

Abstract
Ulvan is a complex sulfated polysaccharide present in the cell wall of green algae of the genus Ulva (Chlorophyta). The first ulvan-degrading polysaccharide lyases were identified several years ago, and more were discovered through genome sequencing of marine bacteria. Ulvan lyases are now grouped in three polysaccharide lyase (PL) families in the CAZy database, PL24, PL25, and PL28. The recently determined structures of the representative lyases from families PL24 and PL25 show that they adopt a seven-bladed β-propeller fold and utilize the His/Tyr catalytic mechanism. No structural information is yet available for PL28 ulvan lyases. NLR48 from Nonlabens ulvanivorans belongs to PL28 together with its close paralog, NLR42. Biochemical studies of NLR42 have revealed that it can cleave ulvan next to both uronic acid epimers. We report the crystal structure of ulvan lyase NLR48 at 1.9-Å resolution. It has a β-jelly roll fold with an extended, deep, and positively charged substrate-binding cleft. Putative active-site residues were identified from the sequence conservation pattern, and their role was confirmed by site-directed mutagenesis. The structure of an inactive K162M mutant with a tetrasaccharide substrate showed the substrate occupying the “−” subsites. Comparison with lyases from other PL families with β-jelly roll folds supported assignment of the active site and explained its ability to degrade ulvan next to either epimer of uronic acid. NLR48 contains the His/Tyr catalytic machinery with Lys162 and Tyr281 playing the catalytic base/acid roles.
Keywords: carbohydrate processing, catalysis, crystal structure, substrate specificity, bacteria, algae, substrate binding, ulvan lyase, β-jelly roll fold, PL28 family
Introduction
Red, brown, and green algae are major members of the marine ecosystem and contribute to the huge aquatic biomass (1). Polysaccharides present in the algal cell wall play a structural role, providing rigidity, and represent a major component of their biomass. They are also a rich source of carbon for cohabiting microorganisms. Carrageenan is the main cell wall polysaccharide of many red algae, alginate is a cell wall constituent of brown algae, and ulvan is present in the green algae belonging to the genus Ulva (Chlorophyta). Carrageenan and alginate have been studied extensively and reported to have many industrial applications (2), and the enzymes degrading these polysaccharides have been well-characterized (3, 4). Ulvan is biosynthesized by marine green algae Ulva spp. and Enteromorpha spp. and contributes to 8–30% of the dry weight of the algae. This sulfated polysaccharide has recently gained attention for a variety of industrial applications in the agriculture, food, pharmaceutical, chemical, and biomaterial industries (1, 5, 6).
Major components of the ulvan polysaccharide are 3-O-sulfated α-l-rhamnose, α-l-iduronic acid, β-d-glucuronic acid, and xylose. Ulvan is made of two main repetitive disaccharide units. Type A ulvanobiourinic 3-sulfate has [→4)-β-d-GlcA-(1→4)-α-l-Rha3S-(1→]2 and type B ulvanobiouronic 3-sulfate has [→4)-α-l-IdoA-(1→4)-α-l-Rha3S(1→] disaccharide units (Scheme 1) (7). The occurrence of a sulfate group in ulvan resembles the glycosaminoglycans present in vertebrates, and the presence of uronic acid makes it subject to depolymerization by a lytic mechanism.
Scheme 1.

Diagram of a tetrasaccharide fragment of ulvan containing either GlcA or IdoA. The product of the cleavage by lyase is shown on the right.
Ulvan lyases degrade ulvan through a β-elimination mechanism, cleaving the 1–4-glycosidic linkage on the nonreducing end of a uronic acid residue with the formation of an unsaturated product at the newly formed nonreducing end of the polysaccharide. The first identified ulvan lyase and the first representative of the PL28 family was NLR42 (NCBI accession number AEN28574; 46 kDa with two domains) isolated from Nonlabens ulvanivorans (synonym, Persicivirga ulvanivorans) (8). The enzyme was able to cleave ulvan next to both GlcA and IdoA. In addition to the catalytic domain, NLR42 also harbors carbohydrate-binding and sorting domains (9).
Genome sequencing of marine microorganisms undertaken in the last few years led to the identification of more genes exhibiting ulvan lyase activity (10–12). Polysaccharide-degrading machineries occur often as coregulated gene clusters called polysaccharide utilization loci (PUL). The PUL regions include genes coding for various transport proteins, highly specific carbohydrate-acting enzymes (glycoside hydrolases, polysaccharide lyases, and sulfatases), and recognition proteins. The complete degradation of the polysaccharides requires the coordination of these different components. We had previously characterized the ulvan PUL from Alteromonas and Pseudoalteromonas species (13), which led to addition of the families PL24 (LOR107) and PL25 (PLSV3936) to the CAZy database (http://www.cazy.org)3 (14). Both LOR107 and PLSV3936 display <14% sequence identity; nevertheless, they both adopt a β-propeller scaffold. LOR107 is reported to degrade ulvan specifically after the IdoA site (15), whereas PLSV3936 can cleave after both GlcA and IdoA sites (16). Recently, a new ulvan polysaccharide family, PL28, was added to the CAZy database.
Here, we report first crystal structure and catalytic mechanism of ulvan lyase NLR48 from N. ulvanivorans grouped in the PL28 family. NLR48 is a 30-kDa protein that was first identified as an ulvan lyase together with its larger homolog, NLR42 (8). NLR48 shows 37% sequence identity with the catalytic module of NLR42, suggesting structural and functional similarity of these enzymes. NLR42 can cleave the ulvan polysaccharide next to both GlcA and IdoA (8). We have determined the crystal structure of NLR48 at 1.9-Å resolution. Unlike the previously characterized ulvan lyases LOR107 and PLSV3936, NLR48 has a β-jelly roll fold. The structure of the NLR active-site mutant K162M in complex with a tetrasaccharide substrate facilitated the identification of active-site residues and allowed us to propose a catalytic mechanism for ulvan degradation.
Results and discussion
We have determined the three-dimensional structure of NLR48, the first representative of the polysaccharide lyase family PL28, which has recently been added to the CAZy database. Several constructs were cloned and tested for expression. Expression yields of His-NLR48 and a shorter construct starting at Asp53, called His-NLR48(53D), were high, but the proteins aggregated in solution and were not suitable for crystallization. Therefore, the MBP and GST fusions were tested. The expression level of His-MBP-NLR48 was low in BL21(DE3), but that of the GST-NLR48 fusion was moderate, and no aggregation was observed. It was essential to purify the latter construct at 4 °C to prevent aggregation. This protein yielded well-diffracting crystals.
