

Abstract
For more than a decade numerous evidence has been reported on the mechanisms of toxicity of α-synuclein (αS) oligomers and aggregates in α-synucleinopathies. These species were thought to form freely in the cytoplasm but recent reports of αS multimer conformations when bound to synaptic vesicles in physiological conditions, have raised the question about where αS aggregation initiates. In this review we focus on recent literature regarding the impact on membrane binding and subcellular localization of αS toxic species to understand how regular cellular function of αS contributes to pathology. Notably αS has been reported to mainly associate with specific membranes in neurons such as those of synaptic vesicles, ER/Golgi and the mitochondria, while toxic species of αS have been shown to inhibit, among others, neurotransmission, protein trafficking and mitochondrial function. Strategies interfering with αS membrane binding have shown to improve αS-driven toxicity in worms and in mice. Thus, a selective membrane binding that would result in a specific subcellular localization could be the key to understand how aggregation and pathology evolves, pointing out to αS functions that are primarily affected before onset of irreversible damage.
Keywords: alpha-synuclein, oligomers, aggregates, subcellular localization, membranes binding, Parkinson's disease, neurodegeneration, alpha-synucleinopathies
Introduction
Parkinson’s disease (PD) is the most frequent neurodegenerative disease among α-synucleinopathies, a family of illnesses that share as a common feature the accumulation of intracellular proteinaceous inclusions made mainly of α-synuclein (αS) [for a review (Goedert et al., 2012)]. In PD, αS inclusions are predominantly present in the soma, named Lewy bodies (LB), or in neurites, named Lewy neurites (LN), of neurons of the central nervous system. Although PD has been previously considered a motor disease, the involvement of peripheral neurons, both sympathetic and parasympathetic bearing LBs/LNs has been shown in recent years (Braak et al., 2003) and has been furthermore suggested to correlate with the presence of numerous non-motor dysfunctions, which represents an important aspect of PD symptomatology and negatively impacts the quality of life of patients.
Pathological accumulation of αS inclusions has been shown to correlate with the degree of neurodegeneration and dysfunction in a variety of animal models [as example flies, worms, mice (Feany and Bender, 2000; Masliah, 2000; Lakso et al., 2003)] and it is thought to be a cardinal step in the pathogenesis of the disease. Formation of αS inclusions is a complex nucleation reaction where αS, a small soluble protein, becomes trapped in an insoluble β-sheet conformation and tightly packed in long filamentous protofibrils and fibrils (Lashuel et al., 2002; Cremades et al., 2012; Tuttle et al., 2016). Chemical and structural variables such as pH, ionic salts and point mutations can influence fibril formation (Buell et al., 2014) and intermediate multiple high molecular weight (HMW) species with different structures, defined collectively as oligomers, can form during the aggregation process, raising the issue about their relative toxicity in αS driven pathology. While for long time αS toxic species have been thought to have a cytoplasmic localization, αS ability to bind membranes and associate with cellular organelles and synaptic vesicles has prompted the question about how cellular localization impacts on pathology and whether membrane interaction influences aggregation.
In this review we attempt to piece together recent findings regarding the subcellular localization of αS toxic HMW species and their relationship with biological membranes. Initially we will discuss αS conformation in native and physiological conditions as well as during aggregation and then we will focus our attention on the impact of membrane binding on αS structure and cellular localization in vitro and in vivo. Finally we will evaluate the impact of subcellular localization of αS and its membrane binding preference on αS pathology in PD.
αS
αS, together with β-synuclein and γ-synuclein, belongs to a family of proteins called synucleins which were discovered in 1988 (Maroteaux et al., 1988). Initially observed to localize in the nucleus and in the presynaptic terminals of neurons, αS was linked to the autosomal dominant form of PD, when a missense mutation of αS, a threonine substitution to an alanine at position 53 (A53T) was found in a family pedigree with early onset PD (Polymeropoulos, 1997). At the same year, αS was found to be the main constituent of LBs/LNs, providing strong evidence that the αS gene, mutated and wild-type isoforms, is associated to familial and sporadic PD and other α-synucleinopathies (Spillantini et al., 1997).
The SNCA gene, which encodes for αS protein, in humans is located in the long arm of chromosome 4 at position 22.1. Besides the A53T mutations, which is so far the most frequent and thus better characterized (Polymeropoulos, 1997), several missense mutations linked to a genetic form of PD and dementia with LBs have been mapped in SNCA gene more recently such as A30P (Krüger et al., 1998), E46K (Zarranz et al., 2004), H50Q (Appel-Cresswell et al., 2013; Proukakis et al., 2013), G51D (Lesage et al., 2013) and A53E (Pasanen et al., 2014). Furthermore, duplication or triplication of the SNCA gene have also been found and linked to familial PD, suggesting that increasing the amount of the wild-type protein is also pathogenic (Singleton et al., 2003; Ibáñez et al., 2004). All missense mutations and amplifications of the SNCA gene were associated with a dominant inheritance and an early onset of the disease compared to the sporadic forms. Since the overexpression of wild-type or mutated αS causes neurodegeneration in different animal models [as examples (Feany and Bender, 2000; Masliah, 2000; Lakso et al., 2003)] while its ablation has little or no effect in mice (Abeliovich et al., 2000), αS toxicity has been explained through a gain-of-function mechanism in which a modified version of the protein is responsible for causing neuronal demise.
