

Abstract
Macrophages are essential for innate immunity and inflammatory responses and differentiate into various functional phenotypes. Tribbles homolog 1 (Trib1), a member of the mammalian Tribbles homolog pseudokinase family, has been implicated in regulation of cell differentiation, proliferation, and metabolism, but its role in macrophage biology has not been fully elucidated. Here, we investigated the consequences of Trib1 deficiency on macrophage functions and M1/M2 polarization. Bone marrow–derived macrophages (BMDMs) from Trib1-deficient (Trib1−/−) mice exhibited elevated phagocytic capacity, correlating with up-regulation of several scavenger receptors. Concomitantly, uptake of modified low-density lipoprotein was increased in Trib1−/− BMDMs. Trib1−/− macrophages also exhibited diminished migration in the presence of the chemokine MCP-1, associated with reduced expression of the MCP-1 receptor Ccr2. Furthermore, Trib1 deficiency attenuated the response of BMDMs to both M1 and M2 stimuli; induction of the M1-marker genes Il6, Il1b, and Nos2 upon LPS/IFNγ stimulation and of the M2-marker genes Cd206, Fizz1, and Arg1 upon IL-4 stimulation was reduced. Functionally, Trib1 deficiency decreased secretion of proinflammatory cytokines (IL-6, TNFα, IL-1β, and CXCL1) and reduced nitric oxide and reactive oxygen species production in M1-polarized macrophages. Supporting the attenuated M2 phenotype, IL-4–stimulated Trib1−/− macrophages secreted less IL-10 and TGFβ. Mechanistically, Trib1−/− BMDMs displayed lower levels of Janus kinase 1 (JAK1), resulting in reduced activation of LPS/IFNγ-mediated STAT1 signaling. Likewise, decreased levels of JAK1 along with lower activation of STAT6 and STAT3 were observed in M2-polarized Trib1−/− BMDMs. Our findings suggest that Trib1 extensively controls macrophage M1/M2 polarization via the JAK/STAT signaling pathway.
Keywords: macrophage, signal transduction, migration, phagocytosis, Janus kinase (JAK), STAT transcription factor, M1/M2, polarization
Introduction
Macrophages are an essential part of innate immunity, inflammatory response, and tissue modulation and remodeling. It is well-established that macrophages possess a remarkable plasticity (1). Depending on the microenvironment, macrophages can transform into different functional phenotypes with classically activated (M1) macrophages and alternatively activated (M2) macrophages representing the extremes of the phenotypic spectrum (2). Macrophages are polarized toward the M1 phenotype, when they are exposed to proinflammatory Th1 cytokines (e.g. TNFα, IFNγ)2 or microbial products like lipopolysaccharide (LPS). Phenotypically, M1 macrophages express high levels of major histocompatibility complex II and produce high levels of proinflammatory cytokines (TNFα, IL-1β, IL-6, IL-12), reactive oxygen species (ROS), and NO. Hence, M1 macrophages are efficient in eliminating pathogens, infected cells and tumor cells. In contrast, M2 macrophages are polarized by anti-inflammatory Th2 cytokines such as IL-4 and IL-13 and are characterized by high expression of M2 marker genes like the mannose receptor Cd206 and Resistin-like β (Retnlb, Fizz1). M2 macrophages produce anti-inflammatory cytokines (IL-10) and high levels of arginase 1 and functionally facilitate angiogenesis, tissue remodeling, and parasite defense (3, 4).
M1 and M2 polarization of macrophages is regulated by several distinct signaling pathways. M1 polarization is triggered by activation of Toll-like receptor signaling, especially Toll-like receptor 4, by LPS and other microbial ligands, resulting in the activation of the NF-κB and mitogen-activated protein kinase (JNK, p38, and ERK1/2) signaling cascades (5, 6). Furthermore, IFNγ stimulates the JAK/STAT pathway, resulting in activated STAT1 binding to the promoter regions of the hallmark M1-associated genes Nos2 and Il12 (7–9). On the other hand, macrophages can be driven toward the M2 phenotype by IL-4, IL-10, and IL-13. Binding of IL-4 or IL-13 to IL-4 receptors results in activation and translocation of STAT6 into the nucleus (10, 11) where it induces the transcription of M2 marker genes and critical regulators of alternative activation (e.g. PPARγ, PPARδ) (12–14).
All signaling pathways that promote M1 or M2 polarization are tightly controlled by a multiplicity of post-translational regulators through diverse mechanisms (8). Recently, the Tribbles pseudokinase family was implicated in the regulation of various cellular signaling pathways (15). Instead of direct phosphorylation of target proteins, the three members of the Tribbles family (TRIB1, TRIB2, TRIB3) appear to act as adapter or scaffolding proteins mediating the interaction with signaling molecules and/or protein degradation (16). For example, Tribbles proteins and the related pseudokinase STK40 contain binding motifs for the E3-ubiquitin ligase COP1 and can thereby facilitate the proteasome-mediated degradation of specific substrates (17, 18).
In particular, TRIB1 was shown to interact with C/EBP transcription factors (C/EBPα and C/EBPβ) and regulate their stability in a COP1 dependent manner (18, 19). TRIB1 was also shown to interact with MAP kinases and mediate signaling changes to MAPK cascades through mechanisms that are likely independent of COP1 (15, 16, 20–23). Although we and others have previously demonstrated a function for Trib1 in lipid metabolism (24–26), Trib1 has also been connected to inflammation (27, 28) and macrophage biology (29, 30). With regards to macrophages, Trib1 deficiency altered the expression of inflammation induced genes in peritoneal macrophages in a C/EBPβ-dependent manner (30). Additionally, Satoh et al. (29) reported an essential role of Trib1 in the development of tissue resident M2-like macrophages (F4/80+CD206+). Mice lacking Trib1 exhibited a reduced number of M2-like macrophages in several organs. However, the molecular mechanisms leading to impaired M2 polarization have not been elucidated in detail, and a putative function of Trib1 in M1 polarization has not been addressed as of now.
Here, we investigated whether Trib1 deficiency alters the main macrophage functions. Therefore we analyzed the capability for phagocytosis, migration, and polarization. We report increased phagocytosis and diminished migration in Trib1−/− BMDMs. Furthermore, Trib1−/− BMDMs showed an impaired M1 and M2 polarization phenotype with a reduced activity of the JAK/STAT signaling pathway. Collectively, our data indicate an important role for Trib1 in controlling both M1 and M2 polarization.
