

Abstract
Our previous work has established that the metabolic sensor AMP-activated protein kinase (AMPK) inhibits the epithelial Na+ channel (ENaC) by promoting its binding to neural precursor cell–expressed, developmentally down-regulated 4-2, E3 ubiquitin protein ligase (Nedd4-2). Here, using MS analysis and in vitro phosphorylation, we show that AMPK phosphorylates Nedd4-2 at the Ser-444 (Xenopus Nedd4-2) site critical for Nedd4-2 stability. We further demonstrate that the Pak-interacting exchange factor β1Pix is required for AMPK-mediated inhibition of ENaC-dependent currents in both CHO and murine kidney cortical collecting duct (CCD) cells. Short hairpin RNA–mediated knockdown of β1Pix expression in CCD cells attenuated the inhibitory effect of AMPK activators on ENaC currents. Moreover, overexpression of a β1Pix dimerization–deficient mutant unable to bind 14-3-3 proteins (Δ602–611) increased ENaC currents in CCD cells, whereas overexpression of WT β1Pix had the opposite effect. Using additional immunoblotting and co-immunoprecipitation experiments, we found that treatment with AMPK activators promoted the binding of β1Pix to 14-3-3 proteins in CCD cells. However, the association between Nedd4-2 and 14-3-3 proteins was not consistently affected by AMPK activation, β1Pix knockdown, or overexpression of WT β1Pix or the β1Pix-Δ602–611 mutant. Moreover, we found that β1Pix is important for phosphorylation of the aforementioned Nedd4-2 site critical for its stability. Overall, these findings elucidate novel molecular mechanisms by which AMPK regulates ENaC. Specifically, they indicate that AMPK promotes the assembly of β1Pix, 14-3-3 proteins, and Nedd4-2 into a complex that inhibits ENaC by enhancing Nedd4-2 binding to ENaC and its degradation.
Keywords: AMP-activated kinase (AMPK), epithelial sodium channel (ENaC), ubiquitin ligase, sodium transport, 14-3-3 protein, kidney, β-Pix
Introduction
ENaC2 is apically expressed in many salt-reabsorbing epithelia (1). In the aldosterone-sensitive distal nephron of the kidney and colonic epithelia, ENaC is a critical regulator of sodium balance, blood volume, and blood pressure (2). ENaC also plays an important role in fluid reabsorption at the air–liquid interface in distal lung airways, which determines the rate of mucociliary transport (2, 3). ENaC appears to form a heterotrimeric channel composed of three subunits (α, β, and γ), each of which contains two transmembrane domains, cytosolic amino and carboxyl termini, and an extracellular loop (4, 5). Abnormal ENaC activity is a characteristic feature in patients with type 1 pseudohypoaldosteronism, Liddle syndrome, and cystic fibrosis (6).
ENaC expression and activity at the apical membrane are regulated by Nedd4-2, a member of the E6-associated protein C Terminus (HECT) family of E3 ubiquitin ligases, which, in turn, is regulated by several kinases, including serum- and glucocorticoid-induced kinase 1 (SGK-1) and PKA (7–9). Nedd4-2 directly binds to PY motifs on the C termini of ENaC subunits, promoting ENaC ubiquitination, internalization, and degradation. SGK1 and PKA activate ENaC largely through a mechanism involving phosphorylation of xNedd4-2 predominantly at residues Ser-338, Thr-363, and Ser-444, which appears to enhance the association of Nedd4-2 with 14-3-3 scaffolding proteins and thereby prevent Nedd4-2-mediated degradation of ENaC (10–14).
AMPK is a Ser/Thr kinase that exists as a heterotrimer comprised of a catalytic α-subunit and regulatory β- and γ-subunits. AMPK is known as a key homeostatic regulator of energy balance at the cellular and systemic levels. A reduction in the energy charge of the cell (increased AMP or ADP or reduced ATP) or increased calcium, osmotic, or oxidative stress elicits AMPK activation during conditions of metabolic and other cellular stresses (15, 16). Phosphorylation of Thr-172 within the α subunit accounts for most of the activation of AMPK by upstream kinases, such as liver kinase B1 (LKB1) or Ca2+-calmodulin-dependent kinase kinase-β (17, 18). Besides being an energy sensor, AMPK also plays an important role in apoptosis, cell growth, gene transcription, and protein synthesis (19). In recent years, reports regarding the roles of AMPK in renal physiology and disease, such as podocyte function, renal hypertrophy, ischemia, inflammation, diabetes, and polycystic kidney disease, have rapidly escalated (20–25). We and others have also demonstrated that AMPK regulates a variety of ion transport proteins (16), including inhibition of ENaC by promoting Nedd4-2 interaction with β-ENaC and enhancing Nedd4-2–dependent ubiquitination, internalization, and degradation of the channel (26–28).
Several small G proteins, including K-Ras, Rho A, and Rab11, have been shown to regulate ENaC activity (29–31). Given that G protein activation relies on the coordinated action of guanine nucleotide exchange factors (GEFs), GEFs could be involved in ENaC modulation. p21-activated kinase (PAK)–interacting exchange factor β (βPix) is a member of the diffuse B cell lymphoma family of Rho-GEFs, existing in two major isoforms, β1Pix and β2Pix. In kidneys, β1Pix has emerged as a main Pix isoform (32, 33) and contains a Dbl homology domain, a pleckstrin homology domain, and an Src homology 3 domain for binding PAK through a proline-rich region. β1Pix also has a GIT1 (G protein–coupled receptor kinase interactor 1) binding domain and a leucine zipper domain responsible for β1Pix dimerization (34, 35). By using tagged 14-3-3 proteins (36) and tandem affinity purification and LC-MS methods (37), dimeric β1Pix was found to bind to dimeric 14-3-3 proteins. Of note, we previously discovered β1Pix to be a critical regulatory co-factor in the endothelin-1 (ET-1)-dependent regulation of ENaC via Nedd4-2 and 14-3-3 proteins (38).
In this study, we show that AMPK inhibits ENaC by direct phosphorylation of xNedd4-2 at Ser-444 (equivalent to Ser-328 of mNedd4-2), a site we previously showed enhances Nedd4-2 stability (14). Through ENaC current recordings in Chinese hamster ovary (CHO) cells and in mouse polarized CCD cells, we also demonstrate that β1Pix expression and function are required for ENaC inhibition by AMPK. Treatment with AMPK activators increases the binding of β1Pix to 14-3-3 proteins in two distinct CCD cell lines. Moreover, β1Pix knockdown inhibits mNedd4-2 phosphorylation at Ser-328, reducing Nedd4-2 protein expression. Overall, our results suggest that functional β1Pix is critical for Nedd4-2 stability and AMPK may enhance Nedd4-2-dependent ENaC degradation by promoting the formation of a β1Pix–Nedd4-2–14-3-3 protein complex. These findings shed new light on the molecular mechanisms by which AMPK regulates ENaC.
Results
AMPK phosphorylates Nedd4-2 at Ser-444
Our previous work has shown that AMPK could phosphorylate Nedd4-2 in vitro and in intact cells (27). To identify the AMPK phosphorylation site(s) on Nedd4-2, purified GST-xNedd4-2 was expressed in Escherichia coli and then subjected to in vitro phosphorylation in the presence of purified active AMPK holoenzyme and [γ-32P]ATP. Phosphorylation site mapping of tryptic fragments was performed by MALDI-TOF MS and solid-phase sequencing as described previously (39). We found that AMPK phosphorylated xNedd4-2 at Ser-444 (Fig. 1, A and B). AMPK phosphorylation at this site was confirmed by comparing [γ-32P]ATP incorporation into the WT with a Ser-to-Ala mutant (S444A) Nedd4-2 in vitro, where significantly reduced 32P incorporation was observed in the S444A mutant compared with WT Nedd4-2 (Fig. 1C).
Figure 1.

