

Abstract
Peripheral vision loss followed by “tunnel vision” and eventual irreversible blindness is the fate of patients afflicted by various forms of glaucoma including primary open-angle glaucoma (POAG) and normotensive glaucoma (NTG). These complex and heterogeneous diseases are characterized by extensive death of retinal ganglion cells (RGCs) accompanied by retraction and severance of their axonal connections to the brain and thus damage to and thinning of the optic nerve. Since patients suffering from this glaucomatous optic neuropathy (GON) first notice visual impairment when they have lost > 40% of their RGCs, early diagnosis is the key to retard the progression of glaucoma. Elevated intraocular pressure (IOP), low cerebrospinal and/or low intracranial fluid pressure, advancing age, and ethnicity are major risk factors associated with POAG. However, retinal vascular abnormalities and a high sensitivity of RGCs and optic nerve head components to neurotoxic, inflammatory, oxidative and mechanical insults also contribute to vision loss in POAG/GON. Current treatment modalities for POAG and NTG involve lowering IOP using topical ocular drugs, combination drug products, and surgical interventions. Two recently approved multi-pharmacophoric drugs (e.g., rho kinase inhibitor, Netarsudil; a drug conjugate, Latanoprostene Bunod) and novel aqueous humor drainage devices (iStent and CyPass) are also gaining acceptance for treating POAG/ NTG. Neuroprotective and regenerative agents, coupled with electroceutical, mechanical support systems, stem cell transplantation and gene therapy are emerging therapeutics on the horizon to help combat GON. The latter techniques and approaches hope to rejuvenate RGCs and repair the optic nerve structures, thereby providing a gain of function of the visual system for the glaucoma patients.
Keywords: glaucoma, ocular hypertension, glaucomatous optic neuropathy, drainage device, optic nerve degeneration, retinal ganglion cells, nerve regeneration, neuroprotection
Introduction to Glaucoma and Ocular Hypertension
The blinding diseases known as “glaucoma” comprise several different forms of optic neuropathies with diverse and complex etiologies. Primary open-angle glaucoma (POAG) is the most common type and is estimated to affect > 80 million people by 2020 (Tham et al. 2014). As with most forms of glaucoma, including normotensive glaucoma (NTG) and pseudo-exfoliation glaucoma (PEXG), POAG is painless and can remain undetected and undiagnosed for decades. POAG, PEXG and NTG develop slowly and their pathology involves defects at multiple locations within the eye-brain visual axes (Figure 1; Weinreb et al., 2014). Whilst the disease-initiating factor(s) remains elusive, it has been chronicled that damage to the retinal ganglion cell (RGC) axons that form the optic nerve causes retrograde and anterograde demise of the corresponding RGC bodies. It is possible that the RGCs themselves are simultaneously and deleteriously impacted by their environment that may contain low oxygen, depleted energy sources, and high levels of damaging neurotoxins and inflammatory cytokines (Ito and Di Polo, 2017). Over time, the RGC axons dissociate from the brain lateral geniculate nucleus (LGN) and superior colliculus (SC) (Yucel et al., 2000). Since the outermost layer of RGCs in the retina are initially the most susceptible to the damaging effects of various mechanical and chemical insults, they die first. Thus, initially the patient’s peripheral vision is diminished followed by gradual but progressive loss of central vision, culminating in total irreversible blindness (Weinreb et al., 2014). Early diagnosis and initiation of treatment are therefore pivotal to preserving vision in POAG and to reduce the harmful effects of this glaucomatous optic neuropathy (GON) on the patient’s quality of life. Unfortunately, since the disease is asymptomatic, the earliest manifestation of visual impairment for the POAG patient occurs when almost half of the total 1 million RGCs have died and the patient begins to notice visual disturbances such as patchy dark visual images, distorted and incomplete images and/or “tunnel vision” (Crabb, 2016). Therapeutic intervention now becomes critical to delay and prevent the loss of additional RGCs and preserve the remaining visual apparatus, thereby preserving the RGC axons and thus reducing the impact on the structure and function of the optic nerve (Figure 2).
Figure 1.
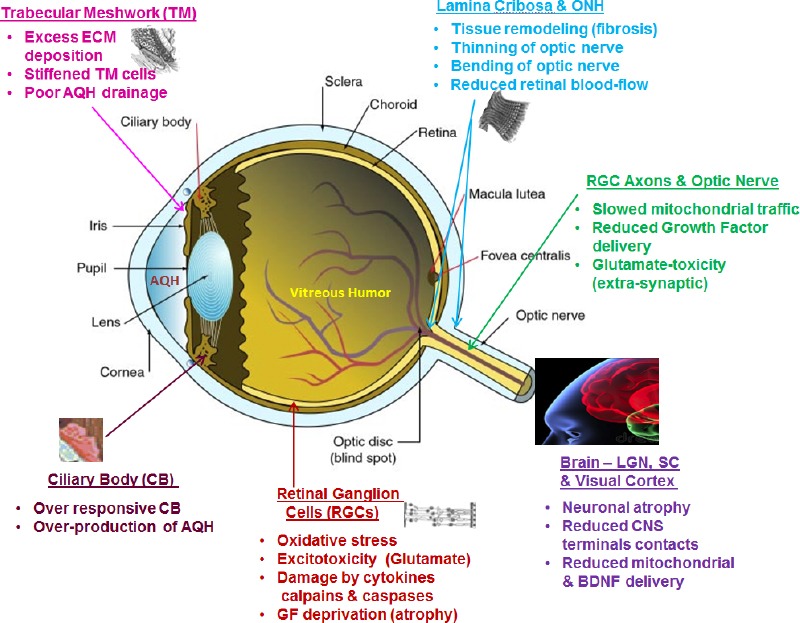
Key vulnerable regions of the ocular-cerebral axis involved in glaucomatous optic neuropathy (GON).