Results
Trib1 deficiency alters macrophage phagocytosis and migration capability
We first characterized the consequences of Trib1 deficiency on major macrophage functions. One of the most important functions of macrophages is the uptake and degradation of pathogens by scavenger receptors. To clarify whether the absence of Trib1 influences phagocytosis capacity, we treated WT and Trib1−/− BMDMs with FITC-labeled Escherichia coli particles and determined the engulfed fluorescence. Trib1-deficient macrophages showed a 1.9-fold mean increase of engulfed E. coli particles as compared with WT cells (Fig. 1A). To examine whether the elevated uptake resulted from higher expression of scavenger receptors, we analyzed the expression profile of 10 common scavenger receptors. We found that seven of the tested scavenger receptors (Cd36, Cd68, Srb1, Scarf1, Cxcl16, Stab1, and Marco) were consistently up-regulated in Trib1−/− BMDMs (Fig. 1B). The expression of Sra, Cd163, and Lox1 was not changed (Fig. S1). To further corroborate the functional significance of increased scavenger receptor expression, we analyzed whether uptake of modified low-density lipoprotein (LDL) was also enhanced in Trib1−/− BMDMs. When WT and Trib1−/− BMDMs were incubated with DiI-labeled acetylated LDL (acLDL), uptake of acLDL was significantly increased by 30% in Trib1−/− as compared with WT BMDMs (Fig. 1C).
Figure 1.

Trib1 deficiency alters macrophage phagocytosis and migration. WT and Trib1−/− BMDMs were treated with FITC-labeled E. coli for 2 h at 37 °C. Intracellular fluorescence was determined after the cells were incubated with trypan blue to quench the extracellular fluorescence (A). mRNA expression of scavenger receptors was analyzed in WT and Trib1−/− BMDMs by qPCR using the ΔCt method and Actb as a housekeeping gene (B). acLDL uptake was determined by incubating BMDMs with DiI-labeled acLDL (25 μg/ml) for 2 h. Fluorescence data from cell lysates was normalized to total cell protein (C). Macrophage migration over 16 h was investigated in medium with or without MCP1 (100 ng/ml) using a modified Boyden chamber (D). The level of Ccr2 mRNA expression under control conditions was determined in WT and Trib1−/− BMDMs (E). The expression of proinflammatory (Il6, Il1b) and anti-inflammatory (Cd206, Arg1) marker genes was determined in naïve WT and Trib1−/− BMDMs (E). The phagocytosis index (A), acLDL uptake (C), and gene expression levels (B, E, and F) were normalized to the values of the WT group and are shown as fold change. In D, all data were expressed as changes compared with the WT control group. All results are from three independent experiments using four mice per genotype. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Activation of macrophages induces migration toward a presenting stimulus. To investigate the role of Trib1 in macrophage migration, we performed cell migration assays in a modified Boyden chamber using monocytotic chemoattractant protein 1 (MCP-1) as the most important chemokine for migration regulation. BMDMs from Trib1−/− mice showed no difference under basal conditions compared with the WT control group. The presence of MCP-1 significantly increased the numbers of migrated WT BMDMs by 57%. In contrast, the migration of Trib1−/− BMDMs was attenuated, with an increase of only 33% (Fig. 1D). Concomitantly, we observed a 2.2-fold lower expression of the MCP-1 receptor (Ccr2) in Trib1−/− BMDMs, which could contribute to the impaired migratory response (Fig. 1E). We further analyzed the expression of pro- and anti-inflammatory genes in naïve BMDMs. Interestingly, we found lower expression levels of proinflammatory Il6 and Il1b, as well as lower expression levels of anti-inflammatory Cd206 and Arg1 in naïve Trib1−/− BMDMs (Fig. 1F), indicating an influence of Trib1 on basal activation status.
Trib1 expression is induced during M1 and M2 polarization of BMDMs
To investigate whether Trib1 is regulated in response to M1 and M2 polarizing stimuli, we treated BMDMs from C57/BL6 mice with either LPS/IFNγ (for M1) or IL-4 (for M2) over a time course of up to 24 h and measured Trib1 mRNA expression levels. Trib1 expression was significantly induced under both M1 and M2 polarizing conditions. In M1-stimulated BMDMs, we found the highest induction (10.6-fold) of Trib1 expression after 1 h (Fig. 2A). Likewise, in M2 stimulated BMDMs, we observed a 5.6-fold increase after 1 h (Fig. 2B).
Figure 2.
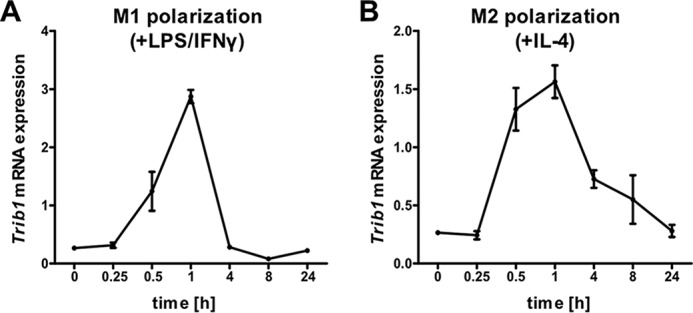
Expression of Trib1 is induced during M1 and M2 polarization. BMDMs from C57BL/6 mice (n = 4) were stimulated with either LPS (100 ng/ml)/IFNγ (20 ng/ml) for M1 polarization (A) or IL-4 (20 ng/ml) for M2 polarization (B). Total RNA was prepared, and Trib1 expression was measured by qPCR over a time course from 0 min (no stimulation) to 24 h using the ΔCt method and Actb as a housekeeping gene. Expression data are presented as means ± S.E. of triplicates.
M1 and M2 polarization is impaired in Trib1-deficient BMDMs
Given the significant induction of Trib1 expression in response to M1 and M2 polarization stimuli, we next investigated the consequences of Trib1 deficiency on M1 and M2 polarization. Macrophage activation in response to LPS/IFNγ (M1) or IL-4 (M2) treatment was validated by substantial shifts in gene expression of M1 (Il1b, Nos) and M2 (Arg1, Fizz1) markers between nonpolarized and M1 and M2 polarized BMDMs (Fig. S2). Comparing the M2 polarization status of WT and Trib1−/− BMDMs after stimulation with IL-4 over 48 h, we observed lower expression of all tested M2 signature marker genes (Arg1, Fizz1, Cd206) in Trib1−/− BMDMs (Fig. 3A). Furthermore, gene and protein expression of PPARγ, which is activated during M2 polarization and important for the acquisition and maintenance of the M2 phenotype (14, 31), was diminished in Trib1−/− BMDMs (Fig. S3, A and B). Impaired M2 polarization of Trib1−/− BMDMs was also functionally corroborated by lower secretion of the M2 associated cytokines TGFβ (1.48 ± 0.06 versus 1.19 ± 0.08 pg/ml, WT versus Trib1−/−; Fig. 3B) and IL-10 (6.30 ± 1.21 versus 2.84 ± 0.31, pg/ml, WT versus Trib1−/−; Fig. 3C).