AMPK phosphorylates xNedd4-2 at Ser-444. A, isolated phosphopeptides were derived from the HPLC system and subjected to MALDI-TOF MS for mass fingerprinting. Peptides with a mass shift of +80 Da (HPO3) were then selected for MS/MS, and phosphorylation was confirmed by a neutral loss of −98 Da (H3PO4 or HPO3 and H2O) during fragmentation. B, solid-phase sequencing of the radiolabeled peptides was performed to verify the phosphosite. Liberated single amino acids were collected and spotted onto a diethylaminoethyl cellulose membrane after each cycle of N-terminal Edman degradation. Autoradiography was then conducted to detect the respective phosphorylated residues. C, S444A reduced the level of AMPK-mediated phosphorylation of xNedd4-2. FLAG-tagged xNedd4-2 (WT or S444A) constructs were transiently transfected into HEK293 cells, immunoprecipitated from cell lysates, and exposed to purified AMPK or buffer alone and [γ-32P]ATP. After SDS-PAGE and transfer to a nitrocellulose membrane, immunoblotting and phosphorimaging were performed on the same membrane.
Functional β1Pix is required for ENaC inhibition by AMPK
It has been reported that phosphorylated Ser-444 on xNedd4-2 could serve as a binding site for 14-3-3 proteins, which may act to sequester Nedd4-2 and thereby prevent its interaction with ENaC (11, 40). Our previous study also showed that a Nedd4-2 mutant (S444A) has a dramatically shorter cellular half-life than WT Nedd4-2, but this property is not dependent on binding to 14-3-3s (14). Because Ser-444 is also phosphorylated by other kinases besides AMPK, including SGK1, PKA, and IκB kinase-β (IKKβ), which have opposite effects on ENaC expression (9–11, 41, 42), we reasoned that AMPK phosphorylation of Nedd4-2 alone could not account for the ENaC regulation. As it has been demonstrated that β1Pix is involved in long-term ENaC inhibition by ET-1 through impairing 14-3-3β binding to Nedd4-2 (38), we hypothesized that β1Pix may also play an important role in AMPK-dependent ENaC inhibition. To examine this hypothesis, ENaC was co-expressed with either WT or a dimerization-deficient deletion tract (Δ602–611) mutant of β1Pix in CHO cells. AICAR treatment (1 mm) was used to activate AMPK (43). Representative current sweeps evoked by a voltage ramp from +60 to −100 mV (holding potential at 40 mV) recorded in whole-cell patch-clamp mode are shown before and after treatment with amiloride (10 μm), an inhibitor of ENaC (Fig. 2A). As summarized in Fig. 2B, either AICAR treatment or co-expression of WT β1Pix significantly decreased ENaC-dependent currents. However, there was no apparent additive effect between WT β1Pix overexpression and AICAR treatment. These effects were prevented with co-expression of the β1Pix-Δ602–611 mutant. Besides using the AMPK activator, we also co-expressed ENaC with either pTracer vector alone (control), a dominant-negative (DN) AMPK-α1–K45R mutant, or a constitutively active (CA) AMPK-γ1–R70Q mutant in CHO cells to modulate AMPK activity. As shown in Fig. 2C, CA-AMPK inhibited the ENaC current relative to DN-AMPK and control, but this inhibition was prevented in cells overexpressing the dimerization-deficient Δ602–611 β1Pix mutant. Together, these findings suggest that AMPK inhibits ENaC activity via β1Pix. This mechanism requires functional dimeric β1Pix, which is known to be required for β1Pix binding to 14-3-3 proteins (44).
Figure 2.

β1Pix is necessary for the inhibition of ENaC by AMPK. A, overlays of typical macroscopic current traces before and after 10-μm amiloride treatment from whole-cell patch-clamped CHO cells expressing mouse ENaC (mENaC) with or without WT β1Pix or β1Pix-Δ602–611 either in the presence or absence of pretreatment with AMPK activator (1 mm AICAR, 12 h). Currents were evoked with a voltage ramp (+60 to −100 mV from a holding potential of 40 mV). B, summary graph of the mean ± S.E. for amiloride-sensitive current density at −80 mV for CHO cells expressing mENaC alone or with β1Pix or β1Pix-Δ602–611 mutant with or without 1 mm AICAR as indicated. pF, picofarad. C, summary graph of the mean ± S.E. for amiloride-sensitive current density for CHO cells expressing ENaC and either pTracer (control), DN AMPK-α1-K45R, or CA AMPK-γ1-R70Q with or without β1Pix-Δ602–611. p values are shown for the indicated comparisons.
Overexpression of WT versus mutant β1Pix modulates ENaC currents in polarized mpkCCDc14 cells
To examine whether the inhibitory effect of β1Pix on ENaC currents can also be observed in more physiologically relevant mouse kidney cortical collecting duct epithelial cells, V5-tagged WT β1Pix or β1Pix-Δ602–611 was stably transduced into mpkCCDc14 cells (45) for inducible overexpression with a Tet-On system. After cells polarized on Transwells were exposed to doxycycline (Dox, 2 μg/ml) for 3 days, amiloride-sensitive ENaC currents were measured using an epithelial volt-ohm–meter (EVOM). Overexpression of β1Pix-Δ602–611, which is unable to bind 14-3-3 proteins, slightly increased ENaC equivalent short-circuit currents in mpkCCDc14 cells, whereas overexpression of WT β1Pix had the opposite effect (Fig. 3A), consistent with the results found in CHO cells. After EVOM measurements, cells were harvested to confirm the expression of V5-tagged WT β1Pix or β1Pix-Δ602–611 by immunoblotting (Fig. 3B). WT β1Pix and β1Pix mutant expression levels were ∼63 and 12% of the endogenous β1Pix levels, respectively, as determined by comparing total cellular β1Pix levels with or without Dox treatment by immunoblotting (data not shown). The lower overexpression level of the β1Pix mutant, which we observed in several different mutant clones generated, may result from its instability because the mutant is unable to dimerize and bind to 14-3-3 proteins.
Figure 3.

Inducible V5-tagged WT β1Pix or β1Pix-Δ602–611 expression modulates ENaC activity in stably transfected mpkCCDc14 cells. A, EVOM studies were performed in the absence (−) or presence (+) of Dox induction (2 μg/ml) for 3 days. We observed a difference in average basal ENaC currents between WT β1Pix and β1Pix-Δ602–611–expressing cells (36.7 ± 1.5 and 72.3 ± 0.7 μA/cm2, respectively). These differences may be because measurements were performed on two separate cell lines isolated after lentiviral transduction with β1Pix constructs, and there appears to be slight “leakage” of WT β1Pix expression even in the absence of doxycycline treatment. B, after current measurements, cells were lysed to confirm the expression of V5-tagged WT β1Pix or β1Pix-Δ602–611 by Western blotting. Data are mean ± S.E., with p values shown for the indicated comparisons.
AMPK-dependent interplay of β1Pix, Nedd4-2, and 14-3-3 proteins in polarized mpkCCDc14 cells with overexpression of WT versus mutant β1Pix
To examine how β1Pix is involved in AMPK-regulated ENaC inhibition, we also tested whether AMPK modulation alters the associations between β1Pix, Nedd4-2, and 14-3-3 proteins. Inducible β1Pix construct–expressing mpkCCDc14 cells were polarized on Transwell plates, followed by exposure to doxycycline for 3 days and then combined treatment with the AMPK activators AICAR (1 mm) and A769662 (100 μm) (AA) versus vehicle for 1 day. Cells were then lysed, and immunoblotting for various proteins was performed on a small sample of the whole-cell lysates (Fig. 4A, top panel, Input), whereas the remaining cell lysate was subjected to immunoprecipitation (IP) with a pan-14-3-3 protein antibody. Immunoblotting was then performed to detect co-immunoprecipitated V5 (β1Pix) and Nedd4-2 (Fig. 4A, bottom panel). Neither overexpression of WT β1Pix nor the β1Pix-Δ602–611 mutant affected AMPK activation (Fig. 4B). In cells overexpressing WT β1Pix, AMPK activators significantly enhanced the binding of β1Pix to 14-3-3 proteins (Fig. 4, A and C). As expected, the β1Pix-Δ602–611 mutant was unable to bind to 14-3-3 proteins. However, the association between Nedd4-2 and 14-3-3 proteins was not significantly altered by AMPK activation and/or overexpression of the WT or the β1Pix-Δ602–611 mutant in these cells (Fig. 4, D and E).