ONH: Optic nerve head; ECM: extracellular matrix; AQH: aqueous humor; LGN: lateral geniculate nucleus; SC: superior colliculus; GF: growth factor.
Figure 2.
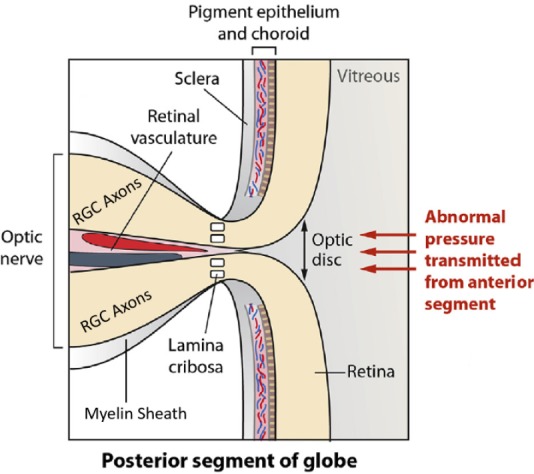
Magnified region of the optic nerve head depicting components of the retina, lamina cribosa, retinal ganglion cell axons (optic nerve) and retinal vasculature that are impacted by ocular hypertension and glaucomatous optic neuropathy.
Years of basic and clinical research have finally yielded some clues as to what may be happening in the eyes of the POAG patients. Intraocular pressure (IOP) is determined by the drainage of aqueous humor (AQH) from the anterior chamber of the eye via the trabecular meshwork (TM), and in POAG patients, the TM cells can become dysfunctional and congested, interfering with proper flow of AQH through the TM. This can cause an increase in (IOP), which epidemiological and pathological studies have shown to be the most damaging risk factor associated with POAG. However, poor ocular blood perfusion (Pasquale, 2016), low inter-cranial and cerebrospinal fluid pressure (CSF) (Jonas et al., 2015), advancing age, and genetics (Aung and Khor, 2016; Danford et al., 2017) also play a part in the disease process. The sequence of these destructive events, that also include heightened TM and RGC apoptosis, and the relative severity of their impact may be patient-dependent, but it is clear that a multitude of factors conspire to undermine the RGCs and their axons leading to their degeneration, dysfunction and demise (Weinreb et al., 2014; Sharif, 2018).
The elevated IOP causes mechanical distortion and stretching of the unmyelinated RGC axons at the lamina cribosa (LC), where they exit the globe to form the optic nerve (Figure 2; Danford et al., 2017). This appears to involve release of matrix metalloproteinases (MMPs) that digest and weaken the LC tissue leading to further bending and stretching of the optic nerve and the associated retinal blood vessels at the back of the eye (Hollander et al., 1995; Xu et al., 2014). The spaces previously occupied by neurons are invaded by glial cells that form a fibrotic scar over time. The ensuing ischemia and local hypoxia causes production of reactive oxygen species, aglycemia, activation of complement system (Tezel et al., 2010), inflammasome activation (Chi et al., 2014), and reduced axonal flow of mitochondria and neurotrophic factors to-and-from the brain LGN/SC and RGC somas (Quigley et al., 2000). As the mitochondrial energy stores diminish (Thomas et al., 2000; Osborne et al., 2014; Li et al., 2015), the LC and RGC cellular machinery maintaining homeostasis becomes dysfunctional (McElnea et al., 2011) and the somal and axonal demise begins. The net effect of these events is the death of some of the RGCs and their neighboring retinal neurons, resulting in the release of their cytoplasmic contents into the extracellular space. Consequently, large amounts of glutamate, ATP, endothelin, and preformed inflammatory cytokines such a tumor necrosis factor-α and numerous interleukins begin bathing the retinal neurons. The ensuing inflammation, excitotoxicity and the prevalent oxidative stress conditions induce senescence of even more RGCs and interneurons, and the vicious cycle continues unabated unless the patient receives suitable treatment(s). The various mechanical, chemical, bioenergetic and local environmental conditions/insults that appear involved in the etiology of POAG/ocular hypertension (OHT)-induced GON are pictorially depicted in Figures 1 and 2 (Nickells et al., 2012) Fortuitously, these areas provide suitable points of therapeutic intervention in order to preserve sight and prevent, or at least slow down, visual impairment in OHT/POAG patients (Jonas et al., 2017; Sharif, 2018).
