

Abstract
The cerebral formation of Amyloid β (Aβ) is a critical pathological feature of Alzheimer’s disease (AD). An accumulation of this peptide as senile plaques (SP) was already reported by Alois Alzheimer, the discoverer of the disease. Yet the exact contribution of Aβ to AD development remains elusive. Moreover, while extensive cerebral Aβ formation leads to fibril formation in many species, AD-like symptoms apparently depend on the highly conserved N-terminal residues R5, Y10 and H13. The amino acids were also shown to lead to the formation of Aβ-heme complexes, which exhibit peroxidase activity in the presence of H2O2. Taking together these observations we propose that the formation and enzymatic activity of the named complexes may represent an essential aspect of AD pathology. Furthermore, Aβ is also known to lead to cerebral micro-vessel destruction (CAA) as well as to hemolytic events. Thus we suggest that the Aβ-derived cerebral accumulation of blood-derived free heme represents a likely precondition for the subsequent formation of Aβ-heme complexes.
Keywords: Alzheimer's disease, amyloid β sequence, amyloid β-heme complexes, peroxidase activity, dityrosine formation, cerebral amyloid angiopathy, hemolysis
Unsolved Pathology of Alzheimer’s Disease (AD)
AD is the most common neurodegenerative disease in Western countries (Armstrong, 2013). Moreover, increasing life expectancies and aging societies make AD a growing global medical and socio-economic challenge. The ultimately fatal disease is characterized by progressive neuronal loss which leads to cognitive impairments and other signs of dementia (Armstrong, 2013). Accordingly, brain shrinkage represents a macroscopic post-mortem sign of AD and was already noticed by Alois Alzheimer at the beginning of the 20th century (Dubois et al., 2016). Yet despite over 100 years of AD research, the underlying mechanisms of the disease are still unknown, including especially the initial pathological events which lead to the development of sporadic AD. The latter is by far the most common form of the disease and develops in elderly people without obvious genetic or environmental risk factors (Dubois et al., 2016). In this review, we suggest cerebral amyloid angiopathy (CAA), amyloid β (Aβ)-derived hemolytic events and the subsequent formation of peroxidase-active Aβ-heme complexes in the brain tissue as critical initial events at AD pathology.
Role of Human-Specific Aβ
Disease-defining biochemical key features of AD are the extracellular fibrillation of Aβ to so-called senile plaques (SP) as well as the intracellular accumulation of hyper-phosphorylated neurofibrillary Tau tangles (NFTs) (Dubois et al., 2016). Yet while e.g., the formation of SP at later disease stages represents a well-known post-mortem AD marker, the exact contribution of Aβ and Tau peptides to the onset and the course of AD pathology remains a matter of debate. Still the rare cases of familial AD are linked either to point mutations in the amyloid precursor protein (APP) itself or to proteins involved in the cleavage of this neuronal membrane protein to Aβ, thus proofing an essential role of the latter at AD pathology (Ringman et al., 2012; Armstrong, 2013; Buss et al., 2016). Aβ is itself a neurotoxic peptide and thus contributes to neurodegeneration at AD. Moreover the peptide may also be involved in abnormal Tau phosphorylation. Regarding both aspects, monomeric and/or oligomeric states rather than final Aβ fibrils are regarded as the most likely pathologically active forms of the peptide (Al-Hilaly et al., 2013). Still, while Aβ apparently represents an important precondition for AD and contributes to its pathology, further aspects are apparently necessary for disease development (Armstrong, 2013).
Most interestingly, the formation of Aβ-derived fibrils and their accumulation as extracellular plaques can be found in many species, most likely due to the evolutionary conserved hydrophobic C-terminal part of the peptide. For instance, rodents also exhibit an age-dependent accumulation of Aβ plaques, however without showing signs of neurodegeneration and dementia (Ghosh et al., 2015). In fact, all currently used mouse models for AD are based on the heterologous expression of human Aβ forms in the animals. Thus, the species-specific differences in the N-terminal part of the peptide may be responsible for the role of Aβ during AD development. In fact, the only sequence differences between human and rodent Aβ are exclusively found in the N-terminal part of the peptide, with R5, Y10 and H13 and G5, F10 and R13, respectively (Atamna et al., 2009). In contrast, many animals with the human-like N-terminus of Aβ are also known to develop AD-like symptoms at higher ages, including dogs, cats and several hominids (Salazar et al., 2016). As shown in Figure 1, while a phylogenetic analysis of the whole APP protein sequence (Figure 1A) clearly reflects the expected evolutionary relationship between animals, the AD-relevant Aβ peptide sequence (Figure 1B) appears to be highly conserved with only sporadic mutations.
Figure 1.
