

Abstract
Aneurysmal subarachnoid hemorrhage remains devastating, and the most important determinant of poor outcome is early brain injury (EBI). In clinical settings, as a surrogate marker of EBI, loss of consciousness at ictus, poor initial clinical grades, and some radiographic findings are used, but these markers are somewhat subjective. Thus, it is imperative to find biomarkers of EBI that have beneficial prognostic and therapeutic implications. In our opinion, an ideal biomarker is a molecule that is implicated in the pathogenesis of both EBI and subsequently developing delayed cerebral ischemia (DCI), being a therapeutic target, and can be measured easily in the peripheral blood in an acute stage. A good candidate of such a biomarker is a matricellular protein, which is a secreted, inducible and multifunctional extracellular matrix protein. There are many kinds of matricellular proteins reported, but only tenascin-C, osteopontin, galectin-3 and periostin are reported relevant to EBI and DCI. Reliable biomarkers of EBI may stratify aneurysmal subarachnoid hemorrhage patients into categories of risk to develop DCI, and allow objective monitoring of the response to treatment for EBI and earlier diagnosis of DCI. This review emphasizes that further investigation of matricellular proteins as an avenue for biomarker discovery is warranted.
Keywords: biomarker, early brain injury, galectin-3, matricellular protein, osteopontin, periostin, subarachnoid hemorrhage, tenascin-C
Impact of Early Brain Injury (EBI) on Poor Outcome after Subarachnoid Hemorrhage (SAH)
Aneurysmal SAH remains a devastating cerebrovascular disease (Suzuki et al., 2017), and the most important determinant of poor outcome is considered to be the deleterious effect of acute SAH on the brain, being called EBI (Okada and Suzuki, 2017). EBI is defined as acute pathophysiological events that occur in brain before onset of cerebral vasospasm within the first 72 hours of SAH, which are mainly induced by elevated intracranial pressure and subsequent transient global cerebral ischemia due to aneurysmal rupture, and subarachnoid blood degradation products (Nishikawa and Suzuki, 2017). Patients who survive EBI are still at risk for delayed cerebral ischemia (DCI), another important and preventable determinant of poor outcome, by cerebral vasospasm or other causes that occurs at days 4 to 14 or later post-SAH (Suzuki and Kawakita, 2016). EBI may also cause DCI, but the mechanisms and mediators linking EBI with DCI remain unknown. If it is true that EBI has a central role in the development of DCI and poor outcome, it is imperative to look for clinical features and biomarkers of EBI that could have beneficial prognostic and therapeutic implications, including more objective assessment and monitoring of response to treatment. The authors consider that an ideal biomarker is a molecule that is implicated in the pathogenesis of both EBI and DCI, being a therapeutic target, and can be measured easily in the peripheral blood in an acute stage. In clinical settings, as a surrogate marker of EBI, loss of consciousness at ictus, poor initial clinical grades, and radiographic markers indicating increased amount of SAH or global cerebral edema can be used (Ahn et al., 2017). These markers are also predictors of global cerebral ischemia (microcirculatory changes), DCI regardless of angiographic vasospasm, and outcomes (Ahn et al., 2017). However, these markers are somewhat subjective, and sedation sometimes hampers accurate evaluation of neurological status. The clinical and radiographic markers of EBI are associated with an increased systemic expression of cytokines and other inflammatory markers, but the evidence for cytokines and inflammatory mediators as biomarkers is often conflicting and inconsistent (Al-Mufti et al., 2017). This may be because various pathophysiological reactions other than EBI affect inflammatory reactions and therefore the interactions cause low specificity of inflammatory mediators and hamper the interpretation of a single parameter’s dynamics, although neuroinflammation is a major aspect of EBI (Okada and Suzuki, 2017). There are also multiple interactions among cytokines, precluding a cytokine from being classified simply damaging or protective, and making it difficult to infer the causality.
Myelin basic protein, glial fibrillary astrocyte protein, the calcium-binding protein S100B, neuron-specific enolase (NSE), phosphorylated axonal neurofilament subunit H, tau protein and ubiquitin carboxyl-terminal hydrolase L1 are mainly released from neurons and/or glia in the central nervous system into the peripheral blood after aneurysmal SAH, and may be used as biomarkers reflecting the extent of EBI (Zheng et al., 2017). Especially, increased levels of serum NSE at an acute phase of SAH, which is a globular glycolytic isoenzyme present in neurons throughout the brain with a half-life of approximately 24 hours, were independently associated with poorer admission clinical grades and worse outcomes (Tawk et al., 2016). Integration of biomarkers that are indicative of the amount of neuronal injury would hold great promise for improving the accuracy of outcome prediction and would help clinicians in therapeutic decisions. However, elevation of these biomarkers may be merely a result of brain injuries, and therefore these biomarkers may not be a therapeutic target.
