

Abstract
The role of albumin reabsorption in proximal tubule (PT) cells has emerged as an important factor in the genesis of albuminuria observed in the early stages of diabetes. Evidence has shown that a decrease in megalin expression could be the key mechanism in this process. In the present work, we investigated the molecular mechanism underlying the modulation of albumin endocytosis in LLC-PK1 cells, a model of PT cells. High glucose concentrations (HG) inhibited megalin expression and albumin endocytosis after 48 h of incubation. This inhibitory effect involves the entrance of glucose into PT cells through SGLT located at the luminal membrane. Once inside PT cells, glucose is diverted to the hexosamine biosynthetic pathway (HBP) increasing O-GlcNAcylation of several intracellular proteins, including PKB. This process promotes the inhibition of PKB activity measured by its phosphorylation at Thr-308 and Ser-473 and phosphorylation of specific substrates, glycogen synthase kinase 3β (GSK3β) and tuberous sclerosis complex 2. The decrease in PKB activity led to a decrease in megalin expression and, consequently, reducing albumin endocytosis in LLC-PK1 cells. HG did not change mammalian target of rapamycin (mTOR) C2 activity, responsible for phosphorylated PKB at Ser-473. In addition, HG activated the mTORC1/S6K pathway, but this effect was not correlated to the decrease in megalin expression or albumin endocytosis. Taken together, our data help to clarify the current understanding underlying the genesis of tubular albuminuria induced by hyperglycemia in the early stage of diabetes pathogenesis.
Keywords: kidney, signal transduction, O-GlcNAcylation, mTOR complex (mTORC), Akt PKB, albumin endocytosis, diabetic nephropathy, high glucose, megalin expression, proximal tubule
Introduction
Diabetes mellitus is a major cause of progression to end-stage renal disease, and albuminuria is a good marker of this process (1, 2). The role of albumin reabsorption in proximal tubule (PT)2 cells has emerged as an important factor in the genesis of albuminuria observed in the early stages of diabetes (3–5). Albumin reabsorption in these cells involves receptor-mediated endocytosis, with megalin as a central receptor (6, 7). It was observed that a high glucose concentration (HG) reduces megalin expression and albumin reabsorption in a streptozotocin-induced diabetes animal model as well as in cultured PT cells (8–10). In addition, Wang et al. (11) showed that an intravenous infusion of HG in Sprague-Dawley rats induced microalbuminuria and reduced megalin expression in PT cells without changes in the glomerular flow rate.
Megalin/LRP2 is a scavenger receptor belonging to the low-density lipoprotein receptor family located at the PT luminal membrane (6, 7, 12, 13). Megalin-mediated endocytosis is a fine-tuning process that involves protein–protein interaction and phosphorylation by specific kinases (12–17). In this context, the phosphatidylinositide 3-kinase/PKB pathway has been shown to play an important role in modulating albumin endocytosis in PT cells (14). In a previous work, our group (15–17) showed that PKB binds to megalin at the luminal membrane of PT cells, promoting modulation of albumin endocytosis associated with cell survival. In addition, PKB is a central molecule linking the activation of mammalian target of rapamycin (mTOR) complexes, mTORC2 and mTORC1 (18–20).
Akt/PKB is a serine/threonine kinase activated by phosphorylation at serine 473 (Ser-473) and threonine 308 (Thr-308) residues mediated by mTORC2 and phosphoinositide-dependent protein kinase 1, respectively (16, 18). Once phosphorylated, PKB can lead to activation of mTORC1 (18–25). Besides phosphorylation, PKB activity has been described to be regulated by different post-translational modifications, including O-GlcNAcylation (26, 27).
O-GlcNAcylation is mainly found in serine (Ser) or threonine (Thr) hydroxyl moieties on nuclear and cytoplasmic proteins. Similar to phosphorylation, O-GlcNAcylation is a dynamic post-translational modification that regulates protein stability, subcellular localization, and activity (28–30). Interestingly, PKB was described containing few O-GlcNAcylation sites including residues Thr-308 (31) and Ser-473 (27). It is well-established that HG induces elevated O-GlcNAcylation by increasing glucose flux through the hexosamine biosynthetic pathway (HBP) (32–34). The end product of the HBP is the UDP-GlcNAc, which is used by O-GlcNAc transferase (OGT) to transfer the GlcNAc residue to proteins. O-GlcNAcase (OGA) is the enzyme responsible for its removal.
Based on these observations, we decided to investigate the modulation of albumin endocytosis in PT cells by HG, identifying the possible correlation among O-GlcNAcylation, PKB, and mTOR pathways. We used LLC-PK1 cells, a model of PT cells, incubated with a normal glucose concentration (NG) or HG. We observed that HG increased O-GlcNAcylation in these cells, and this phenomenon was correlated to the inhibition of albumin endocytosis. The molecular mechanism underlying this effect involves the inhibition of PKB activity as a result of its O-GlcNAcylation, leading to a decrease in megalin expression and, consequently, to the decrease in megalin-mediated albumin endocytosis.
Results
Correlation between inhibition of albumin endocytosis in LLC-PK1 cells by high glucose concentration and O-GlcNAcylation
Initially, we investigated the effect of HG on albumin endocytosis in LLC-PK1 cells, a model of PT cells. The cells were incubated with a normal glucose concentration (5.5 mm, NG) or a high glucose concentration (30.0 mm, Fig. S1). Fig. 1A shows one representative experiment of albumin endocytosis measured with fluorescence microscopy using albumin–FITC. The cell membranes were marked with ZO-1 (red staining). HG inhibited albumin endocytosis after 48 h of incubation (Fig. 1, A and B). To further characterize this effect, the cells were incubated with HG for different periods of incubation (from 1 to 48 h), and albumin endocytosis was measured by cell-associated fluorescence using albumin–FITC as a tracer. Albumin endocytosis was inhibited by HG after 2 h of incubation and was maintained up to 48 h (Fig. 1C). Cell viability, measured by lactate dehydrogenase and propidium iodide staining assays, was not changed under these experimental conditions (Fig. S2, A–C).
Figure 1.

High glucose entry through SGLT reduces albumin endocytosis in proximal tubule cells. A, the effect of HG (30.0 mm glucose) on albumin endocytosis. Albumin endocytosis was measured by fluorescence microscopy. The green color (BSA–FITC) indicates endocytic albumin, and red (Alexa Fluor 568-conjugated antibody) indicates ZO-1 expression. Scale bar, 50 μm (n = 3). Details of the original image are shown in the bottom inset. B, quantitative analysis of fluorescence microscopy. C, time course effect of HG on albumin endocytosis (n = 9). D, effect of HG on albumin endocytosis when added to either the luminal side or the basolateral side (n = 3). E, the effect of 100 μm PLZ (an SGLT inhibitor) on HG-inhibited albumin endocytosis (n = 6). In C–E, albumin endocytosis was measured by cell-associated fluorescence using albumin–FITC. The results are shown as mean ± S.E. *, p < 0.05 versus NG (normal glucose condition used as control).
This effect was specific for HG once the incubation of cells with 30 mm mannitol did not change the albumin endocytosis (Fig. S3A). In another experimental set, we grew the cells in Transwell® inserts and the HG effect was added to the basolateral or luminal membranes (Fig. 1D). HG at the basolateral membrane did not change albumin endocytosis, but did inhibit it at the luminal membrane. Usually, entry of glucose into PT epithelial cells (PTECs) is mediated by the Na+/glucose cotransporters, SGLT1 and SGLT2 (35). Co-incubation of the cells with 100 μm phlorizin (PLZ), an SGLT inhibitor, for 48 h abolished the inhibitory effect of HG on albumin endocytosis (Fig. 1E). Based on these results, we postulate that the inhibition of albumin endocytosis in LLC-PK1 cells by HG involves the entry of glucose through SGLT cotransporters.
