

Abstract
The prevalence of childhood/adolescent obesity and insulin resistance has reached an epidemic level. Obesity’s immediate clinical impacts have been extensively studied; however, current clinical evidence underscores the long-term implications. The current study explored the impacts of brief childhood/adolescent obesity and insulin resistance on cognitive function in later life. To mimic childhood/adolescent obesity and insulin resistance, we exposed 9-week-old C57BL/6J mice to a high-fat diet for 15 weeks, after which the mice exhibited diet-induced obesity and insulin resistance. We then put these mice back on a normal low-fat diet, after which the mice exhibited normal body weight and glucose tolerance. However, a spatial memory test in the forms of the Morris water maze (MWM) and contextual fear conditioning at 85 weeks of age showed that these mice had severe deficits in learning and long-term memory consolidation. Mechanistic investigations identified increased expression of histone deacetylases 5, accompanied by reduced expression of brain-derived neurotrophic factor, in the brains 61 weeks after the mice had been off the high-fat diet. Electrophysiology studies showed that hippocampal slices isolated from these mice are more susceptible to synaptic impairments compared with slices isolated from the control mice. We demonstrated that a 15-week occurrence of obesity and insulin resistance during childhood/adolescence induces irreversible epigenetic modifications in the brain that persist following restoration of normal metabolic homeostasis, leading to brain synaptic dysfunction during aging. Our study provides experimental evidence that limited early-life exposure to obesity and insulin resistance may have long-term deleterious consequences in the brain, contributing to the onset/progression of cognitive dysfunction during aging.
Keywords: obesity, insulin resistance, aging, synaptic plasticity, cognitive function
The prevalence and severity of obesity during childhood and adolescence have reached endemic levels in the United States. It is estimated that one-third of the children in the United States are overweight, and over 15% of children between the ages of 6 and 11 years have a body mass index (BMI) characterized as obese (Hedley et al., 2004). The major causes of childhood/adolescent obesity are an imbalanced caloric intake and a reduced physical activity. Individuals with childhood/adolescent obesity have a high probability of becoming obese in adulthood; it is estimated that the odds range from 10% to 50% depending on the individual’s age, the severity of the obesity, and the child/adolescent’s parents’ obesity status (Whitaker et al., 1997).
Obesity is associated with multiple comorbidities such as diabetes, cancer, and cardiovascular complications (Guh et al., 2009). Similarly, childhood/adolescent obesity is commonly accompanied by insulin resistance, a condition that is associated with an increased risk of developing type 2 diabetes (Li et al., 2009). Childhood/adolescent obesity is also associated with a wide spectrum of adverse outcomes, including increased risk of physiological complications, such as hypertension, atherosclerosis, metabolic disturbances, and nonalcoholic fatty liver disease (Calcaterra et al., 2008; Daniels, 2009), as well as psychological distresses, such as depression and anxiety (Anderson et al., 2007; Daniels, 2009). Most clinical research studies conducted thus far have focused on the immediate health consequences of childhood/adolescent obesity, as mentioned above, and its association with cardiovascular disease (CDV). Knowledge gained over the years regarding the long-term effects of childhood/adolescent obesity on adult morbidity and mortality is limited, partially because it requires decades of longitudinal observation and partially because the analyses are complicated by the persistence of obesity into adulthood, making it difficult to clarify the relative contribution of childhood/adolescent obesity to long-term consequences (Dietz, 1998).
Many epidemiological studies have linked insulin resistance and obesity to dementia, including vascular dementia and Alzheimer’s disease dementia (van den et al., 2007; Muller et al., 2007; Razay et al., 2007; Yaffe et al., 2007; Komulainen et al., 2007; Dik et al., 2007). Obesity may also play an important role in cognitive decline during normal aging (Morley, 2004; McNay, 2005; Biessels et al., 2008). By using experimental models of insulin resistance and obesity, either in the genetic model of obese Zucker rats or obesity induced by Western diet-like chow (high content of saturated fats and simple carbohydrates such as sucrose and fructose), we and others have found that chronic obesity and insulin resistance conditions lead to cognitive impairments and synaptic dysfunction (Gerges et al., 2003; Ho et al., 2004; Stranahan et al., 2008; Pancani et al., 2013; Wang et al., 2013). However, very little is known about the long-term consequences of childhood/adolescent obesity on cognition during aging. Given the increasing awareness of childhood/adolescent obesity and the vigorous campaigns to fight it, many children will revert back to normal weight. Therefore, it is important to understand the effects of transient obesity and insulin resistance on brain wellness during aging. This study uses a high-fat diet treatment paradigm to test whether a short-period occurrence of obesity and insulin resistance during childhood/adolescence may have adverse long-term effects on learning and memory, particularly during aging.
MATERIALS AND METHODS
Mouse model and treatment
Female C57BL6/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the Center for Comparative Medicine and Surgery (CCMS) at the Icahn School of Medicine at Mount Sinai. Mice were randomly grouped into two groups: one group received regular diet (CTRL group, 10 kcal% fat; Research Diets D12450B); the other group was treated with a high-fat diet (HF group, 60 kcal% fat; Research Diets, D12492) starting at 2 months of age until 6 months of age, after which they switched to the regular diet until they reached 22 months of age (see Fig. 1 for the detailed study design). All animals were maintained on a 12:12-hour light/dark cycle with lights on at 07:00 hours in a temperature-controlled (20 C ± 2°C) vivarium, and all procedures were approved by the MSSM IACUC.
Figure 1.