Figure 4.

AMPK activation enhances the interaction between β1Pix and 14-3-3 proteins but does not prevent binding of Nedd4-2 to 14-3-3 in mpkCCDc14 cells. A, representative immunoblot (Input) and co-IP results of β1Pix, Nedd4-2, and 14-3-3 proteins in total lysates from polarized mpkCCDc14 cells with Tet-induced V5-tagged WT β1Pix or β1Pix-Δ602–611 after 3-day doxycycline treatment followed by AA treatment for 24 h. B, AMPK activation in cells overexpressing WT or Δ602–611 mutant β1Pix with AA treatment for 24 h. IB, immunoblot. C, binding of WT β1Pix to 14-3-3 proteins increased with AMPK activation in WT β1Pix-expressing cells. D and E, binding of Nedd4-2 to 14-3-3 proteins with AMPK activation in cells with WT (D) or Δ602–611 mutant (E) β1Pix overexpression. Data are mean ± S.E. (p values shown when significant).
Inhibition of ENaC currents by AMPK is blunted with β1Pix knockdown in mCCDcl1 cells
To further confirm the importance of β1Pix in ENaC inhibition by AMPK, β1Pix was stably knocked down in mCCDcl1 cells (46), which have a higher Nedd4-2 abundance relative to total Nedd4 than mpkCCDc14 cells. Using lentiviral constructs, either scrambled control shRNA or shRNA directed against β1Pix, stable cell lines were achieved with 45% knockdown of β1Pix compared with control cells (Fig. 5D). ENaC currents were measured in control and β1Pix knockdown cells before and during combined treatment with AA versus vehicle (control) for 4 and 24 h (Fig. 5, A and B). Although ENaC current inhibition caused by AA treatment was observed in both control and β1Pix knockdown cells, in β1Pix knockdown cells, the decrease of ENaC current was significantly reduced by 55% and 45% at 4 and 24 h, respectively, relative to control cells (Fig. 5C), without any difference in AA treatment–induced AMPK activation between the two cells lines (see below). Together, these results suggest that β1Pix acts as a negative regulator of ENaC downstream of AMPK.
Figure 5.

β1Pix knockdown blunts ENaC inhibition by AMPK. A and B, mCCDcl1 cells were stably transduced with either a scrambled control shRNA (A) or shRNA directed against β1Pix (B). EVOM studies were performed on polarized cells before and after AA treatment for 4 and 24 h (*, p < 0.05, ENaC current differences of AA-treated cells versus nontreatment controls at the indicated time points). C, comparisons of the percent change in ENaC current following 4 and 24 h of AA treatment between control and β1Pix knockdown cells. β1Pix knockdown significantly reduced the ENaC current inhibition induced by treatment with AMPK activators. D, after equivalent short-circuit current measurements, cells were lysed to verify the knockdown efficiency of β1Pix by immunoblotting. Data shown are mean ± S.E. Asterisks indicate significant difference versus the control treatment at the same time point. p values are also indicated for comparisons in the absence (−) or presence (+) of β1Pix knockdown.
Involvement of β1Pix in Nedd4-2 regulation with AMPK modulation in mCCDcl1 cells
Compared with mpkCCDc14 cells, mCCDcl1 cells have a higher Nedd4-2 abundance relative to total Nedd4, so we reasoned that performing immunoprecipitation and immunoblotting assays with mCCDcl1 cells could provide a better opportunity to detect differences in the interaction between β1Pix, Nedd4-2 and 14-3-3 proteins as a function of AMPK activation and β1Pix knockdown (Fig. 6A). As summarized in Fig. 6C, AA treatment significantly enhanced the binding of both β1Pix and Nedd4-2 to 14-3-3 proteins (p = 0.002 and 0.02, respectively), which was abrogated by β1Pix knockdown, without disturbing AMPK activation (Fig. 6B). Of note, β1Pix knockdown caused a significant down-regulation of cellular Nedd4-2 protein expression without reducing Nedd4-2 mRNA levels, as assessed by qPCR (Fig. 6, D and E). Indeed, there was a trend toward increased Nedd4-2 mRNA levels with β1Pix knockdown, suggesting that these changes in Nedd4-2 gene expression represent a compensatory response to decreased Nedd4-2 protein expression rather than a causal explanation for it. Using a validated phospho-specific antibody (47), we also found that phosphorylation of mNedd4-2 at Ser-328 (equivalent to Ser-444 in xNedd4-2) was concomitantly inhibited with β1Pix knockdown (Fig. 6F). Cycloheximide chase assays were performed to examine the rates of cellular Nedd4-2 protein degradation and demonstrated decreased Nedd4-2 protein stability in the setting of β1Pix knockdown (Fig. 6G). This effect is consistent with the known key importance of phosphorylation at this residue (mNedd4-2–Ser-328 or xNedd4-2–Ser-444) in Nedd4-2 protein stability (14). Together, these results demonstrate that β1Pix plays an important role in Ser-328 phosphorylation and, thus, cellular stability of Nedd4-2 in mCCDcl1 cells. Overall, our findings suggest that AMPK activation, through facilitating the association of β1Pix, 14-3-3 proteins, and Nedd4-2 into a complex, stabilizes and enhances Nedd4-2 binding to ENaC, thereby promoting ENaC ubiquitination and degradation, as described previously (27, 28).
Figure 6.

β1Pix regulates AMPK-dependent Nedd4-2–14-3-3 association and Nedd4-2 cellular stability in mCCDcl1 cells. A, representative immunoblot (Input) and co-IP results of β1Pix, Nedd4-2, and 14-3-3 proteins in total lysates from polarized mCCDcl1 cells with or without β1Pix knockdown after 24-h treatment with or without AMPK activators (AA). Ab, antibody. B, summary graph of the ratio of pAMPK-α (Thr-172) to total AMPK-α and β1Pix protein expression levels. IB, immunoblot. C, summary graph of the co-immunoprecipitated β1Pix (left panel) or Nedd4-2 (right panel) with 14-3-3 proteins under the indicated conditions. D, immunoblot analysis of Nedd4-2 protein expression in cells with or without β1Pix knockdown and with or without AA treatment. E, RT-PCR analysis of Nedd4-2 mRNA in cells with or without β1Pix knockdown and with or without AA treatment. F, phosphorylation of Nedd4-2 (Ser-328) in cells with or without β1Pix knockdown and with or without AA treatment. G, Nedd4-2 cellular stability in control versus β1Pix knockdown cells. Cells were treated with cycloheximide (100 μg/ml) and harvested at different time points (*, p = 0.008 for comparison of control and β1Pix knockdown at the 2-h time point). Immunoblot assays were performed to detect changes in cellular Nedd4-2 protein abundance. Data are mean ± S.E. (p values for the indicated comparisons with significant differences are shown).
Discussion
We and others showed previously that AMPK is a negative regulator of the ENaC activity in oocytes and polarized epithelial cells (26–28). ENaC inhibition is mediated by decreasing its expression level at the plasma membrane rather than through a change of single channel properties (open probability or conductance) (26). AMPK typically controls its downstream effectors through phosphorylation of target proteins. However, there is no evidence that ENaC subunits are directly phosphorylated by or interact with AMPK. With the presence of a Liddle's-type mutation (β-mENaC–Y618A), a role of the C-terminal tail of β-ENaC in AMPK-related inhibition was revealed, implying the involvement of the E3 ubiquitin ligase Nedd4-2 (26).
