

Abstract
The channel-kinase transient receptor potential melastatin 7 (TRPM7) is a bifunctional protein with ion channel and kinase domains. The kinase activity of TRPM7 has been linked to the regulation of a broad range of cellular activities, but little is understood as to how the channel itself is regulated by its own kinase activity. Here, using several mammalian cell lines expressing WT TRPM7 or kinase-inactive variants, we discovered that compared with the cells expressing WT TRPM7, cells in which TRPM7's kinase activity was inactivated had faster degradation, elevated ubiquitination, and increased intracellular retention of the channel. Mutational analysis of TRPM7 autophosphorylation sites further revealed a role for Ser-1360 of TRPM7 as a key residue mediating both TRPM7 stability and intracellular trafficking. Additional trafficking roles were uncovered for Ser-1403 and Ser-1567, whose phosphorylation by TRPM7's kinase activity mediated the interaction of the channel with the signaling protein 14-3-3θ. In summary, our results point to a critical role for TRPM7's kinase activity in regulating proteasome-mediated turnover of the TRPM7 channel and controlling its cellular localization in polarized epithelial cells. Overall, these findings improve our understanding of the significance of TRPM7's kinase activity for functional regulation of its channel activity.
Keywords: transient receptor potential channels (TRP channels), ion channel, protein kinase, phosphorylation, 14-3-3 protein, proteosome-mediated turnover, transient receptor potential cation channel subfamily M member 7, TRPM7
Introduction
Transient receptor potential melastatin 7 (TRPM7)2 is a bifunctional protein composed of a channel domain and an α-kinase domain (1–3). This ubiquitously expressed ion channel is permeable to divalent cations including Ca2+, Mg2+, and Zn2+ (1, 4). Numerous physiological and pathological functions have been attributed to TRPM7. The channel is required for early embryogenesis in mice and Xenopus laevis frogs (5–8). In zebrafish, additional roles for the channel have been uncovered for the proliferation of pigment cells and pancreatic epithelial cells and for the differentiation and function of sensory neurons and kidney (9–14). At the cellular level, TRPM7 is implicated in cellular Mg2+ homeostasis, cell survival, proliferation, as well as cell adhesion and motility (15–21). Dysfunction of TRPM7 channel activity has been linked to defects in platelet biogenesis in humans (22). At the physiological level, TRPM7 is linked to the regulation of vertebrate magnesium homeostasis primarily through its physical association with its homolog TRPM6. Trpm6 is found mutated in human hypomagnesemia with secondary hypocalcemia disease (23–25). TRPM7's impact on whole-body magnesium homeostasis was later confirmed in mice (26). Several studies have further linked the TRPM7 channel to pathological processes such as cell death during organ ischemia, tumor proliferation, and metastasis (16, 27).
A unique aspect of both TRPM7 and TRPM6 is the presence of a functional α-kinase domain on the channels' C termini. TRPM7's kinase is not required for channel gating but is involved in the modulation of the channel by intracellular Mg2+-ATP (28). Autophosphorylation of TRPM7 occurs at multiple serine and threonine residues across the protein, many of which are clustered in a serine/threonine (S/T)-rich domain proximal to the kinase domain (29–31). Phosphorylation of the S/T-rich domain has been proposed to facilitate substrate binding for TRPM7 kinase (29). Several in vitro substrates, including nonmuscle myosin heavy chain IIA, annexin I, phospholipase Cγ2, histones, and elongation factor 2 kinase, have been identified (32–36). The kinase retains its functionality in vivo when separated from the channel by caspase-dependent proteolytic cleavage (37). Liberated TRPM7 kinase has been reported to localize to the nucleus (37, 38), where it contributes to chromatin remodeling (37). Studies using TRPM7 kinase-inactive mutant mice have linked the kinase activity of TRPM7 to the sensing and the coordination of both cellular and systemic responses to magnesium deprivation in mice (39). More recent studies have suggested a requirement for TRPM7's kinase in the regulation of the murine mast cell degranulation, intraepithelial T cell gut homing, ameloblast differentiation, and store-operated Ca2+ entry in platelets (40–43). To fully understand the wide range of cellular and physiological activities that TRPM7's kinase influences, it is essential to understand the mechanisms that regulate its kinase activity and the functional significance of autophosphorylation of the channel.
We recently employed MS to map TRPM7 autophosphorylation sites (30), which led us to propose a novel mechanism by which the kinase activity of TRPM7 can be regulated by autophosphorylation. Here we report that inactivation of TRPM7 kinase affects proteasome-mediated turnover of the channel kinase, its cellular localization in polarized epithelial cells, and its interaction with the phospho-binding protein 14-3-3θ, opening up new avenues for regulation of the channel kinase.
Results
TRPM7 kinase activity affects channel turnover
Although numerous studies have explored the consequences of kinase inactivation or kinase removal on TRPM7 channel activity, the impact of the kinase on the stability of the channel, its cellular localization, and its interaction with other cellular proteins remain comparatively unexplored. In our previous investigations of TRPM7, we observed that protein expression of the channel is affected by mutations that inactivate the catalytic activity of its kinase (20, 30). Indeed, transient expression of the kinase-inactive TRPM7–K1646R mutant in HEK-293T cells has lower protein expression levels than the WT protein (Fig. 1A). To elucidate the role of TRPM7 kinase activity in regulating channel expression, we employed tetracycline-inducible HEK-293 cells expressing WT HA-tagged TRPM7 (293–TRPM7–WT) or a kinase-inactive mutant (293–TRPM7–K1646A) to follow the kinetics of TRPM7 protein turnover (20). Following 24 h of tetracycline induction, the cell medium was replaced with tetracycline-free medium containing cycloheximide (CHX) to inhibit protein synthesis. TRPM7 protein levels were then analyzed at various time points over the following 24 h. The protein levels of TRPM7–WT were significantly more stable than that of the TRPM7–K1646A mutant, with protein levels of TRPM7–WT not appreciably decreasing over 24 h of CHX treatment (Fig. 1B). By contrast, the protein levels of TRPM7–K1646A rapidly declined over time, with an apparent half-life of 8.1 h (Fig. 1B), indicating that the kinase-inactive mutant is subject to increased protein turnover.
Figure 1.

Inactivation of TRPM7 kinase increases channel turnover. A, HA–TRPM7–WT (WT) and a kinase-inactive mutant TRPM7–K1646R (K1646R) were transiently expressed with GFP in HEK-293T cells for 24 h. SDS–PAGE and Western blotting demonstrated reduced protein expression of TRPM7–K1646R compared with the WT channel. GFP and actin served as transfection and loading controls, respectively. B, HEK-293 cells stably expressing HA–TRPM7–WT and HA–TRPM7–K1646A were induced with tetracycline for 24 h and replaced with fresh medium containing 10 μg/ml CHX to arrest protein synthesis. Cell lysates were harvested at various time points after CHX treatment. Equal amounts of total proteins were analyzed by SDS–PAGE and Western blotting. The relative TRPM7 levels were calculated by normalizing TRPM7 protein levels against the vinculin control in each sample. Time courses for TRPM7–WT and TRPM7–K1646A protein turnover are shown on the right (n = 3, means ± S.D.). C, HEK-293 cells stably expressing HA–TRPM7–WT and HA–TRPM7–K1646A were induced with tetracycline (Tet) and co-transfected with FLAG–ubiquitin (FLAG-Ub) for 24 h. Cell lysates were immunoprecipitated with anti-FLAG–agarose. TRPM7 and ubiquitinated proteins in FLAG–IP and lysates were probed by an anti-TRPM7 and anti-FLAG antibody, respectively. D, HEK-293 cells stably expressing HA–TRPM7–WT or TRPM7–K1646A were induced with tetracycline for 24 h and then replaced with fresh medium containing DMSO or MG132 (1 μm) for 24 h. Equal amounts of cell lysates were resolved by SDS–PAGE and Western blotting. The fold of TRPM7 protein level change was calculated by dividing relative TRPM7 protein levels (TRPM7/vinculin) in each MG132-treated sample against DMSO-treated controls. Replicates (n = 4) from two independent experiments were analyzed (means ± S.D.). *, p = 0.0158. E, HEK-293T cells transiently expressing HA–TRPM7–WT, HA–TRPM7–K1646R, and HA–TRPM7–G1618D were treated with DMSO or MG132 (1 μm) for 24 h. Equal amounts of cell lysates were resolved by SDS–PAGE and Western blotting. Protein quantification was performed as described above. Replicates (n = 4–6) from three independent experiments were analyzed (means ± S.D.). ****, p = 0.0001; ***, p = 0.0008.
To assess potential mechanisms controlling TRPM7 protein turnover, we first examined whether TRPM7 is ubiquitinated, because our MS analysis from our previous study identified several lysine residues on mouse TRPM7 (Lys-198, Lys-481, Lys-707, Lys-1019, and Lys-1253) that were modified with ubiquitin (30). We attempted to detect ubiquitinated TRPM7 proteins by co-expressing TRPM7 with ubiquitin in mammalian cells. However, we were unsuccessful in resolving ubiquitinated TRPM7 by SDS–PAGE and Western blotting using an anti-ubiquitin antibody. As an alternative approach, we used an anti-FLAG-agarose to immunoprecipitate ubiquitinated proteins and then probed the immunopurified proteins with an anti-TRPM7 antibody. Only the kinase-inactive TRPM7–K1646A mutant, but not TRPM7–WT, co-immunoprecipitated with FLAG–ubiquitin (Fig. 1C). Interestingly, the immunoprecipitated TRPM7–K1646A proteins appeared as a single band migrating at 220 kDa. This single band could indicate that TRPM7 was co-immunoprecipitated with other ubiquitinated proteins or that TRPM7 is modified by mono- or multiubiquitination. Nevertheless, the observation that only TRPM7–K1646A, but not TRPM7–WT, is strongly associated with ubiquitination, gave evidence that the turnover of the kinase-inactive TRPM7 mutant is mediated by the ubiquitin–proteasome pathway.
