

Abstract
A primary pathological defect in the heritable eye disorder Stargardt disease is excessive accumulation of cytotoxic lipofuscin bisretinoids in the retina. Age-dependent accumulation of lipofuscin in the retinal pigment epithelium (RPE) matches the age-dependent increase in the incidence of the atrophic (dry) form of age-related macular degeneration (AMD) and therefore may be one of several pathogenic factors contributing to AMD progression. Lipofuscin bisretinoid synthesis in the retina depends on the influx of serum retinol from the circulation into the RPE. Formation of the tertiary retinol-binding protein 4 (RBP4)–transthyretin–retinol complex in the serum is required for this influx. Herein, we report the pharmacological effects of the non-retinoid RBP4 antagonist, BPN-14136. BPN-14136 dosing in the Abca4−/− mouse model of increased lipofuscinogenesis significantly reduced serum RBP4 levels and inhibited bisretinoid synthesis, and this inhibition correlated with a partial reduction in visual cycle retinoids such as retinaldehydes serving as bisretinoid precursors. BPN-14136 administration at doses inducing maximal serum RBP4 reduction did not produce changes in the rate of the visual cycle, consistent with minimal changes in dark adaptation. Abca4−/− mice exhibited dysregulation of the complement system in the retina, and BPN-14136 administration normalized the retinal levels of proinflammatory complement cascade components such as complement factors D and H, C-reactive protein, and C3. We conclude that BPN-14136 has several beneficial characteristics, combining inhibition of bisretinoid synthesis and reduction in retinaldehydes with normalization of the retinal complement system. BPN-14136, or a similar compound, may be a promising drug candidate to manage Stargardt disease and dry AMD.
Keywords: drug design, drug development, drug discovery, pharmacokinetics, pharmacology, molecular pharmacology, retinoid-binding protein, retinal degeneration, retinal metabolism, retina, age-related macular degeneration, lipofuscin, RBP4, Stargardt disease, visual cycle
Introduction
Stargardt disease (STGD1;2 MIM 248200) is characterized by the abnormal accumulation of lipofuscin in the retina that triggers degeneration of macular rods and cones (1–4). STGD1 is the most common form of inherited juvenile–onset macular dystrophy with an estimated prevalence of 1:10,000 (5–7) and a carrier frequency for mutant ABCA4 alleles reaching as much as 5% in the general population (8, 9). The primary biochemical defect in STGD1 patients is excessive formation of cytotoxic lipofuscin bisretinoids in the retinal pigment epithelium (1, 2) due to recessive mutations in the ABCA4 gene. The atrophic (dry) form of age-related macular degeneration (AMD) represents a slowly progressing maculopathy that is associated with abnormalities in the RPE (10, 11). Dry AMD is a multifactorial and multigenic disorder with several different pathways contributing to its pathogenesis. Age-dependent accumulation of cytotoxic lipofuscin in the RPE matches the age-dependent increase in the prevalence of dry AMD and thus is frequently cited as one of the potential pathogenic factors contributing to the disease progression (3, 12–17). An emerging complementary view on the role of lipofuscin bisretinoids in dry AMD stresses the causative role of retinal aldehyde toxicity in the disease pathology over the contribution of bisretinoids (18–20). A more established hypothesis of dry AMD etiology and pathogenesis stipulates that dysregulation of the complement system in the retina seems to underlie the most important aspects of the disease (21–24). There are no Food and Drug Administration–approved treatments for Stargardt disease and dry AMD. Developing a drug therapy for these forms of macular degeneration addresses a highly significant unmet medical need in ophthalmology. In addition to inhibiting the formation of lipofuscin bisretinoids, an optimal pharmacological therapy for macular degeneration would normalize complement system dysregulation in the retina and ameliorate symptoms of retinaldehyde toxicity prolonging RPE and photoreceptor survival in a patient's retina. Assuming that accumulation of cytotoxic lipofuscin bisretinoids contributes to the disease pathology, it was hypothesized that pharmacological inhibition of bisretinoid formation by small molecule drugs may provide a means by which to delay or suppress degenerative processes in Stargardt disease and AMD (25–29).
Uptake of serum retinol (vitamin A, Fig. 1) from circulation to the RPE fuels the visual retinoid cycle reactions leading to retinaldehyde and bisretinoid synthesis (10). The primary and specific carrier of retinol in the serum is retinol-binding protein 4 (RBP4), which is essential for the transport of retinol from the liver to extrahepatic tissues. In the serum, the RBP4–retinol holoprotein is present as a tertiary complex with transthyretin (TTR), which increases the molecular weight of the retinol-delivery vehicle, thus protecting the RBP4–retinol complex from rapid glomerular filtration and catabolism in the kidney. Retinol binding to RBP4 is required for the formation of the RBP4–TTR complex; apo-RBP4 (devoid of retinol) has reduced affinity for TTR. The synthetic retinoid drug fenretinide (Fig. 1) displaces all-trans-retinol from RBP4 by competing for retinol-binding sites on RBP4 (30), disrupting the retinol-dependent RBP4–TTR interaction (30, 31). Fenretinide administration inhibited the accumulation of lipofuscin bisretinoids in the RPE of Abca4−/− mice (26). Fenretinide was evaluated in clinical trials for late-stage atrophic AMD (32), where the post hoc analysis revealed a positive correlation between the reduction of serum RBP4 below 1 μm and inhibition of the atrophic lesion growth rate (32). However, only 51% of patients from the highest dose group, 300 mg/day, achieved this level of serum RBP4 reduction, although there was no correlation between the rates of atrophic growth and serum RBP4 levels at RBP4 concentrations higher than 1 μm (32). Because of the insufficient RBP4-lowering efficacy, only a limited number of patients achieved the desired level of RBP4 reduction, which prevented the atrophy growth rate difference between the 300 mg/day and placebo groups to reach statistical significance (32). Theoretically, adequate RBP4 lowering could be achieved by increasing the fenretinide dose. However, similarly to other retinoid drugs, fenretinide is non-specific and toxic at elevated doses. Independent of its action as an antagonist of retinol binding to RBP4, fenretinide induces apoptosis in the RPE (33), promotes hemangiosarcoma in mice, and is teratogenic (34, 35). Teratogenicity makes fenretinide's use problematic in STGD1 patients of childbearing age. Thus, identification of new classes of non-retinoid RBP4 antagonists with adequate potency and acceptable safety profile is of significant importance.
Figure 1.
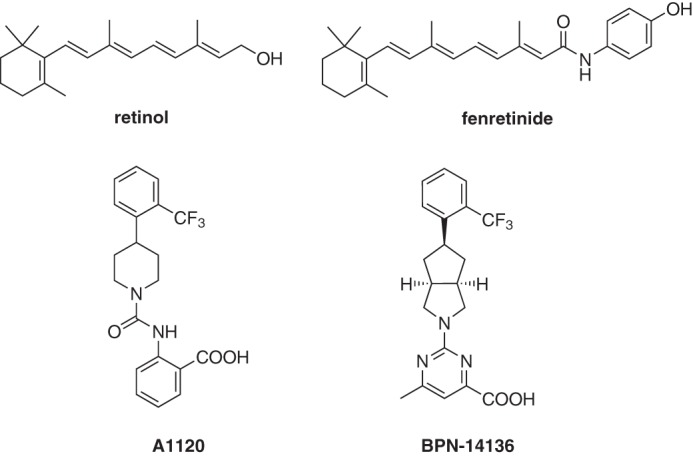
Chemical structure of RBP4 ligands retinol, fenretinide, A1120, and BPN-14136.
We recently reported the discovery and characterization of several classes of non-retinoid RBP4 antagonists disrupting retinol-induced RBP4–TTR interaction (36–38). One of the identified analogs, BPN-14136 (Fig. 1), has excellent drug-like characteristics and demonstrates exceptional in vitro efficacy and selectivity (37). Here, we describe the in vivo efficacy of BPN-14136 in inducing partial reduction of serum RBP4 and visual cycle retinoids such as retinaldehydes, robust inhibition of bisretinoid synthesis, and normalization of complement system dysregulation. Notably, we report that positive attributes of BPN-14136 are not associated with inhibition of the visual cycle or significant suppression of the visual function in dark-adapted eyes, which is consistent with the favorable ocular safety profile of compounds from this pharmacological class.
Results
Compound identification
In our recent reports, we described medicinal chemistry efforts conducted in pursuit of designing novel non-retinoid RBP4 antagonists (36, 37). The starting point for ligand-based rational drug design and optimization was A1120 (Fig. 1), which was previously developed by Amgen for the potential treatment of diabetes (39). Our efforts led to the discovery of the novel RBP4 antagonist BPN-14136, which contains a pyrimidine-4-carboxylic acid appended to a bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo core (Fig. 1) (37). BPN-14136 exhibited excellent in vitro RBP4–binding potency as well as the robust in vitro ability to antagonize retinol-dependent RBP4 interaction with TTR (37). In light of the exceptional in vitro potency, good selectivity, and optimal drug-like characteristics, the compound was selected for further in vivo evaluation.
Pharmacokinetics (PK) and pharmacodynamics (PD) of BPN-14136 in mice
Given that mouse genetic models of enhanced retinal lipofuscinogenesis are widely used for characterization of compounds capable of inhibiting bisretinoid synthesis, it was important to determine mouse pharmacokinetic parameters for BPN-14136 to ensure that adequate compound exposure can be achieved in this animal species. Single-dose PK studies conducted at 2 mg/kg intravenous and 5 mg/kg oral doses showed very low plasma clearance (39.9 ml/h/kg), which is optimal for the compound engaging its target in the systemic blood circulation. BPN-14136 was very well-absorbed resulting in oral bioavailability of about 100%, and it was slowly eliminated from plasma after oral administration with an observed Cmax of 11,027 ng/ml (28.2 μm) and corresponding Tmax at 2.0 h. The greater than 100% oral bioavailability may be related to the low plasma clearance of BPN-14136. The high exposure observed for BPN-14136 in mice (AUCINF of 129,400 ng/ml·h after the oral dose) fully justifies the use of mouse models for characterization of in vivo efficacy of this compound. The mouse PK properties of BPN-14136 are summarized in Table 1. The overall favorable PK profile of BPN-14136 defined in this mouse study (good oral bioavailability, low clearance and high exposure) is similar to the one that we previously reported for this compound in rats (37).