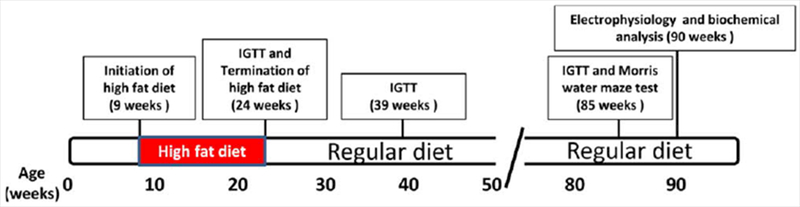
Experimental design and treatment regimen. High-fat diet treatment was initiated when the HF mice were 9 weeks of age and continued until they were 24 weeks of age. The animals were then switched to a normal low-fat diet until the end of the study (90 weeks of age). Glucose tolerance assay was performed at 24, 39, and 85 weeks of age. Animals were tested behaviorally at 85 weeks of age and electrophysiologically at 90 weeks of age. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Intraperitoneal glucose tolerance test and fasting insulin measurements
The intraperitoneal glucose tolerance test (IGTT) was performed as previously reported (Wang et al., 2005, 2007). Specifically, mice were given a single dose of intraperitoneal glucose (2 g/kg BW) postprandially, and blood was collected from the tail vein periodically over a 3-hour period. Blood glucose content was assessed with the Contour blood glucose system (Bayer, Indianapolis, IN).
Cognitive assessment by the Morris water maze test
Spatial learning memory was assessed by the Morris water maze (MWM) behavioral test, as previously described (Morris, 1984; Wang et al., 2007). Mice were tested in a circular pool filled with water mixed with nontoxic white paint (Dick Blick Art Materials). In the initial learning phase, mice were trained for 7 consecutive days, which allowed them to learn to escape from the water onto a hidden/submerged (1.5 cm below the water surface) escape platform (14 \m=x\ 14 cm) in a restricted region of the pool, using the spatial cues provided. Each mouse was subjected to four blocks of training per day, during which the mouse was placed in a different quadrant as the starting point for each block. Spatial learning memory was assessed by recording the latency time for the animal to escape from the water onto the submerged escape platform as a function of the number of learning trials during the learning phase. Twenty-four hours after the last learning session, mice were subjected to a 45-second probe trial, wherein the escape platform was removed. Spatial memory retention was reflected by the percentage of time animals spent within the “target” quadrant of the pool that previously contained the hidden escape platform. Water maze activity during training and probe trials was monitored with the San Diego Instrument Poly-Track video tracking system (San Diego, CA).
Cognitive assessment by contextual fear conditioning test
A contextual fear conditioning test was performed as previously described (Steele et al., 2012). During conditioning, mice were trained and tested in conditioning chambers on 3 consecutive days in the cued and contextual fear conditioning paradigm. On day 1, mice were placed into context A (gray walls, grid floor, houselights at 50% with lemon scent) and allowed to explore for 180 seconds (baseline) prior to three tone/shock pairings (30-second 4.0-kHz pure tone coterminating with a 2-second scrambled 0.6-mA foot shock). Each tone/shock pairing was separated by 30 seconds of exploration time, and animals were given 30 seconds to explore following the final tone/shock pairing (300 seconds total). On day 2, mice were placed into context B (black/white checked walls, black plastic floor, houselights at 100% with vanilla scent) and allowed to explore for 180 seconds in the constant presence of the 4.0 kHz pure tone. On day 3, mice were placed again into context A and allowed to explore for 180 seconds without the tone. Memory for the context (contextual memory) or the tone (cued memory) for each animal was obtained by subtracting the percentage freezing during baseline from the percentage freezing on day 2 or day 3, respectively. Freezing behavior was recorded remotely and analyzed in Video Freeze Fear Conditioning software (MED Associates Inc.).
Electrophysiological recordings
Electrophysiology studies were performed as previously described (Wang et al., 2013). Mice were sacrificed by decapitation, and the brains were quickly removed. Hippocampal slices (350 μm) were placed into oxygenated artificial cerebrospinal fluid (ACSF) at 29°C for a minimum of 90 minutes to acclimatize. Slices were then transferred to a recording chamber and perfused continuously with oxygenated ACSF in the presence or absence of 100 nM oligomeric Aβ (oAβ) at 32°C. For extracellular recordings, CA1 field excitatory postsynaptic potentials (fEPSPs) were recorded by placing stimulating and recording electrodes in CA1 stratum radiatum. For long-term potentiation (LTP) experiments, a 40-minute baseline was recorded every minute at an intensity that evoked a response ~35% of the maximal evoked response. LTP was induced using θ-burst stimulation (four pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including three 10-burst trains separated by 15 seconds), and fEPSPs were monitored for 120 minutes to assess the magnitude of potentiation.
Assessment of gene expression
Total RNA from mouse brain tissues was isolated with a RNeasy Mini Kit (Qiagen, Valencia, CA) and reverse transcribed. A mouse epigenetic chromatin modification enzyme array (Qiagen) was used to survey the epigenetic enzymes’ expression. Real-time (RT) polymerase chain reaction (PCR) was performed to confirm or to identify genes of interest, particularly histone deacetylase genes (HDACs) and synaptic plasticity, mitochondrial, and proinflammatory genes. Gene expression was measured in four replicates by quantitative RT-PCR with Maxima SYBR Green master mix (Fermentas) in an ABI-Prism 7900HT. For primer sequences see Table 1. Hypoxanthine phosphoribosyltransferase (HPRT) expression level was used as an internal control. Data were normalized using the 2−ΔΔCt method (Livak and Schmittgen, 2001). Levels of target gene mRNAs were expressed relative to those in control (CTRL) mice and plotted in GraphPad Prism (GraphPad Software, San Diego, CA).
TABLE 1.