Because of its presynaptic localization (Maroteaux et al., 1988) and its ability to bind biological membrane, it was proposed that αS physiological function was implicated in neurotransmission. More recent findings (Burré et al., 2010; Nemani et al., 2010; Diao et al., 2013; Wang et al., 2014), have strengthened this view and now it is largely accepted that αS can act as a molecular chaperon and promote synaptic transmission by facilitating clustering, recycling and docking of synaptic vesicles to the cell membrane. In addition αS has been involved in intracellular protein trafficking such as vesicles transport from the endoplasmic reticulum (ER) to Golgi (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010; Oaks et al., 2013) and from the Golgi to endosomes/lysosomes (Chung et al., 2013; Volpicelli-Daley et al., 2014; Breda et al., 2015; Mazzulli et al., 2016). An active role in axonal transport has also been reported for αS in which the protein acts as a molecular dynamase, binding directly to microtubule and promoting their assembly and stability (Cartelli et al., 2016).
Native αS Protein Structure
The αS protein, an acidic protein of 140 amino acids with a predicted molecular weight of approximately 14 kDa, is expressed mainly in neurons and possibly oligodendrocytes of the CNS (Asi et al., 2014), but also, under physiological conditions, in the PNS, in circulating blood cells and in hematopoietic cells of the bone marrow (Nakai et al., 2007; Gardai et al., 2013).
Biochemically and functionally the αS protein can be divided into three distinct regions (Figure 1):
Figure 1.

Human α-synuclein (αS) protein sequence.
αS is a small protein of 140 amino acids where point mutations (in red) have been associated with familial forms of Parkinson’s disease (PD) (Polymeropoulos, 1997; Krüger et al., 1998; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013). The protein can be divided in three domains: an N-terminal domain (light blue), important for membrane binding; the non amyloid β-component (NAC) domain (yellow), important for fibril formation (El-Agnaf et al., 1998) and a C-terminal domain (blue) important for protein interaction. Seven 11-amino acids imperfect repeats (purple), a unique motif predicted to form α-helix and highly conserved, are also shown (George et al., 1995). This motif is located within the N-terminal domain and the NAC domain. Notably, missense point mutations that have been found thus far are all located within the N-terminal domain, suggesting that membrane binding may influence αS aggregation.
-
i)
the amphipathic N-terminal domain (residues 1–60), which interacts with phospholipid membranes and micelles;
-
ii)
the hydrophobic non amyloid β-component (NAC) of Alzheimer’s disease (AD) (residues 61–95), which plays a strong role in αS self-aggregation (El-Agnaf et al., 1998);
-
iii)
the acidic C-terminal domain (96–140), a major site for post translational modifications, protein truncation (Li et al., 2005) and interaction with modulators of αS aggregation such as metal cations (Binolfi et al., 2006).
The overall protein contains seven imperfect 11-residues repeats with a conserved KTKEGV sequence: four included in the N-terminal region and three in the NAC core (George et al., 1995).
Soluble cytosolic αS has been described as an intrinsically disorder protein due to an unfolded native conformation (Weinreb et al., 1996). In fact, although αS, purified from mouse brain by gel-filtration, elutes as a single peak with an apparent molecular mass of 63 kDa, close to a folded tetramer, mass spectrometry and circular dichroism analysis reveal a monomer conformation with a single mass of 17 kDa (larger than the expected size, probably due to an in vivo N-terminal acetylation) (Burré et al., 2013). In agreement with these latest data, NMR studies show how acetylated αS, which is the predominant form in physiological conditions, is a disordered monomer but adopts a more compact conformation in solution that shields the NAC domain from other interaction in the cytosol (Theillet et al., 2016). Thus the higher molecular mass obtained previously in native conditions after gel filtration was associated with αS’s tendency to adopt an extended conformation, thereby yielding a larger mass, rather than a tetramer structure. The unfolded and disordered monomer conformation was confirmed in rat, human brain and erythrocytes isolated under denaturing and non-denaturing conditions as well as in bacteria expressed αS, while no oligomer species were found (Fauvet et al., 2012) under physiological conditions.
In contrast with previous evidence, however, Bartels and collaborators have shown how αS extracted in non-denaturating conditions and upon crosslinking in living cells (i.e., human erythrocytes, cell lines and brain tissue), is mainly a metastable homo-tetramer of 58 kDa. This αS tetramer is in a dynamic equilibrium with the unfolded monomer, which on the contrary is more susceptible to aggregation (Bartels et al., 2011). Under conventional extraction protocols, the 58 kDa tetramer disappears resulting in an increase in monomer concentration. Interestingly in the same study 80–100 kDa αS homo-oligomers (i.e., hexamers and octamers) were also detected together with the tetramer in native conditions. A homo-tetramer structure in physiological conditions was also suggested by an independent study (Wang et al., 2011), which found how subunits in the αS tetramer are held together by hydrophobic interactions and each subunit is characterized by two transient α-helices structure in the first 100 N-terminal residues, followed by a disordered C-terminal region. Thus it was postulated that αS tetramer and monomer would co-exist in native conditions and any perturbation of this dynamic equilibrium with an increased accumulation of the monomer would be associated with aggregation and pathology. In agreement with this, it was found that certain missense mutations could decrease the tetramer:monomer ratio and initiate neurotoxicity (Dettmer et al., 2015a, b).
Although the tetramer model was and is still widely debated, more recent work by Burré et al. (2014) showed how αS binds synaptic vesicles in vivo not as a monomer but in a folded α-helical multimer conformation, larger than an octamer. This conformation has a defined structural orientation and occurs only upon binding with vesicles that are docked at the cell membrane. In accordance with this finding, Bartels et al. (2011) described how the αS tetramer isolated upon crosslinking from human erythrocytes had a greater lipid-binding ability than the single monomer although the NMR structure obtained by Wang and coworkers did not show any phospholipid molecule (Wang et al., 2011). Thus while tetramer and membrane-bound multimer might be in reality part of the same complex, more evidence is necessary to fully understand the physiological structure of αS.