Table 1.
In vivo pharmacokinetics data for BPN-14136 following intravenous (i.v.) and oral (p.o.) administration in mice
Dosing cohorts consisted of three groups of drug-naive adult male CD-1 mice. Mean BPN-14136 concentrations per time point were used to calculate the composite PK parameters by non-compartmental analysis using the WinNonlin program.
| Route | Dose | Tmaxa | Cmaxb | T½ c | Vssd | Cle | AUClastf | AUCINFg | %Fh |
|---|---|---|---|---|---|---|---|---|---|
| mg/kg | h | ng/ml | h | ml/kg | ml/h/kg | h·ng/ml | h·ng/ml | ||
| i.v. | 2 | 0.083i | 10,930 | 5.6 | 233 | 39.9 | 49,960 | 50,140 | NAj |
| p.o. | 5 | 2 | 11,027 | 5.0 | NAj | NAj | 129,300 | 129,400 | 103.2 |
a Time of maximum observed concentration of BPN-14136 in plasma.
b Peak plasma concentration of BPN-14136 is shown.
c Apparent half-life of the terminal phase of elimination of compound from plasma is shown.
d Volume of distribution is at steady state.
e Cl is total body clearance.
f Area under the plasma concentration versus time curve from 0 to the last time point that compound was quantifiable in plasma is shown.
g Area under the plasma concentration versus time curve from 0 to infinity is shown.
h Bioavailability is F = (AUCINFpo × doseiv) ÷ (AUCINFiv × dosepo).
i First plasma collection time is shown.
j NA means not applicable.
To confirm the in vivo target engagement in mice, we studied the effect of a single dose of BPN-14136 on the level of plasma RBP4. Aliquots of plasma samples collected during the PK experiments were used to analyze plasma RBP4 concentrations. After a single 5 mg/kg oral dose of BPN-14136, a maximum 90% decrease in plasma RBP4 was observed (Fig. 2A), whereas a 75% plasma RBP4 reduction was achieved following intravenous dosing of 2 mg/kg (Fig. 2B). The dynamics of in vivo RBP4 lowering after oral and intravenous dosing of BPN-14136 showed a correlation between the presence of the compound in circulation (Fig. 2, C and D) and a reduction in serum RBP4 (Fig. 2, A and B). The long exposure and slow clearance of BPN-14136 achieved after a single oral dose (Table 1) correlated very well with the magnitude of the RBP4 reduction (90% reduction at the 8-h time point) and the duration of the RBP4 lowering effect (65% reduction at the 24-h time point). These data provide evidence for a good PK–PD relationship between compound exposure and biological response.
Figure 2.

Pharmacodynamic and pharmacokinetic properties of BPN-14136 in mice. Plasma RBP4 levels in CD-1 mice following a single 5 mg/kg oral (A) and a single 2 mg/kg intravenous (B) administration of BPN-14136 are shown. Plasma compound levels following administration of a single oral 5 mg/kg dose (C) and a 2 mg/kg intravenous dose (D) of BPN-14136 are shown. Data are represented as the mean ± S.D. Three mice per each time point of blood collection were used in the study.
Effects of BPN-14136 on visual cycle retinoids and regeneration rate of visual chromophore
Visual cycle retinoids such as all-trans- and 11-cis-retinaldehyde serve as precursors in the biosynthesis of cytotoxic bisretinoids (40, 41). To prove that BPN-14136–evoked reduction in serum RBP4 can induce the desired partial reduction in levels of visual cycle retinoids, we examined retinoid content in the eyecup extracts of 12-week-old light- or dark-adapted BALB/cJ mice that were orally administered BPN-14136 for 2 weeks at a dose that induced maximal serum RBP4 reduction. The 2-week BPN-14136 treatment induced the desired partial visual cycle retinoid reduction in dark-adapted mice, as well as in light-adapted animals (Table 2). It is noteworthy that despite the maximally possible 90% serum RBP4 reduction induced by BPN-14136, only a partial 40–50% decrease in visual cycle retinoids (retinyl ester, 11-cis-retinal, and all-trans-retinal) was induced by the compound treatment. The concentration of the visual chromophore, 11-cis-retinal, was reduced by 40% in dark-adapted mouse eyes (Table 2). To determine whether BPN-14136 treatment affected the rate of chromophore regeneration (which reflects the rate of the visual cycle), we measured 11-cis-retinal regeneration following the exposure of dark-adapted animals to photobleaching. Dark-adapted mice were subjected to 1000 lx light for 5 min, which bleached ∼90% of rhodopsin. We conducted visual cycle retinoid measurements immediately following bleaching (0 min) as well as at 10, 20, 30, and 40-min time points after returning animals to the dark. Bleaching of dark-adapted mice induced the expected decrease in 11-cis-retinal and a concomitant increase in all-trans-retinal (Table 2). The amount of 11-cis-retinal increased during recovery in the dark environment, and the rate of 11-cis-retinal regeneration was not different in BPN-14136–treated animals compared with vehicle-treated controls (Fig. 3). These data indicate that BPN-14136 treatment does not reduce the rate of the visual cycle while partially reducing the retinoid load in the retina.
Table 2.
Effect of BPN-14136 treatment on concentration of visual cycle retinoids in mouse eyes
Recovered visual cycle retinoids were quantified after their extraction from dark-adapted (DA) and light-adapted (LA) mouse eyes as well as from the eyes collected immediately after exposure to photobleaching (PB) as described under “Experimental procedures.” The number of animals per treatment group is shown in parentheses. The effect of treatment within each group was analyzed using t tests. Statistical significance was determined using the Holm-Sidak method, with α = 0.05. Each retinoid group for a given light condition was analyzed individually, without assuming a consistent S.D. Data are represented as mean ± S.D. ns, p > 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. RAL is retinaldehyde.
| Treatment group | 11-cis-RAL | All-trans-RAL | All-trans-retinyl palmitate |
|---|---|---|---|
| pmol/eye | pmol/eye | pmol/eye | |
| DA | |||
| BPN-14136 (n = 6) | 188.2 ± 27.6**** | 14.5 ± 3.3** | 46.6 ± 7.1ns |
| Control (n = 6) | 315.5 ± 18.8 | 21.1 ± 3.4 | 59.3 ± 13.5 |
| LA | |||
| BPN-14136 (n = 6) | 30.8 ± 7.5*** | 51.9 ± 7.0** | 56.6 ± 18.0** |
| Control (n = 6) | 120.4 ± 43.9 | 90.6 ± 28.8 | 96.2 ± 17.6 |
| PB | |||
| BPN-14136 (n = 6) | 16.9 ± 2.6** | 111.0 ± 7.7**** | 58.7 ± 5.3**** |
| Control (n = 6) | 27.1 ± 5.8 | 182.7 ± 17.5 | 112.4 ± 16.4 |
Figure 3.
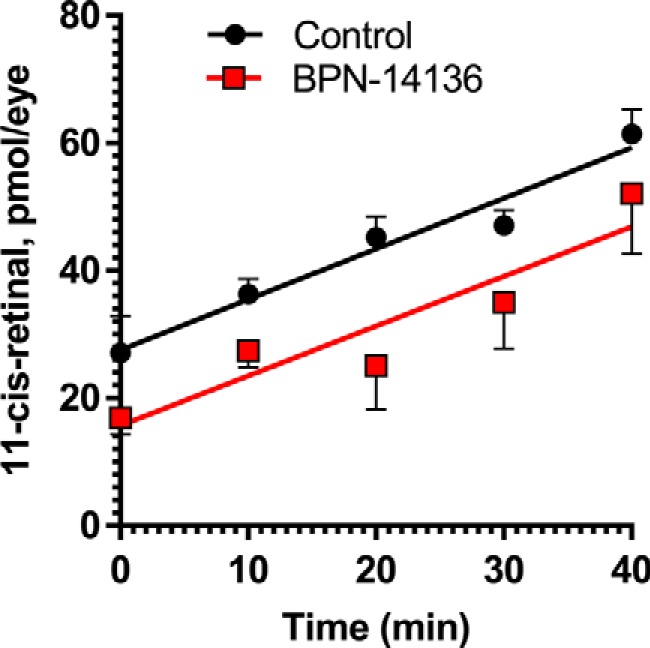
BPN-14136 treatment does not change kinetics of chromophore regeneration in mice. Concentration of 11-cis-retinal was measured in eyes of BALB/cJ mice treated with BPN-14136 at the 20 mg/kg dose for 2 weeks as well as in control eyes from vehicle-treated animals. Chromophore concentration was assessed immediately after photobleaching (0 min) as well as at different time points after the bleach. Whereas the post-bleach level of 11-cis-retinal at 0 min was lower in BPN-14136–treated animals, no difference was observed between the two mouse groups at a rate of 11-cis-retinal regeneration. Slopes were plotted and analyzed using linear regression. The difference between the slopes is not significant (p = 0.9477). Data points are represented as the mean ± S.D. In each treatment group, six mouse eyes per time point were analyzed in the study.
BPN-14136 treatment does not cause delayed dark adaptation in mice
Delayed dark adaptation, which represents a period of reduced light sensitivity following exposure to bright light, can be assessed by measuring recovery of the ERG a- and b-wave amplitude following exposure to the bleaching light (26, 42, 43). Kinetics of the ERG amplitude recovery after bleaching physiologically reflects the rate of the visual retinoid cycle (42). To determine whether BPN-14136 influences the rate of ERG a- and b-wave recovery, BALB/cJ mice were orally administered BPN-14136 for 2 weeks at the dose that induced a maximal serum RBP4 reduction. After the overnight dark adaptation, mice were subjected to photobleaching (1000 lx for 5 min to bleach ∼90% of visual pigment). ERG a- and b-wave amplitudes were recorded using dim light flashes delivered immediately before and after photobleaching, as well as at regular intervals after returning animals to the dark (Fig. 4). There was no difference in pre-bleach ERG a- and b-wave amplitudes in treated and control animals, and we found no significant difference in rates of b-wave recovery in BPN-14136- and vehicle-treated mice in a 12 time-point study (Fig. 4). The lack of delay in return of rod photoresponse after photobleaching in compound-treated animals supports the conclusion that BPN-14136 treatment did not induce physiological inhibition of the visual cycle.