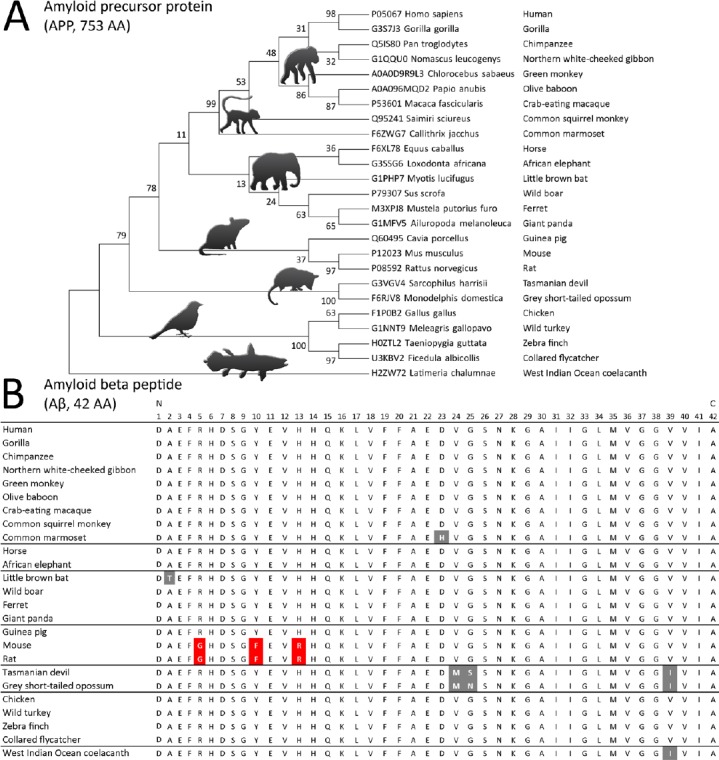
Evolutionary conservation of amyloid precursor protein (APP)-derived amyloid β (Aβ).
(A) Based on the alignment of the APP AA sequence from 25 animal species, a molecular phylogenetic analysis was performed with MEGA7 (Kumar et al., 2016) by using the maximum likelihood method with 100 bootstraps and a JTT matrix-based model after initial tree building with the Maximum Parsimony method. The obtained phylogeny performed on 770 amino acid positions reflects nicely the general evolutionary relationship between the selected species. (B) The manual alignment of the Alzheimer’s disease (AD)-relevant Aβ peptide shows a highly conserved AA sequence among the tested species with only few mutations (grey) in relation to the human-specific sequence. A notable exception is the occurrence of the mutations of R5G, Y10F and Y13R (red) in mice and rats. In the human sequence R5, Y10 and H13 are responsible for the formation and subsequent peroxidase activity of Aβ-heme complexes.
Role of Aβ-Heme Complex Formation
Several further aspects were suggested to also contribute to AD pathology, including oxidative stress (Markesbery, 1997; Chen and Zhong, 2014), free metals (Smith et al., 1997) and the immune system (Armstrong, 2013; Martino Adami et al., 2017). In fact, oxidized neurotransmitters (Van Dam et al., 2013; Mukherjee et al., 2014), lipid peroxides (Chen and Zhong, 2014; Akhter et al., 2017) and neuro-degenerative processes due to oxidative stress (Armstrong, 2013; Bastianetto et al., 2015) are known aspects of AD pathology. Thus amyloidosis apparently represents an essential precondition for disease development while further aspects are required for AD pathology (Armstrong, 2013). Still the local correlation between extracellular Aβ accumulation and oxidative stress strongly suggests a close connection between both aspects. Considering the findings of Atamna et al. (2009), both features may be explained by the formation of Aβ-heme complexes: As verified by a recent publication from our group (Chiziane et al., 2018), the human-specific N-terminal sequence of the named peptide leads to the binding of free heme groups, most likely via H13 (Atamna and Frey, 2004). Histidines are known to serve as heme ligands in many peroxidases (Loughran et al., 2008). Several studies proved the named complex formation e.g., via absorbance measurements (Pramanik and Dey, 2011; Lu et al., 2014). In the additional presence of H2O2 a peroxidase activity of the formed complexes can be observed, whereby again the conserved amino acids R5 and Y10 contribute essentially to this enzymatic activity. While the exact structure of Aβ-heme complexes remains to be solved, several publications suggest a 1:1 ratio upon complex formation (Pramanik et al., 2013; Lu et al., 2014). Yet as shown in Figure 2 based on our studies (Chiziane et al., 2018), we suggest a 2:1 ratio whereby one Aβ molecule binds the heme iron via H13 while a second peptide provides R5 and Y10 for the subsequent peroxidase activity. Accordingly, in the presence of murine Aβ, less complex formation and a considerably lower peroxidase activity were observed (Chiziane et al., 2018). Thus, the named complexes may provide an explanation for the aforementioned species-specific occurrence of AD pathology.