To further test whether TRPM7 protein expression is regulated by the proteasome, TRPM7-expressing 293–TRPM7–WT and 293–TRPM7–K1646A cells were induced with tetracycline for 24 h and then treated with the proteasome inhibitor MG132 for 24 h in the tetracycline-free medium. Proteasome inhibition by MG132 had little effect on TRPM7–WT levels (0.98 ± 0.18-fold change) but caused significant protein accumulation (2.22 ± 0.72-fold) of TRPM7–K1646A (Fig. 1D). By further testing two different kinase-inactive TRPM7 mutants, we confirmed that increased proteasome-mediated TRPM7 turnover is independent of the mechanism by which TRPM7 kinase is rendered catalytically inactive. TRPM7–WT and two kinase-inactive mutants, TRPM7–K1646R and TRPM7–G1618D, were transiently expressed in HEK-293T cells for 24 h and treated with MG132 for 24 h. Both kinase-inactive mutants had a significant amount of protein accumulation (TRPM7–K1646R, 3.19 ± 0.73-fold and TRPM7–G1618D, 2.84 ± 0.75-fold) compared with TRPM7–WT (1.01 ± 0.26-fold) (Fig. 1E). These results collectively support the previous finding that TRPM7–WT is a very long-lived protein, whereas the kinase-inactive mutant is more unstable and subject to protein turnover by the ubiquitin–proteasome pathway. These experimental findings also point to a role for TRPM7 kinase and autophosphorylation in controlling the protein stability and turnover of the channel.
TRPM7 kinase affects cellular localization of the channel in polarized epithelial cells
Our results thus far suggest a role for TRPM7's kinase in controlling protein levels. We next asked whether the kinase influences the cellular localization of the channel. TRPM7 mRNA is widely expressed in adult mice, with the highest expression in the kidney where the channel contributes to Mg2+ reabsorption (1, 2, 23). Protein expression of the native channel, however, was too low for us to detect its cellular localization by immunocytochemistry. Instead, we analyzed the cellular distribution of murine TRPM7 heterologously expressed in opossum kidney (OK) cells, a model proximal tubule epithelial cell line (44). Using confocal microscopy, we found that TRPM7–WT readily localized to the basal–lateral side of OK cells (Fig. 2A). A certain amount of TRPM7 was also located on the apical membrane, albeit at much lower levels, where it co-localized with the apical and microvilli marker NHERF1 (Fig. 2, A and B).
Figure 2.
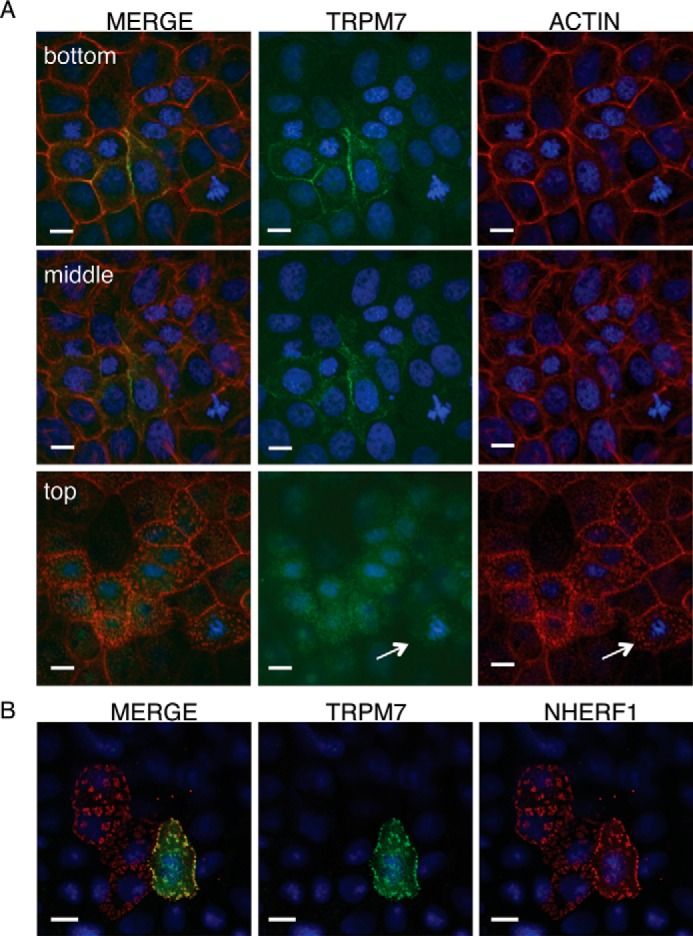
Cellular localization of TRPM7 in polarized epithelial cells. A, representative confocal Z-stack images of transiently expressed HA–TRPM7 (green) and actin (red) in OK proximal tubule epithelial cells 24 h after transfection. Scale bars, 10 μm. TRPM7 prominently localized to the basal–lateral sides of OK cells (bottom and middle rows) and weakly localized to the apical membrane of OK cells in actin patches (white arrow) (top row). B, on the apical membrane, HA–TRPM7 (green) co-localizes with the microvilli marker FLAG–NHERF1 (red) after co-expression for 24 h in OK cells.
In contrast, TRPM7–K1646R poorly localized to basal–lateral or apical membranes in OK cells and was instead retained intracellularly (Fig. 3A). The percentage of OK cells displaying peripheral membrane TRPM7 localization for the kinase-inactive mutant (18.12 ± 7.00%) was significantly lower than that for TRPM7–WT (71.44 ± 6.98%) (Fig. 3A), pointing to a role for autophosphorylation in controlling the cellular localization of the channel. Using the proteasome inhibition assay, we also confirmed that the kinase-inactive TRPM7 in OK cells was more prone to proteasome-mediated degradation compared with the TRPM7–WT (Fig. S1). We further validated TRPM7 cellular localization pattern using another well-established kidney tubule cell line, Madin–Darby canine kidney (MDCK) cells (45). Kinase inactivation of TRPM7 similarly interfered with the channel's localization to the cell border when transiently expressed in MDCK cells: only 5.16 ± 1.92% of TRPM7–K1646R-expressing cells exhibited localization of the channel to cell border compared with 46.95 ± 4.80% of cells expressing TRPM7–WT (Fig. 3B). The discovery that the TRPM7–K1646R fails to efficiently localize to the cell periphery in both OK and MDCK cells provides compelling evidence that TRPM7 kinase activity plays a critical role for localization of the channel in polarized epithelial cells. Because TRPM7's cellular localization is influenced by its kinase activity, we were also motivated to examine the cellular localization of TRPM7's homolog TRPM6. When expressed alone, TRPM6 does not localize to the cell border in OK or MDCK cells. Instead, we found that TRPM6 was primarily located in intracellular compartments (Fig. S2), similar to what was previously reported for TRPM6 expressed in HEK-293 cells (23, 46).
Figure 3.

Inactivation of TRPM7 kinase affects the cellular localization of the channel in polarized epithelial cells. A and C, representative confocal images of HA–TRPM7–WT and HA–TRPM7–K1646R cellular localization in OK (A) and MDCK (C) cells after 48 h of expression. Scale bars, 10 μm. TRPM7–WT readily localizes to the lateral membranes, whereas the kinase-inactive mutant TRPM7–K1646R is retained intracellularly. B and D, quantification of the percentage of cells expressing TRPM7 localized to apical and basal–lateral sides of OK (B) and MDCK (D) cells. Each data point represents a section on the slides where 100–150 cells stained positive for TRPM7 were counted. Two to three nonoverlapping sections per slide were analyzed. Three independent experiments were performed (means ± S.D.). ***, p = 0.0001; ****, p < 0.0001.
TRPM7–S1360 controls proteasome-mediated TRPM7 turnover and localization in epithelial cells
Our previous analysis of TRPM7 autophosphorylation uncovered many sites that are frequently phosphorylated on TRPM7 (Fig. 4A) (30). Because kinase inactivation resulted in a more rapid TRPM7 turnover, we next investigated whether any of the phosphorylation sites we had previously identified are involved in mediating proteasome-dependent degradation of the channel. We mutated phosphorylated TRPM7 residues Ser-1255, Ser-1360, Ser-1403, Ser-1502, Thr-1503, and Ser-1567 to alanine and tested whether these modifications affected the channel's sensitivity to proteasome-mediated degradation (Fig. 4B). In response to MG132 treatment, the protein level of TRPM7–S1360A significantly increased 2.10 ± 0.67-fold, whereas the protein levels of other TRPM7 alanine substitution mutants were unaffected (Fig. 4C).
Figure 4.

Phosphorylation of TRPM7 Ser-1360 controls channel turnover and its cellular localization in polarized epithelial cells. A, schematic representation of a full-length mouse TRPM7 showing the localization of frequently phosphorylated residues. CC, coiled-coil domain; S/T, serine/threonine-rich domain. B, HEK-293T cells expressing TRPM7–WT or phosphorylation site mutants (S1255A, S1360A, S1403A, S1502A/T1503A, and S1567A) were treated with DMSO or MG132 (1 μm) for 24 h. TRPM7 protein levels in cell lysates were analyzed by Western blotting. C, quantification of MG132-induced changes in TRPM7 protein levels for TRPM7–WT and phosphorylation site alanine mutants. Replicates (n = 2–6) from three independent experiments were analyzed (means ± S.D.). **, p = 0.003; ns, not significant. D, HEK-293T cells expressing TRPM7–WT or Ser-1360 mutants (S1360A, S1360D, and S1360E) were treated with DMSO or MG132 (1 μm) for 24 h. TRPM7 protein levels in cell lysates were analyzed by Western blotting. E, quantification of MG132-induced changes in TRPM7 protein levels for TRPM-WT and Ser-1360 mutants. Replicates (n = 4–6) from three independent experiments were analyzed (means ± S.D.). **, p = 0.0011; *, p = 0.011; *, p = 0.024. F, representative confocal images of cellular localization for HA–TRPM7–WT, S1360A, S1360D, and S1360E transiently expressed in OK cells for 48 h. Scale bars, 10 μm. G, quantification of TRPM7 cellular localization at the cell periphery. Data sets (n = 6) from two independent experiments were analyzed (means ± S.D.). ****, p < 0.0001.