αS Aggregation
Besides the controversy about αS native physiological state, it is known that transition to an aggregated β sheet conformation is the necessary step for the formation of insoluble inclusions or LBs. In its amyloid form, αS monomers form antiparallel in-register β-sandwich fold, which in turn stack into a parallel arrangement forming the fibril protofilament (Vilar et al., 2008; Tuttle et al., 2016). Protofilaments further assemble into fully mature fibrils.
The aggregation process (summarized in Figure 2) is a nucleation-type reaction thought to occur in a sequential series of steps, even though “ramifications” of this path are likely to occur. The in vitro characterization of the fibrillation process revealed a precise time course, with an initial lag phase, in which the monomers convert into an oligomer-type of conformation (nucleation), a growth phase and a steady state that terminates with the accumulation of α-sheet rich amyloid fibrils (Cremades et al., 2012). Oligomers are defined in general as low-molecular weight aggregates, soluble or insoluble, that have not acquired a fibrillary organization. Once the seeds are formed, αS fibrils are believed to grow through the addition of monomers rather than oligomers (Buell et al., 2014). At least two different aggregate polymorphs, fibrils and ribbons, that present different biochemical and seeding properties, have been described in vitro, depending on the aggregation protocol used (Bousset et al., 2013; Guo et al., 2013).
Figure 2.
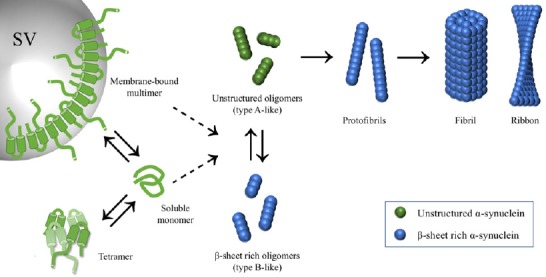
α-Synuclein (αS) fibrils formation.
In physiological conditions, αS has been reported to be a highly disordered monomer in a dynamic equilibrium with a multimer conformation when bound to synaptic vesicles (Weinreb et al., 1996; Burré et al., 2010, 2014; Fauvet et al., 2012; Theillet et al., 2016). Others have suggested that αS native structure is a homo-tetramer and dissolution of this latest conformation gives rise to an increase of the monomer form that drives pathology (Bartels et al., 2011; Wang et al., 2011; Dettmer et al., 2015a, b). Apart from its physiological state, it is still unclear which conformation is more susceptible to aggregation. Formation of αS fibrils is a nucleation reaction in which soluble αS monomer is converted to an insoluble β-sheet rich structure, tightly stacked in a parallel configuration, that give rise to protofibrils and fibrils (Lashuel et al., 2002; Vilar et al., 2008; Tuttle et al., 2016). During this process, a heterogeneous population of intermediate configurations, collectively called oligomers, has been described in vitro (Cremades et al., 2012; Chen et al., 2015). αS fibrils obtained with in vitro fibrillation can have a ribbon or a fibril structure (Bousset et al., 2013; Guo et al., 2013). Both oligomers and fibrils can be toxic though with different mechanisms of pathology (Danzer et al., 2007; Peelaerts et al., 2015; Fusco et al., 2017).
Extensive literature has focused on the role of αS oligomers and aggregates in PD pathology. Despite the presence of fibrillar αS in LBs strongly suggests an involvement of the aggregation process in α-synucleinopathies, it has been proposed that fibrils formation could constitute an innocuous by-product or even a neuroprotective response. For instance, LBs deposition is not always associated with neurological symptoms (Braak et al., 2003), whereas in some forms of familial PD there are no signs of αS aggregation (Schneider and Alcalay, 2017). However, Peelaerts et al. (2015) showed that all the in vitro-generated αS aggregates (fibrils and ribbons) are potentially toxic and can elicit distinct histopathological phenotypes, posing a structural base for heterogeneity among α-synucleinopathies.
Just as with αS fibrils, multiple forms of oligomers have been described in vitro, differing in size and morphology, including spherical, annular and tubular structures (Lashuel et al., 2002). Some of them are described as on-fibrillization pathway, while others generate amorphous, nonfibrillar assemblies. Since the fibrillation process can be influenced by numerous factors, including protein concentration, specific physicochemical conditions, the presence of certain ligands (including dopamine) and cross-linking (Buell et al., 2014), it is still unclear whether this heterogeneity in the oligomers population is due to the aggregation protocol used or has physiological relevance. More recent data obtained by directly following the oligomerization reaction using single molecule fluorescence technique showed how the oligomers population is mainly composed of two different species, named type A and type B, that form during early stages of aggregation (Cremades et al., 2012; Chen et al., 2015). These two oligomer populations seem to differ for chemical, structural and toxic properties. Type B is more resistant to protease K digestion than type A and requires a longer lag time for formation, suggesting that these species could derive from the conversion and rearrangement of type A oligomer. In addition, type B has a higher content in β-sheet structures that is instead negligible in type A (Fusco et al., 2017). In vivo, type B oligomers were shown to induce cell death in neuronal cells, via disruption of cellular ion homeostasis and production of reactive oxygen species with concomitant mitochondrial dysfunction (Danzer et al., 2007; Fusco et al., 2017), while type A were able to enter cells and induce intracellular aggregation, leading indirectly to cell death. Recently the structural basis of these different mechanisms of pathogenicity has been investigated by Fusco et al., who showed that while both oligomers bind biological membranes, only type B form a rigid β-sheet core that is able to insert into the lipid bilayer and induce directly membrane disruption and cellular toxicity (Fusco et al., 2017). Thus based on this model, both αS oligomers and aggregates are toxic. On-pathway type A oligomers are converted in compact protofibrils and fibrils and are responsible for seeding formation of new aggregates and propagate the αS pathology, while off-pathway type B oligomers are still largely detrimental, acting directly on biological membranes but do not self-propagate. In agreement with this, our group found that microsomes-associated αS toxic species behaved differently, in terms of seeding abilities of intracellular aggregates, when isolated from diseased αS transgenic (Tg) mice as opposed to aged presymptomatic littermates, suggesting the presence in vivo of at least two types of αS HMW species depending on the stage of αS pathology (Colla et al., 2018). For instance when isolated from presymptomatic mice, microsomes-associated αS oligomers induce cell death of primary neurons without seeding the formation of intracellular aggregates as opposed to microsomes-associated αS species (probably a mixture of oligomers and aggregates) isolated from diseased mice that had both properties. Thus the heterogeneity of αS toxic species, that is coming to light with recent findings shows a complicated picture of αS aggregation in which both aggregates and oligomers are toxic for cellular functions and the biochemical and functional diversity of αS toxic species is pathologically translated in at least two different and interconnected mechanisms of disease. It becomes evident that targeting one single HMW species of αS is not sufficient to stop successfully aggregation and αS-driven neurodegeneration.