Figure 4.
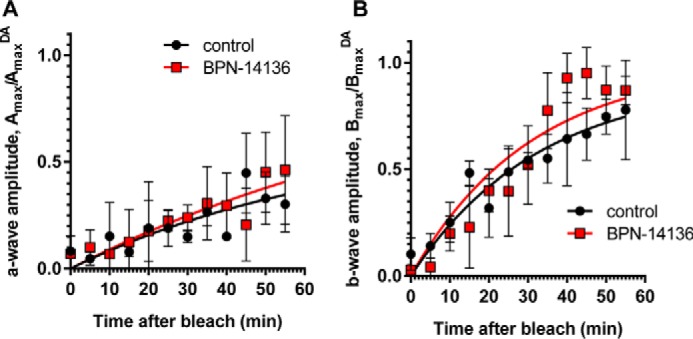
Effect of BPN-14136 administration on kinetics of rod ERG a- and b-wave amplitude recovery after photobleaching. ERG a- and b-wave amplitude measurements (A and B, respectively) were conducted at indicated time points after a photobleach (1000 lux, 5 min). Analysis was performed in BALB/cJ mice following 2 weeks of BPN-14136 administration at the oral dose that induced a maximal 90% serum RBP4 reduction. Six mouse eyes from the control and compound-treated study groups were analyzed. For each time point, Amax and Bmax values were normalized to the corresponding AmaxDA and BmaxDA (dark-adapted) values. Results are averaged from six eyes in each group. Error bars show S.D. A two-way repeated measures ANOVA did not reveal statistically significant differences overall (F(1, 4) = 0.60, p = 0.48) or at any time point (statistical significance was determined using the Holm-Sidak method, α = 0.05. Time course curves were fitted using one-phase exponential association.
Electroretinography and rhodopsin measurements
Significantly reduced concentration or activity of rhodopsin in the retina may lead to decreased rod sensitivity that manifests in humans as nyctalopia (poor or no vision in low-light conditions) (44). Rhodopsin, which mediates low-illumination vision, is a complex of opsin protein and 11-cis-retinaldehyde. Given that 2-week BPN-14136 treatment induced a 40% reduction in dark-adapted levels of 11-cis-retinaldehyde (Table 2), we assessed the effect of compound administration on dark-adapted ERG parameters. After 2 weeks of BPN-14136 administration at a dose that induces 90% serum RBP4 reduction, the ERG waveforms remained normal, and the ERG intensity-response curves overlapped for both vehicle- and BPN-14136-treated BALB/cJ mice (Fig. 5). As ERG gave no physiological evidence of impaired rod function in dark-adapted eyes, we studied the effect of 11-cis-retinaldehyde reduction on rhodopsin levels. Rhodopsin concentration was spectrophotometrically measured in dark-adapted retina excised from BALB/cJ mice following the 2-week oral dosing of BPN-14136. As shown in Fig. 6, we detected a 25% reduction in rhodopsin concentration in a dark-adapted retina in response to BPN-14136 treatment. Interestingly, this 25% rhodopsin reduction did not manifest itself in detectable functional changes as evidenced by normal ERG in dark-adapted compound–treated animals.
Figure 5.

Analysis of visual function by ERG in BPN-14136–treated and control BALB/cJ mice. Three serial single-flash ERG responses of increasing intensity were obtained for BPN-14136–treated and control mice under dark-adapted (scotopic, A–C) and light-adapted conditions (photopic, D–F). Intensity-response is plotted as a function of a- and b-wave amplitude versus varying light intensities under dark-adapted (B and C) and light-adapted (E and F) conditions. Mice were orally administered BPN-14136 for 2 weeks at a dose that induced 90% serum RBP4 reduction. Six mouse eyes from the control and BPN-14136–treated study groups were analyzed. A t test did not reveal a statistically significant difference between the treated and untreated groups at any intensity (statistical significance was determined using the Holm-Sidak method, α = 0.05. Each light intensity was analyzed individually.).
Figure 6.
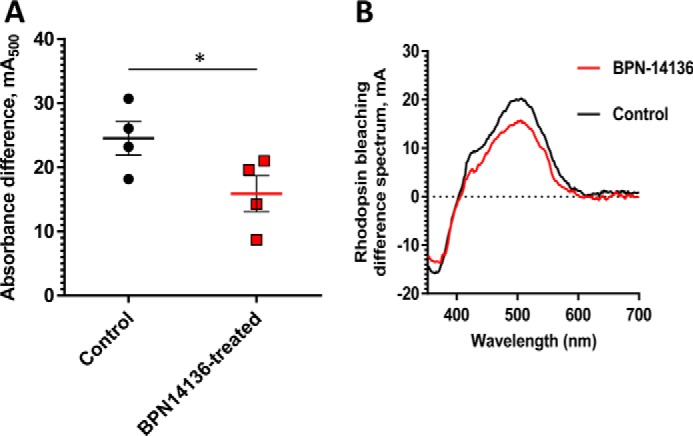
Comparison of rhodopsin levels in BPN-14136–treated and control BALB/cJ mice. The amount of rhodopsin was measured spectrophotometrically in retinal extracts from BPN-14136–treated and control mice (A). Treated mice (n = 4) were orally administered BPN-14136 at 20 mg/kg for 2 weeks. The amount of pigment is expressed as absorbance difference before and after light irradiation at the wavelength of maximum absorption of rhodopsin (λmax = 500 nm). Each data point represents the average of three measurements from one retinal extract prepared from two BPN-14136-treated or control animal retinas. An unpaired t test on the ungrouped data revealed a statistically significant 25% decrease in rhodopsin concentration in treated animals compared with the control group (p < 0.05). B, representative rhodopsin bleaching difference spectra for control and BPN-14136–treated mice. The representative bleaching difference spectra are taken from a single retinal extract prepared from two mouse eyes of control and compound-treated animals.
BPN-14136 inhibits bisretinoid synthesis in Abca4−/− mice
To determine the effect of BPN-14136 on bisretinoid levels in the eye, we administered the compound at a daily dose of 20 mg/kg to Abca4−/− mice for a period of 12 weeks. Blood samples were collected from treatment groups at baseline and weeks 2, 6, and 12 to measure the level of serum RBP4. Chronic oral administration of BPN-14136 induced a 90% decrease in serum RBP4 levels at all time points compared with the baseline levels (Fig. 7A). Levels of lipofuscin fluorophores (A2E) in mouse eyecup extracts were chromatographically determined at the end of the 12-week treatment. The levels of bisretinoid accumulation were significantly higher in the vehicle-treated Abca4−/− mice than in WT controls (Fig. 7B). Administration of BPN-14136 reduced the formation of A2E in BPN-14136–treated Abca4−/− mice compared with vehicle-treated Abca4−/− animals by ∼50%. To confirm the data obtained from the biochemical analysis of lipofuscin fluorophores, we conducted quantification of lipofuscin autofluorescence in mouse retinal cryosections (Fig. 8). We captured and analyzed autofluorescence images from mouse retinal sections prepared from eyes of WT untreated mice as well as from vehicle- and BPN-14136–treated Abca4−/− mice. The images were captured using the excitation wavelength of 488 nm and emission wavelengths of 500–600 nm. The analysis showed significant fluorophore accumulation in the RPE of the vehicle-treated Abca4−/− mice compared with the WT controls (Fig. 8). BPN-14136 administration induced an ∼75% reduction in lipofuscin fluorophores in compound-treated Abca4−/− mice compared with vehicle-treated Abca4−/− animals (Fig. 8G). These data are consistent with the biochemical analysis of A2E (Fig. 7) corroborating the profound inhibitory effect of BPN-14136 on the in vivo accumulation of toxic lipofuscin bisretinoids in the animal model of enhanced lipofuscinogenesis. We did not note any obvious signs of systemic compound toxicity, such as weight loss or reduction in food consumption during the 12-week-long chronic BPN-14136 dosing (Fig. S1). Analysis of the retina structure by light microscopy revealed no changes in retinal morphology or thickness of the outer nuclear layer in relation to the 12-week-long compound treatment (Fig. S2), which is consistent with the lack of obvious retinotoxicity for BPN-14136.
Figure 7.
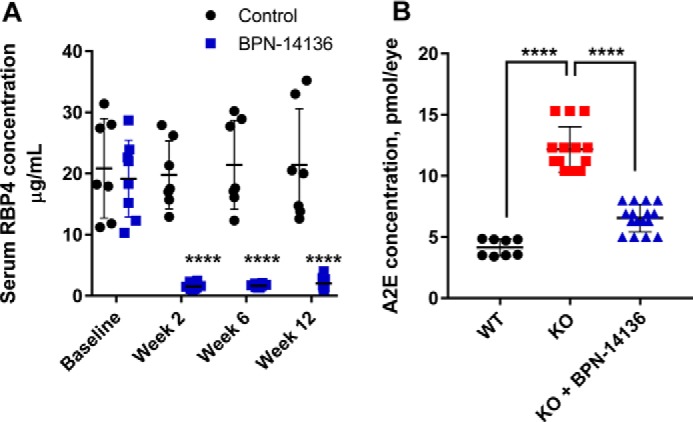
Effect of long-term oral BPN-14136 administration in Abca4−/− mice. A, serum RBP4 levels were measured in vehicle-treated Abca4−/− mice (black circles; n = 7) and BPN-14136–treated Abca4−/− mice (blue squares, n = 8) at the indicated time points. BPN-14136 formulated in a chow was dosed at 20 mg/kg. Compared with day 0, statistically significant 90% RBP4 reduction at weeks 2, 6, and 12 was seen in the BPN-14136 treatment group (two-way ANOVA with Sidak post hoc test, p < 0.0001). Error bars show S.D. Each data point on the graph represents a serum RBP4 concentration from an individual animal. B, effects of BPN-14136 treatment on the level of the lipofuscin fluorophore A2E in eyes of the Abca4−/− mice. Bisretinoids were extracted from the eyecups of vehicle-treated Abca4+/+ WT mice (black circles, 8 eyes), vehicle-treated Abca4−/− knockout mice (red squares, 14 eyes), and BPN-14136–treated Abca4−/− mice (blue triangles, 16 eyes) after 12 weeks of dosing and analyzed by HPLC. A significant 50% reduction in A2E concentration was detected in BPN-14136–treated Abca4−/− mice compared with vehicle-treated knockout controls (one-way ANOVA with Holm-Sidak post hoc test, p < 0.0001 (****)).