Previously, Ser-1360 was identified as one of the most phosphorylated residues on TRPM7 when the channel is constitutively expressed in HEK-293 cells at low levels (Table S3 in Ref. 30). To test whether phosphorylation of Ser-1360 interferes with the proteasome-mediated turnover of TRPM7, we introduced phosphomimetic substitutions at Ser-1360 (TRPM7–S1360D and TRPM7–S1360E). Both TRPM7–S1360D and TRPM7–S1360E mutants were significantly more resistant to proteasome-mediated degradation than the TRPM7–S1360A mutant (Fig. 4, D and E). Interestingly, our previous MS analysis also detected phosphorylation of Ser-1360 on the TRPM7–K1646R kinase-inactive mutant, suggesting that Ser-1360 can be phosphorylated by other cellular kinases. We also investigated whether phosphomimetic substitutions of Ser-1360 could prevent the kinase-inactive forms of TRPM7 from being targeted by proteasome-mediated degradation but found them unable to do so, indicating that other regulatory phosphorylation sites may also be involved in controlling TRPM7 protein stability (Fig. S3). These results suggest that Ser-1360 phosphorylation is an essential post-translational signal for the control of proteasome-mediated turnover of the channel.
Because Ser-1360 phosphorylation affects TRPM7 protein stability, we were motivated to test whether Ser-1360 also affects the cellular localization of the channel (Fig. 4F). When expressed in OK cells, TRPM7–S1360A did not as efficiently localize to the cell border (20.25 ± 5.91%) compared with the WT channel (61.43 ± 4.34%) (Fig. 4G). By contrast, phosphomimetic substitutions of TRPM7–S1360D (51.80 ± 8.70%) and TRPM7–S1360E (50.97 ± 6.33%) were similarly efficient in localizing to the cell periphery as TRPM7–WT (Fig. 4G). Comparable findings were also observed for WT TRPM7 and TRPM7–S1360A/D/E mutants expressed in MDCK cells (Fig. S4). These results suggest a model in which that phosphorylation of TRPM7–S1360 allows the channel to evade proteasome-mediated degradation to allow for effective post-translational trafficking of the channel.
Identification of the phospho-binding protein 14-3-3θ as a binding partner of TRPM7
To gain additional insight into the mechanisms by which TRPM7's kinase affects the stability and localization of the channel, we conducted a yeast two-hybrid screen of a rat brain cDNA library by using the C terminus of the mouse TRPM7 (amino acid 1120–1863) as the bait (47). One of the positive clones identified by the screen was 14-3-3θ. 14-3-3θ belongs to a family of highly conserved and ubiquitously expressed proteins involved in many cell signaling pathways (48). Seven 14-3-3 isoforms are encoded in mammalian genomes, and they exist as homo- and heterodimers with each monomer capable of binding specifically to distinctive phospho-S/T residues on target proteins (49, 50). Binding of 14-3-3 proteins to their cognate protein ligands has been shown to regulate protein enzymatic activity, elicit protein conformational changes, influence the cellular localization proteins, and mediate cross-bridging with other proteins (51).
To confirm the interaction between TRPM7 and 14-3-3θ in mammalian cells, we first conducted GST-pulldown purification assays using GST fused to 14-3-3θ (GST–14-3-3θ) and lysates from cells expressing full-length TRPM7 (HA–TRPM7) or a GFP-fusion protein of the kinase-containing TRPM7–C-terminal tail (GFP–TRPM7–Cterm) (Fig. 5A). A substrate binding-deficient 14-3-3θ–K49E mutant was used as a negative control. The charge reversal of K49E in the 14-3-3θ substrate-binding groove disrupts 14-3-3θ's interaction with phosphorylated ligands (52). Strong interactions between both full-length HA–TRPM7 and GFP–TRPM7–Cterm with GST–14-3-3θ–WT were detected (Fig. 5A), indicating that TRPM7 is a bona fide binding partner of 14-3-3θ and that the strong binding between the two proteins occurs on TRPM7's C terminus. We also tested whether TRPM7 interacts with other 14-3-3 isoforms. The GST-pulldown purification assays demonstrated that all seven isoforms interacted with the GFP–TRPM7–Cterm protein with similar strength (Fig. S5).
Figure 5.

TRPM7 interacts with 14-3-3θ in an autophosphorylation-dependent manner. A, GST-pulldown purification assays using GSH agarose bound with 20 μg of GST–14-3-3θ, GST–14-3-3θ–K49E, or GST proteins were performed to pulldown full-length HA–TRPM7 (left panel) and GFP–TRPM7–Cterm (right panel) expressed in HEK-293T cells. HA–TRPM7 and GFP–TRPM7–Cterm were examined by Western blotting using anti-HA and anti-GFP antibodies respectively. GST proteins were visualized by SDS–PAGE and Coomassie staining. B, cell lysates from HEK-293T cell lysates expressing TRPM7–Cterm–WT and TRPM7–Cterm–K1646R were treated with phosphatase inhibitor (PPI) or a λ-phosphatase (Lamda PP) and subjected to GST-pulldown purification assay. C, HA–TRPM7–WT or HA–TRPM7–K1646R was co-expressed with FLAG–14-3-3θ-WT or binding-deficient mutant FLAG–14-3-3θ–K49E in HEK-293T cells for 48 h. TRPM7 was immunoprecipitated with HA–agarose and blotted with an anti-FLAG antibody to detect 14-3-3θ binding. The total amount of FLAG–14-3-3θ protein in cell lysates and the amount of HA–TRPM7 bound on the HA–agarose were blotted by an anti-FLAG and an anti-TRPM7 antibody, respectively.
Because 14-3-3θ is a phospho-binding protein, we tested whether the interaction between GFP–TRPM7–Cterm and 14-3-3θ is phosphorylation-dependent. A GST-pulldown assay was performed where cell lysates containing GFP–TRPM7–Cterm–WT or GFP–TRPM7–Cterm–K1646R were treated with λ-phosphatase and then subjected to a pulldown with GST–14-3-3θ (Fig. 5B). Both λ-phosphatase treatment of TRPM7 and TRPM7 kinase inactivation abolished the interaction between TRPM7 and 14-3-3θ, indicating that the interaction between the two proteins is autophosphorylation-dependent (Fig. 5B). To further validate the in vivo interaction between TRPM7 and 14-3-3θ, HA–TRPM7–WT, or the kinase-inactive HA–TRPM7–K1646R was transiently co-expressed with FLAG-tagged 14-3-3θ (FLAG–14-3-3θ) or FLAG–14-3-3θ–K49E in HEK-293T cells and subjected to immunoprecipitation. FLAG–14-3-3θ–WT but not the 14-3-3θ–K49E mutant readily co-immunoprecipitated with HA–TRPM7 (Fig. 5C). Likewise, FLAG–14-3-3θ co-immunoprecipitated with the WT channel but failed to do so with HA–TRPM7–K1646R (Fig. 5C), confirming that the in vivo interaction between 14-3-3θ and TRPM7 is autophosphorylation-dependent.
Binding of 14-3-3θ to TRPM7 requires autophosphorylation at S1403
Most of the 14-3-3 binding partners bear variations of consensus recognition motifs RSX(pS/T)XP or RX(Y/F)X(pS/T)XP containing a phospho-S/T residue (53). We screened for potential 14-3-3θ binding sites on TRPM7 by creating GFP–TRPM7–Cterm mutants bearing alanine substitutions at highly conserved serine residues within potential 14-3-3θ binding motifs (Ser-1403, Ser-1567, and Ser-1588) (Fig. 6A) and performed GST-pulldown purification assays with the mutants. The interaction between GFP–TRPM7–Cterm and GST–14-3-3θ was completely abolished in the S1403A mutant and partially reduced in S1567A and S1588A mutants (Fig. 6B). Our recent study of TRPM7 in vivo autophosphorylation found that both Ser-1403 and Ser-1567 are autophosphorylation sites in vivo, whereas Ser-1588 is not (30), thus eliminating Ser-1588 as a 14-3-3θ binding site. There are many examples where dimerized 14-3-3θ proteins bind to their targets using a stronger and a weaker binding site (53). Our data point to TRPM7 Ser-1403 as a strong binding site for 14-3-3θ and Ser-1567 as a potential weaker secondary site. To further explore the requirement of Ser-1403 phosphorylation for 14-3-3θ binding, we created phosphomimetic substitutions of aspartate and glutamate at Ser-1403 (S1403D and S1403E) on GFP–TRPM7–Cterm and tested the mutants' ability to interact with 14-3-3θ (Fig. 6C). It has been reported that 14-3-3 binding requires target site phosphorylation and cannot be replaced with phosphomimetic substitutions (54). Both GFP–TRPM7–Cterm–S1403D and S1403E mutants failed to interact with 14-3-3θ, indicating a specific requirement of Ser-1403 phosphorylation for 14-3-3θ binding (Fig. 6C). To complete our analysis, we also tested for binding of 14-3-3θ to Ser-1502 and Thr-1503, two highly phosphorylated residues of TRPM7 located in a 14-3-3 binding motif 1 (RSX(pS/T)XP) lacking a proline (Fig. S6A). A GST-pulldown purification assay using TRPM7-Cterm carrying S1502A/T1503A double mutation showed robust yet partially reduced interaction with 14-3-3θ (Fig. S6B).
Figure 6.

Binding of 14-3-3θ to TRPM7 requires autophosphorylation of the channel at Ser-1403. A, three highly conserved potential 14-3-3 binding motifs (underlining) were identified on TRPM7's C terminus. Two of the motifs contain TRPM7 autophosphorylation sites (red). B, GFP–TRPM7–Cterm–WT, S1403A, S1567A, and S1588A were expressed in HEK-293T cells and subjected to a GST-pulldown assay using GSH agarose bound with 20 μg of GST–14-3-3θ or GST proteins. GFP–TRPM7–Cterm was examined by Western blotting using an anti-GFP antibody. GST proteins were visualized by SDS–PAGE and Coomassie staining. Ser-1403 was identified as the major 14-3-3θ binding site on GFP–TRPM7–Cterm. C, GFP–TRPM7–Cterm–WT and Ser-1403 mutants (S1403A, S1403D, and S1403E) expressed in HEK-293T cells were subjected to the same GST-pulldown assay. GST–14-3-3θ failed to pulldown GFP–TRPM7–Cterm carrying mutations at Ser-1403, demonstrating a requirement of Ser-1403 phosphorylation for TRPM7 and 14-3-3θ interaction.