αS Binding to Biological Membranes and Subcellular Localization
αS is known to bind lipids and biological membranes in vitro and in vivo. But does it bind them with the same efficiency? The answer is probably no. Physical properties and chemical composition of biological membranes or presence of cationic compounds able to bind lipids (Perni et al., 2017) dictate the strength of αS binding. In vitro αS binding preference is toward membranes composed of negatively charged phospholipids [such as phosphatidylethanolamine (PE), phosphatidic acid (PA) and phosphatidylinositol (PI) over phosphatidylserine (PS), or phosphatidylcholine (PC) (Middleton and Rhoades, 2010; Galvagnion et al., 2016)] or containing lipid packing defects, such as cone-head phospholipids (Ouberai et al., 2013). In addition, αS senses membrane curvature, preferring to bind to small, highly curved vesicles such as synaptic vesicles, rather than large multilamellar bodies (Middleton and Rhoades, 2010).
Membrane interaction is mediated by the αS N-terminal that, upon binding, undergoes a conformational transition from random coil to α-helix, adopting a long extended one-single α-helix (Ferreon et al., 2009) in the presence of big vesicles (diameter larger than 100 nm) or two anti-parallel curved α-helices linked with a short residues chain (Chandra et al., 2003) in the case of small vesicles. In both conformations the amphipathic helices insert between the lipids polar groups at a depth corresponding to that of the glycerol group (Fusco et al., 2016). While both structures seem interchangeable and physiologically relevant (Georgieva et al., 2010), it is not clear if other conditions in vivo, besides vesicles size, would dictate a conformational change toward one structure over the other. Also, of note, is that part of the N-terminal αS region binding lipids contains the NAC domain, which is responsible for αS fibril formation (El-Agnaf et al., 1998). Interaction with membranes is known to modify not only αS conformation, but also the membranes’ physical properties, e.g., inducing changes in melting temperature (Galvagnion et al., 2016) and membrane remodelling (Jiang et al., 2013) such as lateral expansion of membrane lipids and lipid packing modifications (Ouberai et al., 2013). In addition αS binding to membrane promotes clustering of synaptic vesicles (Diao et al., 2013).
How does αS preference for membranes translate in a cell context, in vivo? Physiologically, αS is believed to shift between a free, cytosolic and a membrane-bound state in a dynamic equilibrium with the membrane-bound state accounting for 10–15% of the total protein amount. In line with this, membranes of specific organelle such as the mitochondria and the ER, and synaptic vesicles have been shown to be associated at different extent with αS (Figure 3).
Figure 3.
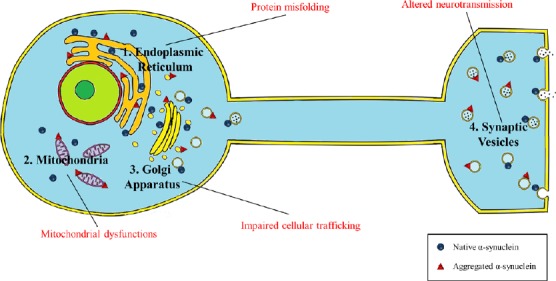
Influence of subcellular localization on α-synuclein (αS) oligomers/aggregates toxicity.
αS has been found to selectively bind to synaptic vesicles, endoplasmic reticulum (ER)/Golgi and the mitochondria and membrane binding seems to be part of its physiological function (Devi et al., 2008; Nakamura et al., 2008; Burré et al., 2010; Colla et al., 2012a). Although it is still unclear if association with membranes interferes with or accelerates αS aggregation, accumulation of αS toxic species selectively along these subcellular locations can primarily impact specific cellular functions such as neurotransmission, protein trafficking and mitochondrial respiration directly linked to the above-mentioned organelles.
αS can bind synaptic vesicles at the synapses and this binding is believed to mediate its synaptic function in neurotransmission. For instance, αS acts as a molecular chaperon to promote SNARE-complex assembly (Burré et al., 2010), which is necessary to regulate docking of synaptic vesicles to the cell membrane. Vesicle binding is mediated not only by the interaction with acidic phospholipids such as PE, PC, PS, or cholesterol, of which the synaptic vesicles are rich, but also by specific proteins such as SNARE-protein synaptobrevin-2/vesicle-associated membrane protein 2 (VAMP2) (Diao et al., 2013). αS interaction with synaptic vesicles that occurs through a multimer conformation, promotes vesicular clustering and thus reducing synaptic vesicles trafficking and recycling (Wang et al., 2014).