Figure 8.

Lipofuscin autofluorescence in mouse retinal sections. A–C, autofluorescence images from mouse retinal sections prepared from eyes of WT untreated mice (A), vehicle-treated Abca4−/− mice (B), and BPN-14136–treated Abca4−/− mice (C). BPN-14136 formulated into a chow was dosed at 20 mg/kg for 12 weeks. The images were captured with confocal microscope under the ×40 oil objective using the excitation wavelength of 488 nm and emission wavelengths of 500–600 nm. D–F, merged images indicating the localization of peak lipofuscin autofluorescence (green) within retinal layers imaged in a differential interference contrast mode. Retinal sections are from WT untreated mice (D), vehicle-treated Abca4−/− mice (E), and BPN-14136–treated Abca4−/− mice (F). Scale bar, 25 μm. G, quantitative analysis of lipofuscin autofluorescence in mouse retinal sections. Three animals per treatment group were used in the study. For each eye, an average of 21 image fields (range: 9–41) from identical area of the retina was used for autofluorescence measurements. Each data point on the graph represents an average of autofluorescence intensities in the RPE layer for images collected from a single animal. Statistical significance was determined using one-way ANOVA with Holm-Sidak post-hoc test, p < 0.0001 (****).
Normalization of complement system dysregulation
Previous studies showed that bisretinoids can induce activation of the complement cascade in RPE cells (45–49). Bisretinoid levels are significantly increased in the retina of Abca4−/− mice where this increase induces secondary complement system dysregulation and inflammation (46). We studied the effect of a 12-week BPN-14136 dosing on complement cascade activation in retinas of Abca4−/− mice. Complement Factor D (CFD) is involved in activation of the alternative complement pathway, whereas complement component C3 plays a central role in activation of all three complement activation pathways. By quantitative immunoblotting, CFD and C3 were significantly increased in Abca4−/− mice compared with WT mice (Fig. 9, A and G). In addition, specific antibodies detected significantly higher CFD and C3 levels in the RPE and inner layers of the retina of Abca4−/− mice compared with normal controls (Fig. 9, B, C, H, and I). BPN-14136 treatment normalized up-regulation of CFD and C3 in the retina as evidenced by the decrease in specific band intensities on immunoblots (Fig. 9, A and G) and by reduced immunofluorescence on retinal sections (Fig. 9, B, C, H, and I). In addition to CFD and C3, complement factor H (CFH), which acts as a down-regulator of the alternative pathway activation, was significantly dysregulated in retinas of Abca4−/− mice. CFH expression was decreased in Abca4−/− mice versus WT mice, whereas BPN-14136 treatment partially restored CFH to normal levels (Fig. 9, D–F). C-reactive protein is an acute-phase reactant and acts as a non-specific serum biomarker for chronic inflammation. Elevated C-reactive protein level is associated with cardiovascular disease, type 2 diabetes, and AMD. We studied the level of C-reactive protein in the eyes of Abca4−/− and WT mice by quantitative immunoblotting and immunocytochemistry. Both analyses showed that the C-reactive protein levels are increased in Abca4−/− mice compared with WT controls and that this up-regulation was partially abolished by BPN-14136 treatment (Fig. 9, J–L).
Figure 9.

Normalization of complement system dysregulation in Abca4−/− mice by BPN-14136 treatment. A, D, G, and J, Western blots and histograms showing expression levels of CFD (A), CFH (D), C3 (G), and C-reactive protein (J) in neurosensory retinas of WT animals (WT) and untreated Abca4−/− mice (KO) compared with BPN-14136–treated knockout animals (KO + BPN-14136). Each treatment group included six animals. The representative blots of at least three independent experiments are presented. The histograms represent pixel volumes of CFD, C3, CFH, and C-reactive protein bands. The band densities were normalized to actin. The vertical axes represent the ratio of pixel volume means ± S.D. of the scanned bands on the immunoblots in arbitrary units. Statistical significance was determined by one-way ANOVA with Holm-Sidak post hoc test; *, p ≤0.05; **, p ≤0.01; ***, p ≤0.001; ****, p ≤0.0001. Representative whole-retina sections (B, E, H, and K) and confocal images of RPE cryosections (C, F, I, and L) from Abca4+/+ WT mice, Abca4−/− knockout mice, and BPN-14136–treated Abca4−/− mice labeled for CFD (B and C), CFH (E and F), C3 (H and I), and C-reactive protein (K and L) are shown. Specific immunofluorescence is in red, and nuclear staining performed using DAPI is in blue. All immunofluorescence images are representative of those collected from three to six individual eyes from WT mice, untreated Abca4−/− mice, and BPN-14136–treated KO mice. Scale bar, 25 μm.
Discussion
Accumulation of lipofuscin bisretinoids in the retina is associated with several retinal degenerative diseases, including dry AMD and Stargardt disease. Excessive production of lipofuscin bisretinoids seems to be the major biochemical trigger of monogenic Stargardt disease caused by recessive mutations in the ABCA4 gene. In contrast, dry AMD has multifactorial etiology with several different pathways contributing to the pathophysiology of the disease. Much of the genetic risk for AMD is associated with DNA polymorphisms in complement cascade genes, and dysregulation of the complement system in the retina is thought to underlie critical aspects of dry AMD (21–24). One other important characteristic of dry AMD is age-dependent accumulation of retinal lipofuscin (3, 12–17) that matches the age-dependent increase in dry AMD prevalence. Pathogenicity of lipofuscin bisretinoids in dry AMD may relate to their well-documented direct cytotoxicity (50–54), as well as to their ability to induce activation of the complement cascade in RPE cells (45–49). Direct retinaldehyde toxicity may also play a role in AMD pathogenesis (18–20). Given that dry AMD is caused by multiple contributing factors, it seems reasonable to suggest that an optimal drug therapy for this condition should combine several positive attributes such as normalization of the complement system dysregulation in the retina, inhibition of bisretinoid synthesis, and attenuation of retinaldehyde toxicity. Here, we describe characterization of the advanced RBP4 antagonist, BPN-14136, as a potential therapy for Stargardt disease and dry AMD. As part of the Blueprint Neurotherapeutics (BPN) Network Project (National Institutes of Health), we used A1120 (Fig. 1) as a starting point in pursuit of designing new classes of non-retinoid RBP4 antagonists (36, 37). BPN-14136 (Fig. 1) was identified as a novel RBP4 antagonist (37) that demonstrated robust in vitro potency, excellent selectivity, and highly favorable other drug-like characteristics (37).
Before advancing BPN-14136 to in vivo characterization in mouse models, we conducted a mouse PK study to ensure adequate compound exposure in this species after oral dosing. One other goal was to demonstrate in vivo target engagement and to establish proof of in vivo activity in mice by assessing the dynamics of plasma RBP4 levels in response to BPN-14136 administration. The mouse PK study confirmed that BPN-14136 is very well-absorbed resulting in high oral bioavailability with slow elimination. Very good PK–PD correlation between the reduction in plasma RBP4 and increase in compound concentration was observed.
Visual cycle retinoids such as all-trans- and 11-cis-retinaldehyde serve as precursors in the biosynthesis of cytotoxic bisretinoids (40, 41). To confirm the mechanistic link between serum RBP4 reduction and inhibition of bisretinoid formation, we studied the effect of BPN-14136 on visual cycle retinoids. BPN-14136 administration elicited the desired 40–50% partial reduction in total concentration of visual cycle retinoids in dark-adapted mice, as well as in light-adapted animals (Table 2). The concentration of retinaldehydes was reduced by 40% in dark-adapted eyes and by 60% in light-adapted mouse eyes. This appreciable retinaldehyde reduction is consistent with the ability of RBP4 antagonists to attenuate aldehyde toxicity in the retina. It is important to stress that despite the maximally possible 90% pharmacological inactivation of the RBP4–TTR delivery route by BPN-14136, only a partial 30–60% decrease in visual cycle retinoids was induced by the compound treatment. This may indicate that the residual RBP4 activity is sufficient for maintaining retinol delivery required to partially sustain visual retinoid synthesis in the retina. Alternatively, this may point to the capacity of non-RBP4 routes of retinoid supply to the retina to provide sufficient amounts of retinoids to maintain proper visual cycle function. There are several alternative RBP4-independent pathways for retinoid delivery to the target organs, including the retina (55–60). Following a consumption of the vitamin A-containing meal, dietary retinoids are packaged in chylomicrons that are delivered predominantly to the liver. However, 25–33% of postprandial retinoid-laden chylomicrons are taken by extrahepatic vitamin A-dependent tissues, such as the retina (55). In addition to chylomicron delivery, retinoic acid can be delivered to the target organs in a complex with serum albumin (56). Moreover, de novo vitamin A biosynthesis from dietary β-carotene has been documented in the RPE cells and other tissues (56, 61) thus exemplifying yet another RBP4-independent route of retinoid supply to the retina. Together, these data provide a possible explanation as to why the visual cycle remains functional in mice despite the maximally possible pharmacological inactivation of the RBP4–TTR delivery route by BPN-14136 and support the view that it would be incorrect to regard pharmacological reduction of serum RBP4 as synonymous to systemic vitamin A deprivation. Published reports indicate that Rbp4−/− mice are phenotypically normal with no systemic abnormalities (55, 57, 58) or histological signs of retinal degeneration (62). At weaning (19–21 days postnatal), Rbp4−/− mice have reduced ERG amplitudes that normalize on a standard vitamin A-sufficient chow (55, 57) after RBP4-independent routes of vitamin A delivery replenish congenitally depleted retinoid stores in the retina (57). TTR knockout mice do not display symptoms of vitamin A deficiency despite low serum RBP4 and the absence of the circulating retinol–RBP4–TTR complex (63). It therefore seems unlikely that partial reduction in serum RBP4 induced by oral administration of RBP4 antagonists will be associated with symptoms of vitamin A deficiency in patients who maintain a standard vitamin A- and β-carotene–sufficient diet.