Primer Sequences Used For Real-Time PCR
| Gene name | Forward | Reverse |
|---|---|---|
| HDAC5 | TTCAACTCCGTAGCCATCAC | GGATCGTTGTAGAATGCTTGC |
| HDAC9 | GCGAGACACAGATGCTCAGAC | TGGGTTTTCCTTCCATTGCT |
| PSD95 | CGGGAGAAAATGGAGAAGGAC | GCATTGGCTGAGACATCAAG |
| Synaptophysin | AGTGCCCTCAACATCGAAG | GCCACGGTGACAAAGAATTC |
| BDNF I | CCTGCATCTGTTGGGGAGAC | GCCTTGTCCGTGGACGTTTA |
| BDNF II | CTAGCCACCGGGGTGGTGTAA | AGGATGGTCATCACTCTTCTC |
| BDNF III | CTTCCTTGAGCCCAGTTCC | CCGTGGACGTTTACTTCTTTC |
| BDNF IV | CAGAGCAGCTGCCTTGATGTT | GCCTTGTCCGTGGACGTTTTA |
| BDNF V | TTGGGGCAGACGAGAAAGCGC | AGGATGGTCATCACTCTTCTC |
| BDNF total | GAGGGCTCCTGCTTCTCAA | GCCTTCATGCAACCGAAGT |
| Arc | GCAGGTGGG TGGCTCTGAAGAATA | TCCCGCTTGCGCCAGAGGAACT |
| C-Fos | TCCTTACGGACTCCCCAC | CTCCGTTTCTCTTCCTCTTCAG |
| Egr1 | AGCGCCTTCAATCCTCAAG | TTTGGCTGGGATAACTCGTC |
| CCL2 | CATCAGTCCTCAGGTATTGGC | TTGTGATTCTCCTGTAGCTCTTC |
| IL1β | TCCTGTGTAATGAAAGACGGC | ACTCCACTTTGCTCTTGACTTC |
| IL-6 | CAAAGCCAGAGTCCTTCAGAG | GTCCTTAGCCACTCCTTCTG |
| IL-10 | TTGAATTCCCTGGGTGAGAAG | ATGGCCTTGTAGACACCTTG |
| TNF-α | CTTCTGTCTACTGAACTTCGGG | CAGGCTTGTCACTCGAATTTTG |
| IFN-γ | ATGCATTCATGAGTATTGCCAAG | ACTCCTTTTCCGCTTCCTG |
| Mouse hypoxanthine phosphoribosyltransferase (HPRT) | CCCCAAAATGGTTAAGGTTGC | AACAAAGTCTGGCCTGTATCC |
Statistical analysis
In these studies, all values are expressed as mean and SEM. Differences between means were analyzed by either two-way repeated-measures ANOVAs or two-tailed student t-test. In all analyses, the null hypothesis was rejected at the 0.05 level. All statistical analyses were performed in the Prism Stat program (GraphPad Software).
RESULTS
High-fat diet during childhood/adolescence in female mice does not have long-term periphery consequences
To establish childhood/adolescent insulin resistance and obesity, we treated the mice with a high-fat (HF) diet. After initiation of the diet treatment at 9 weeks of age (equivalent to 6 years old in humans), the mice started to gain body weight and continued to gain weight until the end of the 15-week treatment, when they reached 24 weeks of age, equivalent to 15 years of age in humans (Fig. 2A). At the end of the treatment, they were severely obese and were 45.9% heavier than the CTRL group (33.7 ± 2.1 vs. 23.1 ± 0.4 g; P < 0.0001). Moreover, the HF mice developed an impaired glucose tolerance response, as reflected by IGTT assay (ANOVA: P < 0.0001, F4,52 = 55.23 for glycemic content over time; P < 0.01, F1,52 = 14.82 for diet; F4,52 = 4.22 for interaction; Fig. 2B) compared with the CTRL mice.
Figure 2.
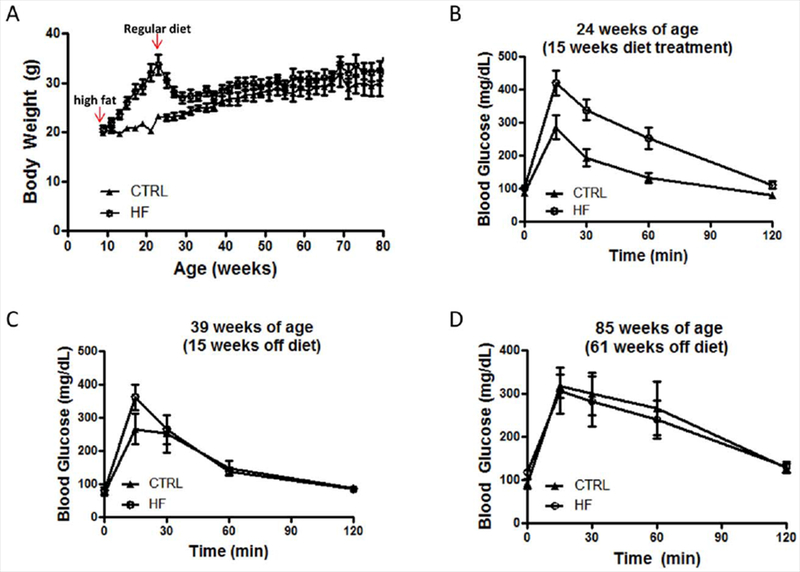
Body weight and glucose tolerance test in HF and CTRL C57BL/6J mice. A: Body weight record: arrows indicate the initiation and the end of high-fat diet treatment. B–D: Intraperitoneal glucose tolerance test following 15 weeks of high-fat diet treatment (B), 15 weeks off of the high-fat diet (C), and 61 weeks off of the high-fat diet (D; n = 7–8 per group). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The HF mice were then taken off of the high-fat diet and resumed a normal low-fat diet, the same diet as the CTRL mice. The HF mice started to lose weight immediately, and at ~10 months of age the HF mice had body weights similar to those of the CTRL mice. Glucose tolerance responses were tested when the mice reached 8 months and 20 months of age. We found that the HF mice had almost normal glucose tolerance responses following 15 weeks off of the high-fat diet (Fig. 2C). Fifteen minutes after the glucose injection, the HF mice had slightly higher glucose content in plasma, but this did not reach statistical significance (Fig. 2C). At 20 months of age, the HF mice were virtually indistinguishable from the CTRL mice, regarding their glucose tolerance responses and body weights (Fig. 2D). Our data suggest that, when given a high-fat diet at a young age, the mice developed features of a noninsulin-dependent form of diabetes mellitus (NIDDM), including hyperinsulinemia, hyperglycemia, increased body weight, and glucose intolerance. However, these features were transient.