We then investigated if the inhibitory effect of HG on albumin endocytosis is due to an increase in the O-GlcNAcylation level. Cells were incubated with HG (from 1 to 48 h) and the O-GlcNAcylation level was assessed by immunoblotting using mAb (clone CTD110.6). HG increased the total O-GlcNAcylation level after 1 h of incubation and this was maintained up to 48 h (Fig. 2, A and B) as observed with albumin endocytosis. Simultaneous incubation of the cells with HG and 100 μm PLZ reduced the O-GlcNAcylation induced by HG (Fig. 2, C and D).
Figure 2.

The inhibitory effect of HG on albumin endocytosis depends on increased O-GlcNAcylation. A, the time course effect of HG on O-GlcNAcylation (n = 3). The cells were incubated with HG for 1, 2, 4, 12, and 48 h for subsequent assessment of O-GlcNAcylation. B, densitometry analysis of the time course effect of HG on O-GlcNAcylation. C and E, the effect of 100 μm PLZ (C) and 10.0 μm DON (E) on HG-induced O-GlcNAcylation measured by immunoblotting (n = 3). D and F, densitometry analyses related to C and E. G and H, the effect of DON (G) and 1.0 μm TMG (H) on HG-inhibited albumin endocytosis (n = 6). PLZ, an SGLT inhibitor; DON, 6a GFAT inhibitor; TMG, an OGA inhibitor. In G and H, albumin endocytosis was measured by cell-associated fluorescence using albumin–FITC. The results are shown as mean ± S.E. *, p < 0.05 versus NG; #, p < 0.05 versus HG.
The rate-limiting step in the production of UDP-GlcNAc, a substrate for O-GlcNAcylation, in the HBP is the conversion of fructose 6-phosphate into glucosamine 6-phosphate mediated by glutamine fructose-6-phosphate amidotransferase (GFAT) (26, 33). As expected, the incubation of HG cells with 10.0 μm 6-diazo-5-oxo-l-norleucine (DON), an inhibitor of GFAT, reduces the O-GlcNAcylation level (Fig. 2, E and F). Interestingly, the reduction of O-GlcNAcylation impaired the inhibitory effect on albumin endocytosis (Fig. 2G). Furthermore, the incubation of LLC-PK1 cells with Thiamet G (TMG), an inhibitor of OGA, for 48 h increased the O-GlcNAcylation level (Fig. S3, B and C) and mimicked the inhibitory effect of HG on albumin endocytosis (Fig. 2H).
So far, these results show that HG increases cellular O-GlcNAcylation, leading to a reduction in albumin endocytosis in PT cells. However, how the increase in the O-GlcNAcylation level is correlated to the decrease in albumin endocytosis is an open question.
Decrease in megalin expression mediates the inhibitory effect of high glucose concentration on albumin endocytosis in PTECs
HG decreased megalin expression in LLC-PK1 cells measured by immunoblotting and immunofluorescence (Fig. 3, A–F). In agreement with a previous result, this effect of HG was only observed when HG was added at the luminal membrane (Fig. 3, C and D). Furthermore, co-incubation of the cells with 100 μm PLZ or 10 μm DON abolished the inhibitory effect of HG on megalin expression (Fig. 3, E and F). In addition to megalin, it has been proposed that CD36 present at the luminal membrane of PT cells could also be involved in albumin endocytosis in PT cells (36). Thus, the effect of HG on CD36 expression was assessed (Fig. 3, G and H). Contrary to the effect on megalin expression and albumin endocytosis, HG increased CD36 expression. These results indicate that the inhibitory effect of HG on albumin endocytosis should be correlated to the decrease in megalin expression due to the increase in the O-GlcNAcylation level.
Figure 3.

O-GlcNAcylation mediates the effects of HG on megalin expression. A, the effect of HG on megalin expression (n = 3). B, densitometry analysis of megalin expression. C, the effect of HG on megalin expression when HG was added to the luminal side or the basolateral side measured by immunofluorescence microcopy (n = 3). D, quantitative analysis of C. E, the effect of 100 μm PLZ and 10.0 μm DON on HG-inhibited megalin expression measured by immunofluorescence microcopy (n = 3). F, quantitative analysis of E. G, the effect of HG on CD36 expression measured by immunofluorescence microcopy (n = 3). H, quantitative analysis of G. The green color (Alexa Fluor 488-conjugated antibody) indicates megalin expression in B and C, and CD36 expression in D. The blue color (DAPI) indicates nuclei. Scale bars, 50 μm. PLZ, an SGLT inhibitor; DON, a GFAT inhibitor. The results are shown as mean ± S.E. *, p < 0.05 versus NG.
Activation of the mTORC1/S6K pathway by high glucose concentration is not involved in the inhibition of albumin endocytosis
Here, it was observed that HG increased mTORC1 activity measured by phosphorylation of mTOR at Ser-2448, a marker of mTORC1 activation, and phosphorylation of S6K at Thr-389, a specific substrate for mTORC1, in LLC-PK1 cells (Fig. 4, A and B). The co-incubation of the cells with 100 μm PLZ or 10 μm DON abolished the phosphorylation of both residues induced by HG (Fig. 4, C and D), indicating the role of O-GlcNAcylation in this activation.
Figure 4.

HG increases mTORC1 activity through O-GlcNAcylation. A and B, the effect of HG on mTOR phosphorylation at Ser-2448 (A) and S6K phosphorylation at Thr-389 (B) (n = 3). C, the effect of 10.0 μm DON on HG-induced mTOR phosphorylation (n = 3). D, the effect of 100 μm PLZ on HG-induced S6K phosphorylation (n = 3). All densitometry analyses are shown at the bottom of each subpanel. Man, mannitol; DON, a GFAT inhibitor; PLZ, an SGLT inhibitor. The results are shown as mean ± S.E. *, p < 0.05 versus NG; #, p < 0.05 versus HG.
mTOR inhibitors, rapamycin and WYE-354, were used to study the possible correlation of the mTORC1/S6K pathway with the inhibitory effect of HG on albumin endocytosis. Co-incubation of the cells with 10−9 m rapamycin and 10−6 m WYE-354 did not change the inhibitory effect of HG on albumin endocytosis (Fig. 5, A–C). In agreement, these mTOR inhibitors also did not block the inhibitory effect of HG on megalin expression (Fig. 5, D and E). Thereby, the inhibitory effect of HG on albumin endocytosis does not involve activation of the mTORC1/S6K pathway.
Figure 5.

mTORC1 overactivation is not involved in the inhibitory effect of HG on megalin-mediated albumin endocytosis. A, the effect of 10−9 m rapamycin and 10−6 m WYE-354 on HG-inhibited albumin endocytosis. Albumin endocytosis was measured by fluorescence microscopy. The green color (BSA–FITC) indicates endocytic albumin, and red (Alexa Fluor 568-conjugated antibody) indicates ZO-1 expression. Details of the original image are shown in the bottom inset. Scale bar, 50 μm (n = 3). B, quantification analysis of A. C, BSA–FITC uptake assay (n = 9). Albumin endocytosis was measured by cell-associated fluorescence using albumin–FITC. D, the effect of 10−9 m rapamycin and 10−6 m WYE-354 (mTOR inhibitors) on HG-inhibited megalin expression measured by immunofluorescence. The green color (Alexa Fluor 488-conjugated antibody) indicates megalin expression and the blue color (DAPI) indicates nuclei. Scale bar, 100 μm (n = 3). E, quantitative analysis of D. The results are shown as mean ± S.E. *, p < 0.05 versus NG.
PKB mediates the inhibitory effect of HG on albumin endocytosis
In previous work (15, 16), it was shown that PKB anchors at the plasma membrane through megalin binding, and this process is important for albumin endocytosis. Thus, we investigated the possible involvement of PKB in the inhibitory effect of HG on albumin endocytosis.
Initially, PKB activity was measured by phosphorylation of Ser-473 and Thr-308 residues and phosphorylation of specific substrates GSK3β and TSC2 at Ser-9 and Thr-1462, respectively. It was observed that PKB activity was decreased by HG (Fig. 6, A and B). Then, to correlate the inhibition of PKB activity with the inhibitory effect of HG on albumin endocytosis, the cells were incubated with 10 nm MK-2206, a PKB inhibitor. Interestingly, the PKB inhibitor mimicked the inhibitory effect of HG on albumin endocytosis (Fig. 6C) and on megalin expression (Fig. 6, D and E). Furthermore, it was observed that incubation of the cells with 10% FBS, which activates PKB, blocked the inhibitory effect of HG on albumin endocytosis (Fig. 6C) as well as on megalin expression (Fig. 6, D–G). Together, our results show that HG inhibited PKB activity, and this effect is involved in the inhibition of albumin endocytosis in PT cells.