Figure 3.

Trib1 deficiency suppresses M1 and M2 macrophage polarization. Gene expression of M2 macrophage markers Arg1, Fizz1, and Cd206 (A) and M1 macrophage markers Il6, Il1β, and Nos2 (D) was measured in BMDMs from WT and Trib1−/− mice after stimulation with either IL-4 (20 ng/ml, A) or LPS (100 ng/ml)/IFNγ (20 ng/ml, D) for 48 h. Gene expression levels were calculated by the ΔCt method using Actb as a housekeeping gene; values are presented as relative changes compared with control group. The levels of secreted cytokines in M2 polarized macrophages (TGFβ, IL-10) (B and C) and M1 polarized macrophages (TNFα, IL-6, IL-1β, CXCL1) (E) were measured by ELISA. Nitrite levels as marker of NO synthase activity were determined using Griess reagent in culture medium of naïve (M0) and M1 polarized BMDMs (F). ROS production was detected by using a ROS sensitive fluorescence probe and was measured in naïve (M0) and M1 polarized (24 h LPS (100 ng/ml)/IFNγ (20 ng/ml)) BMDMs from WT and Trib1−/− mice (G). All results are from two to three independent experiments of four mice per genotype. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.d., not detectable.
To clarify the impact of Trib1 deficiency on M1 polarization, we examined characteristic properties of M1 polarized macrophages in response to LPS/IFNγ stimulation. The expression of M1 marker genes Il6, Il1b, and Nos2 remained significantly lower in Trib1−/− BMDMs as compared with WT controls (Fig. 3D). To ensure that the changes in M1 marker expression were also reflected at a functional level, the secretion of proinflammatory M1 cytokines was quantified. In agreement with the gene expression data, we detected significantly lower levels of TNFα, IL-6, IL-1β, and CXCL1 in the supernatant of Trib1−/− BMDMs (TNFα: 68.2 ± 18.3 versus 14.8 ± 0.9; IL-6: 51.7 ± 1.5 versus 24.7 ± 7.8; IL-1β: 7.3 ± 2.5 versus 1.8 ± 0.2; CXCL1: 22.5 ± 5.5 versus 5.0 ± 0.5; pg/ml, WT versus Trib1−/−; see Fig. 3E). In addition, we verified the effects of Trib1 deficiency to reduce M1 activation by detecting NO production. Production of NO was determined by the accumulation of nitrite (the first stable metabolite of NO) in the supernatant of nonpolarized (M0) and M1 polarized macrophages. Nitrite levels were not detectable in the supernatant of M0 macrophages but were abundantly detectable in M1-stimulated macrophages, reflecting the induction of inducible nitric-oxide synthase enzymatic activity during M1 polarization. In agreement with lower Nos2 mRNA expression (Fig. 3D), nitrite levels were 39% lower in the supernatant of M1 polarized Trib1−/− BMDMs (6.6 ± 0.7 versus 4.0 ± 0.7 μmol/L, WT versus Trib1−/−; Fig. 3F). Likewise, the production of ROS, another hallmark of M1 macrophages, was significantly reduced in M1 stimulated Trib1−/− BMDMs as compared with WT controls (Fig. 3G).
Collectively, these data indicate that Trib1 plays an as yet underappreciated function in both M1 and M2 polarization of macrophages. Because Trib1 deficiency impaired response to both LPS/IFNγ– and IL-4–induced polarization, we tested whether increasing concentrations of stimuli could equalize the induction of M1 and M2 polarization markers. However, the blunted response of Trib1−/− macrophages was not dose-dependent and maintained across the entire range of tested concentrations (Fig. S4). These data suggest that Trib1 does not merely alter the activation threshold to M1 and M2 stimuli.
Trib1 deficiency impairs activation of the JAK/STAT signaling pathway in M1- and M2-activated macrophages
To address potential underlying molecular mechanisms leading to impaired macrophage polarization in Trib1−/− BMDMs, we evaluated the activation of major signaling pathways in response to M1 and M2 stimuli. Given the known interaction of Trib1 and C/EBPs, we first reassessed the C/EBP pathway, which has previously been shown to modulate both M1 and M2 polarization of macrophages (32). C/EBPα was only modestly increased in Trib1−/− BMDMs, whereas C/EBPβ was comparable to WT controls (Fig. S5, A and B). Next, we examined whether Trib1 deficiency alters response to LPS/IFNγ–induced signal transduction: LPS-mediated initiation of M1 polarization activates the MAP kinases p38, ERK1/2, and JNK, as well as the NF-κB cascade. However, Trib1−/− BMDMs did not show differences in the activation of the NF-κB subunit p65 and the MAP kinases p38, ERK1/2, and JNK as compared with controls (Fig. S6, A–D). Furthermore, binding of IFNγ to its receptor activates the JAK/STAT pathway, resulting in the phosphorylation and dimerization of STAT1 molecules, which is crucial for M1 macrophage polarization (7). Interestingly, in response to LPS/IFNγ treatment, Trib1−/− BMDMs exhibited significantly lower levels of phosphorylated STAT1 and a reduced pSTAT1/STAT1 ratio (Fig. 4A).
Figure 4.

Absence of Trib1 impairs JAK-mediated STAT activation. BMDMs from WT and Trib1−/− mice were treated with LPS (100 ng/ml)/IFNγ (20 ng/ml) (M1 polarization; A, D, E, and I) or IL-4 (20 ng/ml) (M2 polarization; B, C, and F–H) for 30 min. Total cell lysates were prepared and analyzed by immunoblotting. Abundance of total and phosphorylated protein was determined for STAT1 (A), STAT6 (B), STAT3 (C), JAK 1 (D and F), JAK2 (E), and JAK3 (G). Protein levels of SOCS1 and SOCS3 were determined under M2 (H) and M1 (I) polarizing conditions. An anti-GAPDH antibody was used to assess equal protein loading (A–I). All panels display a representative blot from two to four independent experiments with two samples per genotype used in each experiment. Relative quantification of phosphorylated and total protein was performed using the ImageJ software and is shown below each blot. For quantitative analysis, data were normalized to the WT group and presented as fold change (mean ± S.D.). *, p < 0.05; **, p < 0.01. The striped columns represent WT control, and the solid white columns represent Trib1−/− BMDMs. The asterisks in A–C indicates that membranes were cut below the 115-kDa marker to allow parallel incubation with different antibodies.