High-fat diet during childhood/adolescence in female mice leads to spatial memory impairment during aging
To test the long-term effects of childhood/adolescent obesity on cognitive function, we used the MWM test when the animals reached ~20 months of age. We found that in the hidden platform learning trial both groups of mice were able to learn to locate the hidden platform following 7-day progressive training. Two-way ANOVA comparison of the escape latencies during 7 days of acquisition revealed that the HF group performed worse than the CTRL group, however, this did not reach statistical significance (F1,12 = 2.74, P = 0.129; Fig. 3A). Individual t-tests revealed that on days 1, 3, and 7 the CTRL group performed significantly better than the HF group (Fig. 3A).
Figure 3.
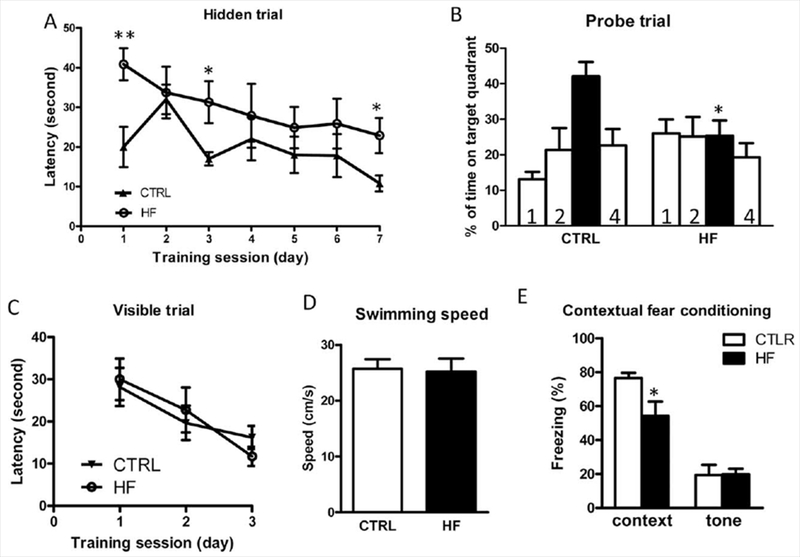
Fifteen weeks of high-fat diet during childhood/adolescence leads to spatial memory impairments during aging, as assessed by the MWM test and contextual fear conditioning test in female mice. A: Hidden platform acquisition: latency score represents time taken to escape to the platform from the water. B: Spatial memory retention in probe trial: percentage of time in quadrant is calculated as the ratio of time spent in the target quadrant area relative to the time spent in the rest of the pool (quadrants: solid bar, target; 1, left; 2, opposite; 4, right). C: Cued platform visible trial. D: Average swimming speed. E: Contextual fear conditioning test. *P < 0.05, **P < 0.01, data represent mean ± SEM (n = 7–8 per group().
During the probe trial, which is used to test memory consolidation and long-term memory formation, we found that the HF mice performed significantly worse, as reflected by limited time spent in the target quadrant (Fig. 3B; P < 0.05), suggesting that transient insulin resistance and obesity during childhood/adolescence may lead to memory impairments in older age. In parallel control studies (Fig. 3C,D), we confirmed that all of the mice performed equally well, excluding the possibility that the HF mice might have impaired nonspatial parameters, such as sensorimotor performance, motivation, or swimming ability, which might have interfered with their performance.
After the MWM test, we performed an independent contextual fear conditioning test to investigate further the effects of high-fat feeding during childhood/adolescence on cognitive function during aging. We found that the HF mice exhibited significant impairment in contextual memory compared with the control mice (Fig. 3E). There was no impairment in the cued memory test (Fig. 3E).
High-fat diet during childhood/adolescence in female mice is associated with increased susceptibility to synaptic insults
Synaptic function is integral for learning and memory. Our previous studies revealed that 6 months of a high-fat diet treatment in C57BL6/J mice leads to increased susceptibility to oAβ-induced synaptic toxicity, and 10 months of high-fat diet treatment leads to significant LTP dysfunction (Wang et al., 2013). Aβ peptides are the major constituents of the senile plaques formed in the brains of Alzheimer’s disease (AD) subjects. A major hypothesis regarding the pathogenesis of AD is that abnormally elevated Aβ peptides, produced through amyloidogenic processes of amyloid precursor protein (APP), lead to the accumulation of extracellular soluble oAβ species in the brain. Oligomeric forms of Aβ, including high-molecular-weight (HMW) oAβ as well as low-n oligomers ranging from dimers to octamers, induce synapse degeneration, synaptic plasticity disruption, and decreases in LTP (Walsh and Selkoe, 2007; Selkoe, 2008), all of which contribute to the mechanisms underlying the onset and progression of dementia in AD (Terry et al., 1991; Selkoe, 2002; Walsh et al., 2002; Scheff and Price, 2003; Lacor et al., 2004; Coleman et al., 2004; Jacobsen et al., 2006; Shankar et al., 2007, 2008). After the MWM and contextual fear conditioning tests, we tested whether synaptic impairments may be responsible for the spatial memory dysfunction seen in the HF mice. LTP was measured in hippocampal slices isolated from the HF mice at ~22 months of age (see Fig. 1 scheme). We found that hippocampal slices derived from the HF mice had normal electrophysiological responses (data not shown). However, these hippocampal slices had a much lower threshold to oAβ-induced LTP deficits. Normally, healthy hippocampal slices show compromised LTP when exposed to 200 nM oAβ (data not shown). However, hippocampal slices derived from HF mice showed limited potentiation following tetanus stimulation in the presence of 100 nM of oAβ, whereas slices from CTLR mice showed robust LTP under the same conditions (157.8% ± 9.08% vs. 210% ± 8.42%, P < 0.05; Fig. 4A), suggesting that childhood/adolescent insulin resistance and obesity conditions might adversely influence synaptic function and eventually lead to increased susceptibility to insults such as oAβ, which is one of the major culprits responsible for cognitive decline in AD patients.