Crystal structure of NLR48
The three-dimensional crystal structure of NLR48 was solved by a single anomalous dispersion method using selenomethionine (SeMet)-incorporated protein crystals. There are two molecules in the asymmetric unit, which are related by a noncrystallographic 2-fold rotation. Both molecules are nearly identical with a root-mean-square deviation (r.m.s.d.) of 0.2 Å for all Cα atoms. NLR48 displays a β-sandwich jelly roll fold with two concave β-sheets stacking on each other (Fig. 1A). Each β-sheet has seven antiparallel, strongly bent β-strands. The inner β-sheet is formed from strands β1-β4-β13-β6-β7-β8-β9, whereas the outer β-sheet contains strands β2-β3-β14-β5-β10-β11-β12 (Fig. 1B). There are seven crossovers between the two sheets. The N-terminal segment, amino acids 21–50, is devoid of secondary structure elements and is fixed to the loop connecting strands β1 (inner sheet) and β2 (outer sheet) by a disulfide bond between Cys32 and Cys59. The inner β-sheet forms a deep cleft extending across the entire β-sheet in a direction nearly perpendicular to the strands. The bottom of the cleft is formed by the centers of the β-stands; one wall is formed from the loops connecting strands β3 to β4 and β5 to β6 (intersheet), whereas the other wall is made from the loops connecting β6 to β7 and β8 to β9 (intra-inner sheet). The cleft is lined predominantly with basic and polar side chains and displays a positive electrostatic surface potential as calculated with APBS software (17) (Fig. 1C). Two phosphate molecules acquired from the crystallization solution are found in equivalent positions in both molecules. This cleft is likely the substrate-binding site of NLR48. A large positive difference map peak was identified in each molecule near the disulfide bond described above. Based on the environment and the coordination geometry, these peaks are assigned as Ca2+ ions. Each ion is heptacoordinated in a pentagonal bipyramid arrangement by Oδ1Asp79, Oδ2Asp79, Oδ1Asn85, O=CGly59, and O=CSer81 in the equatorial plane and Oδ1Asn61 and O=CAla84 in axial positions. The Ca–O distances are in the 2.25–2.50-Å range (Fig. 2A). This Ca2+ ion provides additional stability to the N-terminal region of NLR48. This Ca2+-binding site is separated from the positively charged cleft, suggesting that Ca2+ plays a structural rather than catalytic role in NLR48.
Figure 1.

Structural details of NLR48. A, stereoview of the cartoon representation of the structure. The molecule is rainbow-colored from blue at the N terminus to red at the C terminus. The β-strands are numbered sequentially along the protein sequence. This and other figures were made with PyMOL (Schrödinger, LLC). B, topology diagram showing the connectivity of the strands. The strands are similarly rainbow-colored as in the cartoon figure. C, view of the electrostatic potential projected on the protein surface within the cleft in the concave side of the protein. The cleft displays strongly positive potential matching the negatively charged polysaccharide substrate. D, residue conservation among sequence homologs of NLR48 as calculated by ConSurf (24). The highly conserved residues are colored burgundy. The surface representation shows their concentration within the canyon.
Figure 2.

Ion-binding sites in the native structure of NLR48. A, the coordination sphere of the Ca2+ ion, which plays a structural role in this enzyme. B, the environment of the PO43− ions.
Two phosphate (PO43−) ions are located within the positively charged cleft and make many contacts with the surrounding residues (Fig. 2B). PO43− at site 1 binds in the middle of the cleft and forms hydrogen bonds with NζLys162, Nϵ2Gln160, Oϵ1Gln160, Nη1Arg117, and OηTyr281. The PO43− at site 2 makes H-bonds with Nη2Arg216, OγSer171 and backbone NHAla175. The phosphate in site 1 has lower B-factors, suggesting a somewhat stronger binding than in site 2.
Putative substrate-binding and catalytic sites
The β-sandwich jelly roll fold is not unique within the polysaccharide lyases. It is also observed in alginate lyase families PL7, PL14, and PL18; in heparin lyase family PL13; and in exoglucuronase family PL20 (18). Representative structures from these PL families were compared with NLR48. Representative structures for these families were: PL7, alginate lyase (A1-II′;Aly) complexed with tetrasaccharide, Protein Data Bank (PDB) code 2ZAA (19); PL13, heparin lyase I with octasaccharide, Protein Data Bank code 3INA (20); PL18, alginate lyase aly-SJ02, PDB code 4Q8K (21); PL20, glucuronan lyase, PDB code 2ZZJ (22); and PL14, Chlorella virus enzyme vAL-1, PDB code 3A0N (23). The number of β-strands in the inner and outer sheets varies between these enzymes, but the core structures (∼100 residues; Fig. 3B) superpose well with that of NLR48 (Table 1). They differ mainly in the loop regions (Fig. 3A). The PL14 lyase is the least similar and is excluded from further analysis. Despite sharing the same fold, the sequence identity between NLR48 and these other lyases is less than 14%. Nevertheless, despite low sequence identity, these enzymes have similar active sites and a shared catalytic mechanism belonging to the His/Tyr class (18). The active site is located in the cleft extending along the inner β-sheet.
Figure 3.