We also examined the status of in vivo phosphorylation of the potential 14-3-3θ binding sites on TRPM7 using a mouse mAb that recognizes phosphorylated 14-3-3θ motifs containing phosphoserine residues ((R/K)XXpSXP). TRPM7–WT, TRPM7–S1403A, TRPM7–S1502A/T1503A, and TRPM7–S1567A were transiently expressed in HEK-293T cells, immunoprecipitated, and probed with the phospho-14-3-3θ-motif antibody. The phospho-14-3-3-motif antibody recognized TRPM7–WT strongly but failed to detect TRPM7–S1403A and only weakly recognized TRPM7–S1567A (Fig. S6C). In contrast, alanine substitutions at Ser-1502 and Thr-1503, two residues that fall within a partial motif of the phospho-14-3-3θ-motif antibody, did not affect the recognition of TRPM7 by the 14-3-3θ-motif antibody. These results suggest that TRPM7–S1403 is the primary 14-3-3θ binding site and that other weaker secondary sites such as TRPM7–S1567 may be involved in mediating the interaction between 14-3-3θ and TRPM7 in vivo.
TRPM7 14-3-3–binding motifs influence the distribution of the channel in polarized epithelial cells
Having found that 14-3-3θ binds to the channel, we were motivated to understand 14-3-3θ's functional impact on TRPM7. We did not see evidence of 14-3-3θ affecting TRPM7's kinase activity (Fig. S7) or the channel's protein stability and total protein levels (Fig. 4B and Fig. S8). 14-3-3 proteins have been shown to regulate the cellular localization of ion channels and transporters (55–58). Because TRPM7 kinase-inactivation affected the channel's localization and TRPM7's interaction with 14-3-3θ is autophosphorylation-dependent, we investigated whether mutations in the 14-3-3θ-binding sites could affect the cellular localization of the channel (Fig. 7A). By expressing TRPM7–WT and 14-3-3-binding-deficient mutants in OK cells, we found that the peripheral localizations of TRPM7–S1403A (45.09 ± 7.25%) and TRPM7–S1567A (35.89 ± 6.28%) were significantly lower than TRPM7–WT (63.82 ± 6.17) (Fig. 7B).
Figure 7.

Mutations of 14-3-3θ–binding sites on TRPM7 affects channel localization in OK cells. A, representative confocal images of cellular localization for HA–TRPM7–WT, Ser-1403 mutants, and Ser-1567 mutants expressed in OK cells for 48 h. Scale bars, 10 μm. B, quantification of the peripheral cell localization of TRPM7–WT and indicated mutants. Data sets (n = 5–11) from five independent experiments were analyzed (means ± S.D.). ****, p < 0.0001; ***, p = 0.0001; **, p = 0.0016; *, p = 0.0191; ns, not significant.
We also created phosphomimetic substitutions at Ser-1403 (TRPM7–S1403D and TRPM7–S1403E) and Ser-1567 (TRPM7–S1567D and TRPM7–S1567E) and examined cellular localization of these TRPM7 mutants. If the localization defects of the TRPM7–S1403A and TRPM7–S1567A were due to the mutants' inability to interact with 14-3-3θ, phosphomimetic substitutions at these sites would be predicted not to rescue their intracellular localization, because we previously showed that phosphomimetic substitutions do not reconstitute the phosphorylation-dependent interaction of GFP–TRPM7–Cterm proteins with 14-3-3θ (Fig. 6C). On the other hand, if the localization defects of TRPM7–S1403A and TRPM7–S1567A were due to a lack of phosphorylation at these sites in a 14-3-3θ-binding-independent manner, then the phosphomimetic substitutions at the corresponding residues would be predicted to increase the channel's localization to the cell periphery. Indeed, we found that both TRPM7–S1403D (41.40 ± 3.60%) and TRPM7–S1403E (50.26 ± 8.35%) exhibited similarly reduced peripheral localization in OK cells as compared with the alanine mutant TRPM7–S1403A (45.09 ± 2.738%) (Fig. 7). The inability of the Ser-1403 phosphomimetic substitutions to stimulate the channel's localization to the cell border correlates with the inability of the phosphomimetic substitutions to increase the in vitro interaction of the channel with 14-3-3θ (Fig. 6C). In MDCK cells, similar localization patterns of the TRPM7–S1403 mutants were also observed (Fig. S9), supporting our hypothesis of the role of 14-3-3θ in regulating TRPM7 cellular localization. We attempted to disrupt in vivo binding between 14-3-3θ and TRPM7 by overexpressing difopein, a protein that scavenges 14-3-3θ proteins in preventing them from binding to targets (59). However, overexpression of difopein caused excessive cellular death, which precluded analysis of difopein's effect on TRPM7 localization. Interestingly, we found that phosphomimetic substitution of glutamate at TRPM7–S1567 had a higher level of peripheral localization (53.28 ± 8.93%) compared with the alanine mutant (35.89 ± 6.28%). Oddly, the introduction of aspartate at TRPM7–S1567 produced a severe trafficking defect (1.5 ± 1.52%), which is likely nonspecific (Fig. 7). Nevertheless, the ability of TRPM7–S1567E to localize to the cell border with an efficiency similar to TRPM7–WT suggests that phosphorylation of TRPM7–S1567 contributes to the regulation of TRPM7 trafficking through both 14-3-3θ-dependent and independent mechanisms. Likewise, we also observed that the TRPM7–S1502A/T1503A mutant, which does not strongly interact with 14-3-3θ, had a significant reduction in peripheral membrane localization (51.23 ± 3.8%) compared with the WT protein (63.82 ± 6.17) (Fig. S6D). These data suggest that multiple phosphorylation sites contribute to the trafficking of TRPM7 in vivo and that the 14-3-3θ binding site TRPM7–S1403 plays a vital role in regulating phosphorylation-dependent post-translational trafficking of TRPM7. Collectively these results underscore an important role of TRPM7 kinase activity in regulating the cellular trafficking of the channel.
Discussion
The results of our investigation point to a significant role for TRPM7's kinase in regulating proteasome-mediated turnover of the channel and controlling its cellular localization in polarized epithelial cells. We observed that TRPM7-kinase inactivation led to faster protein turnover and a higher level of protein ubiquitination. Inhibition of the proteasome with MG132 raised the expression levels of kinase-inactive TRPM7 mutants but not the WT protein. These findings suggest that the kinase activity of TRPM7 mediates the post-translational processing of the channel by mediating protein turnover via the ubiquitin–proteasome degradation pathway. We speculate that the ER-associated degradation pathway is at play in mediating the turnover of the TRPM7 kinase-inactive mutants. Our previous MS analysis of TRPM7 uncovered several ubiquitination sites on TRPM7: Lys-198 and Lys-481 for TRPM7–WT and Lys-481, Lys-707, Lys-1019, and Lys-1253 on TRPM7–K1646R (30). In particular, residue Lys-1019, which was exclusively identified on the kinase-inactive TRPM7, is located on the extracellular loop between the membrane-spanning S5 and S6 domains of the channel. Because the ubiquitin–proteasome system is located outside of the ER, ubiquitination of TRPM7–K1019 on the extracellular/ER luminal side is likely achieved after the protein is retrotranslocated into the cytoplasm, an essential step of ER-associated degradation processing for targeted substrates (60). We speculate that critical TRPM7 phosphorylation events occur in the ER during channel assembly. During the synthesis of oligomeric membrane proteins, monomers are first translated and then assembled into the protein's final oligomeric form (61). It was previously shown that TRPM7 kinase remains catalytically inactive until assembled into dimers (62). Dimer formation occurs through domain swapping between the “exchange segment” of one kinase monomer and the catalytic domain of the other monomer (62). Together, these data suggest at least one model for control of TRPM7 kinase activity. When first translated in the ER as a monomer, TRPM7's kinase is predicted to be catalytic inactive. Upon channel assembly, dimer formation occurs, and the TRPM7 kinase becomes active and capable of autophosphorylation. How TRPM7's kinase is regulated after channel assembly remains unknown.
In our previous study, MS analysis identified Ser-1360 as the most frequently phosphorylated residue on constitutively expressed TRPM7 (30). Our experiments point to phosphorylation of Ser-1360 as critical for controlling protein stability and cellular localization of the channel. The TRPM7–S1360A mutant phenotypically resembled the kinase-inactive TRPM7–K1646R mutant in the proteasome inhibition assay and its epithelial cell localization. Phosphomimetic substitutions of Ser-1360 effectively reversed the defects of the alanine mutants, stabilized TRPM7 protein levels, and allowed for efficient trafficking of the channel to the cell periphery. How phosphorylation of Ser-1360 influences channel turnover is not apparent. One possibility is that TRPM7–S1360 phosphorylation may interfere with the ubiquitination of TRPM7 by the preventing the channel from binding to proteasome-related proteins such as E3 ubiquitin ligases. Interestingly, our previous MS analysis detected Ser-1360 phosphorylation on the overexpressed TRPM7 kinase-inactive mutant, indicating that Ser-1360 is not necessarily just an autophosphorylation site but could also be targeted by other kinases. In probing for the functional significance of the TRPM7 kinase, TRPM7 kinase-inactive knockin mice have been generated (39, 63). Studies utilizing TRPM7 kinase-inactive knockin mice have revealed diverse phenotypes (39–43). In light of our discovery that TRPM7 stability and cellular localization are affected by the same kinase-inactivating mutation, extra care must be taken in teasing apart the mechanistic origins of phenotypes attributed to TRPM7's kinase. In this genetic background, it is also possible that other kinases, including TRPM6, may substitute for the loss of TRPM7 kinase catalytic activity.