We previously demonstrated that AMPK phosphorylates Nedd4-2 both in vitro and in an intact cellular milieu (27). The association of Nedd4-2 with β-ENaC was enhanced with AMPK activation. A ubiquitin ligase–deficient Nedd4-2 mutant blocked the AMPK inhibitory effect on ENaC, as did addition of the deubiquitinating enzyme Usp2–45, suggesting that Nedd4-2-mediated ubiquitination is necessary for ENaC inhibition (27, 28). In this study, we found that Ser-444 on xNedd4-2 is an AMPK phosphorylation site (Fig. 1). Phosphorylation at Ser-444 has been revealed previously to be critical for Nedd4-2 cellular stability and its association with 14-3-3 (14). This phosphorylation event by AMPK is the first key step in our current simplified working model for how ENaC regulation by AMPK occurs via Nedd4-2 (Fig. 7). However, Ser-444 is a shared phosphorylation site targeted by several other kinases, such as SGK1, PKA, and IKKβ (10, 42), leading to the hypothesis that, besides Nedd4-2, other molecules may be critical for ENaC regulation by AMPK. Our earlier work studying the mechanism of ENaC regulation by ET-1 demonstrated that the guanine nucleotide exchange factor β1Pix inhibits ENaC expression through the 14-3-3/Nedd4-2 pathway (38). The results from this study show that treatment with an AMPK activator inhibits ENaC-dependent current, an effect that is also observed with overexpression of WT β1Pix (Fig. 2). We also found that there was no synergistic or additive inhibitory effect observed with combined WT-β1Pix overexpression and AICAR treatment, suggesting that β1Pix and AMPK are components of the same regulatory pathway for ENaC. Moreover, ENaC inhibition caused by either AICAR treatment or overexpression of a constitutively active form of AMPK could be abolished with coexpression of the β1Pix-Δ602–611 mutant. The importance of β1Pix in AMPK-dependent ENaC regulation was also demonstrated in two distinct polarized mouse kidney principal cell lines, mpkCCDc14 and mCCDcl1, with either overexpression of WT β1Pix or β1Pix-Δ602–611 or with β1Pix knockdown in this study. Our data further revealed that β1Pix knockdown blunts ENaC inhibition caused by combined AICAR and A769662 treatment without affecting AMPK activation. Taken together, these results indicate that β1Pix is a downstream mediator of AMPK and suggest that its dimerization and binding to 14-3-3 proteins are necessary for ENaC regulation.
Figure 7.
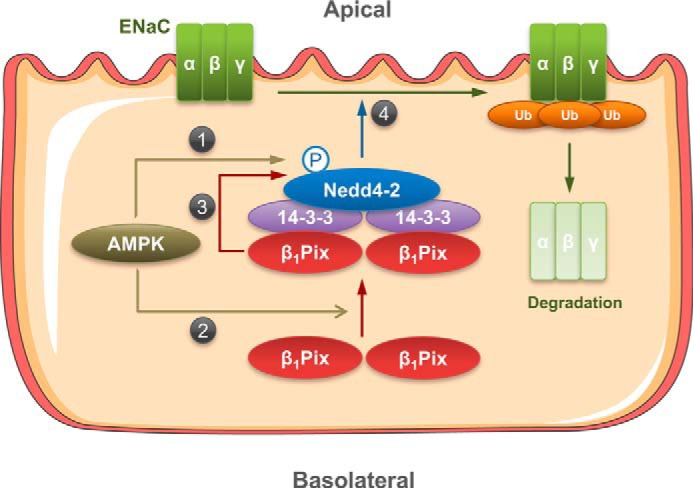
Proposed model for the roles of Nedd4-2 and β1Pix in the regulation of ENaC by AMPK. Activation of AMPK promotes AMPK-mediated phosphorylation of Nedd4-2 at Ser-328 (1) and also promotes the binding of β1Pix to the Nedd4-2–14-3-3 complex (2). β1Pix association with Nedd4-2 enhances Nedd4-2 stability by helping to maintain Nedd4-2 phosphorylation at Ser-328 (3), which ultimately enhances the association of Nedd4-2 with ENaC (4) and thereby induces ENaC ubiquitination and degradation.
As phospho-Ser/phospho-Thr binding proteins, 14-3-3s regulate proliferative, survival, apoptotic, and stress signaling by interacting with a diverse array of binding partners (48). Of note, 14-3-3 proteins are critical for suppressing mTOR complex 1 (mTORC1) activity under conditions of cell stress. AMPK has been shown to inhibit mTORC1 through stimulating the Rheb-GAP (GTPase-activating protein) activity of TSC2 and by phosphorylating Raptor, an mTORC1 scaffolding protein that recruits downstream substrates of mTOR (49, 50). Phosphorylation of Raptor at Ser-722 and Ser-792 by AMPK mediates 14-3-3 binding and is required for mTORC1 inactivation in vivo (50). Recent work has established a new role of 14-3-3 proteins in ENaC regulation. Phosphorylation of Nedd4-2 by SGK1 has been reported to enhance the association between Nedd4-2 and 14-3-3 proteins and maintain Nedd4-2 in an inactive (sequestered) state, thereby causing a phosphorylation-dependent inhibition of the interaction between Nedd4-2 and ENaC (10, 51). In our previous study of ENaC regulation by ET-1, recruitment of 14-3-3β by β1Pix appeared to prevent the association between Nedd4-2 and 14-3-3β, promoting the ubiquitination and degradation of ENaC (38). Although ET-1 has been reported to induce β1Pix translocation and Cdc42 activation via a PKA-dependent pathway in primary human mesangial cells (52), Rac1 and Cdc42 were not involved in β1Pix-dependent ENaC inhibition in CHO cells, suggesting that the GEF activity of β1Pix is not required for ENaC suppression (38). β1Pix is up-regulated in primary human mesangial cells with ET-1 treatment for 24 h (53). However, in this study, there was no significant change in β1Pix expression in polarized renal epithelial cells with AMPK activation for 24 h (Fig. 6B). Importantly, we found that AMPK activation increases the association between β1Pix and 14-3-3 proteins in both CCD cell lines (Fig. 7, step 2) but does not inhibit the binding of Nedd4-2 to 14-3-3 proteins in either cell line (Figs. 4 and 6) under conditions when AMPK inhibits ENaC (Fig. 5). Specifically, in mpkCCDc14 cells, the association between Nedd4-2 and 14-3-3 proteins was not significantly affected either by AMPK activators or by overexpression of WT or mutant β1Pix (Fig. 4, D and E). In contrast, AMPK activation in mCCDcl1 cells promoted Nedd4-2 binding to 14-3-3 proteins, and β1Pix knockdown blunted this increased Nedd4-2 association with 14-3-3s (Fig. 6C). Moreover, as shown in Fig. 6, D–F, β1Pix is required for the phosphorylation and cellular stability of Nedd4-2 (Fig. 7, step 3). Taken together, our findings indicate that AMPK decreases ENaC activity through a β1Pix/Nedd4-2–14-3-3–dependent mechanism that differs from the proposed mechanism of ET-1–dependent ENaC inhibition, although the final regulatory step featuring enhanced Nedd4-2–ENaC interaction with subsequent ENaC ubiquitination and degradation is the same (Fig. 7, step 4).
Pix is well-known as a regulator of cell motility, functioning as a GEF for Cdc42 and Rac1 and a binding partner to the PAK family of Cdc42/Rac1-activated kinases (54). βPix has related roles in promoting membrane ruffling (55), focal complex disassembly facilitating migration (56), and maintenance of epithelial cell polarization (57, 58) and survival (59). Besides cell migration and survival, several studies reported that βPix is critical for cellular transformation and in vivo tumorigenesis, as it can sequester the c-Cbl ubiquitin ligase and prevent the ubiquitination and degradation of various growth factor receptors, including epidermal growth factor receptor, VEGFR2, and IGF1R (60, 61). Here we further demonstrate a novel role of β1Pix in AMPK-dependent ENaC regulation, suggesting the involvement of β1Pix in the regulation of the E3 ubiquitin ligase Nedd4-2 during metabolic stress conditions.