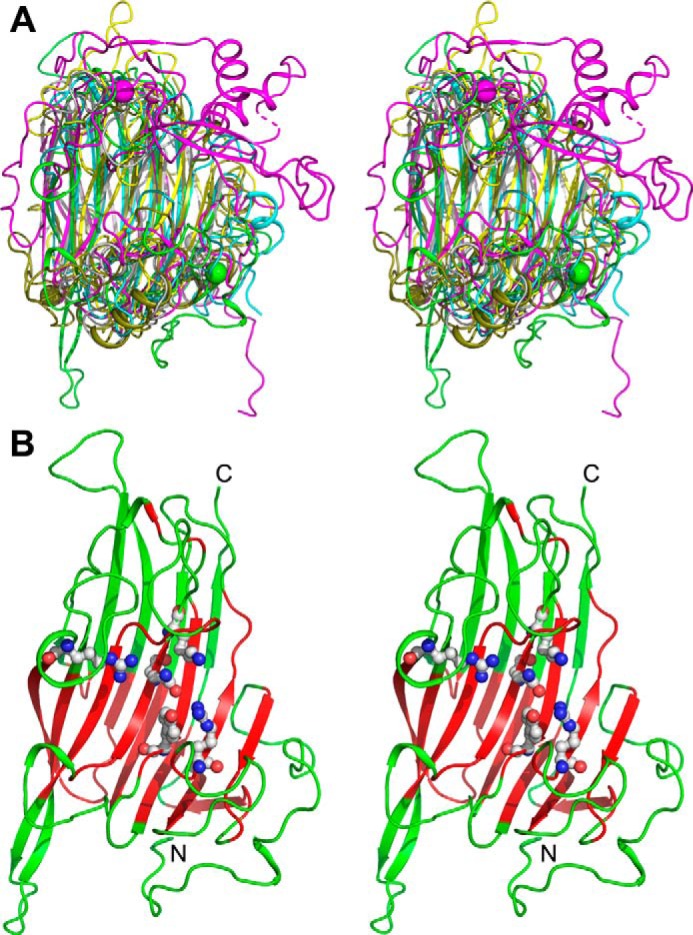
Stereoview of the superposition of polysaccharide lyases that display the β-sandwich jelly roll fold. A, representative structures from families PL7, PL13, PL14, PL18, and PL20 were superimposed on NLR48 using the Swiss-PdbViewer (34). NLR48 is colored green, heparinase I (PL13) is colored magenta, alginate lyase (PL7) is colored cyan, PL18 is colored forest green, and PL20 is colored gray. B, the core ∼100 residues of NLR48 that superimpose well among the PL lyases are colored red, and the less structurally conserved segments are colored green. Residues Arg117, Gln160, Lys162, Arg216, and Tyr281 that were mutagenized are shown explicitly in ball-and-stick representation.
Table 1.
Superposition of polysaccharide lyase with a β-sandwich jelly roll fold on NLR48
| PDB code | PL family | No. of overlapping Cα | r.m.s.d. |
|---|---|---|---|
| Å | |||
| 4Q8K | PL18 | 100 | 1.47 |
| 2ZZJ | PL20 | 97 | 1.55 |
| 3INA | PL13 | 93 | 1.60 |
| 2ZAA | PL17 | 125 | 1.49 |
| 3A0N | PL14 | 83 | 1.84 |
To identify the putative catalytic site, we first utilized ConSurf software (24), which showed the highly conserved residues among ∼30 NLR48 homologs (Fig. 1D). The solvent-accessible and conserved residues located within the cleft are: Glu203, Leu178, Gln160, and Arg277 on the bottom of the cleft; Lys162, Asp173, and Arg216 along one wall; and Tyr157, Tyr281, Arg117, Arg80, and Asp78 along the other wall. Next, we compared the structure of NLR48 with the lyases from the aforementioned PL families (excluding PL14). The structural alignment of these four lyases showed that within the cleft only Glu119, Gln160, and Tyr281 had counterparts in four other lyase families, whereas Arg117 is replaced by a histidine in PL20; Lys162 is a histidine in PL7, PL13, and PL18 but a leucine in PL20; and Arg277 has lysine counterparts in PL7, PL13, and PL18 (Fig. S1). PL14 lyases differ in this respect, and their putative active-site residues are located in a different region of the cleft (23).
Site-directed mutagenesis of putative active-site and substrate-binding residues
Based on these observations the cleft region was considered as a substrate-binding site. As such, Arg117, Gln160, Tyr281, and Lys162 were deemed potential candidates for active-site residues with Arg216 likely being involved in substrate binding (H-bonds to phosphates in the native structure). These residues were mutated as follows: R117N, Q160A, K162M, R216N, and Y281F. The single mutants were expressed and purified, and their activity on the ulvan substrate was measured. The enzymatic activity was monitored as a function of time by following the appearance of unsaturated sugars in the reaction product with a characteristic absorbance at 232 nm. Although the native NLR48 showed easily measurable activity, no measurable activity was observed for the mutants (Fig. 4). This confirmed their involvement in either catalysis or substrate binding.
Figure 4.
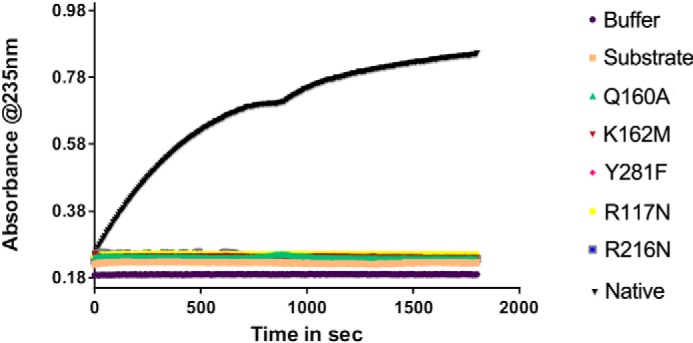
The activity measurements of the WT NLR48 and several mutants on ulvan polysaccharide. Only the naive NLR48 showed measurable activity.
Structure of NLR48 mutant with a tetrasaccharide substrate
Three of the aforementioned mutants, K162M, Q160A, and Y281F, were screened for crystallization. Well-diffracting crystals could only be obtained for K162M mutant, and these crystals required high concentration of phosphate ions. These crystals were soaked for 2 h in a solution containing the tetrasaccharide substrate and diffracted to 2.2-Å resolution. They belong to the space group P212121 and are isomorphous to the native NLR48 crystals. The K162M mutation did not introduce any significant changes into the protein structure; the r.m.s.d. between the native and mutant structures for all Cα atoms is 0.3 Å. The electron density map calculated for the complex structure showed a blob of positive density associated with each molecule. The entire tetrasaccharide could be built into this density (Fig. 5A). The tetrasaccharide does not fully enter the cleft but is docked in the entrance and does not reach into the part where the putative active site is located (residues conserved between the three lyases). We adopt the nomenclature of Davies et al. (25) where the sugars are marked as +1, +2, etc. starting from the first sugar on the reducing end of the cleaved bond and −1, −2, etc. on the nonreducing end. Correspondingly, the sugar-binding sites on the protein are referred to as ± subsites. The tetrasaccharide is oriented with its reducing end directed toward the center of the cleft, indicating that it occupies the “−” subsites in the substrate-binding sites.