Figure 4.
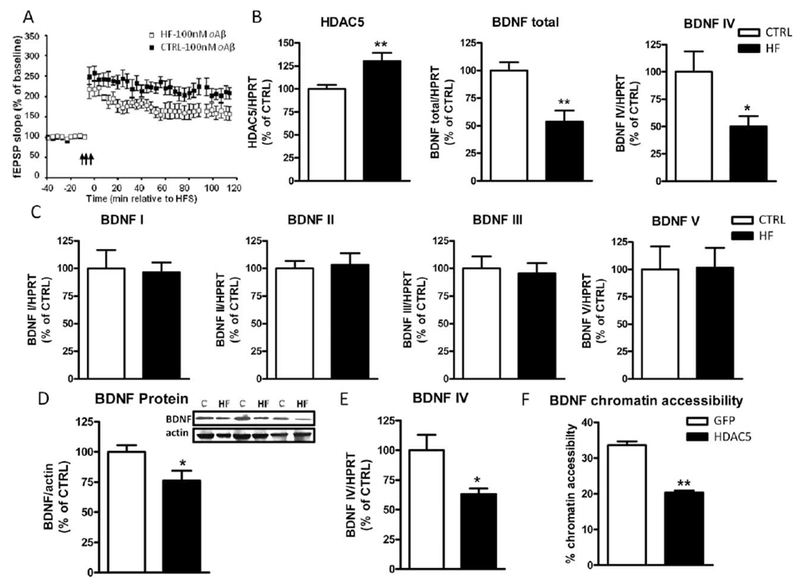
Childhood/adolescent obesity induces epigenetic changes in the brain, leading to reduced BDNF expression and increased synaptic susceptibility. A: The fEPSPs of LTP were recorded from the CA1 region of hippocampal slices from CTRL and HF mice at 22 months of age, following 1 hour of treatment with 100 nM of oAβ. The arrows indicate the beginning of tetanus to induce LTP; n = 8 slices per group from four individual mice. Messenger RNA expression of HDAC5, total BDNF, BDNF IV (B), and variants of BDNF genes (C; BDNF I, II, III, and V) in the brains of CTRL and HF mice. *P < 0.05, **P < 0.01 (n = 6–7 per group). D: Protein levels of total BDNF in the hippocamus of HF mice vs. CTRL mice by Western blot analysis. Inset: Representative micrograph of Western blot. E: Messenger RNA level of BDNF IV in SY5Y cells transfected with control EGFP and HDAC5 plasmid by real-time PCR. F: Chromatin accessibility of BDNF following overexpression of HDAC5, normalized with GAPDH, which is considered to be 100% accessible for transcription.
High-fat diet during childhood/adolescence in female mice induces epigenetic changes in the brain. The HF mice were obese and insulin resistant for only a short period of time during their life span, yet these mice exhibited spatial memory impairment and synaptic dysfunction at an advanced age. Therefore, we hypothesized that childhood obesity may induce some epigenetic changes that lead to long-term adverse effects in the brain. We first used an epigenetic chromatin modification enzyme array to survey the expression of epigenetic enzymes in the brains of HF and CTRL mice. We found that there was an upregulation of HDAC5 in the brains of the HF mice compared with the CTRL mice. Previously, we found that chronic obesity and insulin resistance induce increased expression of HDAC5 in the brain, which is partially responsible for the impaired synaptic plasticity (Wang et al., 2013). Using real-time PCR, we independently confirmed that the expression of HDAC5 was indeed increased in the brains of the HF mice (Fig. 4B, left), although there was no difference in the expression of other HDACs (data not shown). Based on previous evidence that HDAC5 regulates the expression of BDNF (Tsankova et al., 2006; Wang et al., 2013), an important molecule for synaptic function, we next examined the expression of BDNF in the brains by real-time PCR. We found that there was a twofold reduction in the expression of total BDNF gene in the brains of the HF mice (Fig. 4B, middle) and that this reduction is very likely mediated by BDNF IV (Fig. 4B, right). We did not find any changes in the expression of other BDNF variants, including I, II, III, and V (Fig. 4C). We then performed Western blot analyses and confirmed that the BDNF protein levels in the hippocampus were significantly lower in the HF mice compared with the CTRL mice (Fig. 4D). To investigate further the cause-effect relationship, we transfected human SH-SY5Y cells with HDAC5 plasmid and found that HDAC5 overexpression leads to significantly reduced expression of BDNF IV mRNA 48 hours after transfection (Fig. 4E). This reduction was partially mediated by the reduced BDNF chromatin accessibility, as assessed by the nuclease sensitivity assay (Fig. 4F); without transfection, approximately 32% of the BDNF promoters are accessible for transcription, whereas introduction of HDAC5 significantly reduced the accessibility to approximately 20% (Fig. 4F).