Reduced rate of visual chromophore (11-cis-retinal) regeneration after its light-induced isomerization is the underlying mechanism of delayed dark adaptation, which represents a period of reduced light sensitivity following exposure to bright light. The speed of chromophore regeneration after photobleaching reflects the rate of the visual cycle. Given that BPN-14136 reduced the concentration of 11-cis-retinal by 40% in the dark-adapted eye (Fig. 3A), we wanted to determine whether BPN-14136 affects the rate of chromophore regeneration. We conducted visual cycle retinoid measurements immediately after photobleaching as well as at different time points after returning animals to the dark. Photobleaching of dark-adapted BALB/cJ mice induced an expected decrease in 11-cis-retinal and a concomitant increase in all-trans-retinal (Table 2). Indicating the properly functioning visual cycle, the amount of 11-cis-retinal linearly increased during dark recovery in both BPN-14136 and vehicle-treated animals (Fig. 3). Importantly, the rate of 11-cis-retinal regeneration was not different in BPN-14136–treated animals compared with vehicle-treated controls (Fig. 3). This indicates that BPN-14136 treatment does not reduce the rate of the visual cycle in mice while partially reducing the retinoid load in the retina. Given the fact that RBP4 antagonists do not inhibit the rate of the visual cycle, they should not be referred to as visual cycle inhibitors.
The rate of the visual cycle as well as the propensity of compounds to induce delayed dark adaptation can be electrophysiologically assessed by measuring recovery of the ERG b-wave amplitude following exposure to a bleaching light. Kinetics of the ERG b-wave recovery after photobleaching physiologically reflects the rate of the visual retinoid cycle. There was no statistically significant difference in pre-bleach ERG b-wave amplitudes in BPN-14136–treated and control animals, and the rate of the ERG b-wave recovery was identical in BPN-14136- and vehicle-treated animals (Fig. 4). This is consistent with our previous results for another RBP4 antagonist, A1120, which did not induce the delay in the rate of the b-wave amplitude recovery after a photobleach following 6 weeks of compound dosing in Abca4−/− mice or after 3-week dosing in WT 129 p3/J mice (38). The lack of delay in return of rod photoresponse after a photobleach in compound-treated animals supports the conclusion that BPN-14136 treatment did not induce physiological inhibition of the visual cycle in mice. This lack of the visual cycle rate inhibition by RBP4 antagonists is one of the most conspicuous differences between the pharmacological classes of RBP4 antagonists and direct visual cycle enzyme inhibitors, such as RPE65 antagonists. Although the direct inhibitors of visual cycle enzymes, such as isotretinoin (27), retinylamine (64), and emixustat (65), are able to decrease the level of bisretinoids, these inhibitors also can induce a significant delay in the recovery of rod function following exposure to bright light (27, 64, 65).
Given that BPN-14136 induced appreciable reduction of 11-cis-retinal in dark-adapted and light-adapted mouse eyes (Table 2), we examined the effect of such reduction on retinal physiology using electroretinography. After 2 weeks of BPN-14136 administration at a dose that induces 90% serum RBP4 reduction, the ERG waveforms remained normal, and the ERG intensity-response curves overlapped for vehicle- and BPN-14136–treated BALB/cJ mice (Fig. 5). Similarly, no ERG changes in response to fenretinide treatment were reported in Abca4−/− mice (26). As ERG gave no physiological evidence of impaired rod function in dark-adapted (or light-adapted) BPN-14136–treated mice, we studied the effect of compound administration on rhodopsin levels. As shown in Fig. 6, we detected a 25% reduction in rhodopsin concentration in a dark-adapted retina in response to BPN-14136 treatment. Interestingly, this 25% rhodopsin reduction was not manifested as detectable functional changes as evidenced by normal scotopic and photopic ERG in compound-treated animals. A non-linear relationship between the extent of 11-cis-retinal lowering (40% reduction) and a drop in rhodopsin (25% reduction) in dark-adapted mouse eyes in response to the same extent of serum RBP4 lowering may imply that more chromophore is produced in the visual cycle than is required for complete opsin loading. If an appreciable pool of “free” 11-cis-retinal unbound to opsin exists in the dark-adapted retina, then pharmacological lowering of 11-cis-retinal by RBP4 antagonists may potentially reduce aldehyde and bisretinoid toxicity without significant effect on phototransduction.
To study the ability of BPN-14136 to inhibit the formation of lipofuscin fluorophores in the retina, we administered the compound at a daily 20 mg/kg dose to Abca4−/− mice for 12 weeks. BPN-14136 induced a sustained serum RBP4 reduction over the treatment period (Fig. 7A), which translated to the inhibition of A2E formation by ∼50% in compound-treated animals (Fig. 7B). Corroborating the biochemical analysis of A2E, quantification of lipofuscin autofluorescence established that BPN-14136 induced a 75% reduction of lipofuscin fluorophores in compound-treated Abca4−/− mice (Fig. 8). Consistent with our published data for A1120 (38) and similar to previously reported fenretinide results (26), we established that BPN-14136 was able to significantly inhibit in vivo accumulation of lipofuscin bisretinoids in the Abca4−/− knockout mouse model of Stargardt disease.
Dysregulation of the complement system in the retina is one of the critical features of dry AMD. Fortunately, this inflammatory component of dry AMD phenotype can be partially mimicked in the Abca4−/− mouse model where enhanced accumulation of lipofuscin bisretinoids induces dysregulation of the complement system in the retina and complement-mediated inflammation in the RPE (46). We studied the effect of BPN-14136 on dysregulation of complement component C3, C-reactive protein, CFD, and CFH in the Abca4−/− mouse retina. BPN-14136 treatment normalized bisretinoid-induced up-regulation of CFD and C3 in the mouse Abca4−/− retina (Fig. 9, A–C and G–I). Similarly, levels of C-reactive protein up-regulated in the Abca4−/− mouse retina were reduced as a result of the compound treatment (Fig. 9, J–L). Expression of CFH, which was partially suppressed in the Abca4−/− mouse retina (Fig. 9, D–F), was restored following administration of BPN-14136. Given that complement system dysregulation in Abca4−/− mice seems to be secondary to the excessive lipofuscin production, the most likely explanation for the beneficial effect of BPN-14136 on normalization of the complement system in this animal model relates to the ability of this RBP4 antagonist to inhibit bisretinoid synthesis (Fig. 7B). However, emerging evidence suggests that independent of its retinol-trafficking function, RBP4 can act as a direct modulator of inflammation in different tissues (66–68), including cells of the retina (67). In addition to the robust inhibitory effect on bisretinoid synthesis, RBP4 antagonists might potentially modulate direct pro-inflammatory signaling by RBP4 thus conferring additional benefits in reducing local retinal inflammation associated with pathogenesis of dry AMD.
In summary, we have shown that BPN-14136, a highly optimized RBP4 antagonist, can induce partial reduction of serum RBP4 and visual cycle retinoids such as retinaldehydes, which translated to robust inhibition of lipofuscin bisretinoid synthesis and normalization of complement system dysregulation in the retina. Importantly, these positive attributes of BPN-14136 were not associated with inhibition of the visual cycle as measured biochemically or electrophysiologically. In addition, no detectable compound-induced suppression of the visual function in dark- or light-adapted mouse eyes was seen in the ERG experiments that we conducted. BPN-14136 exemplifies a class of compounds that may be advanced for characterization as an oral therapy for dry AMD, Stargardt disease, and other forms of retinal disease associated with accumulation of lipofuscin, retinaldehyde toxicity, or complement system dysregulation in the retina.
Experimental procedures
Chemical synthesis
BPN-14136 was synthesized as described previously (37) using the procedure outlined in supporting information.
Animal experiments
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University and were performed following the guidelines of the Association for Research in Vision and Ophthalmology (ARVO) statement for the “Use of Animals in Ophthalmic and Vision Research.” Albino BALB/cJ mice, homozygous for the Leu-450 allele of RPE65, were used in ERG and visual cycle retinoid experiments. Abca4 null mutant mice (129/SV × C57BL/6J) homozygous for the Rpe65–Leu-450 polymorphism were bred as described previously (69, 70) and used to study the BPN-14136 effect on bisretinoid accumulation. CD-1 mice were used in pharmacokinetic studies. Mice were maintained on a 12-h on/12-h off light/dark cycle. For long-term oral dosing in Abca4−/− mice, BPN-14136 was formulated into Purina 5035 rodent chow at Research Diets, Inc. (New Brunswick, NJ) to ensure consistent 20 mg/kg daily oral dosing over the period of 12 weeks. To test the effect of BPN-14136 on ERG parameters and to study the effect of the compound on visual cycle retinoids, BALB/cJ mice (The Jackson Laboratory) were dosed for 2 weeks with the BPN-14136–containing chow to ensure 8 mg/kg daily oral dosing that induced 90% serum RBP4 reduction. In the rhodopsin measurements experiment, BPN-14136 was administered for 2 weeks at the 20 mg/kg dose that induced 90% serum RBP4 reduction. RBP4-lowering potency for BPN-14136 peaks in mice at the 5 mg/kg oral dose (90% serum RBP4 reduction) and does not increase with further dose escalation. No strain-dependent differences in RBP4 lowering potency for BPN-14136 were noticed.
Pharmacokinetic studies
Pharmacokinetic studies were conducted in CD-1 mice at SRI International, Menlo Park, CA. Mice received a single i.v. dose of a test compound at 2 mg/kg or a single p.o. dose of a test compound of at 5 mg/kg. The i.v. vehicle was 3% N,N-dimethylacetamide, 45% PEG300, 12% ethanol, 40% sterile water; the p.o. vehicle was 2% Tween 80 in 0.9% saline. Blood was collected from mice at pre-dose and at 5, 15, and 30 min and at 1, 2, 4, 8, 12, 24, and 48 h post-dose for processing to plasma. Following protein extraction with acetonitrile, compound levels in plasma were measured by LC-MS/MS. Compound plasma levels after oral administration of the 5 mg/kg dose are presented in Table S1. The mean plasma drug level values were analyzed using WinNonlin package by non-compartmental modeling with the sparse sampling feature. Aliquots of pre-dose plasma samples, as well as aliquots of plasma PK samples, were used for the analysis of the serum biomarker RBP4.