When heterologously expressed in OK cells, TRPM7 is efficiently trafficked to basal–lateral membranes and less efficiently to the apical membrane, where ion exchange occurs between cells and the tubular fluid (44). Using a second kidney epithelial cell line, MDCK cells, we found a similar pattern of TRPM7 cellular localization. Our experiments establish polarized epithelial kidney cell lines as a useful system for investigating the mechanisms involved in regulating the cellular localization of the channel. In both OK and MDCK cells, we found that mutations disrupting TRPM7 kinase catalytic activity led to intracellular retention of the channel, suggesting a role for TRPM7 autophosphorylation in regulating cellular localization of the channel. Also, we discovered that 14-3-3θ, a member of a large phospho-binding signal protein family, interacts with TRPM7 in a TRPM7 kinase-dependent manner. Our biochemical analysis identified Ser-1403 and Ser-1567 as a strong and a weak 14-3-3θ binding site on TRPM7. Having more than one 14-3-3θ binding site potentially allows regulation of the 14-3-3-binding partners by two separate phosphorylation events (64, 65), but whether such a mechanism occurs for TRPM7 remains to be determined. Our study did not find evidence that 14-3-3θ binding to TRPM7 affects protein stability or kinase activity but instead suggested a role for 14-3-3θ in the regulation of the channel's cellular localization. Indeed, 14-3-3 proteins have been found to mediate both forward tracking to and endocytic recycling of membrane proteins from the plasma membrane (66, 67). Thus, it is reasonable to speculate that the autophosphorylation-dependent interaction of 14-3-3θ with TRPM7 affects the intracellular trafficking of the channel. Consistent with this hypothesis, in both OK and MDCK cells we found that alanine mutation at the primary 14-3-3 binding site on TRPM7 (TRPM7–S1403A) significantly interfered with localization of the channel to the cell border. Phosphomimetic substitutions of Ser-1403 (TRPM7–S1403D and –S1403E) did not alleviate defects in peripheral localization of the channel, which is in agreement with our biochemical assays showing that phosphomimetic substitutions at Ser-1403 are not effective in promoting the interaction between TRPM7 and 14-3-3θ. These results suggest that it is the disruption of phosphorylation-dependent 14-3-3θ interaction, not a lack of phosphorylation activity at this residue per se, that contributes to the defect of localization of the TRPM7–S1403 mutant channels. More work is required to evaluate the hypothesis that autophosphorylation-induced 14-3-3θ binding is part of the mechanism by which the kinase activity of TRPM7 regulates the trafficking and localization of the channel. However, it is worth noting that it has been reported that several 14-3-3 isoforms (14-3-3θ, 14-3-3ϵ, and 14-3-3β) are enriched in TRPM7-containing intracellular vesicles, further implicating 14-3-3 proteins in the regulation TRPM7 localization via vesicle trafficking (68).
Our investigation also revealed that there might be 14-3-3θ-independent mechanisms at play in controlling TRPM7's cellular localization. For example, TRPM7 exhibited peripheral localization defects when Ser-1567 was mutated to either alanine or aspartate. However, when Ser-1567 was replaced with a phosphomimetic glutamate substitution, the channel localized to the cell border as efficiently as the WT channel. Ser-1567 itself is a frequently phosphorylated residue of TRPM7, as has been identified by our previous MS analysis (30), and therefore could mediate TRPM7 localization through both 14-3-3θ-dependent and independent means. We also found that mutation of TRPM7–S1502/T1503, which does not strongly bind to 14-3-3θ, also disrupted TRPM7 localization in the OK cells. Thus, additional work is needed to determine how the Ser-1502/Thr-1503 and Ser-1567 TRPM7 phosphorylation sites affect the trafficking of the channel in different cell types and under varied physiological and pathological conditions.
TRPM7-mediated ion conductance appears to be involved in a broad array of cellular and developmental processes. Similarly, evidence points to multifaceted roles for TRPM7's kinase. Our results thus far hint that each of the identified autophosphorylation sites of TRPM7 represents a unique regulatory mechanism to control the activity of the channel kinase in response to diverse cellular signals. The discovery that the kinase activity of TRPM7 functionally regulates the channel protein stability and cellular localization provides us with our first glimpse as to why nature endowed an ion channel its own kinase function. The intrinsic kinase activity of TRPM7 constitutes a mechanism to allow the channel to respond nimbly to different cellular demands and cellular contexts. Also, the effect of kinase-inactivation on TRPM7 protein stability and localization may be cell type-specific, because of the differences in intracellular signaling pathways unique to each cell type. Still not understood is how the kinase itself is regulated in vivo and when and under what conditions phosphorylation of the critical residues we identified occur. A key goal going forward will be to determine signaling pathways that control TRPM7 kinase, which will be needed to fully elaborate the function and regulation of this unique bifunctional ion channel.
Experimental procedures
Constructs
The pcDNA5/FRT/TO-HA-mTRPM7 and the pcDNA6-mfGFP-mTRPM7-Cterm (amino acids 1120–1863) were described previously (47). Site-directed mutations were introduced into HA–TRPM7 and GFP–TRPM7–Cterm using the QuikChange mutagenesis kit (Stratagene). 14-3-3θ cDNA was amplified by PCR using mouse brain RNA after reverse transcription with the following primer pair: 5′-ATGGAGAAGACCGAGCTGATCCAG-3′ and 5′-TTAGTTTTCGGCCCCCTCTGCTG-3′. The ORF of 14-3-3θ was then subcloned into the pGEX-6P3 vector into the BamH1 and EcoR1 sites (pGEX–14-3-3θ) and into the HindIII and BamHI sites of pcDNA6–V5–HisB vector modified to contain the FLAG epitope (pcDNA6–FLAG) (pcDNA6–FLAG–14-3-3θ). The 14-3-3θ–K49E binding-deficient mutant was created using the site-directed mutagenesis system (QuikChange; Stratagene) using the following primers 5′-CTGTCGGTGGCCTACGAAAACGTGGTAGGG-3′ and 5′-CCCTACCACGTTTTCGTAGGCCACCGACAG-3′. cDNAs encoding various 14-3-3 isoforms were acquired from Addgene. GST–14-3-3wt (pGEX) was a gift of Dr. Joseph Avruch (14-3-3ζ, item 1944) (69). We express gratitude to Dr. Michael Yaffe's lab for the following plasmids: pGEX-2TK–14-3-3σ GST (14-3-3σ, item 11944), pGEX-2TK–14-3-3β–GST (14-3-3β, item 13276), pGEX-2TK–14-3-3η–GST (14-3-3η, item 13277), pGEX-4T1–14-3-3γ–GST (14-3-3γ, item 13280), and pGEX-4T1–ϵ-GST (14-3-3 ϵ, item 13279) (70). A pcDNA3/HA–ubiquitin construct (HA–ubiquitin, item 18712) was also purchased from Addgene and was a generous gift from Dr. Edward Yeh (71). The ubiquitin sequence was subcloned into the pcDNA6–FLAG vector between the BamHI and EcoR1 sites (pcDNA6–FLAG–Ub). FLAG–NHERF1 was from Addgene (pCMV–NHERF1–FL, item 28291, generous gift of Dr. Maria-Magdalena Georgescu) (72).
Cell culture and transfection
HEK-293T cells are from ATCC® (CRL-3216TM). 293–TRPM7–WT and 293–TRPM7–K1646A cells have been described previously (20). HEK-293 cells were cultured under standard conditions in Dulbecco's modified Eagle's medium (Thermo Fisher) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals). For biochemical analysis, transient transfections were performed using the Turbofect transfection reagent (Thermo Fisher) according to the manufacturer's protocol. The OK cell line was a generous gift of Dr. Judith A. Cole (Department of Biological Sciences, University of Memphis), cultured in Dulbecco's modified Eagle's medium/F-12 medium supplemented with 5% FBS. The MDCK cell line was a generous gift of Dr. Kenneth Irvine (Waksman Institute of Microbiology, Rutgers University) and maintained in minimal essential media (Thermo Fisher) supplemented with 5% FBS. Transfections of OK cells and MDCK cells were performed with Lipofectamine 3000 (Thermo Fisher) according to the manufacturer's protocol.
Protein stability assay
Approximately 0.6 × 106 293–TRPM7–WT and 293–TRPM7–K1646A cells were seeded onto 6-well plates coated with poly-l-lysine (Sigma–Aldrich). Two hours after seeding, the cells were treated with 5 μg/ml tetracycline to induce protein expression for 24 h. To arrest protein synthesis, the cells were replaced with fresh medium containing 10 μg/ml CHX. At various time points following CHX treatment, the cells were lysed in lysis buffer containing 50 mm Tris (pH 7.4), 150 mm NaCl, 1% IGEPAL CA-630 (Sigma–Aldrich), and the protease inhibitor mixture (Roche Life Sciences). The protein concentrations of cell lysates were measured using the Bradford assay (Thermo Fisher). Equal amounts of protein (40–80 μg/well) were loaded for each sample for SDS–PAGE and Western blotting. The rabbit polyclonal TRPM7 antibody (anti-TRPM7) was used to probe for HA–TRPM7 (20). Vinculin, which was used as loading control, was detected by Western blotting using a monoclonal anti-vinculin antibody (clone hVIN-1; Sigma–Aldrich). Vinculin was chosen as a loading control because it is a very stable protein and also because unlike other focal adhesion proteins (e.g. talin), vinculin's protein expression levels are not effected by overexpression of TRPM7 (20). The protein concentration in the cell lysates were quantified and normalized so that equal amounts of proteins were loaded for analysis by SDS–PAGE and Western blotting. To quantify protein levels, immunochemiluminescence signals of the Western blots with unsaturated bands were detected by X-ray film exposure using horseradish peroxidase secondary antibodies. Intensities of the protein bands on the scanned films were measured using the LI-COR Image Studio software (LI-COR Biotechnology). The relative levels of TRPM7 in each sample were calculated by normalizing TRPM7 protein intensity against vinculin, which was used as a loading control. To calculate protein half-lives, the relative protein levels of TRPM7–WT and TRPM7–K1646A were fitted to a one-phase exponential decay using GraphPad Prism 7 software (GraphPad Software, Inc.).