Matricellular Proteins (MCPs) in SAH
MCPs are an important component of the extracellular matrix (ECM) but different from classical ECM proteins as follows: 1) MCPs do not provide scaffolds for stable cell adhesion, but play regulatory roles in cell-cell and cell-matrix interactions, regulate cellular morphology, differentiation, migration, proliferation, and survival, and induce tissue remodeling; 2) MCPs are not constitutively expressed, but are secreted by diverse types of cells, and readily and transiently upregulated during specific developmental stages and in pathological conditions; 3) MCPs are not present as structural components of the ECM, but can be present as soluble proteins; 4) MCPs bind to ECM proteins, various receptors such as integrins, and biological active molecules including growth factors, chemokines, cytokines and proteases, thereby directly or indirectly modulating their functions or cellular responses to extracellular signals, either at the plasma membrane, intracellularly, in body fluids, or from the ECM, and/or acting as reservoirs of those molecules; and 5) most of MCPs do not produce apparent phenotypes after targeted gene deficiency in steady-state condition, but exhibit various phenotypes in response to diverse insults (Murphy-Ullrich and Sage, 2014). There are many kinds of MCPs reported including SPARC (secreted protein acidic and rich in cysteine), thrombospondins, tenascins, osteopontin, members of the CCN (cysteine-rich protein 61, connective tissue growth factor and nephroblastoma overexpressed gene) family, periostin, R-spondins, the short fibulins such as hemicentin, galectins, small leucine rich proteoglycans, autotaxin, pigment epithelium derived factor, and plasminogen activator inhibitor-1. MCPs are implicated in both paracrine and autocrine cellular regulation, and play key roles in various physiological and pathological conditions such as cell death, inflammation, fibrosis, vascular permeability and angiogenesis (Murphy-Ullrich and Sage, 2014). In addition, as many MCPs are present in body fluids, MCPs can be used to monitor disease progression such as tumor, cardiovascular, inflammatory and fibrotic diseases. In fact, recommendations for the measurement of galectin-3 as a biomarker of myocardial fibrosis are included in the American College of Cardiology Foundation and the American Heart Association guidelines to aid in risk stratification and prognosis prediction in patients with heart failure (Yancy et al., 2017). Understanding the role of MCPs has great potential in their use as prognostic biomarkers and therapeutic targets, but the information is limited in the central nervous system. In SAH, only tenascin-C (TNC), osteopontin (OPN), galectin-3 and periostin are reported relevant to EBI.
TNC in SAH
TNC is a huge molecule of approximately 220–400 kDa as an intact monomer, and forms a typical disulfide-linked hexamer (Midwood and Orend, 2009). Clinically, plasma levels of large-splice variants of TNC containing fibronectin type III C domain were increased after aneurysmal SAH, but not different between patients with and without subsequent angiographic vasospasm at days 1–3 (Suzuki et al., 2010). However, the plasma TNC levels peaked at days 4–6, when the levels were significantly greater in patients with subsequent angiographic vasospasm compared with those with no vasospasm: the peak occurred 2.4 days before the occurrence of transcranial Doppler-determined vasospasm and 3.6 days before the onset of DCI (Suzuki et al., 2010). On the other hand, TNC levels in the cerebrospinal fluid (CSF) peaked immediately after SAH, and worse admission clinical grade, more severe SAH on admission computed tomography, subsequent angiographic vasospasm, DCI, and worse outcomes were associated with higher CSF TNC levels, suggesting that more severe SAH or EBI may induce more TNC, which may cause angiographic vasospasm, and DCI by severe angiographic vasospasm and/or vasospasm-unrelated causes that are supposed to be EBI (Suzuki and Kawakita, 2016). The differences in the time course of TNC levels between plasma and CSF may reflect the time lag that TNC is released from CSF into plasma due to its huge molecular weight, or the differences of TNC-producing cells between plasma and CSF.