Serum and plasma RBP4 measurements
Given the excellent correlation between serum RBP4 and serum retinol across a wide range of retinol concentrations, we used serum RBP4, a surrogate marker for serum retinol in this study. In the Abca4−/− dosing experiment, blood samples were collected from a tail vein at pre-dose and after 2, 6, and 12 weeks of BPN-14136 administration. Blood samples were collected between 10:00 a.m. and 12:00 p.m. In ERG, rhodopsin measurements, and visual cycle retinoid experiments, blood samples were collected from BALB/cJ mice at pre-dose and after 2 weeks of compound treatment. Whole blood was drawn into a centrifuge tube and was allowed to clot at room temperature for 30 min followed by centrifugation at 2000 × g for 15 min at 4 °C to collect serum. Serum RBP4 was measured using the RBP4 (mouse/rat) dual ELISA kit (AdipoGen, Switzerland) following the manufacturer's instructions. Aliquots of plasma samples collected in the mouse pharmacokinetic study were similarly analyzed for the RBP4 concentration.
Retinoid extraction and analysis
For bisretinoid analysis, posterior eyecups were pooled and homogenized in PBS using a tissue grinder. An equal volume of a mixture of chloroform and methanol (2:1) was added, and the sample was extracted three times. Bisretinoids were extracted from the eyecups of untreated WT mice (8 eyes), vehicle-treated Abca4−/− mice (14 eyes), and BPN-14136-treated Abca4−/− mice (16 eyes). The pooled organic phases were dried under a stream of argon and resuspended in 100 μl of methanol. A2E was separated using a mobile-phase gradient of methanol in water (85–96% methanol and 0.1% TFA) through a C18 reverse–phase column. A photodiode array detector was used to monitor the absorbance of A2E at 430 nm. A2E levels were quantified from the area of each peak using a synthetic A2E standard for calibration.
For the analysis of visual cycle retinoids (retinyl ester, 11-cis-retinaldehyde, and all-trans-retinaldehyde), the whole eyes were homogenized with a glass grinder in lysis buffer (10 mm NH2OH, 50% ethanol, 50% MES, pH 6.5), and retinoids were extracted with hexane. Solvent was evaporated under argon gas, and dried samples were resuspended in 200 μl of mobile phase (11.2% ethyl acetate, 2.0% dioxane, 1.4% octanol, 85.4% hexane) and injected into the HPLC at the isocratic flow rate of 1 ml/min for separation using a normal-phase 5-μm column. Individual retinoids were quantified from the area of its corresponding peak, determined by using synthetic purified retinoid standards for calibration. Bisretinoid and visual cycle retinoid extraction and analysis were conducted at EyeCRO, LLC (Oklahoma City, OK).
Quantification of lipofuscin autofluorescence
Eyes from three freshly euthanized untreated WT mice, three vehicle-treated Abca4−/− mice (14 eyes), and three BPN-14136–treated Abca4−/− mice were enucleated and fixed by immersion in cold (−20 °C) 90% methanol, 10% MES (100 mm MES, 1 mm EGTA, 1 mm MgSO4, pH 6.9), rehydrated in PBS, and processed as described previously (71). Cryosections of 10 μm in thickness were prepared on Leica cryostat (Leica CM1850) and mounted. Confocal imaging was performed on a Nikon A1RMP laser-scanning system on an Eclipse Ti stand (Nikon Instruments, Melville, NY) using a Plan Fluor 40×/1.3 oil objective (Nikon). Lipofuscin autofluorescence datasets were acquired at 488 nm excitation and emission, and spectra were collected between 500 and 600 nm as recommended previously (71). The confocal pinhole was set at 3 Airy units to produce an optical section of 1.2-μm thick. A single optical section was collected from the plane with maximum fluorescence. All scanning settings were held constant throughout the experiment. Autofluorescence intensity was determined by using Fiji software (National Institutes of Health). For each mouse eye, autofluorescence was quantified in an average of 21 image fields (range: 9–41) for each eye. Confocal microscopy and the image analysis protocol used in this study were developed in the Confocal and Specialized Microscopy Shared Resource facility of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by National Institutes of Health Grant P30 CA013696 (from NCI). The A1RMP confocal microscope was purchased with National Institutes of Health Grant S10 RR025686.
Rhodopsin measurements
All procedures were carried out under dim red light. Mice were dark-adapted overnight and euthanized. The eyes were removed, and retinas were dissected. Immediately after tissue collection, each retina was homogenized in 50 mm Tris-HCl, pH 7.4, containing 100 mm hydroxylamine, and 0.2% cetyltrimethylammonium chloride. Homogenates were centrifuged at 20,000 × g, and the supernatant absorbance (350–700 nm) was determined before and after the 2-min bleaching. The difference in absorbance between pre- and post-bleached samples at 500 nm was used to represent rhodopsin content. Representative rhodopsin bleaching difference spectra for control and BPN-14136–treated mice are shown in Fig. 6B.
Immunohistochemistry
Eyes from freshly euthanized mice were enucleated and immersed in OCT solution (Tissue Tek, Sakura) and frozen in liquid nitrogen. 10-μm sections were prepared on a Leica cryostat (Leica CM1850). Sections were fixed in 4% paraformaldehyde for 10 min at room temperature. Sections were then washed three times in PBS for 10 min at room temperature. The non-specific sites are blocked using 5% host serum in PBS with 1% BSA and 0.5% Triton X-100 for 1 h at room temperature. Section were then immunostained overnight at 4 °C with primary antibody in PBS with 1% BSA and 0.5% Triton X-100. The following antibodies were used: sheep anti-complement Factor H (1:200; ab8842; Abcam); rabbit anti-complement component C3 (1:200; NBP1-32080; Novusbio); mouse anti-complement Factor D (1:100; sc-373860; Santa Cruz Biotechnology, Inc.); and mouse anti-C-reactive protein (1:100; sc-69770; Santa Cruz Biotechnology, Inc.). Subsequently, sections were washed three times in PBS, and donkey anti-sheep Alexa 594-conjugated secondary antibody (1:500; A11016; Molecular Probes, Life Technologies, Inc.), rhodamine red-X goat anti-mouse antibody (1:500; R6393; Molecular Probes, Life Technologies, Inc.), or rhodamine red-X goat anti-rabbit antibody (1:500; R6394; Molecular Probes, Life Technologies, Inc.) were applied for 1 h at room temperature. Sections were then washed with PBS and covered by Vectashield mounting media with DAPI (Vector Laboratories). The images of the mouse retina sections were captured with a confocal microscope (Nikon Ti Eclipse Confocal Microscopy) under ×60 oil objective using excitation wavelengths of 350 and 570 nm with emission wavelengths of 470 and 590 nm, respectively. Images were processed, and analysis for this work was performed in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by National Institutes of Health Grant P30 CA013696 from NCI.
Immunoblotting
Retinas were collected from Abca4+/+ WT, vehicle-treated Abca4−/−, and BPN-14136–treated Abca4−/− mice. Samples were homogenized in ice-cold isotonic Tris buffer (50 mm, pH 8.0) containing 0.5 mm sodium metavanadate, 1 mm EDTA, and a protease inhibitor mixture (1:1000, Sigma), and protein concentrations were measured using the BCA protein assay (ThermoFisher Scientific). Proteins were separated by NuPAGE BisTris gels (4–12%) (Novex, Life Technologies, Inc.) and transferred to polyvinylidene difluoride Novex, Life Technologies, Inc.). After blocking, membranes were incubated in sheep anti-complement Factor H (1:1000; NB100-62177; Novusbio), rabbit anti-complement C3 (1:1000; NBP1-32080; Novusbio); mouse anti-complement factor D (1:1000; sc-373860; Santa Cruz Biotechnology, Inc.), and mouse anti-C-reactive protein (1:1000; sc-69770; Santa Cruz Biotechnology, Inc.) overnight at 4 °C. Membranes were washed six times for 5 min in TBS, pH 7.5, containing 0.2% Tween (TBST) and then incubated in goat anti-rabbit IgG-HRP secondary antibody (1:2000; sc-2004; Santa Cruz Biotechnology, Inc.), rabbit anti-sheep IgG-HRP (1:2000; sc-2770; Santa Cruz Biotechnology, Inc.), and anti-mouse IgG-HRP (1:2000; 7076; Cell Signaling Technology) for 1 h at room temperature. β-Actin loading control was detected using mouse anti-β-actin antibody (1:2000; A5441; Sigma). The protein bands were visualized with enhanced chemiluminescence labeling using an ECL immunoblotting detection system (Pierce, ThermoFisher Scientific, 32106). The developed films (CL-XPosureTM Film, X-Ray Film; ThermoFisher Scientific) were scanned, and the pixel volumes of the bands were determined by using ImageJ software (National Institutes of Health), with the values in ratios of intensity. Each experiment was repeated a minimum of three times.
Electroretinography
Mice were dark-adapted overnight before ERG experiments, and recordings were carried out under dim red-light illumination. Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). Body temperature was maintained at 37 °C using a heating pad. Mouse pupils were dilated with 1% phenylephrine and 1% cyclopentolate. Gold electrodes were placed on the corneas, and gonioscopic prism solution (Alcon) was applied to each eye. Subcutaneous 30-gauge needles on the forehead and trunk were used as reference and ground electrodes, respectively. The light stimulus was obtained from a Ganzfeld dome. Maximum flash intensity was measured with a calibrated light meter (J16; Tektronics Instruments, Beaverton, OR). ERG responses were recorded simultaneously from both eyes using an Espion Visual Electrophysiological System (Diagnosys) followed by an exposure to 1000 lux of bleaching light for 5 min. Under these conditions, >90% of the rhodopsin was bleached. The ERG was recorded from the cornea with gold electrodes for 50 min immediately after bleaching for b-wave recovery study. During the scotopic and photopic study, rod and mixed rod-cone responses were recorded in dark-adapted mice using pulses of 0.001 and log −2.8 cd × s/m2 (white 6500 K), log −1 cd × s/m2 (white 6500 K), and log 1.9 cd × s/m2 (white 6500 K). Then, mice were light-adapted for photopic responses for at least 5 min in the Ganzfeld dome. Recordings were carried out under rod-suppressing continuous background illumination of 30 cd/m2 (white 6500 K), and cone responses were recorded using pulses of log −0.6 cd × s/m2 (white 6500 K), log 0.4 cd × s/m2 (white 6500 K), and log 1.47 cd × s/m2 (xenon). Results were analyzed by Espion V6 software.