Drug Treatment Options for OHT and Glaucoma
What constitutes the current and future treatment options for the patients afflicted with POAG and OHT, and how may damage to the optic nerve and its components be prevented to help preserve vision? This is a complex problem since many elements are involved in the pathophysiology of the disease process. As elevated (IOP) appears most intimately associated with POAG, clinical medicine has focused on lowering IOP as the first step. Unfortunately, since even normalizing IOP doesn’t stop the ravages of GON, and as ocular normotensive patients’ vision still continues to deteriorate, it has become abundantly clear that direct protection of the RGCs and their axons is also necessary in addition to reducing the OHT. Nevertheless, since every 1 mm Hg IOP-lowering results in 10-13% reduction in progression of POAG, it is important to address elevated IOP in the context of glaucoma optic nerve changes immediately. Pharmacotherapy to treat and lower elevated IOP has constituted drugs to reduce the production of AQH (e.g., carbonic anhydrase inhibitors [CAIs: dorzolamide; brinzolamide]; beta-blockers [e.g., timolol; betaxolol]; α2-adrenergic agonists [e.g., apraclonidine and brimonidine]), and agents that stimulate AQH outflow through the trabecular meshwork (TM) [conventional outflow] (e.g., pilocarpine; brimonidine), and the mainstay first-line uveoscleral outflow stimulator drugs (e.g., FP-receptor agonists latanoprost; travoprost; tafluprost) (Sharif, 2017). Unfortunately, all these drugs have significant ocular and/or systemic side-effects (e.g., stinging, burning, ocular allergy, hyperemia, iris color changes, lethargy, pulmonary and cardiovascular insufficiency, and/or short duration of action) that lead to significant patient non-compliance in administering their eye-drop medicines (Weinreb et al., 2014; Jonas et al., 2017; Sharif, 2017). Whilst certain long-term drug delivery approaches and microsurgeries coupled with AQH drainage shunts may help address the compliance issues, novel drugs that offer longer duration of action, higher potency and efficacy, perhaps involving multiple mechanisms of action, are thus urgently needed. Certain IOP-lowering combination products (e.g., a CAI + a β-blocker + an α2-agonist; Hollo et al., 2014) have partially filled this gap temporarily, but since ocular hypotensive agents alone may be insufficient to combat GON, multi-pharmacophoric drugs with poly-pharmacological properties (including cyto-axon-protective agents) will ultimately be needed to stem the tide of GON damage and to preserve the optic nerve and vision.
Some of the latter features (dual pharmacophoric activities) appear to be present in two recently FDA-approved novel drugs, namely netarsudil 0.02% (Rhopressa®; Serle et al., 2018) and latanoprostene bunod 0.024% (Vyzulta®; Weinreb et al., 2018). Thus, netarsudil inhibits rho kinase and norepinephrine transporter--it relaxes the TM and Schlemm’s canal (SC) cells (thereby helping AQH to drain via the conventional pathway), and it inhibits Na+/K+-ATPase in the ciliary epithelial cells thereby inhibiting AQH production and lowering IOP. In a similar vein, latanoprostene bunod releases latanoprost free acid (LFA) and nitric oxide (NO)--the FP-receptors in ciliary muscle and TM are activated by LFA to cause local release of MMPs that digest extracellular matrix (ECM) to create/enlarge the UVS outflow pathway and promote AQH drainage from both the UVS and TM/SC pathways, while the NO activates soluble guanylate cyclase in TM/SC cells (Dismuke et al., 2009, 2010) that produces cGMP that relaxes TM/SC cells and enhances conventional outflow of AQH. Netarsudil also offers the possibility of adjunctive therapy to further enhance the IOP-lowering activity by combining it with FP-receptor prostaglandin agonist analogs (PGAs) such as latanoprost (Lewis et al., 2016). Indeed, such studies have been conducted in POAG/OHT patients and the results are encouraging for this novel formulation containing both netarsudil and latanoprost (Roclatan™) (Lewis et al., 2016).
Devices and Novel Approaches to Combat OHT and Glaucoma
Since some patients do not respond well to PGAs, in particular to latanoprost, and to other drugs and their IOPs remain higher than desired, such glaucoma patients may be good candidates to receive the AQH drainage devices implanted in the anterior chambers of their eyes. These drainage implants extrude excess AQH down the conventional outflow pathway, into the suprachoroidal space or subconjunctival or into sub-tenon space. Indeed, such novel AQH drainage devices (e.g., iStent; XEN Gel Stent; CyPass Microshunt; Hydrus Microshunt; InnFocus Microshunt) (Batlle et al. 2016; Pillunat et al., 2018) represent some of the most innovative advances in treating OHT/POAG. These devices are already making a big difference in the clinical setting in the management of these disorders by providing long-term ocular hypotensive activity and control of IOP. Some of these novel tools are able to bring the IOP down to 10–13 mmHg and keep it lowered over a 3-year period, which is remarkable efficacy (Batlle et al. 2016; Pillunat et al., 2018). Similarly, the ability of corneal endothelial cells transfected with vectors that continuously release small amounts of endogenous MMPs to digest away ECM accumulated in the TM/SC cells to enhance outflow of AQH appears very exciting (O’Callagan et al., 2017). At any rate, based on the currently reported efficacy of the drainage devices in POAG/OHT patients, lowering IOP down to 10–14 mmHg and maintaining this for multiple years, there may come a time when anti-glaucoma eyedrop medications may be replaced by AQH microshunts and gene therapy in the Western world. Revolutionary as this may appear, patients’ continued poor compliance with topically delivered IOP-decreasing drugs may necessitate these advances.