Figure 5.

Tetrasaccharide substrate bound to NLR48. A, the final 2Fo − DFc map contoured at 1.5 σ with the superimposed model of the tetrasaccharide. B, close-up of the tetrasaccharide and its contacts with NLR48.
In contrast to the native structure, the NLR48 mutant harbors only one phosphate at site 1. The second phosphate, observed in site 2 of the native structure, is replaced by Rha3S of the substrate in the mutant. The phosphate in site 1 was refined to an occupancy of ∼0.7. Nevertheless, a high concentration of phosphate in the crystallization conditions might effectively block the center of the cleft from the incoming tetrasaccharide and prevent it from occupying the +1 subsite. Indeed, a comparison with the lyase–oligosaccharide complexes from PL7 and PL13 confirms that the tetrasaccharide in NLR48 occupies − subsites with the Rha3S on the reducing end occupying the −1 subsite. The first three sugars make multiple contacts with the lyase. Rha3S sugar at the −1 subsite interacts with Tyr281 (at the beginning of the crossover loop following β13), Arg216 (beginning of β9), and Tyr157 (beginning of β6), whereas its sulfate H-bonds with Arg282 (loop following β13). GlcA at the −2 subsite interacts with Lys284 (crossover loop after β13), and its carboxylic group H-bonds with Ser211 (loop connecting β8 and β9). Finally, Rha3S at the −3 subsite interacts through its sulfate group with Ser213 (loop connecting β8 and β9) (Fig. 5B). The last sugar, ΔUA, makes no contacts with this molecule of NLR48 or with symmetry-related molecules, but its rotational freedom is restricted through the constraints imposed by the presence of bulky substituents at C3 and C5 positions of the preceding Rha3S sugar. Consequently, this sugar is more mobile than the others and has higher B-factors. No aromatic stacking interactions are observed in the lyase–tetrasaccharide contacts. Another factor that may restrict passage of the tetrasaccharide into the cleft is the dynamics of walls motion: they may have to move out to allow the substrate to slide in. In particular, the loop 164–174 with the Asp173 protruding into the cleft could play a gating function in solution. However, in the crystal structure, this loop butts against its counterpart in the neighboring molecule related by the noncrystallographic 2-fold axis (Fig. 6) and has no ability to expand the cleft. Because the complex was obtained by soaking the substrate into a preformed crystal, this could be an additional obstacle to the presence of a phosphate in the center of the cleft.
Figure 6.
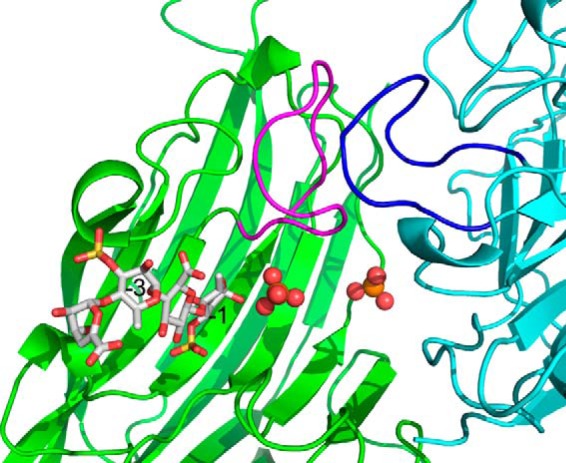
Packing of a neighboring molecule against the loop forming one wall of the substrate-binding site. This tight packing restricts the movement of this loop and partially blocks the entry into the active site.
Interestingly, an additional phosphate ion was observed in this structure near Arg124 and Arg277 that define the “+” subsites. Although these side chains have the same conformation in the native structure, there are only water molecules in this area. This new phosphate-binding site might reflect a subtle change of the electrostatic potential when the substrate in occupies the − sites.
Catalytic mechanism
The β-elimination catalytic mechanism requires neutralization of the negative charge on the C5 carboxylic group. This reduces the pKa of the C5 proton and makes it vulnerable to abstraction by a base. Concomitant donation of a proton by a protonated amino acid or water molecule to the leaving group results in the cleavage of the O–C4 bond. The end product carries an unsaturated molecule at the newly formed nonreducing end (26). Two general mechanisms have been identified among polysaccharide lyases: (i) metal-assisted catalysis and (ii) His/Tyr–dependent catalysis (18, 27). Lyases adopting the β-jelly roll fold utilize the latter mechanism. Based on residue conservation, it is likely that NLR48 utilizes the same mechanism.
To define the roles of residues within the cleft, we superimposed the three lyase–oligosaccharide complexes, NLR48, heparinase I, and alginate lyase (A1-II′;Aly), based only on the backbone atoms of the five conserved residues representing the local environment of the active site (Fig. 7A). In this superposition, the only sugar that has a similar position and orientation is the +1 uronate (Fig. 7B). The +2 and −1 sugars are oriented differently in these structures, reflecting different local shapes of the clefts. Nevertheless, the O1 of Rha3S in the NLR48 complex is in nearly the same place as the corresponding anomeric oxygen atoms in −1 sugars of the other two complexes (Fig. 7B). It is therefore very likely that the +1 GlcA/IdoA sugar is placed in the same way as in the other two lyases. In such a case, Gln160 would serve to neutralize the acidic group on the +1 sugar, promoting protonation through formation of two hydrogen bonds (Fig. 7A). Additional stabilization of the acidic group of the +1 sugar is provided by Arg117, which forms a hydrogen bond to the acidic group of +1 sugar. The Arg117-Gln160 arrangement is further stabilized by the formation of hydrogen bonds with a bridging Glu119 (Fig. 7A). The same arrangement is present in the other two lyases. The hydroxyl group of Tyr281 would point toward the bridging oxygen. With GlcA in the +1 site, the Tyr281 hydroxyl would also be in close proximity to the C5 proton. This residue could therefore perform the proton abstraction and donate it to the leaving group, the oxygen bridging the −1 and +1 sugars (Scheme 2, upper left). In the case of IdoA, the C5 proton is on the opposite side of the sugar ring to Tyr281 (Scheme 2, bottom left). The only side chain close to this proton is Lys162, which was mutated to methionine in the NLR48–tetrasaccharide complex (Fig. 5A). There is a histidine in an equivalent position in PL13 lyases that break the bond next to IdoA and in PL7 lyases that break the bond next to guluronate with the same configuration of the C5 carboxylic group as IdoA. In PL20 lyases, which break the bond next to GlcA, the C5 proton is facing the tyrosine, and the residue in a position equivalent to Lys162 does not participate in the reaction. Indeed, an isoleucine is found in the PL20 family. Because NRL48 can likely cleave the bond next to either epimer of the uronic acid, like its homolog NLR42 (8), Lys162 must be able to act as a base for proton abstraction. This would require an unusual pKa for this side chain in the context of the enzyme. The pKa could be influenced by the proximity to 3-sulfate of +2 Rha3S. In heparinase I, the equivalent residue is a histidine. The role of a lysine residue as a proton acceptor is not unusual; it fulfills the same role in lyases from several other families with different folds, namely PL3, PL9, PL6, and PL11 (18). Overall, both the fold and catalytic machinery are conserved among families PL28, PL7, PL13, PL18, and PL20 (Fig. 7).