Experimental studies demonstrated that blockage of TNC induction prevented post-SAH mitogen-activated protein kinase (MAPK) activation in brain, and suppressed EBI in terms of neuronal apoptosis and blood-brain barrier (BBB) disruption (Suzuki and Kawakita, 2016). In addition, TNC-knockout SAH mice showed fewer inflammatory cell infiltration in the subarachnoid space (Fujimoto et al., 2018) as well as suppression of post-SAH activation of Toll-like receptor 4/nuclear factor-κB/interleukin-1β and interleukin-6 signaling cascades (Liu et al., 2018). TNC may be induced in astrocytes, neurons and brain capillary endothelial cells after experimental SAH (Shiba et al., 2014). Experimental studies also demonstrated that TNC is induced in cerebral arterial wall, causing cerebral vasospasm (Suzuki and Kawakita, 2016). TNC may have the positive feedback mechanisms on upregulation of TNC itself via Toll-like receptor 4 activation in an acute phase of SAH, leading to more activation of the signaling transduction and the development or aggravation of EBI and cerebral vasospasm, potentially leading to DCI (Suzuki and Kawakita, 2016; Okada and Suzuki, 2017). These findings suggested that TNC causes both EBI and cerebral vasospasm. However, plasma TNC may not reflect the severity of EBI but can be a useful biomarker to timely diagnose or predict the development of angiographic vasospasm and vasospasm-induced DCI.
OPN in SAH
OPN is expressed as a 40–80 kDa protein due to varied posttranslational modifications (Uede, 2011). In a clinical setting, plasma full-length OPN levels were significantly increased and peaked at days 4–6 post-SAH (Nakatsuka et al., 2018). Poor-outcome patients had significantly higher plasma OPN levels through all sampling points (days 1–12): delayed cerebral infarction showed higher OPN levels at days 1–3 and 10–12, while DCI had higher OPN levels at days 4–9 (Nakatsuka et al., 2018). Plasma OPN at days 1–3 might increase reflecting the severity of EBI that might cause delayed cerebral infarction as suggested by experimental studies (Nishikawa and Suzuki, 2018). Serum levels of C-reactive protein, a non-specific but sensitive marker of systemic inflammatory reactions, were not correlated with OPN levels at days 1–3 (Nakatsuka et al., 2018). Higher plasma OPN levels associated with DCI at days 4–9 may suggest the existence of non-EBI-related causes of DCI. On the other hand, experimental studies showed that OPN was induced in reactive astrocytes and brain capillary endothelial cells or cerebral arterial smooth muscle cells during the recovery phase of EBI or cerebral vasospasm, respectively, and worked protectively against BBB disruption, neuronal apoptosis, and cerebral vasospasm (Nakatsuka et al., 2018). Recombinant OPN treatment not only prevented EBI and vasospasm, but also stabilized vascular smooth muscle cell phenotype and protected cerebral autoregulation (Liu and Suzuki, 2018). The protective mechanisms may involve multiple pathways including inactivation of MAPK or nuclear factor-κB pathways, activation of focal adhesion kinase-phosphatidylinositol 3-kinase-Akt signaling pathway, integrin-linked kinase-Rac-1 signaling pathway via L-arginyl-glycyl-L-aspartate-dependent integrin receptor, and P-glycoprotein glycosylation signaling via CD44 (Liu and Suzuki, 2018). Taken together, as basic researches suggest that OPN is induced in damaged tissues in a delayed fashion and acts protectively against SAH-induced EBI, vasospasm and therefore DCI, plasma OPN may increase in a delayed fashion reflecting the severity of preceding tissue injuries and/or the extent of the resultant inflammation, and so on. Increased plasma OPN may have healing effects, but may be not enough to recover the injuries, if not OPN is exogenously administered. Thus, acute-phase plasma OPN could be used as a useful prognostic biomarker reflecting post-SAH EBI.
Galectin-3 in SAH
Galectin-3 is the sole member of chimera-type family of galectins with an approximate molecular weight of 30 kDa and can pentamerize (Dings et al., 2018). Clinically, higher plasma galectin-3 levels within 24 hours post-SAH reflected worse clinical grade and more subarachnoid and intraventricular hematoma volume on computed tomography at admission, and predicted 6-month worse outcomes (Nishikawa and Suzuki, 2018). Interestingly, higher plasma galectin-3 levels at days 1–3 post-SAH were associated with subsequent development of DCI and cerebral infarction without cerebral vasospasm in non-severe aneurysmal SAH patients (Nishikawa et al., 2018). Although the functional role of galectin-3 in SAH remain unclear, galectin-3 may be secreted by glial cells, act as a Toll-like receptor 4 ligand or bind to TNC, and activate microglia to induce pro-inflammatory cytokines, causing EBI (Nishikawa et al., 2018).
Periostin in SAH
Periostin is a 90 kDa N-glycoprotein that is expressed as multiple spliced isoforms (Nishikawa and Suzuki, 2017). Although blood concentrations of periostin are clinically reported to be elevated in various diseases, they have been never investigated in aneurysmal SAH patients. However, full-length periostin was upregulated in brain capillary endothelial cells and neurons after experimental SAH in mice and responsible for EBI in terms of BBB disruption through activating MAPK pathways and interacting with TNC (Nishikawa and Suzuki, 2017).