Inflammation, mitochondria function, and energy metabolism play important roles in the pathogenesis of obesity and insulin resistance, so we measured select regulators and genes involved in energy metabolism/mitochondrial function, as well as proinflammatory cytokines in the brains, of the HF mice. We found no gross differences, except for significantly increased expression of Sirt7 (Fig. 5) and an increase in IL-1 β (Fig. 6), which did not reach statistical significance (P = 0.08).
Figure 5.
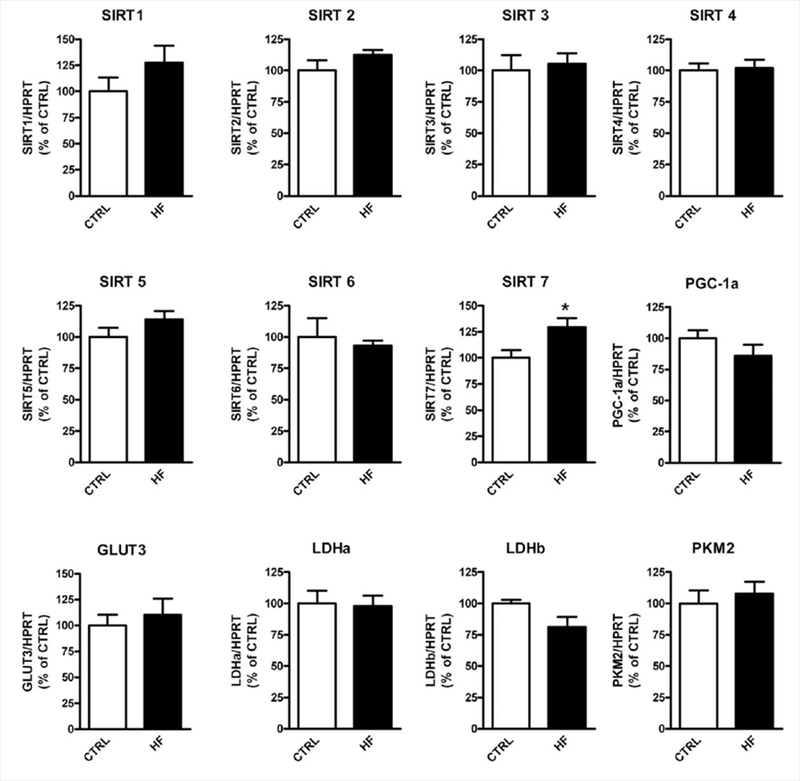
Expression of select mitochondria/energy metabolism-related genes in the brains of HF mice compared with CTRL mice. Messenger RNA expression of select genes in the brain was measured by real-time PCR. *P < 0.05 (n = 6–7 per group).
Figure 6.
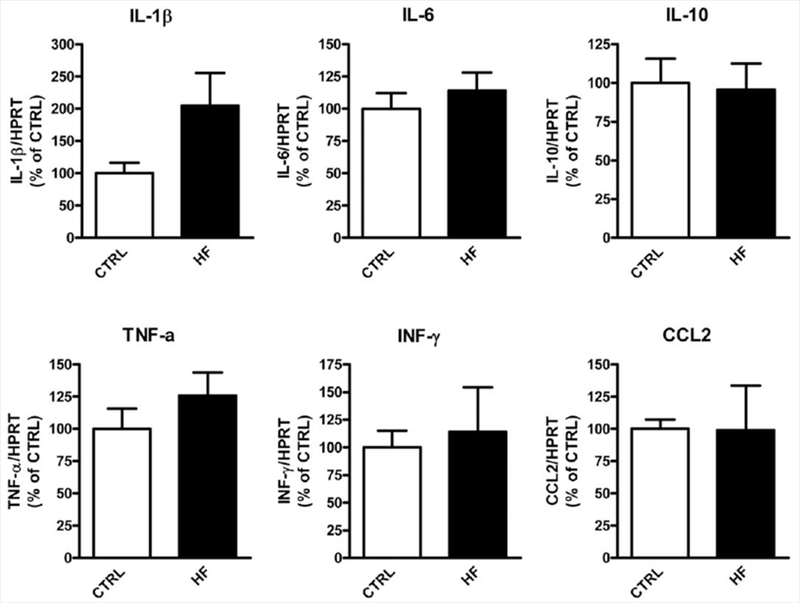
Expression of select proinflammatory genes in the brains of HF mice compared with CTRL mice. Messenger RNA expression of select genes in the brains were measured by real-time PCR (n = 6–7 per group).
DISCUSSION
Although the overall rates of obesity have plateaued or even declined in the United States in recent years, obesity is still one of the greatest health threats for young people. The negative impacts and long-term con-sequences of obesity in adults have been extensively investigated clinically. Obesity is associated with increased risk for a wide spectrum of diseases, including those affecting the cardiovascular, metabolic, pulmonary, gastrointestinal, and skeletal systems. It is well-established that obesity in adults is also associated with increased incidences of cognitive dysfunction.