Figure 6.

Reduction of PKB activity mediates the inhibitory effect of HG on megalin expression and albumin endocytosis. A, the inhibitory effect of HG on PKB phosphorylation and activity. PKB phosphorylation at both Thr-308 and Ser-473 residues (n = 4). PKB substrate phosphorylation: GSK3β at Ser-9 and TSC2 at Thr-1462 (n = 4). B, densitometry analysis of A. PKB at Thr-308 (first bar), PKB at Ser-473 (second bar), GSK3β at Ser-9 (third bar), and TSC2 at Thr-1462 (fourth bar). C, effect of 10−8 m MK-2206 and 10% FBS in HG-inhibited albumin endocytosis (n = 5). Albumin endocytosis was measured by cell-associated fluorescence using albumin–FITC. D, the effect of 10−8 m MK-2206 and 10% FBS on HG-inhibited megalin expression using immunofluorescence analysis (n = 3). E, quantitative analysis of D. F, the effect of 10% FBS on HG-inhibited megalin expression using immunoblotting analysis (n = 3). G, densitometry analysis of F. Man, mannitol. The results are shown as mean ± S.E. *, p < 0.05 versus NG.
PKB O-GlcNAcylation induced by HG is responsible for reduced PKB activity
Because phosphorylation of PKB at Ser-473 is mediated by mTORC2, we decided to investigate if modulation of mTORC2 activity mediates the inhibitory effect of HG on PKB activity. Interestingly, HG did not change the phosphorylation of mTOR at Ser-2481, a marker of mTORC2 activity (Fig. 7A) or the phosphorylation of PKB at Thr-450, a specific substrate for mTORC2 (Fig. 7B). Furthermore, it has been shown that the phosphorylation of RICTOR at Thr-1135 is a negative modulator of mTORC2 activity (20). Here, we observed that this phosphorylation is not changed by HG (Fig. 7C). These results indicate that mTORC2 activity is not changed by HG. Based on these results, we investigated if O-GlcNAcylation could be responsible for PKB inhibition induced by HG.
Figure 7.

The inhibition of PKB activity by HG does not involves changes in mTORC2 activity. A–C, the effect of HG on: (A) mTOR phosphorylation at Ser-2481 residue (n = 4), (B) PKB phosphorylation at Thr-450 residue (n = 4), and (C) RICTOR phosphorylation at Thr-1135 residue (n = 4). All densitometry analyses are shown at the bottom of each subpanel. Man, mannitol. The results are shown as mean ± S.E. n.s., not significant (p > 0.05).
Co-incubation of the cells with 100 μm PLZ (Fig. 8A) or 10 μm DON (Fig. 8B) completely abolished the inhibition of PKB phosphorylation at Ser-473 induced by HG. Following the idea that O-GlcNAcylation could be involved in inhibition of PKB activity by HG, we decided to investigate if HG induces O-GlcNAcylation of PKB in LLC-PK1 cells. Immunoprecipitation of PKB was performed and then probed for O-GlcNAcylation. Clearly, HG induced the O-GlcNAcylation of PKB, which was determined using immunoprecipitation (Fig. 8C) and enzymatic labeling (Fig. 8D). Taken together these results indicate that PKB inactivation is probably induced by its O-GlcNAcylation. This effect leads to a decrease in PKB activity, megalin expression, and PT albumin uptake.
Figure 8.

PKB O-GlcNAcylation induced by HG is responsible for reduced PKB activity. A and B, the effect of 100 μm PLZ (A) and 10.0 μm DON (B) on HG-inhibited PKB phosphorylation at Ser-473 (n = 4). C, the effect of HG on PKB O-GlcNAcylation (n = 4). All densitometry analyses are shown at the bottom of each subpanel. D, PKB O-GlcNAcylation determined by enzymatic labeling (n = 3). Plus sign, the presence of Gal-T1; minus sign, absence of Gal-T1. Man, mannitol; DON, a GFAT inhibitor; PLZ, an SGLT inhibitor. The results are shown as mean ± S.E. *, p < 0.05 versus NG; #, p < 0.05 versus HG. IP, immunoprecipitation.
Discussion
Albuminuria is observed in the early stages of diabetes even before the onset of chronic kidney disease (1, 2). The role of albumin reabsorption in PT cells has been highlighted as an important mechanism in this process and a possible therapeutic target to halt the progression of renal disease (5, 16, 37–39). Some groups showed that HG decreases megalin expression and albumin endocytosis in PT cells (8–10). In this present work, using LLC-PK1 cells, we showed that the mechanism underlying the effect of HG involves the entry of glucose through luminal SGLT, O-GlcNAcylation of PKB, and its inhibition. These effects lead to a decrease in megalin expression and, consequently, in albumin reabsorption in PT cells. Interestingly, the modulatory effect of HG on the mTORC1/S6K pathway is not involved in this process. These results help to clarify the current understanding underlying the genesis of tubular albuminuria induced by HG in the early stage of diabetes pathogenesis.
One possible question concerns the concentration of HG used. The blood glucose concentration in the streptozotocin-induced diabetes animal model ranges from 360 to 600 mg/dl, corresponding to 20–33 mm (8, 9, 40, 41). This blood glucose concentration is close to the concentration used in the present study. Furthermore, the concentration of HG in the present work follows the same protocol used by different authors to study the effect of HG on PT cells (43–48). In addition, Ishibashi (43) showed that incubation of LLC-PK1 cells for 6 days with 16 mm glucose inhibits albumin uptake in LLC-PK1 cells at the same magnitude as 27 mm glucose.
In agreement with our results, Tojo et al. (8), using an early-stage diabetic nephropathy animal model, observed glucosuria and albuminuria without changes in the glomerular flow rate. They observed a decrease in megalin expression leading to a decrease in PT albumin reabsorption. In addition, it has been shown that PT PKB activity is inhibited in the streptozotocin-induced diabetes animal model (49). Here, we observed that HG decreases megalin expression and albumin uptake and inhibits PKB activity in LLC-PK1 cells. These observations indicate that the results obtained for LLC-PK1 cells are compatible with those obtained in the animal model.
It is well-known that the genesis of diabetic nephropathy (DN) comprises deleterious effects of hyperglycemia in renal glomerular and tubular structures (1, 2, 22, 23, 25, 26, 50, 51). During the early stage of diabetes, there is an increase in serum glucose, glomerular glucose filtration, and, consequently, luminal glucose concentration (1, 2, 52). Glucose is reabsorbed at PT cells through coordinate transporter mechanisms located at the luminal and basolateral membranes, avoiding loss of glucose in the urine. At the early stage of diabetes, this mechanism could lead to a dangerous loop aggravating hyperglycemia (1, 2, 52). Therefore, PT cells are exposed to HG at both luminal and basolateral membranes.
One important issue to be considered is the laterality of the HG effect. Here, we observed that the inhibitory effect of HG on albumin endocytosis depends on its entry at the luminal membrane by SGLT decreasing megalin expression and, consequently, inhibition of albumin endocytosis in LLC-PK1 cells. In agreement, some authors showed that the treatment of patients and animal models of DN with SGLT2 inhibitors ameliorates proteinuria, tubule-interstitial injury, and consequently, slows the progression of renal disease (52–55). We showed that glucose inside the LLC-PK1 cells increases O-GlcNAcylation, which is correlated with inhibition of albumin endocytosis, opening the possibility that this mechanism could be involved in albuminuria in the early stage of DN. In agreement, Akimoto et al. (56) showed increased O-GlcNAcylation in specific PT cytoskeleton proteins in a T2DM animal model. Ji et al. (57) suggested that abnormal O-GlcNAcylation of actin and tubulin alters their polymerization, reducing microtubule-dependent reabsorption mechanism in PT cells. Gellai et al. (58) also showed that incubation of HK2 (human kidney cells, a model of PT cells) with HG also increased O-GlcNAcylation, which was correlated to an increase in DN-induced fibrosis.