Future Directions of MCPs
The discovery of reliable biomarkers of EBI may guide neurosurgeons or intensivists in stratifying SAH patients into categories of risk to develop DCI by cerebral vasospasm or other causes, and facilitate improved monitoring of the response to treatment for EBI and earlier diagnosis of DCI. Both EBI and DCI are multifactorial and may involve several distinct but interconnected pathological processes. Thus, it is unlikely that a single protein biomarker will be clinically effective and reliable for assessing EBI and predicting different causes of DCI. Instead, a combination of temporal progression and relative levels of MCPs may be promising (Figure 1), because changes in plasma MCPs can occur rapidly after SAH and can be associated with a wide variety of pathophysiological processes. MCPs also can be easily detected using an enzyme-linked immunosorbent assay test. However, we do not know whether peripherally detected MCP levels accurately reflect the levels in the brain or CSF. We should measure plasma and CSF concentrations of MCPs at the same time, and examine if plasma levels may reflect CSF levels but have different temporal patterns, or plasma levels may represent systemic inflammation independent of brain pathophysiological processes, and so on. In addition, the role of MCPs in EBI and DCI as well as the interactions among MCPs are not completely unveiled, and therefore further meticulous translational studies are needed (Suzuki and Nakano, 2018). A better understanding of the role of MCPs will provide valuable insights into the disease pathogenesis that may ultimately lead to improved therapeutic outcomes for SAH patients with EBI and DCI. Lastly, it is necessary to perform the review and the meta-analysis for all the MCPs described in the manuscripts to further validate whether they are significantly associated with SAH pathophysiological processes. Further investigation of MCPs as an avenue for biomarker discovery is warranted.
Figure 1.
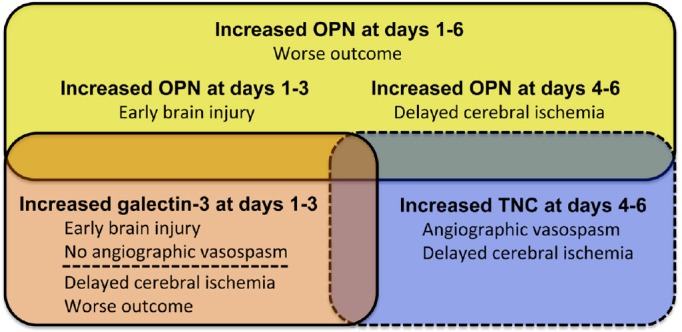
Relationships between increased plasma levels of tenascin-C (TNC), osteopontin (OPN) and galectin-3, and early brain injury, angiographic vasospasm, delayed cerebral ischemia and outcome after aneurysmal subarachnoid hemorrhage.
Footnotes
Conflicts of interest: None declared.
Financial support: This work was supported by a Grant-in-Aid for Scientific Research from Novartis Pharmaceuticals to HS.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Ramon Navarro, Cleveland Clinic Abu Dhabi, United Arab Emirates.
Funding: This work was supported by a Grant-in-Aid for Scientific Research from Novartis Pharmaceuticals to HS.