The immediate and intermediate comorbidities associated with childhood/adolescent obesity are similar, including hypertension, dyslipidemia, and an increased risk of developing insulin resistance and type 2 diabetes (Dietz et al., 1990; Williams et al., 2001). Many clinical studies focus on the psychological impact of childhood/adolescent on mental wellbeing, including its effects on self-esteem and quality of life (De Niet and Naiman, 2011; Janicke et al., 2014; Buttitta et al., 2014; Halliday et al., 2014). However, the long-term impacts of childhood/adolescent obesity on cognitive function have not been explored. Using an experimental model of obesity and insulin resistance, we exposed mice to a 15-week period of high-fat diet, inducing an obesity/glucose intolerance phenotype in mice equivalent to 5–16 years of age in humans. The animals were then switched back to normal diet for the rest of their lives. They exhibited normal periphery homeostasis, including normal body weight and normal glucose utilization capacity. However, cognitive tests performed when the animals reached advanced age demonstrated impairments in learning and memory formation. We found that BDNF protein levels in the brains of HF mice were significantly lower than thjose in the CTRL mice, suggesting that BDNF may play an important role in the cognitive impairment process. BDNF is a key regulator of synaptogenesis and synaptic plasticity, which are essential for learning and memory. We further demonstrated that the reduced expression of BDNF was very likely a result of increased expression of HDAC5, a histone deacetylase whose activity can reduce BDNF chromatin accessibility, leading to reduced transcription of BDNF. Previously, we have shown that the levels of HDAC5 are significantly increased in the brains of diabetic patients and in the brains of mice chronically fed a high-fat diet (Wang et al., 2013). This persistent increase of HDAC5 in the brain, albeit the lack of periphery obesity/insulin resistance phenotype, suggests that, even when the periphery recovers to normal homeostasis, the epigenetic changes in the brain may persist over the long term and continue to induce pathological alterations, both structurally and functionally, leading to a more susceptible, less resilient cognitive machinery (Fig. 7).
Figure 7.
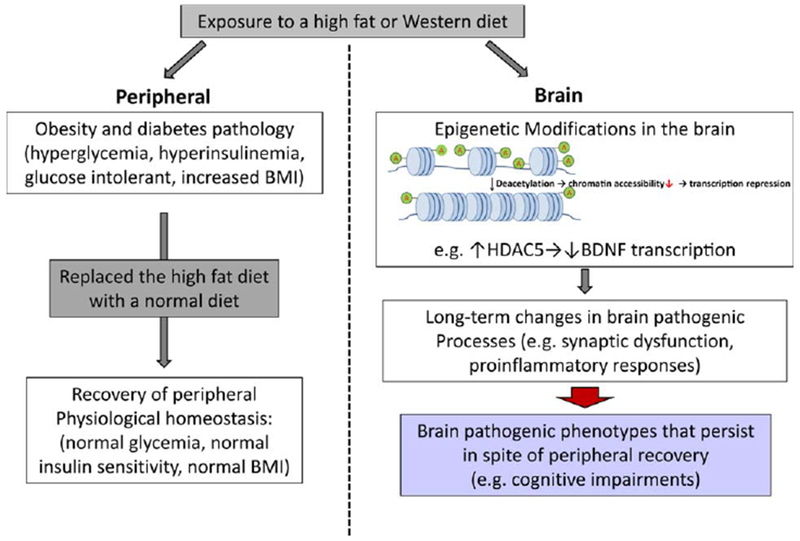
Schematic illustration. Brief exposure to diabetes during adolescence leads to long-term pathogenic consequences in the brain, which persist even after restoration of peripheral physiological homeostasis. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
It is well-established that men and women differ with respect to their regulation of energy homeostasis (Shi et al., 2009). The sex hormone estrogen plays an important role in diabetes and obesity. 17β-Estradiol, the main circulating estrogen in women, can promote energy homeostasis, improve body fat distribution, and enhance insulin sensitivity (Mauvais-Jarvis et al., 2013). In men, the reduced levels of estrogen protection may exacerbate metabolic dysfunction, predisposing men to obesity and type 2 diabetes. Although our studies were conducted in female mice, it is possible that 15 weeks of high-fat diet treatment during childhood/adolescence in male mice may lead to more severe cognitive dysfunction during aging because of their decreased levels of estrogen. Future studies will be carried out to determine whether gender difference can influence the long-term cognitive consequences induced by obesity in young age.
In conclusion, our study demonstrates that a brief occurrence of obesity and insulin resistance during childhood/adolescence may induce irreversible epigenetic modifications in the brain that persist following restoration of normal metabolic homeostasis, leading to brain synaptic dysfunction during aging. Our study provides experimental evidence that limited, early exposure to obesity and insulin resistance may have long-term deleterious consequences in the brain, contributing to the onset/progression of cognitive dysfunction during aging. Although the majority of the clinical studies performed thus far have focused on the cardiometabolic morbidity associated with obesity, our study provides the scientific motivation for clinical investigations of the long-term impacts of childhood/adolescent obesity on cognitive function and brain wellness during aging.
ACKNOWLEDGMENTS
We thank Ms. Lauren Dubner for her help with formatting and revising this article. All those acknowledged have agreed to be listed.
Grant sponsor: Mount Sinai School of Medicine.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no known or potential conflicts of interest.