Statistical analyses
SigmaPlot 14 (Systat Software, Inc.) and GraphPad Prism 7.03 (GraphPad Software, Inc.) software packages were used for statistical analyses. Data are represented as the mean ± S.D. To mark statistical significance on figures, the GraphPad style was used: ns, p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. For the analysis of ERG data, a t test was used. Statistical significance was determined using the Holm-Sidak method, α = 0.05. Data for each light intensity was analyzed individually. Comparison of rhodopsin levels was performed using an unpaired t test. In the long-term oral BPN-14136 administration experiment, serum RBP4 concentrations were analyzed using two-way ANOVA with Sidak post hoc test, and A2E lowering was analyzed using one-way ANOVA with Holm-Sidak post hoc test.
Author contributions
B. R., G. J., C. L. C., and K. P. conceptualization; B. R., C. L. C., and K. P. data curation; B. R., R. A., P. G. P., G. J., C. L. C., and K. P. supervision; B. R. and K. P. validation; B. R., A. V., J. K., R. A., P. G. P., G. J., C. L. C., and K. P. investigation; B. R., A. V., J. K., R. A., P. G. P., G. J., C. L. C., and K. P. methodology; B. R., A. V., J. K., and K. P. writing-original draft; A. V. and J. K. formal analysis; R. A., P. G. P., G. J., C. L. C., and K. P. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Drs. Theresa Swayne and Laura Munteanu for the assistance with confocal microscopy. Research facilities were supported by NEI Grant P30 EY019007 (Core Support for Vision Research) and by unrestricted funds from Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University.
This work was supported by National Institutes of Health Grants U01 NS074476 (to K. P.) and R24 EY019861 (to R. A.). Columbia University licensed some of the technology described in the article to Lin BioScience, Inc. As inventors of the technology, K. P. and R. A. may benefit from this licensing agreement. K. P. is a consultant for Lin Bioscience. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- STGD1
- Stargardt disease
- TTR
- transthyretin
- RPE
- retinal pigment epithelium
- AMD
- age-related macular degeneration
- A2E
- N-retinyl-N-retinylidene ethanolamine
- ERG
- electroretinogram
- ANOVA
- analysis of variance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PK
- pharmacokinetics
- PD
- pharmacodynamics
- CFD
- complement Factor D
- CFH
- complement factor H
- cd
- candela
- lx
- lux
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Birnbach C. D., Järvelainen M., Possin D. E., and Milam A. H. (1994) Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology 101, 1211–1219 10.1016/S0161-6420(13)31725-4 [DOI] [PubMed] [Google Scholar]
- 2. De Laey J. J., and Verougstraete C. (1995) Hyperlipofuscinosis and subretinal fibrosis in Stargardt's disease. Retina 15, 399–406 10.1097/00006982-199515050-00005 [DOI] [PubMed] [Google Scholar]
- 3. Delori F. C. (1995) in Retinal Pigment Epithelium and Macular Disease (Documenta Ophthalmologica) (Coscas G., and Felice C. P., eds) pp. 37–45, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 4. Eagle R. C. Jr., Lucier A. C., Bernardino V. B. Jr., and Yanoff M. (1980) Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology 87, 1189–1200 10.1016/S0161-6420(80)35106-3 [DOI] [PubMed] [Google Scholar]
- 5. Walia S., and Fishman G. A. (2009) Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 30, 63–68 10.1080/13816810802695550 [DOI] [PubMed] [Google Scholar]
- 6. Travis G. H., Golczak M., Moise A. R., and Palczewski K. (2007) Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu. Rev. Pharmacol. 47, 469–512 10.1146/annurev.pharmtox.47.120505.105225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allikmets R., Singh N., Sun H., Shroyer N. F., Hutchinson A., Chidambaram A., Gerrard B., Baird L., Stauffer D., Peiffer A., Rattner A., Smallwood P., Li Y., Anderson K. L., Lewis R. A., et al. (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- 8. Jaakson K., Zernant J., Külm M., Hutchinson A., Tonisson N., Glavac D., Ravnik-Glavac M., Hawlina M., Meltzer M. R., Caruso R. C., Testa F., Maugeri A., Hoyng C. B., Gouras P., Simonelli F., et al. (2003) Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 22, 395–403 10.1002/humu.10263 [DOI] [PubMed] [Google Scholar]
- 9. Roberts L. J., Nossek C. A., Greenberg L. J., and Ramesar R. S. (2012) Stargardt macular dystrophy: common ABCA4 mutations in South Africa–establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 18, 280–289 [PMC free article] [PubMed] [Google Scholar]
- 10. Petrukhin K. (2007) New therapeutic targets in atrophic age-related macular degeneration. Expert Opin. Ther. Targets 11, 625–639 10.1517/14728222.11.5.625 [DOI] [PubMed] [Google Scholar]
- 11. Petrukhin K. (2013) Pharmacological inhibition of lipofuscin accumulation in the retina as a therapeutic strategy for dry AMD treatment. Drug Discov. Today Ther. Strateg. 10, e11–e20 10.1016/j.ddstr.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sparrow J. R., Dowling J. E., and Bok D. (2013) Understanding RPE lipofuscin. Invest. Ophthalmol. Vis. Sci. 54, 8325–8326 10.1167/iovs.13-13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delori F. C., Goger D. G., and Dorey C. K. (2001) Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest. Ophthalmol. Vis. Sci. 42, 1855–1866 [PubMed] [Google Scholar]
- 14. Dorey C. K., Wu G., Ebenstein D., Garsd A., and Weiter J. J. (1989) Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest. Ophthalmol. Vis. Sci. 30, 1691–1699 [PubMed] [Google Scholar]
- 15. Feeney-Burns L., Hilderbrand E. S., and Eldridge S. (1984) Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Invest. Ophthalmol. Vis. Sci. 25, 195–200 [PubMed] [Google Scholar]
- 16. Holz F. G., Bellman C., Staudt S., Schütt F., and Völcker H. E. (2001) Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 42, 1051–1056 [PubMed] [Google Scholar]
- 17. von Rückmann A., Fitzke F. W., and Bird A. C. (1997) Fundus autofluorescence in age-related macular disease imaged with a laser scanning ophthalmoscope. Invest. Ophthalmol. Vis. Sci. 38, 478–486 [PubMed] [Google Scholar]
- 18. Maeda A., Golczak M., Chen Y., Okano K., Kohno H., Shiose S., Ishikawa K., Harte W., Palczewska G., Maeda T., and Palczewski K. (2011) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol. 8, 170–178 10.1038/nchembio.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y., Okano K., Maeda T., Chauhan V., Golczak M., Maeda A., and Palczewski K. (2012) Mechanism of all-trans-retinal toxicity with implications for Stargardt disease and age-related macular degeneration. J. Biol. Chem. 287, 5059–5069 10.1074/jbc.M111.315432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith R. T. (2016) New understanding of age-related macular degeneration through quantitative autofluorescence. JAMA Ophthalmol. 134, 824–826 10.1001/jamaophthalmol.2016.1466 [DOI] [PubMed] [Google Scholar]
- 21. Gehrs K. M., Jackson J. R., Brown E. N., Allikmets R., and Hageman G. S. (2010) Complement, age-related macular degeneration and a vision of the future. Arch. Ophthalmol. 128, 349–358 10.1001/archophthalmol.2010.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weber B. H., Charbel Issa P., Pauly D., Herrmann P., Grassmann F., and Holz F. G. (2014) The role of the complement system in age-related macular degeneration. Dtsch. Arztebl. Int. 111, 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scholl H. P., Charbel Issa P., Walier M., Janzer S., Pollok-Kopp B., Börncke F., Fritsche L. G., Chong N. V., Fimmers R., Wienker T., Holz F. G., Weber B. H., and Oppermann M. (2008) Systemic complement activation in age-related macular degeneration. PLoS ONE 3, e2593 10.1371/journal.pone.0002593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donoso L. A., Kim D., Frost A., Callahan A., and Hageman G. (2006) The role of inflammation in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 51, 137–152 10.1016/j.survophthal.2005.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kennedy C. J., Rakoczy P. E., and Constable I. J. (1995) Lipofuscin of the retinal pigment epithelium: a review. Eye 9, 763–771 10.1038/eye.1995.192 [DOI] [PubMed] [Google Scholar]
- 26. Radu R. A., Han Y., Bui T. V., Nusinowitz S., Bok D., Lichter J., Widder K., Travis G. H., and Mata N. L. (2005) Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest. Ophthalmol. Vis. Sci. 46, 4393–4401 10.1167/iovs.05-0820 [DOI] [PubMed] [Google Scholar]
- 27. Radu R. A., Mata N. L., Nusinowitz S., Liu X., Sieving P. A., and Travis G. H. (2003) Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 100, 4742–4747 10.1073/pnas.0737855100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maeda A., Maeda T., Golczak M., Imanishi Y., Leahy P., Kubota R., and Palczewski K. (2006) Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol. Pharmacol. 70, 1220–1229 10.1124/mol.106.026823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palczewski K. (2010) Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 31, 284–295 10.1016/j.tips.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berni R., and Formelli F. (1992) In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett. 308, 43–45 10.1016/0014-5793(92)81046-O [DOI] [PubMed] [Google Scholar]
- 31. Schaffer E. M., Ritter S. J., and Smith J. E. (1993) N-(4-Hydroxyphenyl)retinamide (fenretinide) induces retinol-binding protein secretion from liver and accumulation in the kidneys in rats. J. Nutr. 123, 1497–1503 10.1093/jn/123.9.1497 [DOI] [PubMed] [Google Scholar]
- 32. Mata N. L., Lichter J. B., Vogel R., Han Y., Bui T. V., and Singerman L. J. (2013) Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina 33, 498–507 10.1097/IAE.0b013e318265801d [DOI] [PubMed] [Google Scholar]
- 33. Samuel W., Kutty R. K., Nagineni S., Vijayasarathy C., Chandraratna R. A., and Wiggert B. (2006) N-(4-Hydroxyphenyl)retinamide induces apoptosis in human retinal pigment epithelial cells: retinoic acid receptors regulate apoptosis, reactive oxygen species generation, and the expression of heme oxygenase-1 and Gadd153. J. Cell. Physiol. 209, 854–865 10.1002/jcp.20774 [DOI] [PubMed] [Google Scholar]