Figure 7.
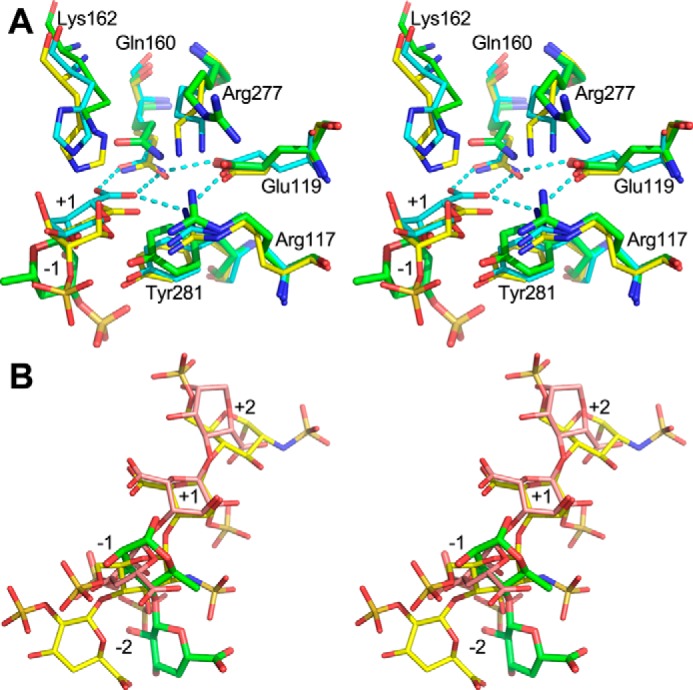
Stereoview of the superposition of enzyme–oligosaccharide complexes based on the backbone of five conserved residues in the cleft (Arg117, Glu119, Gln160, Arg277, and Tyr281 and their equivalences in other lyases). A, the key residues are displayed in a stick representation. NLR48, green carbons; PL7 (PDB code 2ZAA), cyan carbons; PL13 (PDB code 2INA), yellow carbons. Only the +1 sugar in PL7 and PL13 is shown for clarity. The Rha3S in −1 subsite of NLR48 is shown. The hydrogen-bond network among the acidic group of +1 sugar, Gln, Arg, and Glu is shown with cyan dashed lines. B, overlay of the oligosaccharides from the three lyases from the above superposition. Only −2 to +2 sugars are shown. NLR48, green carbons; PL7 (PDB code 2ZAA), pink carbons; PL13 (PDB code 2INA), yellow carbons. Only the +1 sugar has a similar position and orientation as is required for the catalysis. The orientations of the − and + sugars differ to be accommodated well into the clefts of individual enzymes. The O1 of Rha3S in NLR48 is very close to the oxygen bridging −1 and +1 sugars, suggesting that the +1 sugar would take a similar position to that observed in the other two lyases.
Scheme 2.
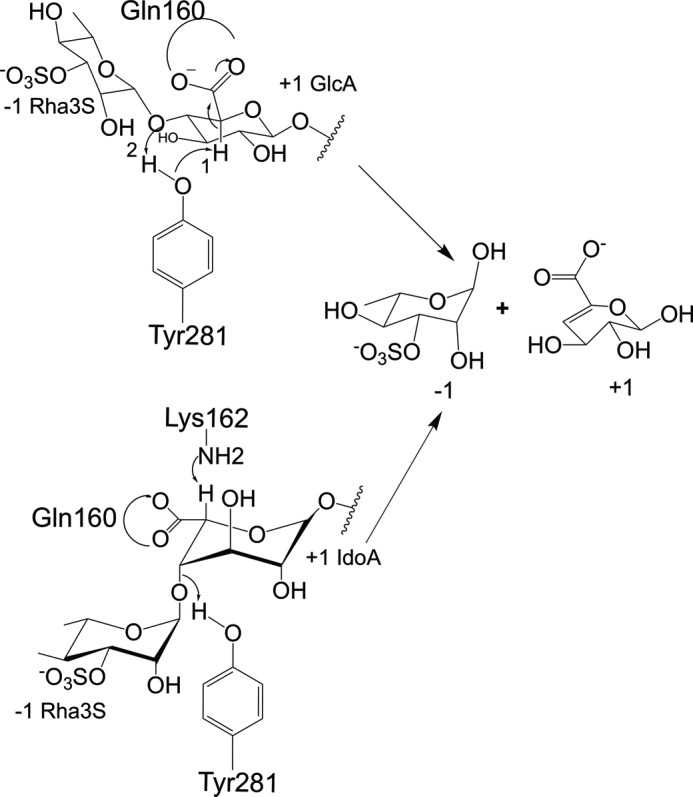
Proposed mechanism of catalysis. Tyr281 acts as a base and an acid for the cleavage near GlcA, whereas for IdoA the base is Lys162, and the acid is Tyr281.
Comparison with other ulvan lyases
Lyases that degrade ulvan polysaccharide have been identified and characterized only in the last several years. They have led to the addition of three new families to the CAZy classification, namely PL24, PL25, and PL28 families, in the CAZy database. We have previously characterized the structures and catalytic mechanisms of LOR107 (PL24) and PLSV3936 (PL25). Both ulvan lyases adopt a seven-bladed β-propeller fold. PLSV3936 utilizes the His/Tyr catalytic machinery with Arg204, Tyr188, and His123 as neutralizer, catalytic base, and acid, respectively (16). LOR107 uses Arg259 to neutralize the C5 carboxylic group, and His146 appears to play both catalytic base/acid roles aided by His167 and Tyr243 (28). Moreover, LOR107 binds the substrate in two stages with Arg320 initiating substrate binding at the top of the cleft followed by substrate sliding deeper into the cleft toward the active site (28).