Activation of the JAK/STAT signaling pathway also promotes M2 macrophage polarization via phosphorylation of STAT3 and STAT6 proteins. To investigate STAT signaling under M2 polarizing conditions, Trib1−/− and WT control BMDMs were stimulated with IL-4. We detected a profound reduction of phosphorylated STAT6 and the pSTAT6/STAT6 ratio in Trib1−/− BMDMs (Fig. 4B). Further, Trib1−/− BMDMs also displayed diminished phosphorylation of STAT3 and a reduced pSTAT3/STAT3 ratio as compared with WT controls (Fig. 4C). Collectively, these data strongly suggest that attenuated activation of STAT signaling pathways may underlie the impaired response to both M1 and M2 stimuli in Trib1−/− BMDMs.
Given the reduced STAT activation in Trib1−/− BMDMs, we hypothesized that the common upstream regulator JAK may cause the diminished STAT activation. Therefore, we analyzed abundance and activation of the different JAKs according to the polarization status. Under M1 polarizing conditions JAK1 and JAK2 are responsible for transmitting the activation signal to STAT1 molecules (33). Interestingly, LPS/IFNγ–treated Trib1−/− BMDMs demonstrated significantly lower levels of total JAK1 protein but a comparable JAK1 activation, evidenced by an equal pJAK1/JAK1 ratio as compared with WT controls (Fig. 4D). Trib1 deficiency did not alter phosphorylated or total JAK2 (Fig. 4E).
In macrophages stimulated with IL-4, M2 polarization is initiated by phosphorylation of JAK1 and JAK3 (34). Similar to the observation in M1-stimulated macrophages, abundance of JAK1 protein was significantly reduced in Trib1−/− BMDMs stimulated with IL-4 (Fig. 4F). Here, we also observed a modest reduction in the pJAK1/JAK1 ratio (Fig. 4F). Trib1−/− BMDMs treated with IL-4 also displayed a tendency to lower total JAK3 levels and lower phosphorylated JAK3 (Fig. 4G). Together, these data support the notion that Trib1 deficiency in macrophages leads to lower JAK abundance, which attenuates subsequent activation of STAT signaling in response to M1 and M2 stimuli.
To investigate whether the lower abundance of JAKs in Trib1−/− BMDMs is due to lower gene expression, we determined mRNA expression of Jak1 and Jak3. Levels of Jak1 and Jak3 mRNA were only modestly reduced in Trib1−/− BMDMs (Fig. S7), which is unlikely to account for the lower protein abundance. Because activation of the JAK/STAT pathway can also be inhibited by elevated suppressors of cytokine signaling (SOCS), we analyzed the protein levels of SOCS1 and SOCS3 in response to M1 and M2 stimuli. In BMDMs treated with IL-4, we observed comparable levels of SOCS1 and SOCS3 between WT and Trib1−/− cells (Fig. 4H). In BMDMs treated with LPS/IFNγ, levels of SOCS1 were also comparable, whereas levels of SOCS3 actually tended to be lower in Trib1−/− BMDMs (Fig. 4I). Therefore, reduced abundance and activation of JAKs/STATs in Trib1−/− BMDMs are unlikely to result from elevated SOCS1 and SOCS3. Collectively, our observations identify Trib1 as a novel modulator of the JAK/STAT signaling pathway in M1 and M2 macrophage polarization. Trib1 modulates the abundance of JAKs and thereby adjusts the activation of STAT signaling molecules in response to stimulation with LPS/IFNγ or IL-4, which subsequently affects pro- and anti-inflammatory gene expression (Fig. 5).
Figure 5.
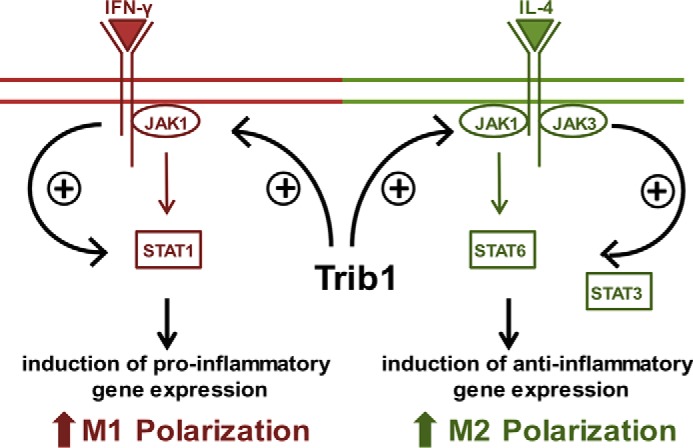
Trib1 modulates the abundance of JAKs and thereby regulates STAT activation-dependent M1 and M2 polarization of murine macrophages.
Discussion
In the present study, we demonstrated that Trib1 contributes to major macrophage functions including phagocytosis, migration, and polarization. Trib1 deficiency in bone marrow–derived macrophages resulted in increased phagocytosis and reduced migratory response to the chemoattractant MCP-1. Furthermore, we observed an impaired induction of M1 marker genes, along with lower secretion of proinflammatory cytokines, ROS and NO production in Trib1−/− BMDMs upon stimulation with LPS/IFNγ. Likewise, induction of M2 markers and secretion of M2 cytokines were reduced upon stimulation with IL-4 in Trib1−/− BMDMs. Moreover, we provide evidence that reduced activation of the JAK/STAT signaling pathway is responsible for the impaired M1 and M2 phenotype in Trib1−/− BMDMs.