Another paradigm shift on the horizon for lowering and controlling IOP involves ability of sustained drug delivery platforms (Barar et al., 2016; Hartman and Kompella, 2018). Included amongst the technologies to enhance patient adherence are: drug-coated contact lenses, PGA-containing silicone insert that encircles the eyeball (Brandt et al., 2017), intracameral injection of a biodegradable poly-lactic-co-glycolic acid-containing PGA implant, poly-ethylene-glycol(PEG)-containing- and PEG-hydrogel-containing-ocular hypotensive drug implant, drugs placed inside bioerodable microspheres (Bertram et al., 2009), dendrimer-(synthetic polymeric nanoparticles)-containing drug, punctal plugs containing IOP-lowering drug (Perera et al., 2016), and a polycaprolactone-device containing a novel non-PG EP2-receptor agonist (DE-117; Omidenepag Isopropyl) that can be intracamrally injected affording months of continued ocular hypotensive treatment due to the extended release characteristics of the delivery technologies (e.g., Barar et al. 2016; Kim et al. 2016). Lastly, transscleral (Nagai et al., 2018), juxtrascleral (Robin et al., 2009) and suprachoroidal (Hartman and Kompella, 2018) drug delivery via specifically designed and fabricated devices and injections could prove extremely useful for providing extended release of IOP-lowering and/or neuroprotective drugs.
Drugs, Electroceuticals, Cell and Gene Therapy as Soma-Axonal Rescue Strategies
It is evident that despite maximum lowering of IOP in POAG/OHT patients, many millions of these patients continue to experience progressive visual impairment and still go blind, as do patients whose long-term ambient IOPs are considered in the normal range and who are classified as “ocular normotensive” glaucoma patients. These facts and observations have led to the conclusion that direct protection of RGC cell bodies and associated axons are paramount, in addition to lowering and controlling IOP. This concept of neuroprotection or soma-axon-protection is now well recognized and accepted in neurodegenerative brain and spinal diseases, and now also in ocular diseases including in POAG. Despite discovery of numerous classes of neuroprotective agents using isolated primary brain neurons, surrogate neuronal cells (neuroblastoma cells), and RGCs subjected to metabolic, excitotoxic, inflammatory and oxidative insults, and confirmation of efficacy in animal models of various diseases (e.g., stroke, Parkinson’s/Alzheimer’s/Huntington’s disease, amyotrophic lateral sclerosis, chronic OHT/glaucoma; Ito and Di Polo, 2017; Jonas et al., 2017; Sharif et al., 2018), we still lack bona fide translation of such neuroprotection in human subjects suffering from these diseases.
Poor translation of laboratory-based efficacy studies of GON to the clinical setting occurs for numerous reasons. First and most importantly, in the animal models the RGC/optic nerve head (ONH)/RGC axon-damaging insult (e.g., high IOP or cytokine or neurotoxic agent injection) used to recapitulate the human disease processes is singular in nature and often an acute one, whereas in the human disease progression at multiple retinal and axonal dysfunctions occur simultaneously and in a chronic manner. Secondly, only a single presumed therapeutic agent is tested against a single ocular insult, and it is administered prior to or during the induced damage, whereas in the human situation the damage has been ongoing for a protracted period of time. Thirdly, in POAG patients with chronically elevated IOP, a significant number of retinal neurons have died, and other RGC bodies and axons may be beyond rescue. Other reasons of clinical failure of neuroprotective agents that show efficacy in vitro and in animals encompass issues of metabolic and or chemical instability of the drugs when administered over long periods of time, drug formulation/delivery/bioavailability, lack of attainment of therapeutic concentration of the drug at target tissues/cells, and unacceptable systemic, central and local ocular side-effects. Obviously, the multifactorial aspects and chronic nature of GON/POAG/OHT disease (Figure 1) also makes it difficult to mitigate the disease processes. Consequently, only a combinatorial amalgam of numerous different classes of drugs targeting different intervention points of the pathological cascade may be necessary to combat the symptoms associated with the optic and brain neuropathies.
Thus, one can envision scenarios where “soft” multiple conjugates or suitable and compatible mixtures of efficacious health-authority-approved drugs [e.g., anti-oxidants: edaravone and resveratrol; Ca2+-channel blockers: lamotrogine and nimodepine; rho kinase (ROCK) inhibitors: netarsudil and ripasudil; PGAs like latanoprost, and other IOP-lowering agents; and marketed drugs that have shown neuroprotective activity in animal models (e.g., α2-agonists: brimonidine; β-blockers: betaxolol; anti-epileptics: valproate, phenytoin; anti-inflammatory agents like ibudilast, aspirin and meloxicam)] could be synthesized, formulated and delivered intravitreally to slow down the death of RGCs and their axons. Perhaps the above approach coupled with gene-therapy (Hines-Beard et al. 2016) to deliver neurotrophins such as brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), nerve-growth factor (NGF) and/or erythropoietin to RGC bodies and axons may be successful. Another approach that may help prevent RCG loss is using immortalized human neural stem cells that can differentiate into neurons and oligodendrocytes to form a “physical platform/substratum/bridge” in vivo to repair and support the optic nerve components in GON/POAG patients (Kador et al., 2013; Kashani et al., 2018; O’Rourke et al., 2018) may be necessary. Physical grafting of human optic nerve components such a delivery of Schwann cells that form myelin (Guo et al., 2014; Smedowski et al., 2016; Auricchio et al., 2017) to the weakened optic nerve could also become a reality in the near future. Likewise, stem cells to replace dying or dead RGCs is another possibility (Venugopalan et al., 2016; Daliri et al., 2017). However, the latter will be ineffective if we cannot first overcome the growth-inhibiting effects of the local chemical environment around the damaged axons and RGC somas. Thus, it is imperative to find ways to induce the phagocytosis, degradation, active uptake and elimination of some or all of these culprits that inhibit axonal/dendritic growth including excess zinc, myelin-associated glycoprotein, Nogo, oligodendrocyte-myelin glycoprotein, chondroitin sulfate proteoglycans and the physical barrier represented by glial scar tissue.