Some authors have proposed that changes in the albumin endocytosis machinery induce a proinflammatory phenotype in the PT environment, contributing to tubule-interstitial injury (16, 39). It was observed that the decrease in megalin expression involves transforming growth factor-β secretion (59–61). In a previous work (16), our group showed that the decrease in megalin expression induced by a high albumin concentration is associated with apoptosis in LLC-PK1 cells. Thus, it is possible to postulate that the decrease in megalin expression due to HG-induced O-GlcNAcylation is also involved with the development of tubule-interstitial injury.
Besides megalin, some evidence indicates that CD36 could also contribute to albumin endocytosis in PTECs (36, 62). However, the significance of this process on total albumin endocytosis is still an open matter. Significant albuminuria was observed in T2DM patients, even with increasing CD36 expression in PT cells (36). We also observed an increase in CD36 expression induced by HG despite the decrease in albumin endocytosis in LLC-PK1 cells. Taken together, these observations indicate that the increase in CD36 expression could be a compensatory mechanism avoiding more severe proteinuria and/or it could be involved in tubule-interstitial injury induced by HG contributing to the progression of DN. Reinforcing the second idea, it has been proposed that CD36 mediates injury in PT cells, including apoptosis of these cells in DN (62).
The idea that mTOR complexes could be involved in the effects of HG on renal function is supported by enhancement of mTORC1 activity under these conditions. This process modifies the fine balance between mTORC1 and mTORC2 activities, which is required to maintain cell homeostasis (18, 21–23). In podocyte cells, the activation of mTORC1 by hyperglycemia mediates the podocyte injury observed in DN (22, 23). In recent work, Grahammer et al. (21), using specific raptor knockout mice in renal tubular cells, observed significant proteinuria due to inhibition of albumin endocytosis in PT cells, although without change in megalin expression. Similarly, we observed that the inhibition of mTORC1 activity decreased albumin endocytosis in PT cells. However, we observed that despite activation of the mTORC1/S6K pathway, HG inhibited albumin endocytosis, indicating that these effects involve independent pathways. Palm et al. (63), using mouse embryonic fibroblasts, showed that mTORC1 activation inhibits albumin proteolysis in lysosomes.
Interestingly, we observed that HG did not change the mTORC2 activity measured by phosphorylation of PKB at Thr-450, a specific site for mTORC2 (18, 27). Supporting our observation, Grahammer et al. (21) showed that inhibition of mTORC2, using a specific RICTOR knockout in renal tubular cells, did not change albumin reabsorption in PTECs. In a previous work, we showed that overactivation of the mTORC1/S6K pathway by higher albumin concentration promotes the inhibition of mTORC2 activity due to the increase in phosphorylation of RICTOR at Thr-1135 (20). Here, it was observed that the phosphorylation of this residue was not changed by HG, even when the mTORC1/S6K pathway was activated. Despite these results, we cannot rule out the possible modulation of albumin endocytosis in PT cells by mTORC2. It has been shown that simultaneous inhibition of mTORC1 and mTORC2 could have additional deleterious effects on podocyte cells and albumin endocytosis in PT cells (21, 22). This view agrees with a proposal that a fine balance between mTORC1 and mTORC2 activities is required to maintain the fine-tuning in cell homeostasis.
The following data obtained in our study show that the inhibition of PKB activity mediates the inhibitory effect of HG on megalin expression and, consequently on albumin endocytosis: 1) HG inhibits PKB activity; 2) PKB inhibitor mimics the effects of HG on albumin endocytosis and megalin expression; 3) serum reconstitution, an activator of PKB, reversed the effects of HG. In agreement with this result, a strict relationship between albumin endocytosis and PKB activity has been shown (14–17). Here, we show that the mechanism underlying the inhibitory effect of HG on PKB activity involves O-GlcNAcylation. The increase of O-GlcNAc levels induced by HG often results in deregulation of signaling pathways that are important for the development of pathologies such diabetes, chronic kidney disease, cancer, among others (33, 64–69). Recent evidence indicates that O-GlcNAcylation of kinases play an important role in regulating cell signaling (70). In accordance, it has been shown that O-GlcNAcylation of PKB in different cell types decreases phosphorylation at Ser-473 and Thr-308 by steric hindrance or competition, and consequently, inhibition of PKB (27, 31). Once O-GlcNAcylated, PKB is inhibited, leading to a decrease in total megalin level. How this happens is still an open matter. One possibility is the change in the half-life of megalin due to its degradation (71). Another possibility could be a decrease in megalin synthesis. Zhou et al. (10) showed that the incubation of NRK52E cells, a tubule epithelial cell line, with HG decreases mRNA for megalin, as well as megalin synthesis. In addition, some studies have shown that PKB can modulate transcription factors such as FOXO1 and nuclear factor-κB, changing the protein synthesis (18, 72). However, more experiments will be necessary to clarify this issue.
Taken together, our results allow us to build up a model describing the molecular mechanism underlying the inhibition of megalin-mediated albumin endocytosis triggered by HG (Fig. 9). Glucose enters PT cells through SGLT, increasing UDP-GlcNAc levels and O-GlcNAcylation of several intracellular proteins, including PKB, promoting its inhibition. This process leads to a decrease in megalin levels and, consequently a decrease in albumin endocytosis in PT cells.
Figure 9.
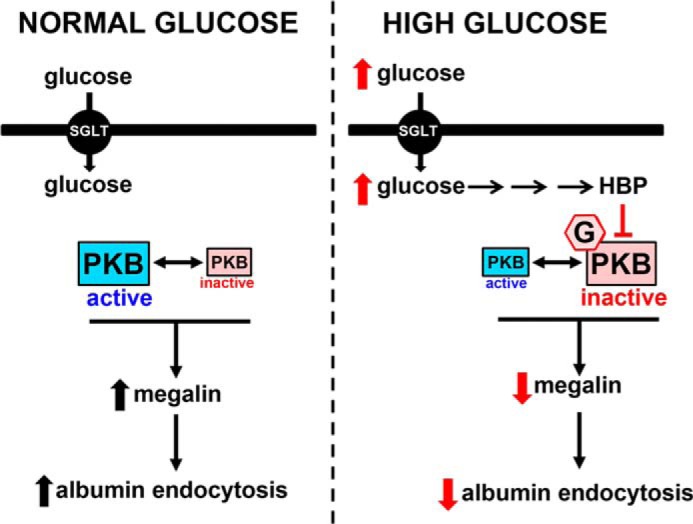
Schematic overview of the effects of HG on albumin endocytosis in PT cells. The scheme shows PKB in two different states (“active” shown in blue and “inactive” shown in red), which lead to modulation of megalin expression and albumin endocytosis. Under a normal glucose concentration (5.5 mm), PKB is in the active state, whereas under a high glucose concentration (30 mm), PKB is in an inactive state induced by O-GlcNAcylation. G, GlcNAc residue transferred to PKB. HBP, hexosamine biosynthetic pathway. Black arrows indicate activation and red arrows indicate inhibition.
The results obtained in the present study together with those previously published indicate that PT cells could play a more important role in DN than has been proposed previously. This hypothesis is well-supported in a recent revision published by Gilbert (73). The author pointed out the central role of PT cells in DN, suggesting that a paradigm shift in “glomerulocentric” is necessary and pointing out the role of the PT segment in the genesis and development of so-called diabetic kidney disease.