Trib1, together with Trib2 and Trib3, constitutes the mammalian tribbles family of genes, which encode well-conserved pseudokinase proteins involved in the regulation of intracellular signaling networks (16). Previous studies indicate that functional properties of tribbles are highly context-dependent and tissue-specific, which may explain the pleiotropic role in several diseases (15, 16, 27). However, to date, the physiologic roles of tribbles proteins have remained largely unknown. Notably, Trib1 has been associated with atherosclerosis, dyslipidemia, and obesity (24–26, 28), all of which are characterized by chronic inflammatory states and the involvement of macrophages in disease pathology. Trib1 was shown to impair cytokine gene expression in white adipose tissue and suggested to serve as a functional receptor in the communication between adipocytes and immune cells (28). Further, Trib1 deficiency was reported to result in a severe reduction of F4/80+MR (mannose receptor) + tissue-resident M2-like macrophages in various organs (29). Our findings in bone marrow–derived macrophages corroborate a role of Trib1 in M2 polarization, because a lack of Trib1 impaired M2 polarization. In addition, we provide evidence for the first time that Trib1 has a functional role in M1 polarization of BMDMs, suggesting that Trib1 acts as a bidirectional regulator of macrophage M1 and M2 activation. The impaired response of Trib1−/− BMDMs to LPS/IFNγ or IL-4 treatment was also maintained at higher concentrations, which implies that Trib1 does not merely alter the activation threshold to M1 and M2 stimuli.
Macrophage polarization is controlled by multiple signaling pathways, transcription factors, and post-transcriptional regulators. Activation of the JAK/STAT signaling pathway has a pivotal role for M1 and M2 polarization in response to IFNγ or IL-4, respectively (3, 35).
We identified impaired activation of STAT3 and STAT6 in IL-4–stimulated Trib1-deficient BMDMs. IL-4–induced activation of the transcription factor STAT6 is the major signaling pathway in M2 activation, with STAT6 regulating the expression of many M2 associated marker genes including Arg1, Cd206, and Fizz1 (36), all of which showed lower expression in Trib1−/− BMDMS. Interestingly, phosphorylation of STAT3 was also reduced in Trib1−/− BMDMs treated with IL-4. Commonly, STAT3 activation is caused by binding of IL-10 to the IL-10 receptor, but recent work indicates that STAT3 phosphorylation in macrophages may also be induced by IL-4 treatment (37). Furthermore, we observed lower levels of PPARγ mRNA and protein in IL-4–stimulated Trib1-deficient macrophages. Recent studies demonstrated cross-talk between the nuclear receptor PPARγ and the IL-4/STAT6 axis that may coordinately control the M2 phenotype (38). PPARγ is induced and activated by IL-4–mediated signaling and contributes to the acquisition and maintenance of the M2 polarization status (14). Further, Odegaard et al. (31) demonstrated that PPARγ-deficient macrophages are resistant to M2 polarization. Hence, it is conceivable that reduced levels of phosphorylated STAT3, STAT6, and lower levels of PPARγ may collectively underlie the impaired M2 phenotype in Trib1-deficient BMDMs.
Whereas activation of STAT3/STAT6 is crucial in controlling M2 polarization, STAT1 signaling is the major determinant for M1 polarization. STAT1 is activated by the binding of IFNγ to its receptor and induces the expression of many proinflammatory M1-related genes (7, 9). We demonstrated that Trib1−/− BMDMs exhibit reduced levels of activated STAT1 molecules, which may explain lower expression levels of M1-associated genes in Trib1-deficient BMDMs.
In addition to STAT1 signaling, M1 polarization is triggered by the activation of the NF-κB and MAP kinase signaling pathways, including JNK, p38 and ERK1/2 (5, 6, 39). Previous studies indicate that Trib1 modulates signal transduction in the MAPK pathway in HeLa cells and smooth muscles cells (21, 22). Furthermore, Trib1 was implicated as a transcriptional coactivator for NF-κB/RelA in the control of cytokine gene expression in white adipocytes (28). However, we did not detect altered activation of the MAPK and NF-κB pathways in Trib1-deficient macrophages. These discrepancies may be ascribed to context and tissue/cell type–specific functions of Trib1. Further, the use of primary cells versus established cancer cell lines that often show aberrantly activated MAPK signaling may account for these differences.
In light of our data, it appears more likely that reduced activation of STAT molecules underlies the impaired M1 and M2 polarization of Trib1−/− BMDMs. STAT activation is controlled by the receptor-associated JAK phosphorylation (40). In M1 polarization, IFNγ induces the activation of JAK1 and JAK2, and in M2 polarization IL-4 induces the activation of JAK1 and JAK3 (41). Our findings indicate that Trib1 deficiency leads to lower protein levels of JAK1, the common upstream factor in M1 and M2 signaling, as well as a reduction of JAK3. As a consequence of the lower JAK abundance, JAK-mediated phosphorylation of STAT1, STAT3, and STAT6 in response to IFNγ or IL-4 is reduced, which in turn impairs expression of respective pro- and anti-inflammatory STAT target genes (Fig. 5).
Currently, it is not clear how Trib1 controls the abundance of JAK1 and JAK3 in macrophages. Jak1 and Jak3 mRNA expression was only modestly reduced in Trib1−/− BMDMs. Hence, a post-transcriptional regulation of JAKs by Trib1 seems more plausible. This would also be in line with the proposed function of TRIB1 as an adaptor protein that mediates the stability and degradation of binding partners (16). Because TRIB1 interacting proteins are thought to accumulate in the absence of TRIB1, it is tempting to speculate that Trib1 deficiency leads to the accumulation of a negative regulator of JAKs. Future studies aimed at elucidating the precise molecular mechanisms with which TRIB1 regulates the abundance of JAKs, as well as identifying the participating TRIB1 interaction partners, should lead to further understanding of the role of TRIB1 in macrophage biology and inflammatory diseases.
Experimental procedures
Animals
Male Trib1−/− mice on C57BL/6 background and WT littermate control (WT) mice (25) were used for bone marrow isolation at the age of 20 weeks. WT and knockout mice were cohoused in individually ventilated cages on a 12-h light/dark cycle with free access to food (standard chow diet (ssniff)) and water. Experiments were performed in accordance with the rules for animal care of the local government authorities and were approved by the animal care and use committee of Leipzig University, as well as by the animal care committee of the Bezirksregierung Leipzig, Germany (approval N10/13, T10/16).