Electrotherapy that can release endogenous neurotrophins, enhance ocular blood-flow and resuscitate energy-depleted mitochondria of the RGCs (Fujikado et al., 2006; Kurimoto et al., 2010; Morimoto et al., 2011; Ota et al., 2018), coupled with delivery of exogenous re-energizing agents like vitamin B3 (Williams et al., 2017) could be used to rescue failing retinal cellular and axonal components (Ito and Di Polo, 2017). Additional contributors to this cause could include drugs that inhibit formation and deposition of amyloid/tau proteins and complement, and agents that can abrogate the actions of damaging cyto-/chemo-kines, and anti-apoptotic drugs. Likewise, inhibiting p38-MAP kinase, ROCK, dual leucine zipper kinase, Janus kinase, mammalian target of rapamycin (mTOR), phosphoinositide-3 kinase, and suppression of various transcription factors that can induce apoptosis, or generate cytokines or elicit fibrosis [by up-regulating thromospondin-1, periostin and collagen-1A1] (e.g., Bax, PTEN, Klf-4, SOC3, NFκB, NFAT) (He et al., 2018; Sharif, 2018), have the potential to enhance regrowth of damaged RGC axons (Morgan-Warren et al., 2016). Similarly, activation of certain transcription factors and proteins (e.g., Bcl; oncomodulin, Mmnat1, tropomyosin regulated kinase-B) represent beneficial survival elements that could be exploited to elicit regeneration of RGCs and their axons (He et al., 2018; Sharif, 2018). Since Ca2+-overloading of cells due to excitotoxicity underlies cellular demise (Irnaten et al., 2018), mitochondrial targeting of specific Ca2+-channel blockers appears worthy of pursuit to combat RGC/LC cell death (Cheung et al., 2017). Despite all this effort, however, all of the above may still be unsatisfactory and prove non-efficacious because what is really needed are disease-modifying therapeutics and most likely a combination product approach. However, irrespective of the hurdles, all this multifaceted research is helping build the neuroprotectant armamentarium necessary to deal with different aspects of POAG/GON pathology (He et al., 2018; Sharif, 2018).
Conclusions
In conclusion, there are still many challenges that need to be overcome in the arena of optic nerve protection in order to preserve vision. The value of sight requires an enhanced public awareness of the insidious nature of glaucoma so that regular eye exams become routine. Such habits will certainly increase the probability of timely diagnosis of OHT/POAG. The availability of Food and Drug Administration (FDA)-approved Icare HOME tonometer (Icare, Raleigh, NC, USA) and future utility of both temporary contact-lens-based (Sensimed’s “TriggerFish”) IOP detector and implantable IOP-sensor (Implandata’s “Eyemate”) should allow more frequent and remote 24-hour monitoring of IOP possible for patients and their ophthalmologists. With such advances it is hoped that OHT/POAG patients can quickly receive appropriate topical ocular hypotensive drug (or a combination product), or AQH drainage device, and/or low responders with fast progressing visual fields can receive suitable gene therapy, and/or receive suitable neuroprotective therapeutic agent(s) without delay. A combination of the above-mentioned treatment paradigms may be necessary to achieve a successful outcome for the patient. In particular, slowing down the progression of the disease processes associated with POAG/OHT/GON and thus limiting further loss of RGCs and their axons should be the primary goal. As is evident from the above discourse, the neurobiology of the disease processes has advanced greatly in recent years such that full restoration of partial blindness or visual impairment due to OHT/POAG/GON may be feasible. The possible linkage of the disease processes in POAG to genetic targets (Aung and Khor, 2016; Danford et al., 2017), metabolomics (Williamson et al., 2018) and other diagnostic/prognostic biomarkers (Cordeiro et al., 2017) can also enhance our understanding of disease pathology and lead to discovery of better medicines to revitalize sick and dying RGCs and optic nerve components (Williams et al., 2017; Ota et al., 2018). Of course, this will eventually depend on how well treatment modalities unearthed by laboratory science can be applied to the human condition, including deployment of gene therapy (Hines-Beard et al., 2016) and perhaps stem cell therapies (Venugopalan et al., 2016; Daliri et al., 2017). This may be a bridge too far at present, but we should not surrender to the threat of burgeoning GONs. The pathways to preserving vision should not be allowed to be obscured or abandoned, and the hope and promise of achieving such audacious goals need to be kept alive as we strive to find solutions for those patients afflicted by these blinding diseases.
Additional file (4.2KB, pdf) : Open peer review report 1.
Footnotes
Conflicts of interest: None declared.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Jiaxing Wang, Emory University, USA.