Experimental procedures
Materials and reagents
d(+)-Glucose, BSA, BSA–FITC, sodium chloride, potassium chloride, magnesium chloride, calcium chloride, sodium orthovanadate, sodium pyrophosphate, sodium fluoride, sodium β-glycerophosphate, sodium azide, sodium carbonate, sodium hydroxide, ammonium persulfate, glycine, TritonTM X-100, Tween® 20, MOPS, EGTA, Hepes, Folin & Ciocalteu's phenol reagent, protease inhibitor mixture (number I3786), tetramethylethylenediamine, acrylamide, bromphenol blue, 2-mercaptoethanol, Ponceau S, PMSF, normal goat serum, normal donkey serum, phlorizin, DON, and rapamycin were purchased from Sigma. Mannitol, sodium/potassium tartrate, copper(II) sulfate, glycerol, and acetic acid were purchased from Reagen (Colombo, PR, Brazil). PVDF membranes, WYE-354, and TMG, methanol, DMSO, and monoclonal AKT/PKB-pleckstrin homology domain (clone SKB1) antibody were purchased from Merck Millipore (Barueri, SP, Brazil). ECLTM prime, SDS, and Tris were purchased from GE Healthcare. Lactate dehydrogenase Liquiform (number 86-2/30) was purchased from Labtest (Lagoa Santa, MG, Brazil). MK-2206 was purchased from Selleckchem (Houston, TX). Monoclonal O-GlcNAc (RL-2 or CTD110.6) and HRP-conjugated anti-mouse IgM antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Polyclonal phospho-mTOR (Ser-2481), monoclonal phospho-mTOR (Ser-2448) (clone D9C2), monoclonal mTOR (clone 7C10), polyclonal mTOR, monoclonal phospho-RICTOR (Thr-1135) (clone D30A3), monoclonal RICTOR (clone D16H9), polyclonal phospho-PKB (Ser-473), monoclonal phospho-PKB (Thr-308) (clone 244F9), polyclonal phospho-PKB (Thr-450), polyclonal PKB, polyclonal phospho-GSK-3β (Ser-9), monoclonal S6K (Thr-389) (clone 108D2), polyclonal S6K, monoclonal phospho-TSC2 (Thr-1462) (clone 5B12), monoclonal TSC2 (clone D93F12), HRP-conjugated anti-rabbit IgG, and HRP-conjugated anti-mouse IgG antibodies were purchased from Cell Signaling Technology (Danvers, MA). Polyclonal Lrp2/megalin and monoclonal CD36 (clone FA6–152) antibodies were purchased from Abcam (Cambridge, MA). Polyclonal ZO-1 antibody, DMEM, PBS, DAPI, FBS, UltraPureTM N,N′-methylenebisacrylamide, Gal-T1 Y289L, UDP-GalNAz, biotin alkyne, Alexa Fluor® 488-conjugated anti-rabbit IgG, Alexa Fluor® 546-conjugated anti-rabbit IgG and Alexa Fluor® 488-conjugated anti-mouse IgG were purchased from Thermo Fisher Scientific (Waltham, MA). Vectashield® antifade mounting medium was purchased from Vector Laboratories (Burlingame, CA). LLC-PK1 cells were obtained from the American Type Culture Collection (Rockville, MD). All other reagents were of the highest purity available.
Cell culture
LLC-PK1 cells, a PT cell line, were cultured in six-well plates or in Transwell inserts when indicated. The cells were grown at 37 °C in 5% CO2 in DMEM (glucose 5.5 mm, NG) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin for 3 consecutive days, usually reaching 95–98% confluence (15, 16, 19, 20, 74–76). When indicated, a high glucose concentration (final concentration, 30 mm) was added to the cells 24 h after seeding and then maintained for 48 h. The cells were serum starved in the last 12 h before they were used in the experiments. The scheme of the experimental design is indicated in Fig. S1. The serum starvation was done to keep several signaling proteins triggered by growth factors under a basal state of activation. After treatment, the cells were used to carry out different experimental approaches as shown below, such as albumin endocytosis, immunoprecipitation, immunoblotting, and immunofluorescence assays.
Albumin endocytosis assay
Albumin endocytosis was measured by BSA–FITC uptake assay as described previously (15, 75). Briefly, cells were incubated with Ringer solution (20 mm Hepes-Tris, pH 7.4, 140 mm NaCl, 2.7 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm d(+)-glucose) containing 30 μg/ml of BSA–FITC at 37 °C for 30 min. Unbound BSA–FITC was removed by washing 11 times with ice-cold Ringer solution. Parallel experiments were performed on cells held at 4 °C to abolish endocytosis and verify possible nonspecific bound BSA–FITC. BSA uptake was assessed by cell-associated fluorescence using albumin–FITC or fluorescence microscopy.
In the cell-associated fluorescence method, the cells were lysed using detergent solution (0.1% Triton X-100 in 20 mm MOPS) and the cell-associated fluorescence was measured using a microplate spectrofluorimeter (SpectraMax M2, Molecular Devices, Sunnyvale, CA). BSA-specific uptake was calculated as the difference between the BSA–FITC uptake in the absence and presence of 20 mg/ml of unlabeled BSA. The blank in the experiment was defined as the fluorescence measured in the absence of BSA–FITC; its value was less than 10% of the total fluorescence.
For fluorescence microscopy, the cells were fixed with 4% paraformaldehyde at 25 °C for 15 min, permeabilized with 0.1% Triton X-100 for 1–2 min, and blocked with 3% nonfat milk in PBS2+ for 30 min (15). To stain the plasma membrane, the cells were incubated with specific mouse primary antibodies, ZO-1 and Alexa Fluor 546-conjugated IgG. Then, the cells were mounted in Vectashield medium. To quantify albumin endocytosis, images of fluorescent-labeled markers were acquired by a Nikon 80i fluorescence microscope using a ×40 oil immersion objective lens, and the average intensity per cell was calculated using Image-Pro Plus software version 7.0.1.658 (Media Cybernetics, Rockville, MD) from multiple images after subtracting the background.
Immunoprecipitation and immunoblotting assay
The immunoprecipitation and immunoblotting assays were performed as described previously (20, 74). Briefly, the cells were incubated in lysis buffer (20 mm Hepes, pH 7.4, 2 mm EGTA, 1% TritonTM X-100, 0.01% SDS, 50 mm sodium fluoride, 5 mm sodium pyrophosphate, 5 mm sodium orthovanadate, 10 mm sodium β-glycerophosphate, 1 mm PMSF, 2× complete protease inhibitor) and cleared by centrifugation at 15,000 × g for 10 min at 4 °C. After centrifugation, the concentration of protein in the supernatant was determined by the Folin phenol method using BSA as standard (77). When indicated, PBS was immunoprecipitated using polyclonal PKB antibody according to the manufacturer's instructions. Then, proteins were then resolved on SDS-PAGE and transferred to PVDF membranes, according to the manufacturer's instructions. After antibody labeling, detection was performed using ECLTM prime.
Enzymatic labeling of O-GlcNAc sites
The enzymatic labeling of O-GlcNAc sites was determined as described previously (42). PKB was immunoprecipitated, washed with lysis buffer, and washed twice with reaction buffer containing 20 mm Hepes, pH 7.9, 50 mm NaCl, 1 μm PUGNAc, and 5 mm MnCl2 with protease and phosphatase inhibitors. Next, 3 μl of Gal-T1 Y289L and 3 μl of 0.5 mm UDP-GalNAz were added to a reaction volume of 30 μl. The reaction was performed overnight at 4 °C. The beads were washed twice with reaction buffer to remove excess UDP-GalNAz. The samples were then reacted with biotin alkyne according to the manufacturer's instructions. The enzymatic reactions were eluted with Laemmli buffer, resolved in SDS-PAGE. 12%, and then transferred to PVDF membranes according the manufacturer's instructions. After HRP-conjugated streptavidin labeling, detection was performed using ECLTM prime.
Immunofluorescence assay
The immunofluorescence assay was performed as described (16, 20, 74). Immunofluorescent staining for megalin, CD36, and ZO-1 was performed using specific antibodies as described by the manufacturer. Briefly, cells were grown in coverslips following the treatment protocols described above. After washing with PBS, the cells were fixed and permeabilized appropriately. The nonspecific binding sites for antibodies were blocked using 5% normal donkey or goat serum for 1 h, both at room temperature. Then, cells were incubated with the primary and secondary antibodies for 1 h at room temperature as indicated by the manufacturer's protocol. The nucleus was stained using DAPI. After staining, the coverslips were washed thoroughly in PBS and mounted with antiquenching medium (Vector Laboratories, Burlingame, CA), and then the slides were sealed. The fluorescence label was examined with a microscope (model Eclipse 80i, Nikon Corporation, Chiyoda-ku, Tokyo, Japan). Images were acquired using the manufacturer's software. Contrast and brightness settings were chosen to ensure that all pixels were within the linear range. Images were prepared for publication with Adobe Photoshop (Adobe Systems, San Jose, CA).