Detection of ubiquitination-associated TRPM7
Approximately 3 × 106 293–TRPM7–WT and 293–TRPM7–K1646A cells were seeded onto 10-cm culture plates. After attachment overnight, the cells were treated with 5 μg/ml tetracycline to induce TRPM7 expression and transfected with 5 μg of FLAG-tagged ubiquitin (pcDNA6–FLAG–Ub). After 24 h of protein expression, the cells were then treated with 20 μm of the proteasome inhibitor MG132 (Sigma–Aldrich) for 8 h and lysed in 1 ml of lysis buffer containing 10 mm of deubiquitinase inhibitor N-ethylmaleimide (Sigma–Aldrich). To detect ubiquitination-associated TRPM7, ubiquitinated proteins from cell lysates were immunoprecipitated using anti-FLAG® M2 agarose affinity gel (Sigma–Aldrich), washed three times with lysis buffer, and eluted to 50 μl of SDS sample buffer. Cell lysates and immunoprecipitated samples were resolved by SDS–PAGE and Western blotting following standard protocols. TRPM7 was detected by the rabbit anti-TRPM7 antibody, and ubiquitinated proteins were recognized by an anti-FLAG antibody (Sigma–Aldrich).
Proteasome inhibition assay with MG132
Approximately 0.7 × 106 293–TRPM7–WT and 293–TRPM7–K1646A cells were seeded on 6-well plates coated with Poly-l-lysine and induced with μg/ml tetracycline 2 h after seeding. After 24 h of expression, cell culture medium was replaced with fresh medium containing DMSO or 1 μm MG132 to inhibit proteasome activity. The cells were harvested 24 h after MG132 treatments in 200 μl of lysis buffer containing protease inhibitor mixture (Roche Life Sciences). For transient transfections, HEK-293T cells (0.3 × 106 cells/well) or OK cells (0.2 × 106 cells/well) were seeded onto 12-well plates with poly-l-lysine coating. The cells were transfected with HA–TRPM7–WT and mutants (0.5 μg for HEK-293T cells and 0.8 μg for OK cells) the next day at 70% confluence. After 24 h of expression, cell culture medium was replaced with fresh medium containing DMSO or 1 μm MG132. After MG132 treatment (24 h for HEK-293T cells and 9 h for OK cells), the cells were lysed in 100 μl of lysis buffer containing protease inhibitor mixture (Roche Life Sciences). Protein concentrations of the cell lysates were measured using Bradford assays (Thermo Fisher). Relative TRPM7 protein levels in each sample were calculated by normalizing TRPM7 protein levels against vinculin. The fold of protein level change induced by MG132 treatment was measured by dividing the relative TRPM7 protein levels in the MG132-treated group with that in DMSO-treated controls. Statistical analysis (Student's t test) was performed using GraphPad Prism 7 software (GraphPad Software, Inc.).
Immunocytochemistry
OK or MDCK cells (0.1 × 106 cells) were seeded on glass coverslips in 24-well plates and transfected the next day at 70% confluence. To confirm TRPM7 apical localization in OK cells, 1 μg of HA–TRPM7–WT and 0.5 μg of FLAG–NHERF1 were co-expressed in OK cells for 24 h. To compare the cellular localization between TRPM7–WT and mutant HA–TRPM7 (0.5 μg for OK cells and 0.8 μg for MDCK cells), the proteins were allowed to express for 48 h. For immunochemical staining, the cells were fixed with 4% paraformaldehyde at room temperature for 30 min, permeabilized in PBS with 0.1% Triton X-100 for 20 min, and blocked with 5% FBS//PBS at 30 °C for 1 h. A rabbit monoclonal anti-HA antibody (C29F4; Cell Signaling Technology) was used to detect the HA-tagged TRPM7, and a mouse anti-FLAG antibody (Sigma–Aldrich) was used to identify FLAG–NHERF1. Alexa FluorTM 488 and Alexa FluorTM 568 goat antibodies raised against rat and mouse were used as secondary antibodies (Thermo Fisher). F-actin was detected with Alexa FluorTM 568 phalloidin (Thermo Fisher). Images were obtained at the Rutgers RWJMS CORE confocal facility using a Yokogawa CSUX1-5000 microscope under 63× magnification using 488- and 561-nm excitation wavelengths. To quantify levels of TRPM7 peripheral localization, the cells were examined under an inverted fluorescent microscope (40× magnification for OK cells and 100× magnification for MDCK cells). On each slide, two to three nonoverlapping sections each containing 100–150 TRPM7 positive-staining cells were counted. The level of peripheral localization was measured as the percentage of cells exhibiting TRPM7 localization to cell periphery against the total number of TRPM7-expressing cells. Each data point represents one section of the slides. Statistical analysis (Student's t test) was performed using GraphPad Prism 7 software (GraphPad Software, Inc.).
GST-pulldown assay
GST–14-3-3 fusion proteins were expressed in transformed BL21-DE3 cells (Stratagene, CA). Bacterial cells were lysed by sonication in ice-cold PBS containing 1% Triton X-100 and protease inhibitor phenylmethylsulfonyl fluoride (Sigma–Aldrich). The bacterial cell lysates were then incubated with GSH agarose (Sigma–Aldrich) overnight at 4 °C with rotation. The agarose beads were washed with PBS with 1% Triton X-100. Concentrations of the GST proteins were measured by Coomassie stain on the SDS–PAGE gel using serial diluted BSA proteins as controls. To expressed TRPM7 proteins, 10 μg of HA–TRPM7 or GFP–TRPM7–Cterm plasmids were transiently transfected into HEK-293T cells plated in a 10-cm dish. After 24 h, the cells were lysed in 1 ml of mild lysis buffer containing protease inhibitor mixture (Roche Life Sciences) and phosphatase inhibitor mixture (EMD Millipore), and spun down at 14,000 × g for 10 min at 4 °C. For GST-pulldown assays, the cell lysate supernatants were incubated with GSH-agarose bound with 20 μg of GST–14-3-3θ or GST proteins overnight at 4 °C with rotation. The bound proteins were washed with 1 ml of PBS with 0.1% Triton X-100 three times by rotation and eluted into 50 μl of SDS sample buffer. Lysates input (20 μl) and pulldown samples were separated on SDS–PAGE gels and analyzed by immunoblotting. The rat monoclonal anti-HA (3F10) and a mouse monoclonal anti-GFP antibody (sc-9996; Santa Cruz Biotechnology, CA) were used to probe HA–TRPM7 and GFP–TRPM7–Cterm proteins, respectively. GST–14-3-3θ and GST proteins were visualized by SDS–PAGE and Coomassie staining. For experiments where protein dephosphorylation was conducted, a λ-phosphatase (New England Biolabs) was added to the lysis buffer in the absence of phosphatase inhibitor.
Immunoprecipitation
Approximately 1.0 × 106 HEK-293T cells were seeded onto 60-mm dishes. 2.5 μg of HA–TRPM7–WT or HA–TRPM7–K1646R plasmids were co-transfected with 1 μg of FLAG–14-3-3 the next day. After 48 h of expression, cell lysates were collected in 1 ml of mild lysis buffer containing protease inhibitor mixture (Roche Life Sciences) and phosphatase inhibitor mixture (EMD Millipore), immunoprecipitated by 20 μl of HA–agarose (Sigma–Aldrich) overnight at 4 °C. Immunoprecipitated (IP) samples were washed three times with lysis buffer and eluted into 50 μl of SDS sample buffer. Lysate input (20 μl) and IP samples were separated on SDS–PAGE gels and analyzed by immunoblotting. The rabbit polyclonal anti-TRPM7 antibody was used to detect HA–TRPM7, and a mouse anti-FLAG antibody was used to detect FLAG–14-3-3θ. To detect potential 14-3-3 binding motifs on TRPM7, HA–TRPM7 was immunopurified from HEK-293T cells by HA–agarose (Sigma–Aldrich) and resolved by SDS–PAGE. The in vivo 14-3-3 binding motifs were detected with a mouse monoclonal phosphoserine–14-3-3 binding Motif (4E2) antibody (9606; Cell Signaling Technology). The total amounts of TRPM7 proteins in the IP samples were detected by a rat anti-HA antibody (3F10; Roche Life Science).
In vitro kinase assay
HA–TRPM7 and GFP–TRPM7–Cterm proteins were transiently expressed in HEK-293T cells and immunoprecipitated using HA–agarose (Sigma–Aldrich) and streptavidin–agarose (to target the streptavidin-binding protein epitope in the GFP tag) (Stratagene). In vitro kinase assays were performed in a kinase buffer containing 50 mm MOPS (pH 7.2), 100 mm NaCl, 2.5 mm MnCl2, and 0.5 mm ATP. The kinase reactions were performed at 30 °C for 20 min in the presence of 4 μCi of [γ-32P]ATP with 5 μg of myelin basic protein as a substrate. Where indicated, 5 μg of GSH-eluted GST or GST–14-3-3θ proteins were added into the kinase reaction. The reactions were stopped by the addition of SDS sample buffer, and proteins in the reaction mix were resolved by SDS–PAGE. Proteins were detected by Coomassie Blue staining, and the gels were dried entirely using a gel dryer (Bio-Rad). Incorporation of [γ-32P]ATP into substrates was analyzed by autoradiography using Cyclone Plus phosphorimaging device (PerkinElmer Life Sciences).
Author contributions
N. C., L. L., N. A.-S., S. T., and L. W. R. investigation; N. C. and L. W. R. project administration; N. C. and L. W. R. writing-review and editing.
Supplementary Material
This work was supported by NIGMS, National Institutes of Health Grant GM080753, New Jersey Commission on Brain Injury Research Grant CBIR13IRG025 (to L. W. R.), and American Heart Association Predoctoral Fellowships 15PRE24890008 (to N. C.) and 17PRE33410172 (to L. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
- TRPM7
- transient receptor potential melastatin 7
- TRPM6
- transient receptor potential melastatin 6
- ER
- endoplasmic reticulum
- HEK
- human embryonic kidney
- OK
- opossum kidney
- MDCK
- Madin–Darby canine kidney
- S/T
- serine/threonine
- CHX
- cyclohexamide
- FBS
- fetal bovine serum
- IP
- immunoprecipitation
- GST
- glutathione S-transferase.