The effects of β1Pix on 14-3-3 proteins and Nedd4-2 downstream of AMPK likely have broader implications for the regulation of additional membrane transport proteins besides ENaC. Specifically, Nedd4-2 is known to regulate a growing list of transport proteins through a direct binding and ubiquitination mechanism similar to that originally characterized for ENaC, including voltage-gated Na+, K+, and Ca2+ channels; Cl− channels; human organic anion transporters; and glutamate transporters in the brain (62). Moreover, it has already been shown that many of these transport proteins are also regulated by AMPK, so it is reasonable to propose that the mechanisms uncovered in this study may be generalizable to the numerous transport proteins that are regulated by AMPK and Nedd4-2. Additional studies to test whether β1Pix is a critical component in the regulation of these transport proteins in the kidney and other important organs like the heart, lung, and brain are thus warranted.
In summary, this study describes a novel function of β1Pix as a positive regulator of Nedd4-2 stability and a β1Pix/Nedd4-2–14-3-3 protein–dependent mechanism of AMPK-regulated ENaC inhibition. Our findings support a model whereby AMPK activation enhances Nedd4-2 cellular stability via β1Pix and induces ENaC degradation by enhancing the association of β1Pix, 14-3-3 proteins, and Nedd4-2 into a complex and further strengthening the phosphorylation of Nedd4-2 (Fig. 7). Future work will focus on investigating the mechanisms of how AMPK promotes the binding of β1Pix to 14-3-3, leading to an increase of ENaC ubiquitination by Nedd4-2.
Experimental procedures
Reagents and chemicals
All chemicals used were purchased from Sigma or Thermo Fisher Scientific unless otherwise noted. [γ-32P]ATP was obtained from MP Biomedicals (Santa Ana, CA). Recombinant active human AMPK holoenzyme (α1-T172D, β1, γ1) was synthesized and purified as described previously (63). 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and A769662 were obtained from Toronto Research Chemicals, Inc. and Selleck Chemicals LLC (Houston, TX), respectively.
Cell culture
CHO, human embryonic kidney (HEK-293), mpkCCDc14, and mCCDcl1 cells were maintained and cultured as described previously (27, 38, 64). mCCDcl1 cells were a kind gift from Dr. Bernard Rossier (University of Lausanne, Lausanne, Switzerland) (46). Immortalized CCD principal cells were grown in defined medium on permeable supports (Costar Transwells, 0.4-μm pore, 6.5- or 24-mm diameter), allowing them to polarize and form monolayers with high resistance and avid Na+ reabsorption.
Plasmids and transfection
For whole-cell patch clamp experiments, CHO cells were seeded on sterile 4 × 4 mm coverglasses in 35-mm Petri dishes, and transfection was performed with Polyfect reagent (Qiagen, Valencia, CA) as described previously (38). Mouse ENaC was reconstituted by co-expressing α-, β-, and γ-channel subunits (38). The expression vectors encoding GSH S-transferase (GST)–tagged WT Xenopus Nedd4-2 (xNedd4-2), rat WT β1Pix, β1Pix-Δ602–611, AMPK-α1-K45R, and AMPK-γ1-R70Q were also described previously (12, 44, 65). WT and mutant β1Pix plasmids were a gift from Dr. Andrey Sorokin (Medical College of Wisconsin).
β1Pix shRNA lentiviral particles (Santa Cruz Biotechnology) were used to knock down β1Pix expression in mCCDcl1 cells. Control shRNA lentiviral particles (Santa Cruz Biotechnology) were used to confirm the transduction efficiency. mCCDcl1 cells were seeded on 60-mm Petri dishes and transduced by replacing growth medium with medium containing lentiviral particles and Polybrene (5 μg/ml) according to the manufacturer's recommendations. The infection medium was removed and replaced with fresh growth medium after overnight incubation. The stable cell line expressing the shRNA was isolated via selection with puromycin (2 μg/ml).
To generate mpkCCDc14 cells with stable conditional overexpression of WT β1Pix or β1Pix-Δ602–611, V5-tagged WT β1Pix and β1Pix-Δ602–611 cDNAs were obtained from FLAG pCMV plasmids followed by tag switching from FLAG to the V5 epitope. The cDNAs were then cloned into the pDONR221 entry vector and integrated into pCW57.1 vector containing a tetracycline response element (Addgene) to generate a conditional pCW57.1 V5-tagged WT β1Pix or β1Pix-Δ602–611 plasmid (66). All constructs were confirmed by DNA sequencing. Lentiviral vector particles were generated by co-transfecting the pCW57.1 V5-tagged β1Pix plasmid and a mixture of packaging vectors (pCMV-ΔR8.2 and pCMV-VSV-G) into HEK293T packaging cells. Vector particles were collected by filtering and concentrating the conditioned medium at 72 h post-transfection. mpkCCDc14 cells were seeded and infected in 6-well plates with lentiviral vector particles and Polybrene. Stable clones were isolated via selection with puromycin for 2 weeks, and expression of recombinant proteins was verified by immunoblotting with a V5 tag antibody.
Mass spectrometry and Edman sequencing
WT GST-xNedd4-2 was expressed in E. coli Rosetta 2 cells (EMD Millipore). The bacteria were harvested after 24 h for elution of GST-xNedd4-2. Purified GST-xNedd4-2 was phosphorylated in vitro with active AMPK holoenzyme, separated by SDS-PAGE, and stained with colloidal Coomassie Blue, and radioactive gel bands were excised. Tryptic digestion, peptide extraction, purification by reverse-phase chromatography, and MALDI-TOF MS analysis were performed by following the reported procedures (67). Selected radioactive peptides were sequenced by solid-phase Edman degradation (68).
In vitro phosphorylation
HEK-293 cells were transiently transfected according to the manufacturer's protocol using Lipofectamine 2000 (Invitrogen) to express FLAG-tagged WT xNedd4-2 or xNedd4-2 S444A. Cells were lysed 2 days after transfection, and the FLAG-tagged xNedd4-2 (WT and S444A) were immunoprecipitated from cell lysates using the M2 anti-FLAG mAb (Sigma) coupled to protein A/G agarose. In vitro phosphorylation was performed using purified active AMPK holoenzyme with [γ-32P]ATP labeling, as described previously (27). After SDS-PAGE and transfer to nitrocellulose membranes, immunoblotting for expression of the FLAG-tagged xNedd4-2 was first performed and quantified using a Versadoc Imager with Quantity One software (Bio-Rad). After the chemiluminescent signal decayed, phosphorylated bands on the membrane were identified by exposure of the same membrane to a phosphoscreen, and the detected bands were quantitated using the same image analysis software. The intensity of each phosphoscreen band was corrected by subtracting the local background in the same lane.
Electrophysiology
A portable epithelial volt-ohm–meter (World Precision Instruments, Sarasota, FL) was used to measure equivalent short-circuit currents across polarized cell monolayers. The electrode was calibrated by placing it into growth medium for 90 min prior to measurement of the potential difference and resistance across the filter. The current was calculated by using the potential difference across the filter divided by the resistance normalized to the surface area to obtain readings measured in microamperes per square centimeter (69). Whole-cell macroscopic patch clamp current recordings of mENaC expressed in CHO cells were made under voltage clamp conditions using our previously described methods (38). Cells were seeded on glass coverslips and then transiently transfected with the bicistronic pTracer plasmid to express GFP and AMPK-α1–K45R, AMPK-γ1–R70Q, or empty vector (65). Whole-cell patch clamping was performed on GFP-positive cells 1–3 days after transfection.
Co-immunoprecipitation assays
To examine the association between β1Pix, Nedd4-2, and 14-3-3 proteins with AMPK modulation, cells were harvested and lysed in ice-cold IP lysis buffer (1% Triton X-100, 2 mm EDTA (pH 8.0), in Dulbecco's PBS with Ca2+ and Mg2+) after AICAR and A769662 treatment versus vehicle, as indicated, for 24 h. Pre-cleared lysates were incubated with the pan-14-3-3 antibody (1:100) coupled to protein A/G beads (Thermo Fisher Scientific) overnight at 4 °C. Immunoprecipitation in the absence of the pan-14-3-3 antibody was also performed as a no-antibody control. After three washes with lysis buffer, the immunoprecipitation samples were eluted in sample buffer and, along with the cell lysate samples, subjected to immunoblotting to detect β1Pix, Nedd4-2, and 14-3-3 proteins. Relative binding was quantified by dividing the co-IP protein signal by the signal for that protein in the cell lysate, which was then corrected for the amount of immunoprecipitated protein for that condition.