References
- 1.Aung T, Khor CC. Glaucoma genetics: recent advances and future directions. Asia Pac J Ophthalmol (Phila) 2016;5:256–259. doi: 10.1097/APO.0000000000000229. [DOI] [PubMed] [Google Scholar]
- 2.Auricchio F, Scavone C, Cimmaruta D, Di Mauro G, Capuano A, Sportiello L, Rafaniello C. Drugs approved for the treatment of multiple sclerosis: review of their safety profile. Expert Opin. Drug Saf. 2017;16:1359–1371. doi: 10.1080/14740338.2017.1388371. [DOI] [PubMed] [Google Scholar]
- 3.Barar J, Aghanejad A, Fathi M, Omidi Y. Advanced drug delivery and targeting technologies for the ocular diseases. BioImpacts. 2016;6:49–67. doi: 10.15171/bi.2016.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle JF, Fantes F, Riss I, Pinchuk L, Alburquerque R, Kato YP, Arrieta E, Peralta AC, Palmberg P, Parrish RK, 2nd, Weber BA, Parel JM. Three-year follow-up of a novel aqueous humor microshunt. J Glaucoma. 2016;25:e58–e65. doi: 10.1097/IJG.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 5.Bertram J, Saluja SS, McKain JA, Lavik EB. Sustained delivery of timolol maleate from poly (lactic-co-glycolic Novel drug delivery systems acid)/poly (lactic acid) microspheres for over 3 months. J Microencapsul. 2009;26:18–26. doi: 10.1080/02652040802095250. [DOI] [PubMed] [Google Scholar]
- 6.Brandt JD, DuBiner HB, Benza R, Sall KN, Walker GA, Semba CP Collaborators. Long-term safety and efficacy of a sustained-release bimatoprost ocular ring. Ophthalmol. 2017;124:1565–1566. doi: 10.1016/j.ophtha.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 7.Cheung LTY, Manthey AL, Lai JSM, Chiu K. Targeted delivery of mitochondrial calcium channel regulators: the future of glaucoma treatment? Front Neurosci. 2017;11:648. doi: 10.3389/fnins.2017.00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, Zhu J, Wu F, Ouyang H, Ge J, Weinreb RN, Zhang K, Zhuo Y. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc Natl Acad Sci U S A. 2014;111:1118–1116. doi: 10.1073/pnas.1402819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordeiro MF, Normando EM, Cardoso MJ, Miodragovic S, Jeylani S, Davis BM, Guo L, Ourselin S, A’Hern R, Bloom PA. Real-time imaging of single neuronal cell apoptosis in patients with glaucoma. Brain. 2017;140:1757–1767. doi: 10.1093/brain/awx088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabb DP. A view on glaucoma--are we seeing it clearly? Eye (Lond) 2016;30:304–313. doi: 10.1038/eye.2015.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danford ID, Verkuil LD, Choi DJ, Collins DW, Gudiseva HV, Uyhazi KE, Lau MK, Kanu LN, Grant GR, Chavali VRM, O’Brien JM. Characterizing the “POAGome”: A bioinformatics-driven approach to primary open-angle glaucoma. Progress Ret Eye Res. 2017;58:89–114. doi: 10.1016/j.preteyeres.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalari K, Ljubimov AV, Hekmatimoghaddan S. Glaucoma, stem cells, and gene therapy: where are we now? Int J Stem Cells. 2017;10:119–128. doi: 10.15283/ijsc17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dismuke WM, Sharif NA, Ellis DZ. Human trabecular meshwork cell volume decrease by NO-independent soluble guanylate cyclase activators YC-1 and BAY-58-2667 involves the BKCa ion channel. Invest Ophthalmol Vis Sci. 2009;50:3353–3359. doi: 10.1167/iovs.08-3127. [DOI] [PubMed] [Google Scholar]
- 14.Dismuke WM, Sharif NA, Ellis DZ. Endogenous regulation of human Schlemm’s canal cell volume by nitric oxide signaling. Invest Ophthalmol Vis Sci. 2010;51:5817–5824. doi: 10.1167/iovs.09-5072. [DOI] [PubMed] [Google Scholar]
- 15.Fujikado T, Morimoto T, Matsushita K, Shimojo H, Okawa Y, Tano Y. Effect of transcorneal electrical stimulation in patients with non-arteritic ischemic optic neuropathy or traumatic optic neuropathy. Jpn J Ophthalmol. 2006;50:266–273. doi: 10.1007/s10384-005-0304-y. [DOI] [PubMed] [Google Scholar]
- 16.Guo L, Davis B, Nizari S, Normando EM, Shi H, Galvao J, Turner L, Shi J, Clements M, Parrinello S, Cordeiro MF. Direct optic nerve sheath (DONS) application of Schwann cells prolongs retinal ganglion cell survival in vivo. Cell Death Dis. 2014;5:e1460. doi: 10.1038/cddis.2014.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartman RR, Kompella UB. Intravitreal, subretinal, and suprachoroidal injections: evolution of microneedles for drug delivery. J Ocular Pharmacol Ther. 2018;34:141–153. doi: 10.1089/jop.2017.0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S, Stankowska DL, Ellis DZ, Krishnamoorthy RR, Yorio T. Targets of neuroprotection in glaucoma. J Ocular Pharmacol Ther. 2018;34:85–106. doi: 10.1089/jop.2017.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hines-Beard J, Bond WS, Backstrom JR, Rex TS. Virus-mediated EpoR76E gene therapy preserves vision in a glaucoma model by modulating neuroinflammation and decreasing oxidative stress. J Neuroinflam. 2016;13:39. doi: 10.1186/s12974-016-0499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollander H, Makarov F, Stefani FH, Stone J. Evidence of constriction of optic axons at the lamina cribrosa in the normotensive eye in humans and other mammals. Ophthalmic Res. 1995;27:296–309. doi: 10.1159/000267739. [DOI] [PubMed] [Google Scholar]