Moreover, αS has been found to be associated with the ER and Golgi in mice and human cells cultures (Colla et al., 2012a). Protease K sensitivity assays have shown how microsomes-associated αS is partially protected from digestion, therefore associating with the lumenal side of the microsomes (a membrane fraction including ER/Golgi and synaptic vesicles) in mice and human cell lines (Lee, 2005; Colla et al., 2012a). While no lipid binding involvement has been described yet, αS was found to bind, in αS Tg mice and human cell lines overexpressing αS, to gpr78/binding immunoglobulin protein (BIP), an ER chaperone bound to the luminal side of the ER, transiently associated with the ER translocon import pore, and directly implicated as a sensor of protein misfolding and initiator of the unfolded protein response (Bellucci et al., 2011; Colla et al., 2012a). Moreover overexpression of αS was shown to impair vesicular trafficking at the ER-Golgi level in yeast and other organisms (Cooper et al., 2006) causing accumulation of ER proteins with induction of ER stress, Golgi fragmentation and depletion of lysosomal enzymes (Oaks et al., 2013; Mazzulli et al., 2016). Interestingly, this transport defect was rescued by overexpression of proteins implicated in vesicles transit from the ER to the cell membrane such as Rab1 (ER-Golgi), Rab8 (Golgi) and Rab3A (post-Golgi) (Gitler et al., 2008) but also from endolysosomal pathway such as Rab-11A (recycling endosomal) (Breda et al., 2015), implicating a major role for αS in vesicle trafficking and recycling, outside the synapses.
In addition, αS has been found to bind mitochondrial outer and inner membrane [(Devi et al., 2008; Nakamura et al., 2008) and our lab (unpublished results)]. Since most of the data were obtained from in vivo observations, it is not clear whether this binding was based on lipids, according to αS preference to cardiolipin, which is rich in the mitochondria membranes, or was also mediated by specific proteins. Interestingly a translocase of the mitochondria outer membrane has been described as responsible for the import of αS into the mitochondria and one of its subunit, TOM20, has been shown to bind αS in vivo (Di Maio et al., 2016). Moreover overexpression of αS was found to promote mitochondria dysfunction in αS Tg mice (Martin et al., 2006, 2014) and mitochondria fragmentation in vitro and in primary neurons. This last effect was dependent on the direct interaction of αS with mitochondria since disruption of αS N-terminal membrane-binding domain, rescued mitochondria morphology (Nakamura et al., 2011).
Impact of Membrane Binding and Subcellular αS Localization In Vivo on αS Pathology
While αS cytosolic and membrane bound-state are both physiologically relevant, it still unclear how their localization affects αS pathology and where aggregation initiates. Accumulating evidence has shown that membrane binding and lipid interaction can stimulate but also attenuate αS fibrillation (Narayanan and Scarlata, 2001; Lee et al., 2002; Jo et al., 2004; Burre et al., 2015; Galvagnion et al., 2016).
In line with this controversial aspect is the observation that point mutations in αS associated with familial PD are located in the N-terminal lipid-binding domain, suggesting that lipid binding can be implicated in αS acquired pathogenicity. This is somewhat true for some point mutations such as A30P, where membrane binding is reduced while aggregation increased, but not others. In fact pathological amino acid substitutions in αS such as A53T, E46K and H50Q, lead to an increase in fibril formation without affecting lipid binding (Bussell and Eliezer, 2004; Fredenburg et al., 2007; Khalaf et al., 2014) while other mutations such as G51D, attenuate both membrane binding and aggregation (Fares et al., 2014). Thus while membrane binding and aggregation may not always be directly correlated, other factors, besides point mutations, such as intramolecular interaction between the N and C termini or protein binding to the C-terminal can influence the propensity of αS to aggregate and compensate for amino acid substitutions (Ulrih et al., 2008; Burré et al., 2010). In addition the presence of oxidative stress-induced posttranslational modifications of αS [such as nitrosylation, metal ion-catalyzed oxidation and presence of dopamine (or its oxidative metabolites) adducts] has been shown to increase oligomerization and, possibly, influence αS ability to bind vesicle membranes as a monomer or in an oligomer conformation (Binolfi et al., 2006; Xiang et al., 2013; Follmer et al., 2015; Plotegher et al., 2017).
More recently, two independent papers proposed how two different small compounds, one of synthesis, NPT100-18A and the other naturally produced, squalamine, could reduce αS oligomers/aggregates content and subsequently rescue behavioural deficits in mice and worms, by interfering with αS binding to membranes (Wrasidlo et al., 2016; Perni et al., 2017). Notably, while NPT100-18A and squalamine have a different origin, they both bind only to membrane-associated αS, inducing a rearrangement of the protein structure that would lead to a displacement from the lipids, therefore reducing the amount of aggregation-prone αS. Thus while these results greatly suggest that aggregation might initiate from a pathological conversion of the membrane-bound αS fraction, because αS membrane-binding is required for αS function, development of therapeutic strategy that would block aggregation by interfering with αS membrane binding has to be taken with caution.