Figure 2.
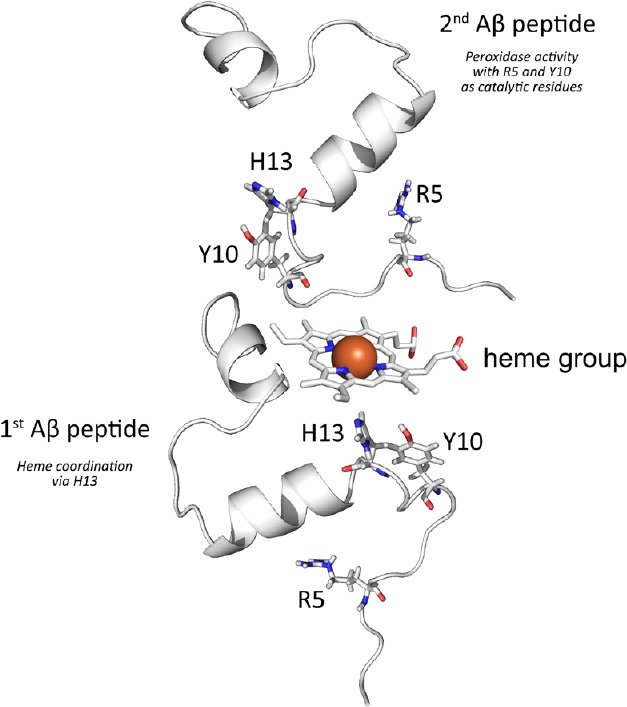
Schematic representation of amyloid β (Aβ)-heme complex formation.
Based on published investigations on the complexation of free heme by Aβ we propose that the interaction takes mainly place between heme and the N-terminal part of the peptide Thereby we suggest that while one peptide is mainly involved in the coordination of the heme iron at the distal heme site via H13 a second peptide may coordinate at the proximal heme site to provide R5 and Y10, which are responsible for the subsequent peroxidase activity of the complex. The model was created by using the PDB entries 1aml and 2dn1 and modified with the PyMOL Molecular Graphics System, Version 0.99 (Schrödinger, LLC, Mannheim, Germany).
In fact, the peroxidase activity of Aβ-heme complexes was already clearly shown to contribute to AD pathology: For instance, the named enzymatic activity leads to neurotransmitter oxidation, a pathological key feature at AD (Atamna et al., 2009). Moreover, Aβ represents also an endogenous substrate for Aβ-heme complex-based peroxidase activity, resulting in peptide dimer formation via di-tyrosine which in turn may influence Aβ fibrillation (Al-Hilaly et al., 2013). In fact, our studies clearly showed that free heme inhibits Aβ aggregation, most likely due to the interaction with F19/20 in the hydrophobic part of the peptide. In striking contrast, under conditions of heme-derived peroxidase activity, either by Aβ-heme complexes or by an exogenous peroxidase, we observed strong effects on the kinetics of peptide aggregation and on the morphology of the formed Aβ fibrils (Chiziane et al., 2018). Comparable studies also showed a positive correlation between peroxidase activity and Aβ aggregation (Al-Hilaly et al., 2013). Thus, while it is still a matter of debate which aggregation state of Aβ contributes most to AD pathology, we strongly recommend further studies on the aggregation behavior of the peptide under conditions of heme-derived peroxidase activity.
At any rate, recent NMR studies on the structure of Aβ fibrils leave open the possibility that free heme may also bind to the aggregated peptide (Tycko, 2016; Wälti et al., 2016; Wang et al., 2017). In fact, in line with others we observed an even stronger peroxidase activity of Aβ-heme complexes by applying the full-length peptide instead of the N-terminal part (Aβ1–16) (Chiziane et al., 2018). Thus, the auto-catalytic structuration of the C-terminal Aβ part in fibrils may contribute considerably to the pathological impact of Aβ-heme complex-derived peroxidase activity at AD. Preliminary docking studies in our group indeed strongly suggest a binding of free heme also to final Aβ fibrils. Yet despite a possibly huge role of Aβ-heme interaction at AD pathology, both the exact stoichiometry and the structure of those complexes are still unknown. The same holds for the precise active site structure and the biochemical mechanisms which lead to a peroxidase activity of these complexes. Thus further studies are mandatory to reveal the role of Aβ-heme complex-derived peroxidase activity at AD pathology.