- 21.Hollo G, Topouzis F, Fetchner RD. Fixed-combination intraocular pressure-lowering therapy for glaucoma and ocular hypertension: advantages in clinical practice. Expert Opin Pharmacother. 2014;15:1737–1747. doi: 10.1517/14656566.2014.936850. [DOI] [PubMed] [Google Scholar]
- 22.Irnaten M, Zhdanov A, Brennan D, Crotty T, Clark A, Papkovsky D, O’Brien C. Activation of the NFAT-calcium signaling pathway in human lamina cribosa cells in glaucoma. Invest Ophthalmol Vis Sci. 2018;59:831–842. doi: 10.1167/iovs.17-22531. [DOI] [PubMed] [Google Scholar]
- 23.Ito YA, Di Polo A. Mitochondrial dynamics, transport, and quality control: a bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochond. 2017;36:186–192. doi: 10.1016/j.mito.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 24.Jonas JB, Aung T, Bourne RR, Bron AM, Ritch R, Panda-Jonas S. Glaucoma. Lancet. 2017;390:2183–2193. doi: 10.1016/S0140-6736(17)31469-1. [DOI] [PubMed] [Google Scholar]
- 25.Jonas JB, Ritch R, Panda-Jonas S. Cerebrospinal fluid pressure in the pathogenesis of glaucoma. Prog Brain Res. 2015;221:33–47. doi: 10.1016/bs.pbr.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Kador KE, Montero RB, Venugopalan P, Hertz J, Zindell AN, Valenzuela DA, Uddin MS, Lavik EB, Muller KJ, Andreopoulos FM, Goldberg JL. Tissue engineering the retinal ganglion cell nerve fiber layer. Biomaterials. 2013;34:4242–4250. doi: 10.1016/j.biomaterials.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kashani AH, Lebkowski JS, Rahhal FM, Avery RL, Salehi-Had H, Dang W, Lin CM, Mitra D, Zhu D, Thomas BB, Hikita ST, Pennington BO, Johnson LV, Clegg DO, Hinton DR, Humayun MS. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci Transl Med. 2018;10:eaao4097. doi: 10.1126/scitranslmed.aao4097. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Kudisch M, Mudumba S, Asada H, Aya-Shibuya E, Bhisitkul RB, Desai TA. Biocompatibility and pharmacokinetic analysis of an intracameral polycaprolactone drug delivery implant for glaucoma. Invest Ophthalmol Vis Sci. 2016;57:4341–4346. doi: 10.1167/iovs.16-19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurimoto T, Oono S, Oku H, Tagami Y, Kashimoto R, Takata M, Okamoto N, Ikeda T, Mimura O. Transcorneal electrical stimulation increases chorioretinal blood flow in normal human subjects. Clin Ophthalmol. 2010;4:1441–1446. doi: 10.2147/OPTH.S14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis RA, Levy B, Ramirez N, Kopczynski CC, Usner DW, Novack GD PG324-CS201 Study Group. Fixed-dose combination of AR-13324 and latanoprost: a double-masked, 28-day, randomised controlled study in patients with open-angle glaucoma or ocular hypertension. Br J Ophthalmol. 2016;100:339–344. doi: 10.1136/bjophthalmol-2015-306778. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Li D, Ying X, Khaw PT, Raisman G. An energy theory of glaucoma. Glia. 2015;63:1537–1552. doi: 10.1002/glia.22825. [DOI] [PubMed] [Google Scholar]
- 32.McElnea EM, Quill B, Docherty NG, Irnaten M, Siah WF, Clark AF, O’Brien CJ, Wallace DM. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol Vis. 2011;17:1182–1189. [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan-Warren PJ, O’Neill J, de Cogan F, Spivak I, Ashush H, Kalinski H, Ahmed Z, Berry M, Feinstein E, Scott RA, Logan A. siRNA-mediated knockdown of the mtor inhibitor rtp801 promotes retinal ganglion cell survival and axon elongation by direct and indirect mechanisms. Invest Ophthalmol Vis Sci. 2016;57:429–443. doi: 10.1167/iovs.15-17511. [DOI] [PubMed] [Google Scholar]
- 34.Morimoto T, Kamei M, Nishida K, Sakaguchi H, Kanda H, Ikuno Y, Kishima H, Maruo T, Konoma K, Ozawa M, Nishida K, Fujikado T. Chronic implantation of newly developed suprachoroidal-transretinal stimulation prosthesis in dogs. Invest Ophthalmol Vis Sci. 2011;52:6785–6792. doi: 10.1167/iovs.10-6971. [DOI] [PubMed] [Google Scholar]
- 35.Nagai N, Yamada S, Kawasaki J, Koyanagi E, Saijo S, Kaji H, Nishizawa M, Nakazawa T, Abe T. Pharmacokinetic and safety evaluation of a transscleral sustained unoprostone release device in monkey eyes. Invest Ophthalmol Vis Sci. 2018;59:644–652. doi: 10.1167/iovs.17-22429. [DOI] [PubMed] [Google Scholar]
- 36.Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–179. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- 37.O’Callaghan J, Crosbie DE, Cassidy PS, Sherwood JM, Flügel-Koch C, Lütjen-Drecoll E, Humphries MM, Reina-Torres E, Wallace D, Kiang AS, Campbell M, Stamer WD, Overby DR, O’Brien C, Tam LCS, Humphries P. Therapeutic potential of AAV-mediated MMP-3 secretion from corneal endothelium in treating glaucoma. Hum Mol Genet. 2017;26:1230–1246. doi: 10.1093/hmg/ddx028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Rourke C, Day AGE, Murray-Dunning C, Thanabalasundaram L, Cowan J, Stevanato L, Grace N, Cameron G, Drake RAL, Sinden J, Phillips JB. An allogeneic “Off the shelf” therapeutic strategy for peripheral nerve tissue engineering using clinical grade human neural stem cells. Sci Rep. 2018;8:2951. doi: 10.1038/s41598-018-20927-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osborne NN, Álvarez CN, del Olmo Aguado S. Targeting mitochondrial dysfunction as in aging and glaucoma. Drug Discov Today. 2014;19:1613–1622. doi: 10.1016/j.drudis.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Ota Y, Ozeki N, Yuki K, Shiba D, Kimura I, Tsunoda K, Shinoda K, Ohde H, Tsubota K. The efficacy of transcorneal electrical stimulation for the treatment of primary open-angle glaucoma: a pilot study. 2018 doi: 10.2302/kjm.2017-0015-OA. doi: 10.2302/kjm.2017-0015-OA. [DOI] [PubMed] [Google Scholar]