Statistical analysis
The results are expressed as mean ± S.E. Statistical significance was assessed by one-way analysis of variance, considering the treatments as factors. The significance of the differences was verified by the Newman-Keuls test for multiple comparisons. Statistical analysis was performed using absolute values. Significance was determined as p < 0.05.
Author contributions
D. d. B. P. and C. C.-N. conceptualization; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. data curation; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. formal analysis; D. d. B. P. and C. C.-N. validation; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. investigation; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. visualization; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. methodology; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. writing-original draft; D. d. B. P., R. P. S.-A., G. M. S., W. B. D., and C. C.-N. writing-review and editing; C. C.-N. resources; C. C.-N. supervision; C. C.-N. funding acquisition; C. C.-N. project administration.
Supplementary Material
Acknowledgments
We thank Douglas Esteves Teixeira (FAPERJ technical training fellowship) for excellent technical support and Lorna O'Brien for editorial assistance.
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico Grant 456997/2014-8 and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro-FAPERJ Grants E-26/110.085/2014, E-26/201.197/2014, and E-26/202.950/2016. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
- PT
- proximal tubule
- Akt/PKB
- protein kinase B
- CD36
- cluster of differentiation 36
- DAPI
- 4′,6-diamidino-2-phenylindole
- DMEM
- Dulbecco's modified Eagle's medium
- DN
- diabetic nephropathy
- DON
- 6-diazo-5-oxo-l-norleucine
- FBS
- fetal bovine serum
- FOXO1
- Forkhead box protein O1
- Gal-T1 Y289L
- mutant β-1,4-galactosyltransferase (Y289L)
- GFAT
- glutamine fructose-6-phosphate amidotransferase
- GSK3β
- glycogen synthase kinase 3β
- HBP
- hexosamine biosynthetic pathway
- HG
- high glucose
- HRP
- horseradish peroxidase
- LRP2
- low density lipoprotein-related protein 2
- mTORC1/2
- mammalian target of rapamycin complex 1 and 2
- NG
- normal glucose
- OGA
- O-GlcNAcase
- OGT
- O-GlcNAc transferase
- PLZ
- phlorizin
- PMSF
- phenylmethylsulfonyl fluoride
- PTECs
- proximal tubule epithelial cells
- PUGNAc
- O-(2-acetamido-2-deoxy-d-glucopyranosylidenamino) N-phenylcarbamate
- PVDF
- polyvinylidene fluoride
- RICTOR
- rapamycin-insensitive companion of mammalian target of rapamycin
- S6K
- S6 kinase
- SGLT1 and -2
- Na+/glucose co-transporter isoforms 1 and 2
- T2DM
- type 2 diabetes mellitus
- TMG
- Thiamet G
- TSC2
- tuberous sclerosis complex 2
- UDP-GalNAz
- uridine diphosphate azido-modified galactose
- UDP-GlcNAc
- uridine diphosphate N-acetylglucosamine
- ZO-1
- Zonula occludens-1.
References
- 1. Doshi S. M., and Friedman A. N. (2017) Diagnosis and management of type 2 diabetic kidney disease. Clin. J. Am. Soc. Nephrol. 12, 1366–1373 10.2215/CJN.11111016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campion C.G., Sanchez-Ferras O., and Batchu S.N. (2017) Potential role of serum and urinary biomarkers in diagnosis and prognosis of diabetic nephropathy. Can. J. Kidney Health Dis. 4, 2054358117705371 10.1177/2054358117705371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Comper W. D. (2014) Albuminuria is controlled primarily by proximal tubules. Nat. Rev. Nephrol. 10, 180 10.1038/nrneph.2013.58-c2, , , [DOI] [PubMed] [Google Scholar]
- 4. Moeller M. J., and Tanner G. A. (2014) Reply: podocytes are key-although albumin never reaches the slit diaphragm. Nat. Rev. Nephrol. 10, 180 10.1038/nrneph.2013.58-c2 [DOI] [PubMed] [Google Scholar]
- 5. Russo L. M., Sandoval R. M., Campos S. B., Molitoris B. A., Comper W. D., and Brown D. (2009) Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J Am. Soc. Nephrol. 20, 489–494 10.1681/ASN.2008050503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dickson L. E., Wagner M. C., Sandoval R. M., and Molitoris B. A. (2014) The proximal tubule and albuminuria: really! J. Am. Soc. Nephrol. 25, 443–453 10.1681/ASN.2013090950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Christensen E. I., and Birn H. (2013) Proteinuria: tubular handling of albumin-degradation or salvation? Nat. Rev. Nephrol. 9, 700–702 10.1038/nrneph.2013.212 [DOI] [PubMed] [Google Scholar]
- 8. Tojo A., Onozato M. L., Ha H., Kurihara H., Sakai T., Goto A., Fujita T., and Endou H. (2001) Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem. Cell Biol. 116, 269–276 10.1007/s004180100317 [DOI] [PubMed] [Google Scholar]
- 9. Tojo A., Onozato M. L., Kurihara H., Sakai T., Goto A., and Fujita T. (2003) Angiotensin II blockade restores albumin reabsorption in the proximal tubules of diabetic rats. Hypertens. Res. 26, 413–419 10.1291/hypres.26.413 [DOI] [PubMed] [Google Scholar]
- 10. Zhou L., Liu F., Huang X. R., Liu F., Chen H., Chung A. C., Shi J., Wei L., Lan H. Y., and Fu P. (2011) Amelioration of albuminuria in ROCK1 knockout mice with streptozotocin-induced diabetic kidney disease. Am. J. Nephrol. 34, 468–475 10.1159/000332040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J. Y., Yang J. H., Xu J., Jia J. Y., Zhang X. R., Yue X. D., Chen L. M., Shan C. Y., Zheng M. Y., Han F., Zhang Y., Yang X. Y., and Chang B. C. (2015) Renal tubular damage may contribute more to acute hyperglycemia induced kidney injury in non-diabetic conscious rats. J. Diabetes Complications 29, 621–628 10.1016/j.jdiacomp.2015.04.014 [DOI] [PubMed] [Google Scholar]
- 12. Nielsen R., Christensen E. I., and Birn H. (2016) Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 89, 58–67 10.1016/j.kint.2015.11.007 [DOI] [PubMed] [Google Scholar]
- 13. Gekle M. (2005) Renal tubule albumin transport. Annu. Rev. Physiol. 67, 573–594 10.1146/annurev.physiol.67.031103.154845 [DOI] [PubMed] [Google Scholar]
- 14. Brunskill N. J., Stuart J., Tobin A. B., Walls J., and Nahorski S. (1998) Receptor-mediated endocytosis of albumin by kidney proximal tubule cells is regulated by phosphatidylinositide 3-kinase. J. Clin. Invest. 101, 2140–2150 10.1172/JCI1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caruso-Neves C., Kwon S. H., and Guggino W. B. (2005) Albumin endocytosis in proximal tubule cells is modulated by angiotensin II through an AT2 receptor-mediated protein kinase B activation. Proc. Natl. Acad. Sci. U.S.A. 102, 17513–17518 10.1073/pnas.0507255102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caruso-Neves C., Pinheiro A. A., Cai H., Souza-Menezes J., and Guggino W. B. (2006) PKB and megalin determine the survival or death of renal proximal tubule cells. Proc. Natl. Acad. Sci. U.S.A. 103, 18810–18815 10.1073/pnas.0605029103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koral K., and Erkan E. (2012) PKB/Akt partners with Dab2 in albumin endocytosis. Am. J. Physiol. Renal Physiol. 302, F1013–F1024 10.1152/ajprenal.00289.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arnaud-Batista F. J., Peruchetti D. B., Abreu T. P., do Nascimento N. R., Malnic G., Fonteles M. C., and Caruso-Neves C. (2016) Uroguanylin modulates (Na2+K+)ATPase in a proximal tubule cell line: interactions among the cGMP/protein kinase G, cAMP/protein kinase A, and mTOR pathways. Biochim. Biophys. Acta 1860, 1431–1438 10.1016/j.bbagen.2016.04.012 [DOI] [PubMed] [Google Scholar]