In vivo subcellular localization of αS and association with specific membranes can determine pathobiology processes connected to aggregation and neuronal degeneration. Our group and others (Colla et al., 2012a, b; Fagerqvist et al., 2013) have shown that αS aggregates can be selectively associated with the secretory pathway including the ER and Golgi, in pathogenic conditions in αS Tg mice but not with other organelles such as the mitochondria. Notably in absence of pathology, aged Tg mice already accumulated specifically oligomer species associated with the ER, Golgi and synaptic vesicles before neuronal degeneration appearance. When compared to other αS species that co-precipitate at lower g, microsomes-associated αS oligomers (i.e., HMW species associated with membranes from the secretory pathway) obtained from healthy aged Tg mice with no overt accumulation of αS insoluble aggregates, were increasingly harmful and able to induce apoptosis, after exogenous administration to murine neuronal cultures (Colla et al., 2018). Additionally, αS oligomers have been found to decrease axonal transport and influence microtubule stability, a condition that could exacerbate synaptic vesicles traffic dysfunction (Prots et al., 2013).
At the synapse, exogenous administration of large oligomers of recombinant αS was shown to bind synaptic vesicles through synaptobrevin-2 causing vesicle docking inhibition to the membrane (Choi et al., 2013) and to lower synapsin-I/II abundance (Larson et al., 2017). Although direct measurement of ER-Golgi traffic was not assessed in these conditions, it is plausible that accumulation of toxic species of αS might affect the whole protein transport system from the ER to the membrane. Moreover, electrophysiology studies showed that αS oligomers impair long-term potentiation (Diogenes et al., 2012; Martin et al., 2012) and reduces neuronal excitability (Kaufmann et al., 2016).
αS oligomers have been shown to be particularly toxic to mitochondria. Mitochondrial damage such as inhibition of complex I activity with subsequently increase of reactive oxygen radical production and oxidative stress (Devi et al., 2008; Cremades et al., 2012), alteration of membrane potential and Ca2+ homeostasis, induction of mitochondrial fragmentation (Nakamura et al., 2011), mitochondrial protein import impairment (Di Maio et al., 2016), and, more recently externalization of cardiolipin to the outer mitochondrial membrane, a process that stimulates mitophagy in response to cellular stress (Ryan et al., 2018), has been associated with accumulation of toxic αS. High-ordered αS structure such as a small oligomer but not the monomer was found to be able to associate and cluster artificial mitochondria and induced fragmentation, a process similar to that described in the case of synaptic vesicles (Diao et al., 2013). In addition cardiolipin has been shown to associate and promote refolding of αS fibrils in vitro, a process negatively affected and reduced by the presence of PD-related αS point mutations (Ryan et al., 2018).
Because of most of those evidence were obtained in vitro, it is not clear whether mitochondria could be a primary site of aggregation or if mitochondrial dysfunction, due to toxic αS, might be the result of a secondary generalized spreading of αS aggregates in the neuron. Thus while more evidence will be necessary to fully understand the influence of membrane binding on αS pathology, it is plausible to hypothesize that the initial pathologic transition of αS toward a toxic conformation might occur in proximity of the membranes in the above mentioned specific locations and then spread to other sites in neurons.
Conclusions
αS has been found to shift between a free native conformation and a membrane-bound state in a dynamic equilibrium. What dictates this transition is not clear but αS has been found to associate with specific membranes in neurons such as that of synaptic vesicles and some cellular organelles, like the ER/Golgi and the mitochondria. Although it is still not clear whether membrane-bound αS is more susceptible to aggregation or binding to membrane prevents the pathologic conversion to toxic species, initial phases of αS aggregation might begin selectively along those specific membranes and compromise, as has been reported, processes linked to these sites such as synaptic transmission, protein folding and trafficking, energy production. Initial damage from these sites would spread to other cellular functions, exacerbating αS aggregation and lead ultimately to neuronal demise. In line with this, compounds that would modify αS interaction with membranes might inhibit the initiation of αS aggregation and clarify whether membranes are necessary for the initiation of αS aggregation. At the same time though, a widespread inhibition of αS binding to membranes might result in a decrease in cellular functions mediated by αS, therefore the use of such strategies in mammals might be difficult and not directly result in a pathology improvement. A way to bypass this problem could be to implement strategies that would act on the initial phases of cellular dysfunction described above. New data will be necessary in the near future to clarify the impact that membrane binding and subcellular localization have on αS toxicity and to understand how to intervene in the early phases of the αS aggregation process before a generalized damage occurs.
Footnotes
Conflicts of interest: The authors declare that they have no competing interests.
Financial support: This work has been supported by the Italian Ministry of University and Research (MIUR) through the Career Reintegration grant scheme (RLM Program for Young Researcher) and from Scuola Normale Superiore.
Copyright transfer agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review reports:
Reviewer 1: Hailong Song, University of Missouri Columbia, USA.
Comments to authors: The authors provided a comprehensive review with nice illustrations and detailed explanation of some current research. The scientific significance, quality, and novelty of this review are high. In this manuscript, the authors reviewed αS’s association with membrane binding and its subcellular localization to further understand the regulation of αS contributing to PD pathophysiology. Specifically, this review included αS conformation, the impact of membrane binding on alpha-synuclein structure and cellular localization, and the subsequent alpha-synuclein pathology in PD. Overall, the authors provided a comprehensive review with nice illustrations and detailed explanation of some current research. The scientific significance, quality, and novelty of this review are high.
Reviewer 2: Darrin Jason Lee, University of Toronto, Canada.
Funding: This work has been supported by the Italian Ministry of University and Research (MIUR) through the Career Reintegration grant scheme (RLM Program for Young Researcher) and from Scuola Normale Superiore.