Despite its different fold, NLR48 shares similarities with the PL24 and PL25 lyases in their catalytic mechanisms. All of them utilize the His/Tyr mechanism; however, the neutralization of the C5 acidic group is achieved by different means. NLR48 uses glutamine that faces the carboxyl and forms a two H-bonds and is supported by a glutamate and an arginine, whereas PL24/25 utilize an arginine to form two H-bonds with the carboxyl.
Conclusions
The marine genome sequencing project resulted in the identification of many ulvan-degrading enzymes. These are classified under PL24, PL25, and PL28 families in the CAZy database. Although PL24 and PL25 ulvan lyases have a seven-bladed β-propeller fold, PL28 lyases have a β-jelly roll fold, adding another family to the list of PL enzymes with this architecture. The catalytic mechanism of the PL28 representative lyase, NLR48 from N. ulvanivorans, is very similar to that of other lyase families with a β-jelly roll fold despite very low sequence identity and is a modification of the His/Tyr catalytic mechanism. The fold similarity of PL28 to several other families of polysaccharide lyases indicates their common evolutionary origin with adaptation to different substrates. Finally, the three families of ulvan lyases utilize a different mode of neutralization of the carboxylic group of the uronic acid.
Materials and methods
Cloning
The gene encoding NLR48 (NCBI accession number KEZ94336), lacking the first 25 residues as they are predicted to be a signal peptide, was cloned into three expression vectors with different tags to improve solubility and to aid in purification. NLR48(26–303) was inserted into pET28 vector using the restriction cloning method, whereas the ligation-independent cloning protocol (29) was used to insert NLR48(26–303) into pRL652 (modified version of pGEX with N-terminal GST tag followed by TEV cleavage site). All mutants used in this study were based on GST-NLR48(26–303) as a template by following the QuikChange site-directed mutagenesis protocol. All constructs were verified by DNA sequencing.
Expression and purification of NLR48
GST-NLR48(26–303) was expressed in BL21(DE3) strain. Overnight inoculum was subcultured into 6 liters of Terrific Broth medium. The cells were induced at ∼1.5 A600 with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. After 16 h of growth at 18 °C, cells were spun down at 6200 × g for 20 min. The cell pellet was resuspended in lysis buffer (20 mm Tris, pH 8, 400 mm NaCl, 5% glycerol) and disrupted by sonication. The supernatant was separated from cell debris by centrifugation at 39,000 × g for 55 min. The clarified supernatant was incubated for 2 h with pre-equilibrated GST resin. Unbound proteins were removed by washing with high-salt buffer (lysis buffer supplemented with 200 mm NaCl). All purification steps were performed at 4 °C. The GST tag was cleaved on the column by incubation for ∼10 h with His-TEV protease, added at a mass ratio of 1:50. The protein eluted from the column was loaded on a nickel-nitrilotriacetic acid column to remove the TEV protease. The flow-through containing highly pure NLR48 was loaded on a size-exclusion column for final purification.
Crystallization of NLR48
NLR48 was concentrated to 16 mg/ml and screened for crystallization by the sitting-drop vapor-diffusion method in 96-well plate format by mixing 300 nl of the protein with 300 nl of well solution using the Gryphon crystallization robot (ArtRobbins Instruments, Sunnyvale, CA). The duplicate plates were maintained at temperatures of 9 and 15 °C. The best crystals grew from solution containing 0.056 m sodium phosphate monobasic, 1.344 m potassium phosphate dibasic, pH 8.2. Crystals grown at 9 °C were somewhat bigger than crystals grown at 15 °C.
Expression, purification, and crystallization of selenomethionine-labeled NLR48
Expression plasmid carrying GST-NLR48 fusion construct was transformed into methionine auxotroph cells (B834(DE3), Novagen). Overnight inoculum grown in 50 ml of M9 medium supplemented with methionine was subcultured into 1 liter of M9 medium. For SeMet labeling, the cells were grown at 37 °C until A600 reached 1.0. The culture was spun gently at 3000 × g for 20 min, the cell pellet was resuspended in 1 liter of M9 medium, and the culture continued to grow at 37 °C for another 4 h to deplete the remaining methionine. Subsequently, 50 mg/liter SeMet was added to the cell culture 30 min prior to the addition of isopropyl β-d-1-thiogalactopyranoside. The cells were further grown at 18 °C. After ∼16 h of growth, the cells were collected by centrifugation, and SeMet-incorporated protein was purified following the same protocol as used for the native enzyme purification. SeMet-incorporated NLR48 was concentrated to 16 mg/ml for crystallization experiments using the hanging-drop vapor-diffusion method. The crystals grown from 0.49 m sodium phosphate monobasic monohydrate, 0.91 m potassium phosphate dibasic, pH 6.9, at 9 °C were cryoprotected with 30% ethylene glycol, and diffraction data were collected at the 08BM beamline at the Canadian Light Source. Data were processed with the XDS software package (30) to 1.9-Å resolution, and the data collection statistics are shown in Table 2. These crystals diffracted significantly better and showed better spot shape than the native crystals and were used for structure determination and refinement.
Table 2.
Data collection and refinement statistics
| Crystal form |
||
|---|---|---|
| NLR48(SeMet) | NLR48(K162M)–tetrasaccharide | |
| Data collection | ||
| Space group | P21 21 21 | P21 21 21 |
| a, b, c (Å) | 82.5, 102.1, 103.7 | 82.5, 102.7, 103.0 |
| Wavelength (Å) | 0.98013a | 0.97949 |
| Resolution (Å) | 50–1.9 | 50–2.2 |
| Observed hkl | 1,001,093 | 432,911 |
| No. of unique hkl | 131,854 | 44,398 |
| Completeness (%) | 99.8 (97.7)b | 99.8 (97) |
| Redundancy | 7.6 | 9.8 |
| Rmeas | 0.143 (1.25) | 0.100 (1.16) |
| I/(σI) | 10.01 (1.82) | 14.86 (2.0) |
| Refinement statistics | ||
| Rwork | 0.169 | 0.189 |
| Rfree | 0.201 | 0.220 |
| Wilson B (Å2) | 26.28 | 49.57 |
| Ramachandran plot (%) | ||
| Favored | 96.57 | 95.35 |
| Allowed | 2.67 | 3.88 |
| Outliers | 0.76 | 0.78 |
| r.m.s.d. | ||
| Bonds (Å) | 0.015 | 0.008 |
| Angles (°) | 1.214 | 1.091 |
| PDB code | 6D2C | 6D3U |
a The data set for structure determination was collected at the wavelength of 0.97864. The data set used for refinement was collected at the wavelength 0.98013 as shown in the table.
b The numbers in parentheses refer to the highest resolution shell.