- 20. Peruchetti D. B., Cheng J., Caruso-Neves C., and Guggino W. B. (2014) Mis-regulation of mammalian target of rapamycin (mTOR) complexes induced by albuminuria in proximal tubules. J. Biol. Chem. 289, 16790–16801 10.1074/jbc.M114.549717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grahammer F., Ramakrishnan S. K., Rinschen M. M., Larionov A. A., Syed M., Khatib H., Roerden M., Sass J. O., Helmstaedter M., Osenberg D., Kühne L., Kretz O., Wanner N., Jouret F., Benzing T., Artunc F., Huber T. B., and Theilig F. (2017) mTOR regulates endocytosis and nutrient transport in proximal tubular cells. J. Am. Soc. Nephrol. 28, 230–241 10.1681/ASN.2015111224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fantus D., Rogers N. M., Grahammer F., Huber T. B., and Thomson A. W. (2016) Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat. Rev. Nephrol. 12, 587–609 10.1038/nrneph.2016.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lieberthal W., and Levine J. S. (2012) Mammalian target of rapamycin and the kidney: II. pathophysiology and therapeutic implications. Am. J. Physiol. Renal Physiol. 303, F180–F191 10.1152/ajprenal.00015.2012 [DOI] [PubMed] [Google Scholar]
- 24. Mariappan M. M., Prasad S., D'Silva K., Cedillo E., Sataranatarajan K., Barnes J. L., Choudhury G. G., and Kasinath B. S. (2014) Activation of glycogen synthase kinase 3β ameliorates diabetes-induced kidney injury. J. Biol. Chem. 289, 35363–35375 10.1074/jbc.M114.587840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong Q., and Hou F. (2016) Silencing of angiotensin II type-1 receptor inhibits high glucose-induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells via inactivation of mTOR/p70S6K signaling pathway. Biochem. Biophys. Res. Commun. 469, 183–188 10.1016/j.bbrc.2015.11.092 [DOI] [PubMed] [Google Scholar]
- 26. Ferrer C. M., Sodi V. L., and Reginato M. J. (2016) O-GlcNAcylation in cancer biology: linking metabolism and signaling. J. Mol. Biol. 428, 3282–3294 10.1016/j.jmb.2016.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Risso G., Blaustein M., Pozzi B., Mammi P., and Srebrow A. (2015) Akt/PKB: one kinase, many modifications. Biochem. J. 468, 203–214 10.1042/BJ20150041 [DOI] [PubMed] [Google Scholar]
- 28. Wang Z., Pandey A., and Hart G. W. (2007) Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol. Cell. Proteomics 6, 1365–1379 10.1074/mcp.M600453-MCP200 [DOI] [PubMed] [Google Scholar]
- 29. Whelan S. A., Dias W. B., Thiruneelakantapillai L., Lane M. D., and Hart G. W. (2010) Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-linked β-N-acetylglucosamine in 3T3-L1 adipocytes. J. Biol. Chem. 285, 5204–5211 10.1074/jbc.M109.077818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X., Molina H., Huang H., Zhang Y. Y., Liu M., Qian S. W., Slawson C., Dias W. B., Pandey A., Hart G. W., Lane M. D., and Tang Q. Q. (2009) O-Linked N-acetylglucosamine modification on CCAAT enhancer-binding protein β: role during adipocyte differentiation. J. Biol. Chem. 284, 19248–19254 10.1074/jbc.M109.005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi J., Gu J. H., Dai C. L., Gu J., Jin X., Sun J., Iqbal K., Liu F., and Gong C. X. (2015) O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci. Rep. 5, 14500 10.1038/srep14500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hart G. W., Slawson C., Ramirez-Correa G., and Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 10.1146/annurev-biochem-060608-102511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bond M. R., and Hanover J. A. (2015) A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol. 208, 869–880 10.1083/jcb.201501101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Degrell P., Cseh J., Mohás M., Molnár G. A., Pajor L., Chatham J. C., Fülöp N., and Wittmann I. (2009) Evidence of O-linked N-acetylglucosamine in diabetic nephropathy. Life Sci. 84, 389–393 10.1016/j.lfs.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 35. Kawanami D., Matoba K., Takeda Y., Nagai Y., Akamine T., Yokota T., Sango K., and Utsunomiya K. (2017) SGLT2 Inhibitors as a therapeutic option for diabetic nephropathy. Int. J. Mol. Sci. 18, E1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baines R. J., Chana R. S., Hall M., Febbraio M., Kennedy D., and Brunskill N. J. (2012) CD36 mediates proximal tubular binding and uptake of albumin and is upregulated in proteinuric nephropathies. Am. J. Physiol. Renal Physiol. 303, F1006–F1014 10.1152/ajprenal.00021.2012 [DOI] [PubMed] [Google Scholar]
- 37. Zoja C., Abbate M., and Remuzzi G. (2015) Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol. Dial. Transplant. 30, 706–712 10.1093/ndt/gfu261 [DOI] [PubMed] [Google Scholar]
- 38. Abbate M., Zoja C., and Remuzzi G. (2006) How does proteinuria cause progressive renal damage? J. Am. Soc. Nephrol. 17, 2974–2984 10.1681/ASN.2006040377 [DOI] [PubMed] [Google Scholar]
- 39. Theilig F., Kriz W., Jerichow T., Schrade P., Hähnel B., Willnow T., Le Hir M., and Bachmann S. (2007) Abrogation of protein uptake through megalin-deficient proximal tubules does not safeguard against tubulointerstitial injury. J. Am. Soc. Nephrol. 18, 1824–1834 10.1681/ASN.2006111266 [DOI] [PubMed] [Google Scholar]
- 40. Furman B. L. (2015) Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 70, 5.47.1–20 [DOI] [PubMed] [Google Scholar]
- 41. Ali M. A. M., Heeba G. H., and El-Sheikh A. A. K. (2017) Modulation of heme oxygenase-1 expression and activity affects streptozotocin-induced diabetic nephropathy in rats. Fundam. Clin. Pharmacol. 31, 546–557 10.1111/fcp.12296 [DOI] [PubMed] [Google Scholar]
- 42. Dias W. B., Cheung W. D., Wang Z., and Hart G. W. (2009) Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J. Biol. Chem. 284, 21327–21337 10.1074/jbc.M109.007310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishibashi F. (2004) High glucose reduces albumin uptake in cultured proximal tubular cells (LLC-PK1). Diabetes Res. Clin. Pract. 65, 217–225 10.1016/j.diabres.2004.02.003 [DOI] [PubMed] [Google Scholar]
- 44. Ishibashi F. (2006) Chronic high glucose inhibits albumin reabsorption by lysosomal alkalinization in cultured porcine proximal tubular epithelial cells (LLC-PK1). Diabetes Res. Clin. Pract. 72, 223–230 10.1016/j.diabres.2005.10.019 [DOI] [PubMed] [Google Scholar]
- 45. Allen D. A., Harwood S., Varagunam M., Raftery M. J., and Yaqoob M. M. (2003) High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 17, 908–910 10.1096/fj.02-0130fje [DOI] [PubMed] [Google Scholar]
- 46. Harwood S. M., Allen D. A., Raftery M. J., and Yaqoob M. M. (2007) High glucose initiates calpain-induced necrosis before apoptosis in LLC-PK1 cells. Kidney Int. 71, 655–663 10.1038/sj.ki.5002106 [DOI] [PubMed] [Google Scholar]
- 47. Tong Y., Chuan J., Bai L., Shi J., Zhong L., Duan X., and Zhu Y. (2018) The protective effect of shikonin on renal tubular epithelial cell injury induced by high glucose. Biomed. Pharmacother. 98, 701–708 10.1016/j.biopha.2017.12.112 [DOI] [PubMed] [Google Scholar]
- 48. Jiao X., Li Y., Zhang T., Liu M., and Chi Y. (2016) Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 480, 387–393 10.1016/j.bbrc.2016.10.060 [DOI] [PubMed] [Google Scholar]