Immunoblotting
Lysis and immunoblotting of cell lysates for Nedd4-2 (EMD Millipore), pNedd4-2 (S328) (Abcam), V5 tag (Cell Signaling Technology), β1Pix (EMD Millipore), phospho-AMPK α (Thr-172, Cell Signaling Technology), AMPK pan α (Cell Signaling Technology), 14-3-3 proteins (Santa Cruz Biotechnology), and β-actin (Sigma) were performed as described previously (25, 38). Gradient gels (4–12%) were used for SDS-PAGE. IRDye® goat anti-rabbit and anti-mouse IgG Dylight 800 and 680 were obtained from LI-COR Biotechnology (Lincoln, NE). Membranes were scanned with an Odyssey Fc imaging system (LI-COR). Bands were quantified by using Image Studio Lite software (LI-COR) on unprocessed raw data files. To compare immunoblotting and co-IP results for specific conditions across multiple replicate experiments, we analyzed the data through the “normalization by sum of the replicate” technique, as described previously (70). Briefly, each data point from an individual group on a blot was divided by the sum of all raw data on the same blot. The data were then further normalized to the average calculated with all of the control group values from all experiments.
RNA extraction and real-time quantitative reverse transcriptase PCR
Total RNA was extracted using the PureLinkTM RNA Mini Kit (Invitrogen), and 0.5-μg aliquots were reverse-transcribed using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer's instructions. First-strand cDNA, 1 μl of the 40-μl reverse transcriptase reaction product, was amplified using PowerUp SYBR Green Master Mix (Applied Biosystems) with primers of Nedd4-2 (forward, 5′-AATGACCTGGGCCCTCTT-3′; reverse, 5′-GTAAAACGTGCGGCCATC-3′) or glyceraldehyde-3-phosphate dehydrogenase (forward, 5′-TCAAGCTCATTTCCTGGTATGACA-3′; reverse, 5′-TAGGGCCTCTCTTGCTCAGT-3′). Quantitative real-time RT-PCR was conducted with the ViiATM 7 real-time PCR system (Applied Biosystems) at 95 °C for 20 s, followed by 40 cycles at 95 °C for 1 s and 60 °C for 20 s. All samples were run in triplicate. The expression of Nedd4-2 was defined from the threshold cycle, and relative expression levels were calculated using the 2−ΔΔCT method. Glyceraldehyde-3-phosphate dehydrogenase was used as a reference gene for normalization.
Cycloheximide chase assay
mCCDcl1 cells with or without β1Pix knockdown were treated with 100 μg/ml cycloheximide and then chased at 37 °C for 0 to 4 h. At the appropriate chase time, cells were rinsed twice with cold PBS and lysed in radioimmune precipitation assay buffer supplemented with phosphatase and protease inhibitors. Proteins were resolved by SDS-PAGE and immunoblotted with anti-Nedd4 WW2 antibody. The detected bands were then quantitated by using Image Studio Lite software (LI-COR).
Statistical analysis
All summarized data are reported as mean ± S.E. Statistical analyses were performed using corresponding t tests and one-way analysis of variance with post hoc Bonferroni corrections for multiple comparisons. In all cases, p < 0.05 was considered significant.
Author contributions
P.-Y. H., H. L., R. B., D. N., A. S., and K. R. H. conceptualization; P.-Y. H., H. L., T. S. P., R. D. T., D. N., and A. S. data curation; P.-Y. H., H. L., T. S. P., R. D. T., R. B., D. N., and A. S. formal analysis; P.-Y. H., H. L., R. B., D. N., A. S., and K. R. H. supervision; P.-Y. H. validation; P.-Y. H., H. L., T. S. P., R. D. T., D. T., D. N., and K. R. H. investigation; P.-Y. H. visualization; P.-Y. H. and R. B. methodology; P.-Y. H., D. T., and K. R. H. writing-original draft; P.-Y. H., H. L., T. S. P., R. D. T., D. T., R. B., D. N., A. S., and K. R. H. writing-review and editing; K. R. H. resources; K. R. H. funding acquisition; K. R. H. project administration.
Acknowledgments
We thank Dr. Nuria Pastor-Soler for feedback. We also thank Dr. Bernard Rossier (University of Lausanne, Lausanne, Switzerland) for providing mCCDcl1 cells and Dr. Andrey Sorokin (Medical College of Wisconsin) for providing the β1Pix plasmids used in this study.
This work was supported by NIDDK, National Institutes of Health Grant R01 DK075048 (to K. R. H.); NHLBI, National Institutes of Health Grants K99/R00 HL116603 (to T. S. P.) and R01 HL108880 and R35 HL135749 (to A. S.); EU FP6 Contract LSHM-CT-2004–005272 (EXGENESIS) (to D. N. and R. D. T.); and Netherlands Organization for Scientific Research VIDI Grant 864.10.007 (to D. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial sodium channel
- PKA
- protein kinase A
- GEF
- guanine nucleotide exchange factor
- PAK
- p21-activated kinase
- CHO
- Chinese hamster ovary
- CCD
- cortical collecting duct
- GST
- glutathione S-transferase
- DN
- dominant-negative
- AMPK
- AMP-activated protein kinase
- CA
- constitutively active
- Dox
- doxycycline
- EVOM
- epithelial volt-ohm–meter
- AA
- AICAR (1 mm) and A769662 (100 μm)
- IP
- immunoprecipitation
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- HEK
- human embryonic kidney
- shRNA
- short hairpin RNA
- cDNA
- complementary DNA
- mENaC
- mouse epithelial sodium channel.