Structure solution and refinement of WT NLR48
The structure of NLR48 was solved by the single anomalous dispersion method with the SeMet data set using the AutoSol procedure in Phenix software (31). There are two molecules in the asymmetric unit. All six expected selenium sites were identified. The initial model contained ∼90% of the expected residues. The remaining residues were built manually using Coot (32). Because the SeMet-containing crystal diffracted better than WT NLR48, this data set was used not only for solving the structure but also for refinement. The refinement was performed with Phenix software interspersed with manual rebuilding using Coot. The final Rwork was 0.169, and Rfree was 0.201. Several N-terminal residues are not visible in the electron density. The model contains residues 44–303 in both chains, two Ca2+ ions, four phosphates, and 710 solvent molecules. The validation by MolProbity (33) identified Arg282 and Val283 as Ramachandran outliers. However, inspection of electron density map confirms the correct modeling of these residues. Data collection and statistics are shown in Table 2. The coordinates and structure factors have been deposited to the Protein Data Bank with accession code 6D2C.
Site-directed mutagenesis
Selected residues were mutated to assess their role in catalysis. The mutants R117A, Q160A, K162M, R217N, and Y281F were made using the GST-NLR48 fusion construct as a template by following the QuikChange site-directed mutagenesis protocol. All the constructs were confirmed by sequencing. The mutants were expressed in BL21(DE3) cells and purified using the same protocol as for the WT protein.
Enzyme activity
The activity of NLR48 and its various mutants was measured by monitoring the absorption at 232 nm, indicative of the formation of an unsaturated double bond in the sugar on the nonreducing end of one of the two reaction products. The assay solution contained 0.5 mg/ml ulvan polysaccharide in 20 mm Hepes, pH 7.5, 150 mm NaCl. 1 μg of the enzyme was added directly to the assay solution with constant stirring, and the absorbance was measured as a function of time. The absorbance of the substrate alone was used as a control.
Expression, purification, and crystallization of NLR48 mutants
Mutants showing no activity, namely K162M, Y281F, and R117N, were expressed and purified. The GST fusion constructs of the mutants were expressed and purified using the same protocol as for WT NLR48. Of these, only K162M was expressed in high yield and was screened for crystallization at 17 mg/ml. Crystals were obtained in the same conditions as for WT NLR48 (0.49 m sodium phosphate monobasic monohydrate, 0.91 m potassium phosphate dibasic, pH 6.9, at 9 °C).
Structure determination of NLR48–tetrasaccharide complex
The K162M mutant crystals were used for soaking experiments with an ulvan tetrasaccharide, which contained a mixture of ΔUA-Rha3S-GlcA-Rha3Sα/β and ΔUA-Rha3S-IdoA-Rha3Sα/β. Briefly, a drop containing 3 μl of substrate solution (∼50 mm tetrasaccharide solution in water) mixed with 3 μl of crystallization well solution on a coverslip was equilibrated against 1 ml of well solution. The tetrasaccharide was prepared as described before (28). The crystals were soaked in the substrate solution for ∼2 h, cryoprotected in well solution supplemented with 30% ethylene glycol, and flash frozen in liquid N2. Diffraction data were collected at the 08ID beamline, Canadian Light Source. These crystals diffracted to 2.2-Å resolution and were isomorphous with the WT NLR48 crystals. One round of rigid-body refinement was done on the complex dataset with the Phenix refinement program. The tetrasaccharide was built into the positive difference electron density with Coot. Further refinement was done using Phenix software. The complex structure was evaluated for stereochemistry using MolProbity. The final model has 44–303 residues in each molecule, 212 water molecules, two calcium ions, two tetrasaccharides, and four phosphate ions. Data collection and statistics are shown in Table 2. The coordinates and structure factors have been deposited to the Protein Data Bank with accession code 6D3U.
Author contributions
T. U. and M. C. conceptualization; T. U. data curation; T. U. and M. C. validation; T. U. investigation; T. U., E. B., and W. H. methodology; T. U. writing-original draft; E. B. and W. H. resources; W. H. and M. C. writing-review and editing; M. C. supervision; M. C. funding acquisition.
Supplementary Material
Acknowledgments
We thank Kevin Voth for comments. We acknowledge the Protein Characterization and Crystallization Facility, College of Medicine, University of Saskatchewan for access to the crystallization robot. Research described in this paper was performed using beamline 08ID-1 at the Canadian Light Source synchrotron, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research.
This work was supported by Natural Science and Engineering Council Grant 155375-2012-RGPIN (to M. C.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
The atomic coordinates and structure factors (codes 6D2C and 6D3U) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- GlcA
- glucuronic acid
- IdoA
- iduronic acid
- PUL
- polysaccharide utilization loci
- PL
- polysaccharide lyase
- Rha3S
- 3-O-sulfated rhamnose
- MBP
- maltose-binding protein
- r.m.s.d.
- root-mean-square deviation
- ΔUA
- 4-deoxy-l-threo-hex-4-enopyranosiduronic acid
- PDB
- Protein Data Bank
- TEV
- tobacco etch virus.