- 49. Lungkaphin A., Arjinajarn P., Pongchaidecha A., Srimaroeng C., Chatsudthipong L., and Chatsudthipong V. (2014) Impaired insulin signaling affects renal organic anion transporter 3 (Oat3) function in streptozotocin-induced diabetic rats. PLoS One 9, e96236 10.1371/journal.pone.0096236 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50. Tang S. C., Leung J. C., and Lai K. N. (2011) Diabetic tubulopathy: an emerging entity. Contrib. Nephrol. 170, 124–134 10.1159/000325647 [DOI] [PubMed] [Google Scholar]
- 51. Tang S. C., and Lai K. N. (2012) The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol. Dial. Transplant. 27, 3049–3056 10.1093/ndt/gfs260 [DOI] [PubMed] [Google Scholar]
- 52. Bertinat R., Nualart F., and Yáñez A. J. (2016) SGLT2 Inhibitors: glucotoxicity and tumorigenesis downstream the renal proximal tubule? J. Cell. Physiol. 231, 1635–1637 10.1002/jcp.25286 [DOI] [PubMed] [Google Scholar]
- 53. Vallon V., and Thomson S. C. (2017) Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60, 215–225 10.1007/s00125-016-4157-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kojima N., Williams J. M., Slaughter T. N., Kato S., Takahashi T., Miyata N., and Roman R. J. (2015) Renoprotective effects of combined SGLT2 and ACE inhibitor therapy in diabetic Dahl S rats. Physiol. Rep. 3, e12436 10.14814/phy2.12436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ojima A., Matsui T., Nishino Y., Nakamura N., and Yamagishi S. (2015) Empagliflozin, an inhibitor of sodium-glucose cotransporter 2 exerts anti-inflammatory and antifibrotic effects on experimental diabetic nephropathy partly by suppressing AGEs-receptor axis. Horm. Metab. Res. 47, 686–692 10.1055/s-0034-1395609 [DOI] [PubMed] [Google Scholar]
- 56. Akimoto Y., Miura Y., Toda T., Wolfert M. A., Wells L., Boons G. J., Hart G. W., Endo T., and Kawakami H. (2011) Morphological changes in diabetic kidney are associated with increased O-GlcNAcylation of cytoskeletal proteins including α-actinin 4. Clin. Proteomics 8, 15 10.1186/1559-0275-8-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ji S., Kang J. G., Park S. Y., Lee J., Oh Y. J., and Cho J. W. (2011) O-GlcNAcylation of tubulin inhibits its polymerization. Amino Acids 40, 809–818 10.1007/s00726-010-0698-9 [DOI] [PubMed] [Google Scholar]
- 58. Gellai R., Hodrea J., Lenart L., Hosszu A., Koszegi S., Balogh D., Ver A., Banki N. F., Fulop N., Molnar A., Wagner L., Vannay A., Szabo A. J., and Fekete A. (2016) Role of O-linked N-acetylglucosamine modification in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 311, F1172–F1181 10.1152/ajprenal.00545.2015 [DOI] [PubMed] [Google Scholar]
- 59. Mazzocchi L. C., Vohwinkel C. U., Mayer K., Herold S., Morty R. E., Seeger W., and Vadász I. (2017) TGF-β inhibits alveolar protein transport by promoting shedding, regulated intramembrane proteolysis and transcriptional downregulation of megalin. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L807–L824 10.1152/ajplung.00569.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gekle M., Knaus P., Nielsen R., Mildenberger S., Freudinger R., Wohlfarth V., Sauvant C., and Christensen E. I. (2003) Transforming growth factor-β1 reduces megalin- and cubilin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. J. Physiol. 552, 471–481 10.1113/jphysiol.2003.048074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chang A. S., Hathaway C. K., Smithies O., and Kakoki M. (2016) Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Renal Physiol. 310, F689–F696 10.1152/ajprenal.00502.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Susztak K., Ciccone E., McCue P., Sharma K., and Böttinger E. P. (2005) Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2, e45 10.1371/journal.pmed.0020045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Palm W., Park Y., Wright K., Pavlova N. N., Tuveson D. A., and Thompson C. B. (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270 10.1016/j.cell.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Banerjee P. S., Lagerlöf O., and Hart G. W. (2016) Roles of O-GlcNAc in chronic diseases of aging. Mol. Aspects Med. 51, 1–15 10.1016/j.mam.2016.05.005 [DOI] [PubMed] [Google Scholar]
- 65. de Queiroz R. M., Carvalho E., and Dias W. B. (2014) O-GlcNAcylation: the sweet side of the cancer. Front. Oncol. 4, 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Peterson S. B., and Hart G. W. (2016) New insights: a role for O-GlcNAcylation in diabetic complications. Crit. Rev. Biochem. Mol. Biol. 51, 150–161 10.3109/10409238.2015.1135102 [DOI] [PubMed] [Google Scholar]
- 67. Akan I., Olivier-Van Stichelen S., Bond M. R., and Hanover J. A. (October 19, 2017) Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 10.1111/jnc.14242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lima V. V., Giachini F. R., Matsumoto T., Li W., Bressan A. F., Chawla D., Webb R. C., Ergul A., and Tostes R. C. (2016) High-fat diet increases O-GlcNAc levels in cerebral arteries: a link to vascular dysfunction associated with hyperlipidaemia/obesity? Clin. Sci. (Lond) 130, 871–880 10.1042/CS20150777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lima V. V., Spitler K., Choi H., Webb R. C., and Tostes R. C. (2012) O-GlcNAcylation and oxidation of proteins: is signalling in the cardiovascular system becoming sweeter? Clin. Sci. (Lond) 123, 473–486 10.1042/CS20110638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dias W. B., Cheung W. D., and Hart G. W. (2012) O-GlcNAcylation of kinases. Biochem. Biophys. Res. Commun. 422, 224–228 10.1016/j.bbrc.2012.04.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gena P., Calamita G., and Guggino W. B. (2010) Cadmium impairs albumin reabsorption by down-regulating megalin and ClC5 channels in renal proximal tubule cells. Environ. Health Perspect. 118, 1551–1556 10.1289/ehp.0901874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Salminen A., and Kaarniranta K. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-κB signaling. Cell. Signal. 22, 573–577 [DOI] [PubMed] [Google Scholar]
- 73. Gilbert R. E. (2017) Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes 66, 791–800 10.2337/db16-0796 [DOI] [PubMed] [Google Scholar]
- 74. Peruchetti D. B., Pinheiro A. A., Landgraf S. S., Wengert M., Takiya C. M., Guggino W. B., and Caruso-Neves C. (2011) (Na2+,K+)-ATPase is a target for phosphoinositide 3-kinase/protein kinase B and protein kinase C pathways triggered by albumin. J. Biol. Chem. 286, 45041–45047 10.1074/jbc.M111.260737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Landgraf S. S., Silva L. S., Peruchetti D. B., Sirtoli G. M., Moraes-Santos F., Portella V. G., Silva-Filho J. L., Pinheiro C. S., Abreu T. P., Takiya C. M., Benjamin C. F., Pinheiro A. A., Canetti C., and Caruso-Neves C. (2014) 5-Lypoxygenase products are involved in renal tubulointerstitial injury induced by albumin overload in proximal tubules in mice. PLoS One 9, e107549 10.1371/journal.pone.0107549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Landgraf S. S., Wengert M., Silva J. S., Zapata-Sudo G., Sudo R. T., Takiya C. M., Pinheiro A. A., and Caruso-Neves C. (2011) Changes in angiotensin receptors expression play a pivotal role in the renal damage observed in spontaneously hypertensive rats. Am. J. Physiol. Renal Physiol. 300, F499–F510 10.1152/ajprenal.00384.2010 [DOI] [PubMed] [Google Scholar]
- 77. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.