- 41.Pasquale LR. Vascular and autonomic dysfunction in primary open-angle glaucoma. Curr Opin Ophthalmol. 2016;27:94–101. doi: 10.1097/ICU.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perera SA, Ting DS, Nongpiur ME, Chew PT, Aquino MC, Sng CC, Ho SW, Aung T. Feasibility study of sustained-release travoprost punctum plug for intraocular pressure reduction in an Asian population. Clin Ophthalmol. 2016;10:757–764. doi: 10.2147/OPTH.S102181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillunat LE, Erb C, Junemann AGM, Kimmich F. Micro-invasive glaucoma surgery (MIGS): a review of surgical procedures using stents. Clin Ophthalmol. 2018;11:1583–1600. doi: 10.2147/OPTH.S135316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41:3460–3466. [PubMed] [Google Scholar]
- 45.Robin AL, Clark AF, Covert DW, Krueger S, Bergamini MV, Landry TA, Dickerson JE, Jr, Scheib SA, Realini T, Defaller JM, Cagle GD. Anterior juxtascleral delivery of anecortave acetate in eyes with primary open-angle glaucoma: a pilot investigation. Am J Ophthalmol. 2009;147:45–50.e2. doi: 10.1016/j.ajo.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 46.Serle JB, Katz LJ, McLaurin E, Heah T, Ramirez-Davis N, Usner DW, Novack GD Kopczynski CC; ROCKET-1 and ROCKET-2 Study Groups. Two phase-3 clinical trials comparing the safety and efficacy of netarsudil to timolol in patients with elevated intraocular pressure: rho kinase elevated IOP treatment trial 1 and 2 (ROCKET-1 and ROCKET-2) Am J Ophthalmol. 2018;186:116–127. doi: 10.1016/j.ajo.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Sharif NA. Ocular hypertension and glaucoma: a review and current perspectives. Int J Ophthalmol Vis Sci. 2017;2:22–36. [Google Scholar]
- 48.Sharif NA. idrugs and idevices discovery and development- preclinical assays, techniques and animal models studies for ocular hypotensives and neuroprotectants. J Ocular Pharmacol Ther. 2018;34:7–39. doi: 10.1089/jop.2017.0125. [DOI] [PubMed] [Google Scholar]
- 49.Smedowski A, Liu X, Pietrucha-Dutczak M, Matuszek I, Varjosalo M, Lewin-Kowalik J. Predegenerated Schwann cells--a novel prospect for cell therapy for glaucoma: neuroprotection, neuroregeneration and neuroplasticity. Sci Rep. 2016;6:23187. doi: 10.1038/srep23187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tezel G, Yang X, Luo C, Kain AD, Powell DW, Kuehn MH, Kaplan HJ. Oxidative stress and the regulation of complement activation in human glaucoma. Invest Ophthalmol Vis Sci. 2010;51:5071–5082. doi: 10.1167/iovs.10-5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmol. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 52.Thomas D, Papadopoulo O, Doshi R, Kapin MA, Sharif NA. Retinal ATP and phosphorus metabolites: reduction by hypoxia and recovery with MK-801 and diltiazem. Med Sci Res. 2000;28:87–91. [Google Scholar]
- 53.Venugopalan P, Wang Y, Nguyen T, Huang A, Muller KJ, Goldberg JL. Transplanted neurons integrate into adult retinas and respond to light. Nat Commun. 2016;7:10472. doi: 10.1038/ncomms10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–1911. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinreb RN, Liebmann JM, Martin KR, Kaufman PL, Vittitow JL. Latanoprostene bunod 0.024% in subjects with open-angle glaucoma or ocular hypertension: pooled phase 3 study findings. J Glaucoma. 2018;27:7–15. doi: 10.1097/IJG.0000000000000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756–760. doi: 10.1126/science.aal0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williamson BK, Hawkey NM, Blake DA, Frenkel JW, McDaniel KP, Davis JK, Satija C, Beazer A, Dhungana S, Carlson J, McRitchie S, Ayyala RS. The effects of glaucoma drainage devices on oxygen tension, glycolytic metabolites, and metabolomics profile of aqueous humor in the rabbit. Transl Vis Sci Technol. 2018;7:14. doi: 10.1167/tvst.7.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu G, Weinreb RN, Leung CK. Optic nerve head deformation in glaucoma: the temporal relationship between optic nerve head surface depression and retinal nerve fiber layer thinning. Ophthalmol. 2014;121:2362–2370. doi: 10.1016/j.ophtha.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 59.Yucel YH, Zhang Q, Gupta N, Kaufman PL, Weinreb RN. Loss of neurons in magnocellular and parvocellular layers of the lateral geniculate nucleus in glaucoma. Arch. Ophthalmol. 2000;118:378–384. doi: 10.1001/archopht.118.3.378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.