References
- 1. Shalaby E. A. (2011) Algae as promising organisms for environment and health. Plant Signal. Behav. 6, 1338–1350 10.4161/psb.6.9.16779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sudha P. N., Aisverya S., Nithya R., and Vijayalakshmi K. (2014) Industrial applications of marine carbohydrates. Adv. Food Nutr. Res. 73, 145–181 10.1016/B978-0-12-800268-1.00008-1 [DOI] [PubMed] [Google Scholar]
- 3. Hehemann J.-H., Boraston A. B., and Czjzek M. (2014) A sweet new wave: structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr. Opin. Struct. Biol. 28, 77–86 10.1016/j.sbi.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 4. Michel G., and Czjzek M. (2013) Polysaccharide-degrading enzymes from marine bacteria, in Marine Enzymes for Biocatalysis (Trincone A., ed) pp. 429–464, Woodhead Publishing Ltd., Cambridge, UK [Google Scholar]
- 5. Cunha L., and Grenha A. (2016) Sulfated seaweed polysaccharides as multifunctional materials in drug delivery applications. Mar. Drugs 14, E42 10.3390/md14030042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Popa E. G., Reis R. L., and Gomes M. E. (2015) Seaweed polysaccharide-based hydrogels used for the regeneration of articular cartilage. Crit. Rev. Biotechnol. 35, 410–424 10.3109/07388551.2014.889079 [DOI] [PubMed] [Google Scholar]
- 7. Lahaye M., and Robic A. (2007) Structure and functional properties of ulvan, a polysaccharide from green seaweeds. Biomacromolecules 8, 1765–1774 10.1021/bm061185q [DOI] [PubMed] [Google Scholar]
- 8. Nyvall Collén P., Sassi J. F., Rogniaux H., Marfaing H., and Helbert W. (2011) Ulvan lyases isolated from the flavobacteria Persicivirga ulvanivorans are the first members of a new polysaccharide lyase family. J. Biol. Chem. 286, 42063–42071 10.1074/jbc.M111.271825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melcher R. L., Neumann M., Fuenzalida Werner J. P., Gröhn F., and Moerschbacher B. M. (2017) Revised domain structure of ulvan lyase and characterization of the first ulvan binding domain. Sci. Rep. 7, 44115 10.1038/srep44115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kopel M., Helbert W., Henrissat B., Doniger T., and Banin E. (2014) Draft genome sequence of Pseudoalteromonas sp. strain PLSV, an ulvan-degrading bacterium. Genome Announc. 2, e01257–14 10.1128/genomeA.01257-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopel M., Helbert W., Henrissat B., Doniger T., and Banin E. (2014) Draft genome sequences of two ulvan-degrading isolates, strains LTR and LOR, that belong to the Alteromonas genus. Genome Announc. 2, e01081–14 10.1128/genomeA.01081-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kopel M., Helbert W., Henrissat B., Doniger T., and Banin E. (2014) Draft genome sequence of Nonlabens ulvanivorans, an ulvan-degrading bacterium. Genome Announc. 2, e00793–14 10.1128/genomeA.00793-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foran E., Buravenkov V., Kopel M., Mizrahi N., Shoshani S., Helbert W., and Banin E. (2017) Functional characterization of a novel “ulvan utilization loci” found in Alteromonas sp. LOR genome. Algal Res. 25, 39–46 10.1016/j.algal.2017.04.036 [DOI] [Google Scholar]
- 14. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kopel M., Helbert W., Belnik Y., Buravenkov V., Herman A., and Banin E. (2016) New family of ulvan lyases identified in three isolates from the Alteromonadales order. J. Biol. Chem. 291, 5871–5878 10.1074/jbc.M115.673947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ulaganathan T., Boniecki M. T., Foran E., Buravenkov V., Mizrachi N., Banin E., Helbert W., and Cygler M. (2017) New ulvan-degrading polysaccharide lyase family: structure and catalytic mechanism suggests convergent evolution of active site architecture. ACS Chem. Biol. 12, 1269–1280 10.1021/acschembio.7b00126 [DOI] [PubMed] [Google Scholar]
- 17. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., et al. (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 10.1002/pro.3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garron M. L., and Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 10.1093/glycob/cwq122 [DOI] [PubMed] [Google Scholar]
- 19. Ogura K., Yamasaki M., Mikami B., Hashimoto W., and Murata K. (2008) Substrate recognition by family 7 alginate lyase from Sphingomonas sp. A1. J. Mol. Biol. 380, 373–385 10.1016/j.jmb.2008.05.008 [DOI] [PubMed] [Google Scholar]
- 20. Han Y. H., Garron M. L., Kim H. Y., Kim W. S., Zhang Z., Ryu K. S., Shaya D., Xiao Z., Cheong C., Kim Y. S., Linhardt R. J., Jeon Y. H., and Cygler M. (2009) Structural snapshots of heparin depolymerization by heparin lyase I. J. Biol. Chem. 284, 34019–34027 10.1074/jbc.M109.025338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong S., Wei T.-D., Chen X.-L., Li C.-Y., Wang P., Xie B.-B., Qin Q.-L., Zhang X.-Y., Pang X.-H., Zhou B.-C., and Zhang Y.-Z. (2014) Molecular insight into the role of the N-terminal extension in the maturation, substrate recognition, and catalysis of a bacterial alginate lyase from polysaccharide lyase family 18. J. Biol. Chem. 289, 29558–29569 10.1074/jbc.M114.584573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konno N., Ishida T., Igarashi K., Fushinobu S., Habu N., Samejima M., and Isogai A. (2009) Crystal structure of polysaccharide lyase family 20 endo-β-1,4-glucuronan lyase from the filamentous fungus Trichoderma reesei. FEBS Lett. 583, 1323–1326 10.1016/j.febslet.2009.03.034 [DOI] [PubMed] [Google Scholar]
- 23. Ogura K., Yamasaki M., Yamada T., Mikami B., Hashimoto W., and Murata K. (2009) Crystal structure of family 14 polysaccharide lyase with pH-dependent modes of action. J. Biol. Chem. 284, 35572–35579 10.1074/jbc.M109.068056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 10.1093/nar/gki370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 10.1042/bj3210557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gacesa P. (1987) Alginate-modifying enzymes. A proposed unified mechanism of action for the lyases and epimerases. FEBS Lett. 212, 199–202 10.1016/0014-5793(87)81344-3 [DOI] [Google Scholar]
- 27. Garron M. L., and Cygler M. (2014) Uronic polysaccharide degrading enzymes. Curr. Opin. Struct. Biol. 28, 87–95 10.1016/j.sbi.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 28. Ulaganathan T., Helbert W., Kopel M., Banin E., and Cygler M. (2018) Structure-function analyses of a PL24 family ulvan lyase reveal key features and suggest its catalytic mechanism. J. Biol. Chem. 293, 4026–4036 10.1074/jbc.RA117.001642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 10.1093/nar/18.20.6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.