Cell culture
BMDMs were generated from bone marrow cells of Trib1−/− and WT mice as previously described (42). Briefly, the mice were sacrificed, and the cells were collected by flushing tibias and femurs of 4–8 mice/genotype with PBS/heparin, passed through a 40-μm strainer (BD Biosciences), and centrifuged for 5 min at 1000 rpm. Subsequently, the cells were differentiated into macrophages in cultured Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 20% L929 cell conditioned medium as a source of colony-stimulating factor-1. For each genotype (Trib1−/− and WT), BMDMs from 3–5 different mice were differentiated separately, and afterward the cells were pooled and used for the experiments. For M1 and M2 polarization, 7.5 × 105 cells (for RNA isolation) or 1 × 106 cells (for protein isolation) were incubated with either 100 ng/ml LPS (R&D Systems) with 20 ng/ml IFNγ (R&D Systems) or 20 ng/ml IL-4 (R&D Systems) over a time course of 0.5 to 48 h (as described in the figure legends). For dose-response experiments, BMDMs were treated with 0, 1, 10, 100, 500, or 1000 ng/ml LPS; 0, 1, 10, 20, 50, or 100 ng/ml IFNγ; or 0, 1, 10, 20, 50, or 100 ng/ml IL-4 for 48 h.
Cytokine secretion and NO synthase activity
BMDMs were polarized over 48 h. Then the media were replaced by serum-free Dulbecco's modified Eagle's medium and harvested after 6 h to determine secreted cytokines. Secreted TNFα, IL-6, IL-1β, CXCL1, and IL-10 were determined by multiplex cytokine assays (V-PLEX proinflammatory panel 1 kit; Mesoscale Discovery). TGFβ was measured by an ELISA (mouse TGF-β 1 DuoSet ELISA; R&D Systems). The level of nitrite as a marker of NO synthase activity was measured by the Griess Reagent System (Promega).
Phagocytosis assay
To assay phagocytotic ability, the Vybrant phagocytosis assay (Molecular Probes) was employed according to the manufacturer's instructions. Briefly, 100,000 WT and Trib1−/− BMDMs were seeded in 96-well plates and incubated with FITC-labeled E. coli particles for 2 h at 37 °C. After quenching the extracellular fluorescence with trypan blue, the intracellular fluorescence was measured with a fluorescence plate reader at the wavelengths of 480 nm (excitation) and 520 nm (emission). Per assay, six replicates were analyzed per genotype; the mean values of Trib1−/− BMDMs were calculated as changes compared with control group (phagocytosis index).
acLDL uptake
To determine scavenger receptor-mediated uptake of acLDL, 750,000 WT and Trib1−/− BMDMs were seeded in 12-well plates and incubated with 25 μg/ml DiI-labeled acLDL (Kalen Biomedical) at 37 °C for 5 h. After incubation, the cells were washed two times with PBS, 0.4% BSA and three times with PBS. Then cells were lysed with lysis buffer (0.1 m NaOH, 0.1% SDS) for 1 h in the dark. Fluorescence intensity was determined with a fluorescence plate reader at 480 nm (excitation) and 520 nm (emission). Protein concentration was measured using the DC protein assay (Bio-Rad) according to the manufacturer's instructions. The results were expressed as μg of absorbed DiI-labeled acLDL per mg of cell protein. Three replicates were analyzed per genotype in three independent experiments.
Migration assay
Migration assays were performed in 24-well plates with 8-μm pore-size membrane inserts. WT and Trib1−/− BMDMs (2 × 105 cells) were added to the upper chamber and 600 μl of serum-free medium, with or without the chemoattractant factor MCP1 (100 ng/ml, R&D Systems), were added in the lower chamber. The cells were incubated for 16 h at 37 °C, and afterward, migrated cells were fixed on the inserts. Nonmigrated cells on the upper side of the membrane were removed with a cotton swab. The inserts with the migrated cells were stained with 4′,6′-diamino-2-phenylindole (Dako). Migration was determined by visualization and counting using a fluorescence microscope (Axio Observer; Zeiss). The number of migrated cells in five randomly chosen fields were counted in five replicates for each treatment. The migration index was calculated by the mean of the counted cells for each treatment. The migration index of the WT group in the absence of MCP1 (control) was set to 1, and all other values were related to that group.
ROS production assay
The total ROS/superoxide detection kit (Enzo Life Sciences) was employed according to the manufacturer's instructions. Briefly, 100,000 WT and Trib1−/− BMDMs were seeded in 96-well plates and cultured over 24 h with or without M1 or M2 stimuli. Subsequently, cells were washed and incubated with the provided oxidative stress detection reagent (1:2500) for 45 min at 37 °C in the dark. Negative control cells were preincubated with 5 mm N-acetyl-l-cysteine, a ROS inhibitor, for 30 min before incubation with the detection reagent. Positive control cells were coincubated with 200 μm pyocyanin in the detection reagent. Following the incubation period, the intracellular fluorescence was measured with a fluorescence plate reader at 488 nm (excitation) and 520 nm (emission). Per assay, six replicates were analyzed per genotype.
RNA isolation, RT-PCR, and qPCR
Total RNA was extracted using the RNeasy micro kit (Qiagen) according to the manufacturer's protocol. cDNA synthesis was performed from 1 μg of total RNA with SuperScript II reverse transcriptase (Invitrogen), random hexamer primers p(dN)6 (Roche Diagnostic), and dNTPs (Promega). Quantitative real-time PCR was performed using a SYBR Green Master Mix and primers for Il6, Il1b, Nos2, Arg1, Cd206, Fizz1, Cd68, Cd36, Srb1, Scarf1, Cxcl16, Stab1, Marco, Sra, Lox1, Cd163, Ccr2, Trib1, Pparg1, and Actb (β-actin) (primer sequences are listed in Table S1). Gene expression levels were calculated by the ΔCt method using Actb as housekeeping gene.
Western blotting analysis
Total cell protein was isolated with radioimmune precipitation assay lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Igepal, 0.1% SDS) containing protease and phosphatase inhibitor mixture (Roche) from WT and Trib1−/− BMDMs. Protein concentration was measured with the DC protein assay (Bio-Rad) according to the manufacturer's instructions. For Western blot analysis, 25 μg of cell protein were loaded, and primary antibodies were incubated for 16 h. The following primary antibodies were purchased from Cell Signaling: phospho-NF-κB p65 (Ser-536, catalog no. 8242), NF-κB p65 (catalog no. 3033), phospho-p38 (Thr-180/Tyr-182, catalog no. 4511), p38 (catalog no. 9212), phospho-ERK1/2 (Thr-202/Tyr-294, catalog no. 4370), ERK1/2 (catalog no. 4695), phospho-JNK (Thr-183/Tyr-185, catalog no. 4668), JNK (catalog no. 9258), phospho-STAT3 (Ser-727, catalog no. 9134), STAT3 (catalog no. 9139), phospho-STAT1 (Tyr-701, catalog no. 9167), STAT1 (catalog no. 9172), phospho-JAK1 (Tyr-1022/Tyr-1023, catalog no. 3331), JAK1 (catalog no. 3344), phospho-JAK2 (Tyr-1007/Tyr-1008, catalog no. 3771), JAK2 (catalog no. 3230), phospho-JAK3 (Tyr-980/Tyr-981, catalog no. 5031), JAK3 (catalog no. 8863), PPARγ (catalog no. 2435), SOCS1 (catalog no. 3950), SOCS3 (catalog no. 2932), and C/EBPα (catalog no. 8178). Phospho-STAT6 (Tyr-641, catalog no. 54461) and STAT6 (catalog no. 44718) were purchased from Abcam, C/EBPβ from Santa Cruz (catalog no. 150) and GAPDH from Fitzgerald (catalog no. 10R-G109a). For assessing total abundance of respective proteins, the blots were stripped with Western blotting stripping buffer (Thermo Fisher Scientific) according to the manufacturer's instruction and reprobed with the total antibody or the loading control antibody against GAPDH, respectively. Immunoblotting was performed at three times for each antibody, and the displayed blots represent one of each with similar results.