References
- 1. Nadler M. J., Hermosura M. C., Inabe K., Perraud A. L., Zhu Q., Stokes A. J., Kurosaki T., Kinet J. P., Penner R., Scharenberg A. M., and Fleig A. (2001) LTRPC7 is a Mg-ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595 10.1038/35079092 [DOI] [PubMed] [Google Scholar]
- 2. Runnels L. W., Yue L., and Clapham D. E. (2001) TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291, 1043–1047 10.1126/science.1058519 [DOI] [PubMed] [Google Scholar]
- 3. Ryazanova L. V., Pavur K. S., Petrov A. N., Dorovkov M. V., and Ryazanov A. G. (2001) Novel type of signaling molecules: protein kinases covalently linked with ion channels. Mol. Biol. 35, 271–283 10.1023/A:1010499720185 [DOI] [PubMed] [Google Scholar]
- 4. Monteilh-Zoller M. K., Hermosura M. C., Nadler M. J., Scharenberg A. M., Penner R., and Fleig A. (2003) TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 121, 49–60 10.1085/jgp.20028740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin J., Desai B. N., Navarro B., Donovan A., Andrews N. C., and Clapham D. E. (2008) Deletion of trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760 10.1126/science.1163493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin J., Wu L. J., Jun J., Cheng X., Xu H., Andrews N. C., and Clapham D. E. (2012) The channel kinase, TRPM7, is required for early embryonic development. Proc. Natl. Acad. Sci. U.S.A. 109, E225–E233 10.1073/pnas.1120033109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu W., Su L. T., Khadka D. K., Mezzacappa C., Komiya Y., Sato A., Habas R., and Runnels L. W. (2011) TRPM7 regulates gastrulation during vertebrate embryogenesis. Dev. Biol. 350, 348–357 10.1016/j.ydbio.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Komiya Y., Bai Z., Cai N., Lou L., Al-Saadi N., Mezzacappa C., Habas R., and Runnels L. W. (2017) A nonredundant role for the TRPM6 channel in neural tube closure. Sci. Rep. 7, 15623 10.1038/s41598-017-15855-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McNeill M. S., Paulsen J., Bonde G., Burnight E., Hsu M. Y., and Cornell R. A. (2007) Cell death of melanophores in zebrafish trpm7 mutant embryos depends on melanin synthesis. J. Invest. Dermatol. 127, 2020–2030 10.1038/sj.jid.5700710 [DOI] [PubMed] [Google Scholar]
- 10. Yee N. S., Zhou W., and Liang I. C. (2011) Transient receptor potential ion channel Trpm7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis. Model Mech. 4, 240–254 10.1242/dmm.004564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Low S. E., Amburgey K., Horstick E., Linsley J., Sprague S. M., Cui W. W., Zhou W., Hirata H., Saint-Amant L., Hume R. I., and Kuwada J. Y. (2011) TRPM7 is required within zebrafish sensory neurons for the activation of touch-evoked escape behaviors. J. Neurosci. 31, 11633–11644 10.1523/JNEUROSCI.4950-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Decker A. R., McNeill M. S., Lambert A. M., Overton J. D., Chen Y. C., Lorca R. A., Johnson N. A., Brockerhoff S. E., Mohapatra D. P., MacArthur H., Panula P., Masino M. A., Runnels L. W., and Cornell R. A. (2014) Abnormal differentiation of dopaminergic neurons in zebrafish trpm7 mutant larvae impairs development of the motor pattern. Dev. Biol. 386, 428–439 10.1016/j.ydbio.2013.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elizondo M. R., Arduini B. L., Paulsen J., MacDonald E. L., Sabel J. L., Henion P. D., Cornell R. A., and Parichy D. M. (2005) Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr. Biol. 15, 667–671 10.1016/j.cub.2005.02.050 [DOI] [PubMed] [Google Scholar]
- 14. Elizondo M. R., Budi E. H., and Parichy D. M. (2010) Trpm7 regulation of in vivo cation homeostasis and kidney function involves stanniocalcin 1 and fgf23. Endocrinology 151, 5700–5709 10.1210/en.2010-0853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schmitz C., Perraud A. L., Johnson C. O., Inabe K., Smith M. K., Penner R., Kurosaki T., Fleig A., and Scharenberg A. M. (2003) Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114, 191–200 10.1016/S0092-8674(03)00556-7 [DOI] [PubMed] [Google Scholar]
- 16. Aarts M., Iihara K., Wei W. L., Xiong Z. G., Arundine M., Cerwinski W., MacDonald J. F., and Tymianski M. (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877 10.1016/S0092-8674(03)01017-1 [DOI] [PubMed] [Google Scholar]
- 17. Wei W. L., Sun H. S., Olah M. E., Sun X., Czerwinska E., Czerwinski W., Mori Y., Orser B. A., Xiong Z. G., Jackson M. F., Tymianski M., and MacDonald J. F. (2007) TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc. Natl. Acad. Sci. U.S.A. 104, 16323–16328 10.1073/pnas.0701149104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sahni J., and Scharenberg A. M. (2008) TRPM7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 8, 84–93 10.1016/j.cmet.2008.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark K., Langeslag M., van Leeuwen B., Ran L., Ryazanov A. G., Figdor C. G., Moolenaar W. H., Jalink K., and van Leeuwen F. N. (2006) TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 25, 290–301 10.1038/sj.emboj.7600931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su L. T., Agapito M. A., Li M., Simonson W. T., Huttenlocher A., Habas R., Yue L., and Runnels L. W. (2006) TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J. Biol. Chem. 281, 11260–11270 10.1074/jbc.M512885200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su L. T., Liu W., Chen H. C., González-Pagán O., Habas R., and Runnels L. W. (2011) TRPM7 regulates polarized cell movements. Biochem. J. 434, 513–521 10.1042/BJ20101678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stritt S., Nurden P., Favier R., Favier M., Ferioli S., Gotru S. K., van Eeuwijk J. M., Schulze H., Nurden A. T., Lambert M. P., Turro E., Burger-Stritt S., Matsushita M., Mittermeier L., Ballerini P., et al. (2016) Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat. Commun. 7, 11097 10.1038/ncomms11097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chubanov V., Waldegger S., Mederos y Schnitzler M., Vitzthum H., Sassen M. C., Seyberth H. W., Konrad M., and Gudermann T. (2004) Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc. Natl. Acad. Sci. U.S.A. 101, 2894–2899 10.1073/pnas.0305252101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlingmann K. P., Weber S., Peters M., Niemann Nejsum L., Vitzthum H., Klingel K., Kratz M., Haddad E., Ristoff E., Dinour D., Syrrou M., Nielsen S., Sassen M., Waldegger S., Seyberth H. W., et al. (2002) Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat. Genet. 31, 166–170 10.1038/ng889 [DOI] [PubMed] [Google Scholar]
- 25. Walder R. Y., Landau D., Meyer P., Shalev H., Tsolia M., Borochowitz Z., Boettger M. B., Beck G. E., Englehardt R. K., Carmi R., and Sheffield V. C. (2002) Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 31, 171–174 10.1038/ng901 [DOI] [PubMed] [Google Scholar]
- 26. Ryazanova L. V., Rondon L. J., Zierler S., Hu Z., Galli J., Yamaguchi T. P., Mazur A., Fleig A., and Ryazanov A. G. (2010) TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 1, 109 10.1038/ncomms1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Middelbeek J., Kuipers A. J., Henneman L., Visser D., Eidhof I., van Horssen R., Wieringa B., Canisius S. V., Zwart W., Wessels L. F., Sweep F. C., Bult P., Span P. N., van Leeuwen F. N., and Jalink K. (2012) TRPM7 is required for breast tumor cell metastasis. Cancer Res. 72, 4250–4261 10.1158/1538-7445.AM2012-4250,10.1158/0008-5472.CAN-11-3863 [DOI] [PubMed] [Google Scholar]
- 28. Demeuse P., Penner R., and Fleig A. (2006) TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J. Gen. Physiol. 127, 421–434 10.1085/jgp.200509410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark K., Middelbeek J., Morrice N. A., Figdor C. G., Lasonder E., and van Leeuwen F. N. (2008) Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS One 3, e1876 10.1371/journal.pone.0001876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai N., Bai Z., Nanda V., and Runnels L. W. (2017) Mass spectrometric analysis of TRPM6 and TRPM7 phosphorylation reveals regulatory mechanisms of the channel-kinases. Sci. Rep. 7, 42739 10.1038/srep42739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim T. Y., Shin S. K., Song M. Y., Lee J. E., and Park K. S. (2012) Identification of the phosphorylation sites on intact TRPM7 channels from mammalian cells. Biochem. Biophys. Res. Commun. 417, 1030–1034 10.1016/j.bbrc.2011.12.085 [DOI] [PubMed] [Google Scholar]
- 32. Clark K., Middelbeek J., Lasonder E., Dulyaninova N. G., Morrice N. A., Ryazanov A. G., Bresnick A. R., Figdor C. G., and van Leeuwen F. N. (2008) TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J. Mol. Biol. 378, 790–803 10.1016/j.jmb.2008.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dorovkov M. V., and Ryazanov A. G. (2004) Phosphorylation of annexin I by TRPM7 channel-kinase. J. Biol. Chem. 279, 50643–50646 10.1074/jbc.C400441200 [DOI] [PubMed] [Google Scholar]
- 34. Deason-Towne F., Perraud A. L., and Schmitz C. (2012) Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase Cγ2 (PLCγ2) using TRPM7-kinase. Cell Signal. 24, 2070–2075 10.1016/j.cellsig.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ryazanova L. V., Dorovkov M. V., Ansari A., and Ryazanov A. G. (2004) Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J. Biol. Chem. 279, 3708–3716 10.1074/jbc.M308820200 [DOI] [PubMed] [Google Scholar]