References
- 1.Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 2.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 3.Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, Houlden H, Holton JL. α-synuclein mRNA expression in oligodendrocytes in MSA: αSyn mRNA Expression in MSA Oligodendrocytes. Glia. 2014;62:964–970. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, Battistin L, Spillantini M, Missale C, Spano P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease: α-Synuclein accumulation induces the UPR. J Neurochem. 2011;116:588–605. doi: 10.1111/j.1471-4159.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- 6.Binolfi A, Rasia RM, Bertoncini CW, Ceolin M, Zweckstetter M, Griesinger C, Jovin TM, Fernández CO Interaction of α-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc. 2006;128:9893–9901. doi: 10.1021/ja0618649. [DOI] [PubMed] [Google Scholar]
- 7.Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two α-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 9.Breda C, Nugent ML, Estranero JG, Kyriacou CP, Outeiro TF, Steinert JR, Giorgini F. Rab11 modulates α-synuclein-mediated defects in synaptic transmission and behaviour. Hum Mol Genet. 2015;24:1077–1091. doi: 10.1093/hmg/ddu521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TPJ, Linse S, Dobson CM. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci. 2014;111:7671–7676. doi: 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burré J, Sharma M, Südhof TC α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci. 2014;111:E4274–E4283. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burre J, Sharma M, Sudhof TC. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci. 2015;35:5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native brain α-synuclein. Nature. 2013;498:E4–E6. doi: 10.1038/nature12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bussell R, Jr, Eliezer D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry. 2004;43:4810–4818. doi: 10.1021/bi036135+. [DOI] [PubMed] [Google Scholar]
- 16.Cartelli D, Aliverti A, Barbiroli A, Santambrogio C, Ragg EM, Casagrande FV, Cantele F, Beltramone S, Marangon J, De Gregorio C, Pandini V, Emanuele M, Chieregatti E, Pieraccini S, Holmqvist S, Bubacco L, Roybon L, Pezzoli G, Grandori R, Arnal I, et al. α-Synuclein is a novel microtubule dynamase. Sci Rep. 2016;6:33289. doi: 10.1038/srep33289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken α-helix in folded α-Synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 18.Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TPJ, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM, Cremades N. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci. 2015;112:E1994–E2003. doi: 10.1073/pnas.1421204112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci. 2013;110:4087–4092. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schüle B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342:983–987. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci. 2012a;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci. 2012b;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colla E, Panattoni G, Ricci A, Rizzi C, Rota L, Carucci N, Valvano V, Gobbo F, Capsoni S, Lee MK, Cattaneo A. Toxic properties of microsome-associated α-synuclein species in mouse primary neurons. Neurobiol Dis. 2018;111:36–47. doi: 10.1016/j.nbd.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cremades N, Cohen SIA, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TPJ, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015a;6:7314. doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D. KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci. 2015b;112:9596–9601. doi: 10.1073/pnas.1505953112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α-synuclein impair complex i in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. 2016;8:342ra78. doi: 10.1126/scitranslmed.aaf3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diao J, Burré J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Südhof TC, Brunger AT. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife. 2013;2:e00592. doi: 10.7554/eLife.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Nasstrom T, Franquelim HG, Oliveira LMA, Castanho MARB, Lannfelt L, Bergstrom J, Ingelsson M, Quintas A, Sebastiao AM, Lopes LV, Outeiro TF. Extracellular α-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci. 2012;32:11750–11762. doi: 10.1523/JNEUROSCI.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Agnaf OM, Bodles AM, Guthrie DJ, Harriott P, Irvine GB. The N-terminal region of non-A beta component of Alzheimer’s disease amyloid is responsible for its tendency to assume beta-sheet and aggregate to form fibrils. Eur J Biochem. 1998;258:157–163. doi: 10.1046/j.1432-1327.1998.2580157.x. [DOI] [PubMed] [Google Scholar]
- 34.Fagerqvist T, Lindström V, Nordström E, Lord A, Tucker SM, Su X, Sahlin C, Kasrayan A, Andersson J, Welander H, Näsström T, Holmquist M, Schell H, Kahle PJ, Kalimo H, Möller C, Gellerfors P, Lannfelt L, Bergström J, Ingelsson M. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J Neurochem. 2013;126:131–144. doi: 10.1111/jnc.12175. [DOI] [PubMed] [Google Scholar]
- 35.Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovi i A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, Lashuel HA. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet. 2014;23:4491–4509. doi: 10.1093/hmg/ddu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 38.Ferreon ACM, Gambin Y, Lemke EA, Deniz AA. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Follmer C, Coelho-Cerqueira E, Yatabe-Franco DY, Araujo GDT, Pinheiro AS, Domont GB, Eliezer D. Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of α-synuclein with the toxic dopamine metabolite 3, 4-dihydroxyphenylacetaldehyde (DOPAL) J Biol Chem. 2015;290:27660–27679. doi: 10.1074/jbc.M115.686584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, Lansbury PT., Jr The impact of the e46k mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry. 2007;46:7107–7118. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- 41.Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM, De Simone A. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science. 2017;358:1440–1443. doi: 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- 42.Fusco G, De Simone A, Arosio P, Vendruscolo M, Veglia G, Dobson CM. Structural ensembles of membrane-bound α-synuclein reveal the molecular determinants of synaptic vesicle affinity. Sci Rep. 2016;6:27125. doi: 10.1038/srep27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galvagnion C, Brown JWP, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc Natl Acad Sci. 2016;113:7065–7070. doi: 10.1073/pnas.1601899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardai SJ, Mao W, Schüle B, Babcock M, Schoebel S, Lorenzana C, Alexander J, Kim S, Glick H, Hilton K, Fitzgerald JK, Buttini M, Chiou SS, McConlogue L, Anderson JP, Schenk DB, Bard F, Langston JW, Yednock T, Johnston JA. Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS One. 2013;8:1–21. doi: 10.1371/journal.pone.0071634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 46.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. The lipid-binding domain of wild type and mutant a-synuclein: compactness and interconversion between the broken and extended helix forms. J Biol Chem. 2010;285:28261, 28274. doi: 10.1074/jbc.M110.157214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2012;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 49.