S., C.K., A.I., K.K., K.O., S.F., M.O., T.I., A.M., E.S., R.F., Koichi Nakanishi, Y.S., M.J.Y., and Y.N. collected and interpreted the data. T.O. interpreted the data and conducted the statistical analyses. M.M., N.M., and H.K. critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018030228/-/DCSupplemental.
References
- 1.Kashtan CE: Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol 9: 1736–1750, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al.: X-linked Alport syndrome: Natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 11: 649–657, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al.: Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 21: 876–883, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M: Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol Dial Transplant 17: 1218–1227, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM: An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2: 90–95, 1988 [DOI] [PubMed] [Google Scholar]
- 6.Nozu K, Vorechovsky I, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al.: X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin J Am Soc Nephrol 9: 1958–1964, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashimura Y, Nozu K, Kaito H, Nakanishi K, Fu XJ, Ohtsubo H, et al.: Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int 85: 1208–1213, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al.: Natural history and genotype-phenotype correlation in female X-linked alport syndrome. Kidney Int Rep 2: 850–855, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozu K, Iijima K, Kawai K, Nozu Y, Nishida A, Takeshima Y, et al.: In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Hum Genet 126: 533–538, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Sado Y, Kagawa M, Kishiro Y, Sugihara K, Naito I, Seyer JM, et al.: Establishment by the rat lymph node method of epitope-defined monoclonal antibodies recognizing the six different alpha chains of human type IV collagen. Histochem Cell Biol 104: 267–275, 1995 [DOI] [PubMed] [Google Scholar]
- 11.Naito I, Kawai S, Nomura S, Sado Y, Osawa G; Japanese Alport Network : Relationship between COL4A5 gene mutation and distribution of type IV collagen in male X-linked Alport syndrome. Kidney Int 50: 304–311, 1996 [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi K, Iijima K, Kuroda N, Inoue Y, Sado Y, Nakamura H, et al.: Comparison of alpha5(IV) collagen chain expression in skin with disease severity in women with X-linked Alport syndrome. J Am Soc Nephrol 9: 1433–1440, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Knox KL, Bajorska A, Feng C, Tang W, Wu P, Tu XM: Survival analysis for observational and clustered data: An application for assessing individual and environmental risk factors for suicide. Shanghai Jingshen Yixue 25: 183–194, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu XJ, Nozu K, Eguchi A, Nozu Y, Morisada N, Shono A, et al.: X-linked Alport syndrome associated with a synonymous p.Gly292Gly mutation alters the splicing donor site of the type IV collagen alpha chain 5 gene. Clin Exp Nephrol 20: 699–702, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Wang F, Wang Y, Ding J, Yang J: Detection of mutations in the COL4A5 gene by analyzing cDNA of skin fibroblasts. Kidney Int 67: 1268–1274, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Said SM, Fidler ME, Valeri AM, McCann B, Fiedler W, Cornell LD, et al.: Negative staining for COL4A5 correlates with worse prognosis and more severe ultrastructural alterations in males with alport syndrome. Kidney Int Rep 2: 44–52, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi K, Nozu K, Hiramoto R, Minamikawa S, Yamamura T, Fujimura J, et al.: A comparison of splicing assays to detect an intronic variant of the OCRL gene in Lowe syndrome. Eur J Med Genet 60: 631–634, 2017 [DOI] [PubMed] [Google Scholar]
- 18.Malone AF, Funk SD, Alhamad T, Miner JH: Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome. Pediatr Nephrol 32: 997–1003, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havens MA, Hastings ML: Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res 44: 6549–6563, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drug Approval Package: Exondys 51 Injection (eteplirsen), https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_TOC.cfm, FDA, 2016
- 21.Black DL: Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Ramanouskaya TV, Grinev VV: The determinants of alternative RNA splicing in human cells. Mol Genet Genomics 292: 1175–1195, 2017. [DOI] [PubMed] [Google Scholar]
- 23.Martin PH, Tryggvason K: Two novel alternatively spliced 9-bp exons in the COL4A5 gene. Pediatr Nephrol 16: 41–44, 2001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.