- 34. Cohen S. M., Storer R. D., Criswell K. A., Doerrer N. G., Dellarco V. L., Pegg D. G., Wojcinski Z. W., Malarkey D. E., Jacobs A. C., Klaunig J. E., Swenberg J. A., and Cook J. C. (2009) Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol. Sci. 111, 4–18 10.1093/toxsci/kfp131 [DOI] [PubMed] [Google Scholar]
- 35. Kenel M. F., Krayer J. H., Merz E. A., and Pritchard J. F. (1988) Teratogenicity of N-(4-hydroxyphenyl)-all-trans-retinamide in rats and rabbits. Teratog. Carcinog. Mutagen. 8, 1–11 10.1002/tcm.1770080102 [DOI] [PubMed] [Google Scholar]
- 36. Cioffi C. L., Dobri N., Freeman E. E., Conlon M. P., Chen P., Stafford D. G., Schwarz D. M., Golden K. C., Zhu L., Kitchen D. B., Barnes K. D., Racz B., Qin Q., Michelotti E., Cywin C. L., et al. (2014) Design, synthesis, and evaluation of non-retinoid retinol binding protein 4 antagonists for the potential treatment of atrophic age-related macular degeneration and Stargardt disease. J. Med. Chem. 57, 7731–7757 10.1021/jm5010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cioffi C. L., Racz B., Freeman E. E., Conlon M. P., Chen P., Stafford D. G., Schwarz D. M., Zhu L., Kitchen D. B., Barnes K. D., Dobri N., Michelotti E., Cywin C. L., Martin W. H., Pearson P. G., Johnson G., and Petrukhin K. (2015) Bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo antagonists of retinol binding protein 4: potential treatment of atrophic age-related macular degeneration and Stargardt disease. J. Med. Chem. 58, 5863–5888 10.1021/acs.jmedchem.5b00423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dobri N., Qin Q., Kong J., Yamamoto K., Liu Z., Moiseyev G., Ma J.-X., Allikmets R., Sparrow J. R., and Petrukhin K. (2013) A1120, a non-retinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest. Ophthalmol. Vis. Sci. 54, 85–95 10.1167/iovs.12-10050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Motani A., Wang Z., Conn M., Siegler K., Zhang Y., Liu Q., Johnstone S., Xu H., Thibault S., Wang Y., Fan P., Connors R., Le H., Xu G., Walker N., Shan B., and Coward P. (2009) Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem. 284, 7673–7680 10.1074/jbc.M809654200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sparrow J. R., Fishkin N., Zhou J., Cai B., Jang Y. P., Krane S., Itagaki Y., and Nakanishi K. (2003) A2E, a byproduct of the visual cycle. Vis. Res. 43, 2983–2990 10.1016/S0042-6989(03)00475-9 [DOI] [PubMed] [Google Scholar]
- 41. Boyer N. P., Higbee D., Currin M. B., Blakeley L. R., Chen C., Ablonczy Z., Crouch R. K., and Koutalos Y. (2012) Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11-cis-retinal. J. Biol. Chem. 287, 22276–22286 10.1074/jbc.M111.329235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sieving P. A., Chaudhry P., Kondo M., Provenzano M., Wu D., Carlson T. J., Bush R. A., and Thompson D. A. (2001) Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc. Natl. Acad. Sci. U.S.A. 98, 1835–1840 10.1073/pnas.98.4.1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kolesnikov A. V., Orban T., Jin H., Brooks C., Hofmann L., Dong Z., Sokolov M., Palczewski K., and Kefalov V. J. (2017) Dephosphorylation by protein phosphatase 2A regulates visual pigment regeneration and the dark adaptation of mammalian photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 114, E9675–E9684 10.1073/pnas.1712405114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baylor D. A., and Burns M. E. (1998) Control of rhodopsin activity in vision. Eye (Lond) 12, 521–525 [DOI] [PubMed] [Google Scholar]
- 45. Radu R. A., Hu J., Jiang Z., and Bok D. (2014) Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J. Biol. Chem. 289, 9113–9120 10.1074/jbc.M114.548669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Radu R. A., Hu J., Yuan Q., Welch D. L., Makshanoff J., Lloyd M., McMullen S., Travis G. H., and Bok D. (2011) Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 286, 18593–18601 10.1074/jbc.M110.191866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou J., Jang Y. P., Kim S. R., and Sparrow J. R. (2006) Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 103, 16182–16187 10.1073/pnas.0604255103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou J., Kim S. R., Westlund B. S., and Sparrow J. R. (2009) Complement activation by bisretinoid constituents of RPE lipofuscin. Invest. Ophthalmol. Vis. Sci. 50, 1392–1399 10.1167/iovs.08-2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brandstetter C., Holz F. G., and Krohne T. U. (2015) Complement component C5a primes retinal pigment epithelial cells for inflammasome activation by lipofuscin-mediated photooxidative damage. J. Biol. Chem. 290, 31189–31198 10.1074/jbc.M115.671180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sparrow J. R., Nakanishi K., and Parish C. A. (2000) The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest. Ophthalmol. Vis. Sci. 41, 1981–1989 [PubMed] [Google Scholar]
- 51. Sparrow J. R., Parish C. A., Hashimoto M., and Nakanishi K. (1999) A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest. Ophthalmol. Vis. Sci. 40, 2988–2995 [PubMed] [Google Scholar]
- 52. Finnemann S. C., Leung L. W., and Rodriguez-Boulan E. (2002) The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 99, 3842–3847 10.1073/pnas.052025899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vives-Bauza C., Anand M., Shiraz A. K., Magrane J., Gao J., Vollmer-Snarr H. R., Manfredi G., and Finnemann S. C. (2008) The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem. 283, 24770–24780 10.1074/jbc.M800706200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eldred G. E., and Lasky M. R. (1993) Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature 361, 724–726 10.1038/361724a0 [DOI] [PubMed] [Google Scholar]
- 55. Vogel S., Piantedosi R., O'Byrne S. M., Kako Y., Quadro L., Gottesman M. E., Goldberg I. J., and Blaner W. S. (2002) Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry 41, 15360–15368 10.1021/bi0268551 [DOI] [PubMed] [Google Scholar]
- 56. Biesalski H. K., Chichili G. R., Frank J., von Lintig J., and Nohr D. (2007) Conversion of β-carotene to retinal pigment. Vitam. Horm. 75, 117–130 10.1016/S0083-6729(06)75005-1 [DOI] [PubMed] [Google Scholar]
- 57. Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., and Gottesman M. E. (1999) Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633–4644 10.1093/emboj/18.17.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Quadro L., Hamberger L., Colantuoni V., Gottesman M. E., and Blaner W. S. (2003) Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol. Aspects Med. 24, 421–430 10.1016/S0098-2997(03)00038-4 [DOI] [PubMed] [Google Scholar]
- 59. Wolf G. (1999) A case of human vitamin A deficiency caused by an inherited defect in retinol-binding protein without clinical symptoms except night blindness. Nutr. Rev. 57, 258–260 [DOI] [PubMed] [Google Scholar]
- 60. Seeliger M. W., Biesalski H. K., Wissinger B., Gollnick H., Gielen S., Frank J., Beck S., and Zrenner E. (1999) Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest. Ophthalmol. Vis. Sci. 40, 3–11 [PubMed] [Google Scholar]
- 61. Chichili G. R., Nohr D., Schäffer M., von Lintig J., and Biesalski H. K. (2005) β-Carotene conversion into vitamin A in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 46, 3562–3569 10.1167/iovs.05-0089 [DOI] [PubMed] [Google Scholar]
- 62. Thompson C. L., Blaner W. S., Van Gelder R. N., Lai K., Quadro L., Colantuoni V., Gottesman M. E., and Sancar A. (2001) Preservation of light signaling to the suprachiasmatic nucleus in vitamin A-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 11708–11713 10.1073/pnas.201301498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wolf G. (1995) Retinol transport and metabolism in transthyretin-“knockout” mice. Nutr. Rev. 53, 98–99 [DOI] [PubMed] [Google Scholar]
- 64. Golczak M., Kuksa V., Maeda T., Moise A. R., and Palczewski K. (2005) Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 8162–8167 10.1073/pnas.0503318102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bavik C., Henry S. H., Zhang Y., Mitts K., McGinn T., Budzynski E., Pashko A., Lieu K. L., Zhong S., Blumberg B., Kuksa V., Orme M., Scott I., Fawzi A., and Kubota R. (2015) Visual cycle modulation as an approach toward preservation of retinal integrity. PLoS One 10, e0124940 10.1371/journal.pone.0124940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kotnik P., Fischer-Posovszky P., and Wabitsch M. (2011) RBP4: a controversial adipokine. Eur. J. Endocrinol. 165, 703–711 10.1530/EJE-11-0431 [DOI] [PubMed] [Google Scholar]
- 67. Farjo K. M., Farjo R. A., Halsey S., Moiseyev G., and Ma J. X. (2012) Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor κB-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32, 5103–5115 10.1128/MCB.00820-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moraes-Vieira P. M., Yore M. M., Dwyer P. M., Syed I., Aryal P., and Kahn B. B. (2014) RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 19, 512–526 10.1016/j.cmet.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maiti P., Kong J., Kim S. R., Sparrow J. R., Allikmets R., and Rando R. R. (2006) Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry 45, 852–860 10.1021/bi0518545 [DOI] [PubMed] [Google Scholar]
- 70. Kim S. R., Fishkin N., Kong J., Nakanishi K., Allikmets R., and Sparrow J. R. (2004) Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc. Natl. Acad. Sci. U.S.A. 101, 11668–11672 10.1073/pnas.0403499101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gibbs D., Cideciyan A. V., Jacobson S. G., and Williams D. S. (2009) Retinal pigment epithelium defects in humans and mice with mutations in MYO7A: imaging melanosome-specific autofluorescence. Invest. Ophthalmol. Vis. Sci. 50, 4386–4393 10.1167/iovs.09-3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.