References
- 1. Garty H., and Palmer L. G. (1997) Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 77, 359–396 10.1152/physrev.1997.77.2.359 [DOI] [PubMed] [Google Scholar]
- 2. Rossier B. C., Pradervand S., Schild L., and Hummler E. (2002) Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu. Rev. Physiol. 64, 877–897 10.1146/annurev.physiol.64.082101.143243 [DOI] [PubMed] [Google Scholar]
- 3. Rossier B. C. (2004) The epithelial sodium channel: activation by membrane-bound serine proteases. Proc. Am. Thorac. Soc. 1, 4–9 10.1513/pats.2306007 [DOI] [PubMed] [Google Scholar]
- 4. Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., and Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 10.1038/367463a0 [DOI] [PubMed] [Google Scholar]
- 5. Kellenberger S., and Schild L. (2002) Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol. Rev. 82, 735–767 10.1152/physrev.00007.2002 [DOI] [PubMed] [Google Scholar]
- 6. Bhalla V., and Hallows K. R. (2008) Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 19, 1845–1854 10.1681/ASN.2008020225 [DOI] [PubMed] [Google Scholar]
- 7. Kamynina E., and Staub O. (2002) Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na+ transport. Am. J. Physiol. Renal Physiol. 283, F377–F387 10.1152/ajprenal.00143.2002 [DOI] [PubMed] [Google Scholar]
- 8. Snyder P. M., Steines J. C., and Olson D. R. (2004) Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J. Biol. Chem. 279, 5042–5046 10.1074/jbc.M312477200 [DOI] [PubMed] [Google Scholar]
- 9. Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Münster C., Chraïbi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., and Staub O. (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 20, 7052–7059 10.1093/emboj/20.24.7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., and Isobe T. (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 10.1074/jbc.M412884200 [DOI] [PubMed] [Google Scholar]
- 11. Bhalla V., Daidié D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., and Pearce D. (2005) Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084 10.1210/me.2005-0193 [DOI] [PubMed] [Google Scholar]
- 12. Hallows K. R., Bhalla V., Oyster N. M., Wijngaarden M. A., Lee J. K., Li H., Chandran S., Xia X., Huang Z., Chalkley R. J., Burlingame A. L., and Pearce D. (2010) Phosphopeptide screen uncovers novel phosphorylation sites of Nedd4-2 that potentiate its inhibition of the epithelial Na+ channel. J. Biol. Chem. 285, 21671–21678 10.1074/jbc.M109.084731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Snyder P. M., Olson D. R., Kabra R., Zhou R., and Steines J. C. (2004) cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 279, 45753–45758 10.1074/jbc.M407858200 [DOI] [PubMed] [Google Scholar]
- 14. Chandran S., Li H., Dong W., Krasinska K., Adams C., Alexandrova L., Chien A., Hallows K. R., and Bhalla V. (2011) Neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J. Biol. Chem. 286, 37830–37840 10.1074/jbc.M111.293233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hardie D. G., Carling D., and Carlson M. (1998) The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 67, 821–855 10.1146/annurev.biochem.67.1.821 [DOI] [PubMed] [Google Scholar]
- 16. Pastor-Soler N. M., and Hallows K. R. (2012) AMP-activated protein kinase regulation of kidney tubular transport. Curr. Opin. Nephrol. Hypertens. 21, 523–533 10.1097/MNH.0b013e3283562390 [DOI] [PubMed] [Google Scholar]
- 17. Stein S. C., Woods A., Jones N. A., Davison M. D., and Carling D. (2000) The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 345, 437–443 10.1042/bj3450437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., and Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 10.1016/j.cub.2003.10.031 [DOI] [PubMed] [Google Scholar]
- 19. Hardie D. G., Scott J. W., Pan D. A., and Hudson E. R. (2003) Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 546, 113–120 10.1016/S0014-5793(03)00560-X [DOI] [PubMed] [Google Scholar]
- 20. Sharma K., Ramachandrarao S., Qiu G., Usui H. K., Zhu Y., Dunn S. R., Ouedraogo R., Hough K., McCue P., Chan L., Falkner B., and Goldstein B. J. (2008) Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Invest. 118, 1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee M. J., Feliers D., Mariappan M. M., Sataranatarajan K., Mahimainathan L., Musi N., Foretz M., Viollet B., Weinberg J. M., Choudhury G. G., and Kasinath B. S. (2007) A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Renal Physiol. 292, F617–F627 10.1152/ajprenal.00278.2006 [DOI] [PubMed] [Google Scholar]
- 22. Mount P. F., Hill R. E., Fraser S. A., Levidiotis V., Katsis F., Kemp B. E., and Power D. A. (2005) Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am. J. Physiol. Renal Physiol. 289, F1103–1115 10.1152/ajprenal.00458.2004 [DOI] [PubMed] [Google Scholar]
- 23. Peairs A., Radjavi A., Davis S., Li L., Ahmed A., Giri S., and Reilly C. M. (2009) Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin. Exp. Immunol. 156, 542–551 10.1111/j.1365-2249.2009.03924.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cammisotto P. G., Londono I., Gingras D., and Bendayan M. (2008) Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. Am. J. Physiol. Renal Physiol. 294, F881–F889 10.1152/ajprenal.00373.2007 [DOI] [PubMed] [Google Scholar]
- 25. Takiar V., Nishio S., Seo-Mayer P., King J. D. Jr., Li H., Zhang L., Karihaloo A., Hallows K. R., Somlo S., and Caplan M. J. (2011) Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 2462–2467 10.1073/pnas.1011498108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carattino M. D., Edinger R. S., Grieser H. J., Wise R., Neumann D., Schlattner U., Johnson J. P., Kleyman T. R., and Hallows K. R. (2005) Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616 10.1074/jbc.M501770200 [DOI] [PubMed] [Google Scholar]
- 27. Bhalla V., Oyster N. M., Fitch A. C., Wijngaarden M. A., Neumann D., Schlattner U., Pearce D., and Hallows K. R. (2006) AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 281, 26159–26169 10.1074/jbc.M606045200 [DOI] [PubMed] [Google Scholar]
- 28. Almaça J., Kongsuphol P., Hieke B., Ousingsawat J., Viollet B., Schreiber R., Amaral M. D., and Kunzelmann K. (2009) AMPK controls epithelial Na+ channels through Nedd4-2 and causes an epithelial phenotype when mutated. Pflugers Arch. 458, 713–721 10.1007/s00424-009-0660-4 [DOI] [PubMed] [Google Scholar]
- 29. Karpushev A. V., Levchenko V., Pavlov T. S., Lam V., Vinnakota K. C., Vandewalle A., Wakatsuki T., and Staruschenko A. (2008) Regulation of ENaC expression at the cell surface by Rab11. Biochem. Biophys. Res. Commun. 377, 521–525 10.1016/j.bbrc.2008.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pochynyuk O., Medina J., Gamper N., Genth H., Stockand J. D., and Staruschenko A. (2006) Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J. Biol. Chem. 281, 26520–26527 10.1074/jbc.M603716200 [DOI] [PubMed] [Google Scholar]
- 31. Staruschenko A., Patel P., Tong Q., Medina J. L., and Stockand J. D. (2004) Ras activates the epithelial Na+ channel through phosphoinositide 3-OH kinase signaling. J. Biol. Chem. 279, 37771–37778 10.1074/jbc.M402176200 [DOI] [PubMed] [Google Scholar]
- 32. Koh C. G., Manser E., Zhao Z. S., Ng C. P., and Lim L. (2001) β1PIX, the PAK-interacting exchange factor, requires localization via a coiled-coil region to promote microvillus-like structures and membrane ruffles. J. Cell Sci. 114, 4239–4251 [DOI] [PubMed] [Google Scholar]
- 33. Staruschenko A., and Sorokin A. (2012) Role of βPix in the kidney. Front. Physiol. 3, 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bagrodia S., Bailey D., Lenard Z., Hart M., Guan J. L., Premont R. T., Taylor S. J., and Cerione R. A. (1999) A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J. Biol. Chem. 274, 22393–22400 10.1074/jbc.274.32.22393 [DOI] [PubMed] [Google Scholar]
- 35. Kim S., Lee S. H., and Park D. (2001) Leucine zipper-mediated homodimerization of the p21-activated kinase-interacting factor, β Pix: implication for a role in cytoskeletal reorganization. J. Biol. Chem. 276, 10581–10584 10.1074/jbc.C000806200 [DOI] [PubMed] [Google Scholar]
- 36. Jin J., Smith F. D., Stark C., Wells C. D., Fawcett J. P., Kulkarni S., Metalnikov P., O'Donnell P., Taylor P., Taylor L., Zougman A., Woodgett J. R., Langeberg L. K., Scott J. D., and Pawson T. (2004) Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 14, 1436–1450 10.1016/j.cub.2004.07.051 [DOI] [PubMed] [Google Scholar]