Statistics
The data in figures are presented as box-and-whisker plots showing minimum to maximum values. Otherwise, the data are expressed as means ± S.D. Fold changes were calculated based on mean values. The data sets were analyzed for statistical significance using unpaired two-tailed Student's t test. p values of <0.05 were considered statistically significant. Levels of statistical significance are indicated by asterisks: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). All experiments were carried out with at least six mice/genotype in three independent approaches.
Author contributions
L. A. and R. B. conceptualization; L. A. and M. G. formal analysis; L. A., C. E. K., S. M., and F. J. investigation; L. A., J. D., M. G., C. E. K., S. M., and F. J. methodology; L. A. and R. B. writing-original draft; J. D. resources; J. D., J. T., and R. B. funding acquisition; J. T. and R. B. writing-review and editing; R. B. supervision.
Supplementary Material
Acknowledgments
We thank Professors Shizuo Akira and Takashi Satoh (Immunology Frontier Research Center, Osaka University, Osaka, Japan) for initially providing the Trib1−/− mice on a mixed background. We thank Dr. Michael Schaab for assistance with cytokine analysis.
This study was supported by Deutsche Forschungsgemeinschaft, Collaborative Research Centre “Obesity Mechanisms” Grant SFB1052-B07 (to R. B.) and a grant from Stiftung für Pathobiochemie und Molekulare Diagnostik (to J. D. and R. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S7.
- TNF
- tumor necrosis factor
- BMDM
- bone marrow–derived macrophage
- JAK
- Janus kinase
- IFN
- interferon
- LPS
- lipopolysaccharide
- NO
- nitric oxide
- ROS
- reactive oxygen species
- PPAR
- peroxisome proliferator-activated receptor
- C/EBP
- CCAAT enhancer-binding protein
- FITC
- fluorescein isothiocyanate
- LDL
- low-density lipoprotein
- acLDL
- acetylated LDL
- MCP
- monocytotic chemoattractant protein
- TGF
- transforming growth factor
- SOCS
- suppressors of cytokine signaling
- qPCR
- quantitative PCR.
References
- 1. Mosser D. M., and Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sica A., and Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang N., Liang H., and Zen K. (2014) Molecular mechanisms that regulate the macrophage M1/M2 polarization balance. Front. Immunol. 5, 614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Y.-C., Zou X.-B., Chai Y.-F., and Yao Y.-M. (2014) Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 10, 520–529 10.7150/ijbs.8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piras V., and Selvarajoo K. (2014) Beyond MyD88 and TRIF pathways in Toll-like receptor signaling. Front. Immunol. 5, 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y. L., and Dong C. (2005) MAP kinases in immune responses. Cell. Mol. Immunol. 2, 20–27 [PubMed] [Google Scholar]
- 7. Lawrence T., and Natoli G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 10.1038/nri3088 [DOI] [PubMed] [Google Scholar]
- 8. Tugal D., Liao X., and Jain M. K. (2013) Transcriptional control of macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 33, 1135–1144 10.1161/ATVBAHA.113.301453 [DOI] [PubMed] [Google Scholar]
- 9. Darnell J. E. Jr., Kerr I. M., and Stark G. R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 10.1126/science.8197455 [DOI] [PubMed] [Google Scholar]
- 10. Wills-Karp M., and Finkelman F. D. (2008) Untangling the complex web of IL-4– and IL-13–mediated signaling pathways. Sci. Signal. 1, pe55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dhakal M., Hardaway J. C., Guloglu F. B., Miller M. M., Hoeman C. M., Zaghouani A. A., Wan X., Rowland L. M., Cascio J. A., Sherman M. P., and Zaghouani H. (2014) IL-13Rα1 is a surface marker for M2 macrophages influencing their differentiation and function. Eur. J. Immunol. 44, 842–855 10.1002/eji.201343755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stein M., Keshav S., Harris N., and Gordon S. (1992) Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 10.1084/jem.176.1.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raes G., Van den Bergh R., De Baetselier P., Ghassabeh G. H., Scotton C., Locati M., Mantovani A., and Sozzani S. (2005) Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol. 174, 6561–6562 10.4049/jimmunol.174.11.6561 [DOI] [PubMed] [Google Scholar]
- 14. Chawla A. (2010) Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569 10.1161/CIRCRESAHA.110.216523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yokoyama T., and Nakamura T. (2011) Tribbles in disease: signaling pathways important for cellular function and neoplastic transformation. Cancer Sci. 102, 1115–1122 10.1111/j.1349-7006.2011.01914.x [DOI] [PubMed] [Google Scholar]
- 16. Eyers P. A., Keeshan K., and Kannan N. (2017) Tribbles in the 21st century: the evolving roles of Tribbles pseudokinases in biology and disease. Trends Cell Biol. 27, 284–298 10.1016/j.tcb.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durzynska I., Xu X., Adelmant G., Ficarro S. B., Marto J. A., Sliz P., Uljon S., and Blacklow S. C. (2017) STK40 is a pseudokinase that binds the E3 ubiquitin ligase COP1. Structure 25, 287–294 10.1016/j.str.2016.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murphy J. M., Nakatani Y., Jamieson S. A., Dai W., Lucet I. S., and Mace P. D. (2015) Molecular mechanism of CCAAT-enhancer binding protein recruitment by the TRIB1 pseudokinase. Structure 23, 2111–2121 10.1016/j.str.2015.08.017 [DOI] [PubMed] [Google Scholar]
- 19. Yoshida A., Kato J.-Y., Nakamae I., and Yoneda-Kato N. (2013) COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood 122, 1750–1760 10.1182/blood-2012-12-476101 [DOI] [PubMed] [Google Scholar]