- 36. Perraud A. L., Zhao X., Ryazanov A. G., and Schmitz C. (2011) The channel-kinase TRPM7 regulates phosphorylation of the translational factor eEF2 via eEF2-k. Cell Signal. 23, 586–593 10.1016/j.cellsig.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krapivinsky G., Krapivinsky L., Manasian Y., and Clapham D. E. (2014) The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell 157, 1061–1072 10.1016/j.cell.2014.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ogunrinde A., Pereira R. D., Beaton N., Lam D. H., Whetstone C., and Hill C. E. (2017) Hepatocellular differentiation status is characterized by distinct subnuclear localization and form of the chanzyme TRPM7. Differentiation 96, 15–25 10.1016/j.diff.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 39. Ryazanova L. V., Hu Z., Suzuki S., Chubanov V., Fleig A., and Ryazanov A. G. (2014) Elucidating the role of the TRPM7 α-kinase: TRPM7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci. Rep. 4, 7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zierler S., Sumoza-Toledo A., Suzuki S., Dúill F. Ó., Ryazanova L. V., Penner R., Ryazanov A. G., and Fleig A. (2016) TRPM7 kinase activity regulates murine mast cell degranulation. J. Physiol. 594, 2957–2970 10.1113/JP271564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Romagnani A., Vettore V., Rezzonico-Jost T., Hampe S., Rottoli E., Nadolni W., Perotti M., Meier M. A., Hermanns C., Geiger S., Wennemuth G., Recordati C., Matsushita M., Muehlich S., Proietti M., et al. (2017) TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat. Commun. 8, 1917 10.1038/s41467-017-01960-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ogata K., Tsumuraya T., Oka K., Shin M., Okamoto F., Kajiya H., Katagiri C., Ozaki M., Matsushita M., and Okabe K. (2017) The crucial role of the TRPM7 kinase domain in the early stage of amelogenesis. Sci. Rep. 7, 18099 10.1038/s41598-017-18291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gotru S. K., Chen W., Kraft P., Becker I. C., Wolf K., Stritt S., Zierler S., Hermanns H. M., Rao D., Perraud A. L., Schmitz C., Zahedi R. P., Noy P. J., Tomlinson M. G., Dandekar T., et al. (2018) TRPM7 (transient receptor potential melastatin-like 7 channel) kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler. Thromb. Vasc. Biol. 38, 344–352 10.1161/ATVBAHA.117.310391 [DOI] [PubMed] [Google Scholar]
- 44. Courjault F., Gerin B., Leroy D., Chevalier J., and Toutain H. (1991) Morphological and biochemical characterization of the opossum kidney cell line and primary cultures of rabbit proximal tubule cells in serum-free defined medium. Cell Biol. Int. Rep. 15, 1225–1234 10.1016/0309-1651(91)90094-Y [DOI] [PubMed] [Google Scholar]
- 45. Leighton J., Estes L. W., Mansukhani S., and Brada Z. (1970) A cell line derived from normal dog kidney (MDCK) exhibiting qualities of papillary adenocarcinoma and of renal tubular epithelium. Cancer 26, 1022–1028 10.1002/1097-0142(197011)26:5%3C1022::AID-CNCR2820260509%3E3.0.CO%3B2-M [DOI] [PubMed] [Google Scholar]
- 46. Brandao K., Deason-Towne F., Zhao X., Perraud A. L., and Schmitz C. (2014) TRPM6 kinase activity regulates TRPM7 trafficking and inhibits cellular growth under hypomagnesic conditions. Cell Mol. Life Sci. 71, 4853–4867 10.1007/s00018-014-1647-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Overton J. D., Komiya Y., Mezzacappa C., Nama K., Cai N., Lou L., Fedeles S. V., Habas R., and Runnels L. W. (2015) Hepatocystin is essential for TRPM7 function during early embryogenesis. Sci. Rep. 5, 18395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ichimura T., Isobe T., Okuyama T., Takahashi N., Araki K., Kuwano R., and Takahashi Y. (1988) Molecular cloning of cDNA coding for brain-specific 14-3-3 protein, a protein kinase-dependent activator of tyrosine and tryptophan hydroxylases. Proc. Natl. Acad. Sci. U.S.A. 85, 7084–7088 10.1073/pnas.85.19.7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Muslin A. J., Tanner J. W., Allen P. M., and Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 10.1016/S0092-8674(00)81067-3 [DOI] [PubMed] [Google Scholar]
- 50. Ichimura T., Ito M., Itagaki C., Takahashi M., Horigome T., Omata S., Ohno S., and Isobe T. (1997) The 14-3-3 protein binds its target proteins with a common site located towards the C-terminus. FEBS Lett. 413, 273–276 10.1016/S0014-5793(97)00910-1 [DOI] [PubMed] [Google Scholar]
- 51. Dougherty M. K., and Morrison D. K. (2004) Unlocking the code of 14-3-3. J. Cell Sci. 117, 1875–1884 10.1242/jcs.01171 [DOI] [PubMed] [Google Scholar]
- 52. Zhang L., Wang H., Liu D., Liddington R., and Fu H. (1997) Raf-1 kinase and exoenzyme S interact with 14-3-3ζ through a common site involving lysine 49. J. Biol. Chem. 272, 13717–13724 10.1074/jbc.272.21.13717 [DOI] [PubMed] [Google Scholar]
- 53. Johnson C., Crowther S., Stafford M. J., Campbell D. G., Toth R., and MacKintosh C. (2010) Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 427, 69–78 10.1042/BJ20091834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ku N. O., Liao J., and Omary M. B. (1998) Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J. 17, 1892–1906 10.1093/emboj/17.7.1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cho C. H., Kim E., Lee Y. S., Yarishkin O., Yoo J. C., Park J. Y., Hong S. G., and Hwang E. M. (2014) Depletion of 14-3-3γ reduces the surface expression of transient receptor potential melastatin 4b (TRPM4b) channels and attenuates TRPM4b-mediated glutamate-induced neuronal cell death. Mol. Brain 7, 52 10.1186/s13041-014-0052-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moeller H. B., Slengerik-Hansen J., Aroankins T., Assentoft M., MacAulay N., Moestrup S. K., Bhalla V., and Fenton R. A. (2016) Regulation of the water channel aquaporin-2 via 14-3-3θ and -ζ. J. Biol. Chem. 291, 2469–2484 10.1074/jbc.M115.691121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kilisch M., Lytovchenko O., Arakel E. C., Bertinetti D., and Schwappach B. (2016) A dual phosphorylation switch controls 14-3-3-dependent cell surface expression of TASK-1. J. Cell Sci. 129, 831–842 10.1242/jcs.180182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernández-Orth J., Ehling P., Ruck T., Pankratz S., Hofmann M. S., Landgraf P., Dieterich D. C., Smalla K. H., Kähne T., Seebohm G., Budde T., Wiendl H., Bittner S., and Meuth S. G. (2017) 14-3-3 proteins regulate K2P5.1 surface expression on T lymphocytes. Traffic 18, 29–43 10.1111/tra.12455 [DOI] [PubMed] [Google Scholar]
- 59. Masters S. C., and Fu H. (2001) 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200 10.1074/jbc.M105971200 [DOI] [PubMed] [Google Scholar]
- 60. MacGurn J. A., Hsu P. C., and Emr S. D. (2012) Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231–259 10.1146/annurev-biochem-060210-093619 [DOI] [PubMed] [Google Scholar]
- 61. Traxler B., and Beckwith J. (1992) Assembly of a hetero-oligomeric membrane protein complex. Proc. Natl. Acad. Sci. U.S.A. 89, 10852–108526 10.1073/pnas.89.22.10852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crawley S. W., and Côté G. P. (2009) Identification of dimer interactions required for the catalytic activity of the TRPM7 α-kinase domain. Biochem. J. 420, 115–122 10.1042/BJ20081405 Côté [DOI] [PubMed] [Google Scholar]
- 63. Kaitsuka T., Katagiri C., Beesetty P., Nakamura K., Hourani S., Tomizawa K., Kozak J. A., and Matsushita M. (2014) Inactivation of TRPM7 kinase activity does not impair its channel function in mice. Sci. Rep. 4, 5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mohammad D. K., Nore B. F., Hussain A., Gustafsson M. O., Mohamed A. J., and Smith C. I. (2013) Dual phosphorylation of Btk by Akt/protein kinase b provides docking for 14-3-3ζ, regulates shuttling, and attenuates both tonic and induced signaling in B cells. Mol. Cell. Biol. 33, 3214–3226 10.1128/MCB.00247-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Van Der Hoeven P. C., Van Der Wal J. C., Ruurs P., Van Dijk M. C., and Van Blitterswijk J. (2000) 14-3-3 isotypes facilitate coupling of protein kinase C-ζ to Raf-1: negative regulation by 14-3-3 phosphorylation. Biochem. J. 345, 297–306 10.1042/0264-6021:3450297,10.1042/bj3450297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rajan S., Preisig-Müller R., Wischmeyer E., Nehring R., Hanley P. J., Renigunta V., Musset B., Schlichthörl G., Derst C., Karschin A., and Daut J. (2002) Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J. Physiol. 545, 13–26 10.1113/jphysiol.2002.027052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gabriel L., Lvov A., Orthodoxou D., Rittenhouse A. R., Kobertz W. R., and Melikian H. E. (2012) The acid-sensitive, anesthetic-activated potassium leak channel, KCNK3, is regulated by 14-3-3β-dependent, protein kinase C (PKC)-mediated endocytic trafficking. J. Biol. Chem. 287, 32354–32366 10.1074/jbc.M112.391458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abiria S. A., Krapivinsky G., Sah R., Santa-Cruz A. G., Chaudhuri D., Zhang J., Adstamongkonkul P., DeCaen P. G., and Clapham D. E. (2017) TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc. Natl. Acad. Sci. U.S.A. 114, E6079–E6088 10.1073/pnas.1707380114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luo Z. J., Zhang X. F., Rapp U., and Avruch J. (1995) Identification of the 14.3.3 ζ domains important for self-association and Raf binding. J. Biol. Chem. 270, 23681–23687 10.1074/jbc.270.40.23681 [DOI] [PubMed] [Google Scholar]
- 70. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., and Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 10.1016/S0092-8674(00)80487-0 [DOI] [PubMed] [Google Scholar]
- 71. Kamitani T., Kito K., Nguyen H. P., and Yeh E. T. (1997) Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562 10.1074/jbc.272.45.28557 [DOI] [PubMed] [Google Scholar]
- 72. Morales F. C., Takahashi Y., Momin S., Adams H., Chen X., and Georgescu M. M. (2007) NHERF1/EBP50 head-to-tail intramolecular interaction masks association with PDZ domain ligands. Mol. Cell Biol. 27, 2527–2537 10.1128/MCB.01372-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.