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VMY. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Agid Y, Dürr A, Brice A, Pollak P. Causal relation between α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 51.Jiang Z, de Messieres M, Lee JC. Membrane remodeling by α-synuclein and effects on amyloid formation. J Am Chem Soc. 2013;135:15970–15973. doi: 10.1021/ja405993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jo E, Darabie AA, Han K, Tandon A, Fraser PE, McLaurin J. α-Synuclein-synaptosomal membrane interactions: Implications for fibrillogenesis. Eur J Biochem. 2004;271:3180–3189. doi: 10.1111/j.1432-1033.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 53.Kaufmann TJ, Harrison PM, Richardson MJE, Pinheiro TJT, Wall MJ. Intracellular soluble α-synuclein oligomers reduce pyramidal cell excitability: Impact of α-synuclein on neocortical neurons. J Physiol. 2016;594:2751–2772. doi: 10.1113/JP271968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee SJ, Eliezer D, Lashuel HA. The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J Biol Chem. 2014;289:21856–21876. doi: 10.1074/jbc.M114.553297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 56.Lakso M, Vartiainen S, Moilanen A-M, Sirviö J, Thomas JH, Nass R, Blakely RD, Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J Neurochem. 2003;86:165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- 57.Larson ME, Greimel SJ, Amar F, LaCroix M, Boyle G, Sherman MA, Schley H, Miel C, Schneider JA, Kayed R, Benfenati F, Lee MK, Bennett DA, Lesné SE. Selective lowering of synapsins induced by oligomeric α-synuclein exacerbates memory deficits. Proc Natl Acad Sci. 2017;114:E4648–E4657. doi: 10.1073/pnas.1704698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 59.Lee HJ. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee HJ, Choi C, Lee SJ. Membrane-bound -synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 61.Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A for the French Parkinson’s Disease Genetics Study Group. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome: SNCA G51D in Parkinsonism. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 62.Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci Off J Soc Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin LJ, Semenkow S, Hanaford A, Wong M. The mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant α-synuclein transgenic mice. Neurobiol Aging. 2014;35:1132–1152. doi: 10.1016/j.neurobiolaging.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases: Cognitive effects of α-synuclein oligomers. J Neurochem. 2012;120:440–452. doi: 10.1111/j.1471-4159.2011.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masliah E. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 68.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci. 2016;113:1931–1936. doi: 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakai M, Fujita M, Waragai M, Sugama S, Wei J, Akatsu H, Ohtaka-Maruyama C, Okado H, Hashimoto M. Expression of α-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem Biophys Res Commun. 2007;358:104–110. doi: 10.1016/j.bbrc.2007.04.108. [DOI] [PubMed] [Google Scholar]
- 71.Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J Neurosci. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Narayanan V, Scarlata S. Membrane binding and self-association of α-synucleins. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 74.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oaks AW, Marsh-Armstrong N, Jones JM, Credle JJ, Sidhu A. Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS One. 2013;8:e70872. doi: 10.1371/journal.pone.0070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, Welland ME. α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J Biol Chem. 2013;288:20883–20895. doi: 10.1074/jbc.M113.478297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35:1–5. doi: 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 78.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 79.Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MB, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SI, Cascella R, Chen SW, Limbocker R, Sormanni P, Heller GT, Aprile FA, Cremades N, Cecchi C, Chiti F, Nollen EA, Knowles TP, et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci. 2017;114:E1009–E1017. doi: 10.1073/pnas.1610586114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Dalla Serra M, Perego C, Casella L, Bubacco L. DOPAL derived α-synuclein oligomers impair synaptic vesicles physiological function. Sci Rep. 2017;7:40699. doi: 10.1038/srep40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polymeropoulos MH. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 82.Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, Böhm KJ, Winner B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem. 2013;288:21742–21754. doi: 10.1074/jbc.M113.451815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel α-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, Ambasudhan R, Mosser DD, Lipton SA, Harauz G, Ryan SD. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun. 2018;9:817. doi: 10.1038/s41467-018-03241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature: neuropathology of genetic Parkinson‘s disease. Mov Disord. 2017;32:1504–1523. doi: 10.1002/mds.27193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. α-Synuclein locus triplication causes parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 87.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 88.Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. 2010;21:1850–1863. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 90.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, Rienstra CM. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ulrih NP, Barry CH, Fink AL. Impact of Tyr to Ala mutations on α-synuclein fibrillation and structural properties. Biochim Biophys Acta BBA-Mol Basis Dis. 2008;1782:581–585. doi: 10.1016/j.bbadis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 92.Vilar M, Chou H-T, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of α-synuclein fibrils. Proc Natl Acad Sci. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014;25:4010–4023. doi: 10.1091/mbc.E14-02-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S. α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol. 2014;24:2319–2326. doi: 10.1016/j.cub.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 97.Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, Ledolter K, Bonhaus D, Paulino A, Eleuteri S, Skjevik ÅA, Kouznetsova VL, Spencer B, Desplats P, Gonzalez-Ruelas T, Trejo-Morales M, Overk CR, Winter S, Zhu C, Chesselet MF, et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain. 2016;139:3217–3236. doi: 10.1093/brain/aww238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xiang W, Schlachetzki JCM, Helling S, Bussmann JC, Berlinghof M, Schäffer TE, Marcus K, Winkler J, Klucken J, Becker CM. Oxidative stress-induced posttranslational modifications of a-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci. 2013;54:71–83. doi: 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 99.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, Del Ser T, Muñoz DG, De Yebenes JG. The new mutation, E46K, of α-synuclein causes Parkinson and lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]