References
- 1.Ahn SH, Savarraj JP, Pervez M, Jones W, Park J, Jeon SB, Kwon SU, Chang TR, Lee K, Kim DH, Day AL, Choi HA. The subarachnoid hemorrhage early brain edema score predicts delayed cerebral ischemia and clinical outcomes. Neurosurgery. 2017 doi: 10.1093/neuros/nyx364. doi: 10.1093/neuros/nyx364. [DOI] [PubMed] [Google Scholar]
- 2.Al-Mufti F, Amuluru K, Smith B, Damodara N, El-Ghanem M, Singh IP, Dangayach N, Gandhi CD. Emerging markers of early brain injury and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017;107:148–159. doi: 10.1016/j.wneu.2017.07.114. [DOI] [PubMed] [Google Scholar]
- 3.Dings RPM, Miller MC, Griffin RJ, Mayo KH. Galectins as molecular targets for therapeutic intervention. Int J Mol Sci. 2018;19:E905. doi: 10.3390/ijms19030905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H. Effects of tenascin-C knockout on cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol. 2018;55:1951–1958. doi: 10.1007/s12035-017-0466-x. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Fujimoto M, Nakano F, Nishikawa H, Okada T, Kawakita F, Imanaka-Yoshida K, Yoshida T, Suzuki H. Deficiency of tenascin-C alleviates neuronal apoptosis and neuroinflammation after experimental subarachnoid hemorrhage in mice? Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1006-z. doi: 10.1007/s12035-018-1006-z. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Suzuki H. The role of matricellular proteins in experimental subarachnoid hemorrhage-induced early brain injury. In: Springer series in translational stroke research: cellular and molecular approaches to regeneration and repair. In: Lapchak PA, Zhang JH, editors. Cham: Springer Nature; 2018. pp. 397–407. [Google Scholar]
- 7.Midwood KS, Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal. 2009;3:287–310. doi: 10.1007/s12079-009-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakatsuka Y, Shiba M, Nishikawa H, Terashima M, Kawakita F, Fujimoto M, Suzuki H pSEED group. Acute-phase plasma osteopontin as an independent predictor for poor outcome after aneurysmal subarachnoid hemorrhage. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-0893-3. doi: 10.1007/s12035-018-0893-3. [DOI] [PubMed] [Google Scholar]
- 10.Nishikawa H, Nakatsuka Y, Shiba M, Kawakita F, Fujimoto M, Suzuki H pSEED Group. Increased plasma galectin-3 preceding the development of delayed cerebral infarction and eventual poor outcome in non-severe aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2018;9:110–119. doi: 10.1007/s12975-017-0564-0. [DOI] [PubMed] [Google Scholar]
- 11.Nishikawa H, Suzuki H. Implications of periostin in the development of subarachnoid hemorrhage-induced brain injuries. Neural Regen Res. 2017;12:1982–1984. doi: 10.4103/1673-5374.221150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishikawa H, Suzuki H. Possible role of inflammation and galectin-3 in brain injury after subarachnoid hemorrhage. Brain Sci. 2018;8:E30. doi: 10.3390/brainsci8020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada T, Suzuki H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen Res. 2017;12:193–196. doi: 10.4103/1673-5374.200795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiba M, Fujimoto M, Imanaka-Yoshida K, Yoshida T, Taki W, Suzuki H. Tenascin-C causes neuronal apoptosis after subarachnoid hemorrhage in rats. Transl Stroke Res. 2014;5:238–247. doi: 10.1007/s12975-014-0333-2. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Kanamaru K, Suzuki Y, Aimi Y, Matsubara N, Araki T, Takayasu M, Kinoshita N, Imanaka-Yoshida K, Yoshida T, Taki W. Tenascin-C is induced in cerebral vasospasm after subarachnoid hemorrhage in rats and humans: a pilot study. Neurol Res. 2010;32:179–184. doi: 10.1179/174313208X355495. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki H, Kawakita F. Tenascin-C in aneurysmal subarachnoid hemorrhage: deleterious or protective? Neural Regen Res. 2016;11:230–231. doi: 10.4103/1673-5374.177721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki H, Nakano F. To improve translational research in subarachnoid hemorrhage. Transl Stroke Res. 2018;9:1–3. doi: 10.1007/s12975-017-0546-2. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki H, Shiba M, Nakatsuka Y, Nakano F, Nishikawa H. Higher cerebrospinal fluid pH may contribute to the development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2017;8:165–173. doi: 10.1007/s12975-016-0500-8. [DOI] [PubMed] [Google Scholar]
- 19.Tawk RG, Grewal SS, Heckman MG, Rawal B, Miller DA, Edmonston D, Ferguson JL, Navarro R, Ng L, Brown BL, Meschia JF, Freeman WD. The relationship between serum neuron-specific enolase levels and severity of bleeding and functional outcomes in patients with nontraumatic subarachnoid hemorrhage. Neurosurgery. 2016;78:487–491. doi: 10.1227/NEU.0000000000001140. [DOI] [PubMed] [Google Scholar]
- 20.Uede T. Osteopontin, intrinsic tissue regulator of intractable inflammatory diseases. Pathol Int. 2011;61:265–280. doi: 10.1111/j.1440-1827.2011.02649.x. [DOI] [PubMed] [Google Scholar]
- 21.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Failure Society of America. Circulation. 2017;136:e137–e161. doi: 10.1161/CIR.0000000000000509. [DOI] [PubMed] [Google Scholar]
- 22.Zheng YK, Dong XQ, Du Q, Wang H, Yang DB, Zhu Q, Che ZH, Shen YF, Jiang L, Hu W, Wang KY, Yu WH. Comparison of plasma copeptin and multiple biomarkers for assessing prognosis of patients with aneurysmal subarachnoid hemorrhage. Clin Chim Acta. 2017;475:64–69. doi: 10.1016/j.cca.2017.10.009. [DOI] [PubMed] [Google Scholar]