- 20. Guan H., Shuaib A., Leon D. D., Angyal A., Salazar M., Velasco G., Holcombe M., Dower S. K., and Kiss-Toth E. (2016) Competition between members of the Tribbles pseudokinase protein family shapes their interactions with mitogen activated protein kinase pathways. Sci. Rep. 6, 32667 10.1038/srep32667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiss-Toth E., Bagstaff S. M., Sung H. Y., Jozsa V., Dempsey C., Caunt J. C., Oxley K. M., Wyllie D. H., Polgar T., Harte M., O'Neill L. A., Qwarnstrom E. E., and Dower S. K. (2004) Human Tribbles, a protein family controlling mitogen-activated protein kinase cascades. J. Biol. Chem. 279, 42703–42708 10.1074/jbc.M407732200 [DOI] [PubMed] [Google Scholar]
- 22. Sung H. Y., Guan H., Czibula A., King A. R., Eder K., Heath E., Suvarna S. K., Dower S. K., Wilson A. G., Francis S. E., Crossman D. C., and Kiss-Toth E. (2007) Human Tribbles-1 controls proliferation and chemotaxis of smooth muscle cells via MAPK signaling pathways. J. Biol. Chem. 282, 18379–18387 10.1074/jbc.M610792200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yokoyama T., Kanno Y., Yamazaki Y., Takahara T., Miyata S., and Nakamura T. (2010) Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood 116, 2768–2775 10.1182/blood-2009-10-246264 [DOI] [PubMed] [Google Scholar]
- 24. Bauer R. C., Sasaki M., Cohen D. M., Cui J., Smith M. A., Yenilmez B. O., Steger D. J., and Rader D. J. (2015) Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα. J. Clin. Invest. 125, 3809–3818 10.1172/JCI77095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burkhardt R., Toh S. A., Lagor W. R., Birkeland A., Levin M., Li X., Robblee M., Fedorov V. D., Yamamoto M., Satoh T., Akira S., Kathiresan S., Breslow J. L., and Rader D. J. (2010) Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J. Clin. Invest. 120, 4410–4414 10.1172/JCI44213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Douvris A., Soubeyrand S., Naing T., Martinuk A., Nikpay M., Williams A., Buick J., Yauk C., and McPherson R. (2014) Functional analysis of the TRIB1 associated locus linked to plasma triglycerides and coronary artery disease. J. Am. Heart Assoc. 3, e000884 10.1161/JAHA.114.000884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnston J., Basatvat S., Ilyas Z., Francis S., and Kiss-Toth E. (2015) Tribbles in inflammation. Biochem. Soc. Trans. 43, 1069–1074 10.1042/BST20150095 [DOI] [PubMed] [Google Scholar]
- 28. Ostertag A., Jones A., Rose A. J., Liebert M., Kleinsorg S., Reimann A., Vegiopoulos A., Berriel Diaz M., Strzoda D., Yamamoto M., Satoh T., Akira S., and Herzig S. (2010) Control of adipose tissue inflammation through TRB1. Diabetes 59, 1991–2000 10.2337/db09-1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satoh T., Kidoya H., Naito H., Yamamoto M., Takemura N., Nakagawa K., Yoshioka Y., Morii E., Takakura N., Takeuchi O., and Akira S. (2013) Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495, 524–528 10.1038/nature11930 [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto M., Uematsu S., Okamoto T., Matsuura Y., Sato S., Kumar H., Satoh T., Saitoh T., Takeda K., Ishii K. J., Takeuchi O., Kawai T., and Akira S. (2007) Enhanced TLR-mediated NF-IL6–dependent gene expression by Trib1 deficiency. J. Exp. Med. 204, 2233–2239 10.1084/jem.20070183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., and Chawla A. (2007) Macrophage-specific PPAR[ggr] controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee B., Qiao L., Lu M., Yoo H. S., Cheung W., Mak R., Schaack J., Feng G.-S., Chi N.-W., Olefsky J. M., and Shao J. (2014) C/EBPα regulates macrophage activation and systemic metabolism. Am. J. Physiol. Endocrinol. Metab. 306, E1144–E1154 10.1152/ajpendo.00002.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu X., and Ivashkiv L. B. (2009) Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity 31, 539–550 10.1016/j.immuni.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelms K., Keegan A. D., Zamorano J., Ryan J. J., and Paul W. E. (1999) The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17, 701–738 10.1146/annurev.immunol.17.1.701 [DOI] [PubMed] [Google Scholar]
- 35. Biswas S. K., and Mantovani A. (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11, 889–896 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- 36. Ohmori Y., and Hamilton T. A. (1998) STAT6 is required for the anti-inflammatory activity of interleukin-4 in mouse peritoneal macrophages. J. Biol. Chem. 273, 29202–29209 10.1074/jbc.273.44.29202 [DOI] [PubMed] [Google Scholar]
- 37. Bhattacharjee A., Shukla M., Yakubenko V. P., Mulya A., Kundu S., and Cathcart M. K. (2013) IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Rad. Biol. Med. 54, 1–16 10.1016/j.freeradbiomed.2012.10.553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szanto A., Balint B. L., Nagy Z. S., Barta E., Dezso B., Pap A., Szeles L., Poliska S., Oros M., Evans R. M., Barak Y., Schwabe J., and Nagy L. (2010) STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 33, 699–712 10.1016/j.immuni.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wells C. A., Ravasi T., and Hume D. A. (2005) Inflammation suppressor genes: please switch out all the lights. J. Leukocyte Biol. 78, 9–13 10.1189/jlb.1204710 [DOI] [PubMed] [Google Scholar]
- 40. Rawlings J. S., Rosler K. M., and Harrison D. A. (2004) The JAK/STAT signaling pathway. J. Cell Sci. 117, 1281–1283 10.1242/jcs.00963 [DOI] [PubMed] [Google Scholar]
- 41. Martinez F. O., and Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Reports 6, 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teupser D., Kretzschmar D., Tennert C., Burkhardt R., Wilfert W., Fengler D., Naumann R., Sippel A. E., and Thiery J. (2008) Effect of macrophage overexpression of murine liver X receptor-α (LXR-α) on atherosclerosis in LDL-receptor deficient mice. Arterioscler. Thromb. Vasc. Biol. 28, 2009–2015 10.1161/ATVBAHA.108.175257 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.