LITERATURE CITED
- Anderson SE, Cohen P, Naumova EN, Jacques PF, Must A. 2007. Adolescent obesity and risk for subsequent major depressive disorder and anxiety disorder: prospective evidence. Psychosom Med 69:740–747. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. 2008. Cognition and diabetes: a lifespan perspective. Lancet Neurol 7:184–190. [DOI] [PubMed] [Google Scholar]
- Buttitta M, Iliescu C, Rousseau A, Guerrien A. 2014. Quality of life in overweight and obese children and adolescents: a literature review. Qual Life Res 23:1117–1139. [DOI] [PubMed] [Google Scholar]
- Calcaterra V, Klersy C, Muratori T, Telli S, Caramagna C, Scaglia F, Cisternino M, Larizza D. 2008. Prevalence of metabolic syndrome (MS) in children and adolescents with varying degrees of obesity. Clin Endocrinol 68:868–872. [DOI] [PubMed] [Google Scholar]
- Coleman P, Federoff H, Kurlan R. 2004. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63:1155–1162. [DOI] [PubMed] [Google Scholar]
- Daniels SR. 2009. Complications of obesity in children and adolescents. Int J Obes 33(suppl 1):S60–S65. [DOI] [PubMed] [Google Scholar]
- De Niet JE, Naiman DI. 2011. Psychosocial aspects of childhood obesity. Minerva Pediatr 63:491–505. [PubMed] [Google Scholar]
- Dietz WH. 1998. Health consequences of obesity in youth: childhood predictors of adult disease. Pediatrics 101: 518–525. [PubMed] [Google Scholar]
- Dietz WH, Bandini LG, Gortmaker S. 1990. Epidemiologic and metabolic risk factors for childhood obesity. Prepared for the Fourth Congress on Obesity Research, Vienna, Austria, December, 1988. Klin Padiatr 202:69–72. [DOI] [PubMed] [Google Scholar]
- Dik MG, Jonker C, Comijs HC, Deeg DJ, Kok A, Yaffe K, Penninx BW. 2007. Contribution of metabolic syndrome components to cognition in older individuals. Diabetes Care 30:2655–2660. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Aleisa AM, Alkadhi KA. 2003. Impaired long-term potentiation in obese zucker rats: possible involvement of presynaptic mechanism. Neuroscience 120:535–539. [DOI] [PubMed] [Google Scholar]
- Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. 2009. The incidence of comorbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday JA, Palma CL, Mellor D, Green J, Renzaho AM. 2014. The relationship between family functioning and child and adolescent overweight and obesity: a systematic review. Int J Obes 38:480–493. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. 2004. Prevalence of overweight and obesity among US children, adolescents, adults, 1999–2002. JAMA 291:2847–2850. [DOI] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. 2004. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 18:902–904. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. 2006. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 103:5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke DM, Steele RG, Gayes LA, Lim CS, Clifford LM, Schneider EM, Carmody JK, Westen S. 2014. Systematic review and meta-analysis of comprehensive behavioral family lifestyle interventions addressing pediatric obesity. J Pediatr Psychol (in press). [DOI] [PubMed] [Google Scholar]
- Komulainen P, Lakka TA, Kivipelto M, Hassinen M, Helkala EL, Haapala I, Nissinen A, Rauramaa R. 2007. Metabolic syndrome and cognitive function: a population-based follow-up study in elderly women. Dement Geriatr Cogn Disord 23:29–34. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. 2004. Synaptic Targeting by Alzheimer’s-related amyloid β oligomers. J Neurosci 24:10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ford ES, Zhao G, Mokdad AH. 2009. Prevalence of pre-diabetes and its association with clustering of cardiometabolic risk factors and hyperinsulinemia among U.S. adolescents: National Health and Nutrition Examination Survey 2005–2006. Diabetes Care 32:342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-Delta Delta Ct method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Clegg DJ, Hevener AL. 2013. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34:309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC. 2005. The impact of recurrent hypoglycemia on cognitive function in aging. Neurobiol Aging 26(Suppl 1): 76–79. [DOI] [PubMed] [Google Scholar]
- Morley JE. 2004. The metabolic syndrome and aging. J Gerontol A Biol Sci Med Sci 59:139–142. [DOI] [PubMed] [Google Scholar]
- Morris R 1984. Developments of a water-maze procedure for studying spatial-learning in the rat. J Neurosci Methods 11:47–60. [DOI] [PubMed] [Google Scholar]
- Muller M, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. 2007. Metabolic syndrome and dementia risk in a multiethnic elderly cohort. Dement Geriatr Cogn Disord 24:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Anderson KL, Brewer LD, Kadish I, DeMoll C, Landfield PW, Blalock EM, Porter NM, Thibault OL. 2013. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol Aging 34:1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razay G, Vreugdenhil A, Wilcock G. 2007. The metabolic syndrome and Alzheimer disease. Arch Neurol 64:93–96. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. 2003. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging 24:1029–1046. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 2002. Alzheimer’s disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 2008. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. 2007. Natural oligomers of the alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. 2008. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Seeley RJ, Clegg DJ. 2009. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 30:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, Delgado LM, Alfaro IE, Bernales S, Verdile G, Bharadwaj P, Gupta V, Barr R, Friss A, Dolios G, Wang R, Ringe D, Fraser P, Westaway D, George-Hyslop PH St., Szabo P, Relkin NR, Buxbaum JD, Glabe CG, Protter AA, Martins RN, Ehrlich ME, Petsko GA, Yue Z, Gandy S. 2012. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol Psychiatry 18:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. 2008. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18:1085–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, Deteresa R, Hill R, Hansen LA, Katzman R. 1991. Physical basis of cognitive alterations in Alzheimers-disease—synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. 2006. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 9:519–525. [DOI] [PubMed] [Google Scholar]
- van den Berg E, Biessels GJ, de Craen AJ, Gussekloo J, Westendorp RG. 2007. The metabolic syndrome is associated with decelerated cognitive decline in the oldest old. Neurology 69:979–985. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. 2007. A beta oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. 2002. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, Maniar K, Dolios G, Wang R, Hof PR, Pasinetti GM. 2005. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J 19:659–661. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. 2007. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest 117:3393–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gong B, Zhao W, Tang C, Varghese M, Nguyen T, Bi W, Bilski A, Begum S, Vempati P, Knable L, Ho L, Pasinetti GM. 2013. Epigenetic mechanisms linking diabetes and synaptic impairments. Diabetes 63:645–654. [DOI] [PubMed] [Google Scholar]
- Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. 1997. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med 337:869–873. [DOI] [PubMed] [Google Scholar]
- Williams CL, Gulli MT, Deckelbaum RJ. 2001. Prevention and treatment of childhood obesity. Curr Atheroscler Rep 3: 486–497. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Haan M, Blackwell T, Cherkasova E, Whitmer RA, West N. 2007. Metabolic syndrome and cognitive decline in elderly Latinos: findings from the Sacramento Area Latino Study of Aging study. J Am Geriatr Soc 55:758–762. [DOI] [PubMed] [Google Scholar]