- 37. Angrand P. O., Segura I., Volkel P., Ghidelli S., Terry R., Brajenovic M., Vintersten K., Klein R., Superti-Furga G., Drewes G., Kuster B., Bouwmeester T., and Acker-Palmer A. (2006) Transgenic mouse proteomics identifies new 14-3-3-associated proteins involved in cytoskeletal rearrangements and cell signaling. Mol. Cell. Proteomics 5, 2211–2227 [DOI] [PubMed] [Google Scholar]
- 38. Pavlov T. S., Chahdi A., Ilatovskaya D. V., Levchenko V., Vandewalle A., Pochynyuk O., Sorokin A., and Staruschenko A. (2010) Endothelin-1 inhibits the epithelial Na+ channel through βPix/14-3-3/Nedd4-2. J. Am. Soc. Nephrol. 21, 833–843 10.1681/ASN.2009080885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alzamora R., Al-Bataineh M. M., Liu W., Gong F., Li H., Thali R. F., Joho-Auchli Y., Brunisholz R. A., Satlin L. M., Neumann D., Hallows K. R., and Pastor-Soler N. M. (2013) AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am. J. Physiol. Renal Physiol. 305, F943–F956 10.1152/ajprenal.00303.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Staub O., and Verrey F. (2005) Impact of Nedd4 proteins and serum and glucocorticoid-induced kinases on epithelial Na+ transport in the distal nephron. J. Am. Soc. Nephrol. 16, 3167–3174 10.1681/ASN.2005050454 [DOI] [PubMed] [Google Scholar]
- 41. Liang X., Peters K. W., Butterworth M. B., and Frizzell R. A. (2006) 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. J. Biol. Chem. 281, 16323–16332 10.1074/jbc.M601360200 [DOI] [PubMed] [Google Scholar]
- 42. Edinger R. S., Lebowitz J., Li H., Alzamora R., Wang H., Johnson J. P., and Hallows K. R. (2009) Functional regulation of the epithelial Na+ channel by IκB kinase-β occurs via phosphorylation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 284, 150–157 10.1074/jbc.M807358200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corton J. M., Gillespie J. G., Hawley S. A., and Hardie D. G. (1995) 5-Aminoimidazole-4-carboxamide ribonucleoside: a specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 10.1111/j.1432-1033.1995.tb20498.x [DOI] [PubMed] [Google Scholar]
- 44. Chahdi A., and Sorokin A. (2008) Protein kinase A-dependent phosphorylation modulates β1Pix guanine nucleotide exchange factor activity through 14-3-3β binding. Mol. Cell. Biol. 28, 1679–1687 10.1128/MCB.00898-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., and Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 46. Gaeggeler H. P., Gonzalez-Rodriguez E., Jaeger N. F., Loffing-Cueni D., Norregaard R., Loffing J., Horisberger J. D., and Rossier B. C. (2005) Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J. Am. Soc. Nephrol. 16, 878–891 10.1681/ASN.2004121110 [DOI] [PubMed] [Google Scholar]
- 47. Gleason C. E., Frindt G., Cheng C. J., Ng M., Kidwai A., Rashmi P., Lang F., Baum M., Palmer L. G., and Pearce D. (2015) mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J. Clin. Invest. 125, 117–128 10.1172/JCI73935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morrison D. K. (2009) The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 19, 16–23 10.1016/j.tcb.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Inoki K., Zhu T., and Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- 50. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nagaki K., Yamamura H., Shimada S., Saito T., Hisanaga S., Taoka M., Isobe T., and Ichimura T. (2006) 14-3-3 mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry 45, 6733–6740 10.1021/bi052640q [DOI] [PubMed] [Google Scholar]
- 52. Chahdi A., Miller B., and Sorokin A. (2005) Endothelin 1 induces β 1Pix translocation and Cdc42 activation via protein kinase A-dependent pathway. J. Biol. Chem. 280, 578–584 10.1074/jbc.M411130200 [DOI] [PubMed] [Google Scholar]
- 53. Chahdi A., and Sorokin A. (2008) Endothelin-1 couples βPix to p66Shc: role of βPix in cell proliferation through FOXO3a phosphorylation and p27kip1 down-regulation independently of Akt. Mol. Biol. Cell 19, 2609–2619 10.1091/mbc.e07-05-0424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manser E., Loo T. H., Koh C. G., Zhao Z. S., Chen X. Q., Tan L., Tan I., Leung T., and Lim L. (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1, 183–192 10.1016/S1097-2765(00)80019-2 [DOI] [PubMed] [Google Scholar]
- 55. Lee S. H., Eom M., Lee S. J., Kim S., Park H. J., and Park D. (2001) βPix-enhanced p38 activation by Cdc42/Rac/PAK/MKK3/6-mediated pathway: implication in the regulation of membrane ruffling. J. Biol. Chem. 276, 25066–25072 10.1074/jbc.M010892200 [DOI] [PubMed] [Google Scholar]
- 56. Zhao Z. S., Manser E., Loo T. H., and Lim L. (2000) Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 20, 6354–6363 10.1128/MCB.20.17.6354-6363.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Valdes J. L., Tang J., McDermott M. I., Kuo J. C., Zimmerman S. P., Wincovitch S. M., Waterman C. M., Milgram S. L., and Playford M. P. (2011) Sorting nexin 27 protein regulates trafficking of a p21-activated kinase (PAK) interacting exchange factor (β-Pix)-G protein-coupled receptor kinase interacting protein (GIT) complex via a PDZ domain interaction. J. Biol. Chem. 286, 39403–39416 10.1074/jbc.M111.260802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iden S., and Collard J. G. (2008) Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 9, 846–859 10.1038/nrm2521 [DOI] [PubMed] [Google Scholar]
- 59. Frank S. R., Bell J. H., Frödin M., and Hansen S. H. (2012) A βPIX-PAK2 complex confers protection against Scrib-dependent and cadherin-mediated apoptosis. Curr. Biol. 22, 1747–1754 10.1016/j.cub.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu W. J., Tu S., and Cerione R. A. (2003) Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 114, 715–725 10.1016/S0092-8674(03)00688-3 [DOI] [PubMed] [Google Scholar]
- 61. Stevens B. M., Folts C. J., Cui W., Bardin A. L., Walter K., Carson-Walter E., Vescovi A., and Noble M. (2014) Cool-1-mediated inhibition of c-Cbl modulates multiple critical properties of glioblastomas, including the ability to generate tumors in vivo. Stem Cells 32, 1124–1135 10.1002/stem.1644 [DOI] [PubMed] [Google Scholar]
- 62. Goel P., Manning J. A., and Kumar S. (2015) NEDD4-2 (NEDD4L): the ubiquitin ligase for multiple membrane proteins. Gene 557, 1–10 10.1016/j.gene.2014.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Neumann D., Woods A., Carling D., Wallimann T., and Schlattner U. (2003) Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr. Purif. 30, 230–237 10.1016/S1046-5928(03)00126-8 [DOI] [PubMed] [Google Scholar]
- 64. Mukherjee A., Wang Z., Kinlough C. L., Poland P. A., Marciszyn A. L., Montalbetti N., Carattino M. D., Butterworth M. B., Kleyman T. R., and Hughey R. P. (2017) Specific palmitoyltransferases associate with and activate the epithelial sodium channel. J. Biol. Chem. 292, 4152–4163 10.1074/jbc.M117.776146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hallows K. R., McCane J. E., Kemp B. E., Witters L. A., and Foskett J. K. (2003) Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J. Biol. Chem. 278, 998–1004 10.1074/jbc.M210621200 [DOI] [PubMed] [Google Scholar]
- 66. Hartley J. L., Temple G. F., and Brasch M. A. (2000) DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795 10.1101/gr.143000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tuerk R. D., Auchli Y., Thali R. F., Scholz R., Wallimann T., Brunisholz R. A., and Neumann D. (2009) Tracking and quantification of 32P-labeled phosphopeptides in liquid chromatography matrix-assisted laser desorption/ionization mass spectrometry. Anal. Biochem. 390, 141–148 10.1016/j.ab.2009.04.015 [DOI] [PubMed] [Google Scholar]
- 68. Djouder N., Tuerk R. D., Suter M., Salvioni P., Thali R. F., Scholz R., Vaahtomeri K., Auchli Y., Rechsteiner H., Brunisholz R. A., Viollet B., Mäkelä T. P., Wallimann T., Neumann D., and Krek W. (2010) PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. EMBO J. 29, 469–481 10.1038/emboj.2009.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mohan S., Bruns J. R., Weixel K. M., Edinger R. S., Bruns J. B., Kleyman T. R., Johnson J. P., and Weisz O. A. (2004) Differential current decay profiles of epithelial sodium channel subunit combinations in polarized renal epithelial cells. J. Biol. Chem. 279, 32071–32078 10.1074/jbc.M405091200 [DOI] [PubMed] [Google Scholar]
- 70. Degasperi A., Birtwistle M. R., Volinsky N., Rauch J., Kolch W., and Kholodenko B. N. (2014) Evaluating strategies to normalise biological replicates of Western blot data. PLoS ONE 9, e87293 10.1371/journal.pone.0087293 [DOI] [PMC free article] [PubMed] [Google Scholar]