Role of Micro-Angiopathy and Hemolysis
The assumption that Aβ-heme complex formation may represent an important step during the initiation of AD pathology still leaves open the question which mechanisms are involved in the accumulation of free heme in the brain tissue. As the analysis of brain tissue from AD patients showed a specific elevation of hemoglobin-derived peptides (Slemmon et al., 1994), we deduce that blood-derived hemoglobin may represent the major source for free heme. In fact, the aforementioned accumulative nature of AD pathology is also reflected by the progressive destruction of small blood vessels in the brain (Armstrong, 2013), leading to micro-hemorrhages of small brain vessels in about 90% of all AD cases. Accordingly the disease was already suggested to represent a thrombohemorrhagic disorder (Schmaier, 2016). Other erythrocyte-derived heme proteins like catalase may also contribute to cerebral accumulation of free heme.
Most strikingly, regarding the reasons for this cerebral angiopathy again a clear correlation to the deposition of Aβ in brain tissue can be drawn (Mendel et al., 2013). The peptide actively accumulates around small vessels, leading to blood flow defects and ultimately to vessel destruction, a pathological feature known as CAA (Cullen et al., 2006). Furthermore, Aβ-derived cerebral micro-vessel destruction leads to micro-bleedings which result in up to micro-molar concentrations of free heme in the brain tissue of AD patients due to subsequent Aβ-derived hemolytic events. In fact, the peptide is known to quickly bind to erythrocytes and to impair the stability of the cells (Jayakumar et al., 2003). Consequently, Aβ-enriched red blood cells are more prone to hemolysis, especially during the passage through small cerebral capillaries. Most interestingly, Aβ-derived phospholipid oxidation in erythrocytes also leads to a higher endothelial binding of the cells, which may further promote CAA at AD. Moreover, the peptide disturbs catalase activity in erythrocytes (Habib et al., 2010). Thus Aβ-derived accumulation of erythrocytes in the brain tissue may lead not only to the accumulation of free heme for Aβ-heme complex formation but also to higher H2O2 concentrations as a precondition for a subsequent peroxidase activity of the named complexes.
In summary, Aβ-derived CAA and hemolytic effects provide a likely pathway for the accumulation of free heme, and maybe also H2O2, in the brain tissue of AD patients. The co-localization of heme-rich deposits (HRDs) with Aβ at AD was already reported (Cullen et al., 2006) and it may even be speculated that the well-known local correlation of senile plaques with free iron may represent a clinical end-stage of Aβ-heme complex formation during AD pathology. Also the up-regulation of hemolysis-associated acute phase proteins like hemopexin or heme oxygenase is a common clinical feature of the disease. Both the increasing extracellular accumulation of Aβ and decreasing blood vessel stability are well-known age-dependent processes. Thus, Aβ-derived CAA, cerebral hemolysis and Aβ-heme complex formation may provide an explanation for the strong age-dependence of AD pathology. Conversely, early disease onset in patients with familial AD may be attributed to considerably higher cerebral Aβ concentrations and result in much stronger micro-hemorrhagic pathologies.
Alternatively, the neuronal loss at AD may also contribute to the accumulation of free heme in the brain tissue, e.g., due to cytochrome c release. A disturbed heme metabolism and mitochondrial dysfunction of neuronal cells were already suggested to contribute to Aβ-heme complex formation at AD (Atamna et al., 2009). Furthermore, inflammatory reactions are also discussed to play a role at AD pathology (Martino Adami et al., 2017). Thus we guess that the activation of cerebral immune cells like astrocytes, which results in elevated H2O2 production and heme peroxidase activity, may support, or even replace, the pathological effects of Aβ-heme complex-derived peroxidase activity at AD. Moreover, the aforementioned micro-hemorrhagic effects of Aβ leave open the possibility that also blood-derived immune cells like myeloperoxidase-containing neutrophil granulocytes may accumulate in the brain tissue and take a part in AD pathology. Thus, the destruction of neuronal cells as well as the activation of cerebral and/or blood derived heme peroxidase-containing immune cells may, at least at later stages, enhance AD pathology. Still, as illustrated in Figure 2, we suggest Aβ-derived CAA, hemolysis and the formation and peroxidase activity of Aβ-heme complexes as the major initial events during the preclinical onset of AD pathology.
Summary and Perspectives
The rare cases of familial AD clearly show a central role of cerebral Aβ at AD. In fact, many pathological features of this neurodegenerative disease are known to be linked to this peptide, including neuronal oxidative stress and cell degeneration, CAA and hemolytic events in the brain. As ageing still represents the major risk factor for the development of sporadic AD we suggest, that the initial cause for the onset of the disease is a disturbed cerebral Aβ cleavage, which, especially in age- related pre-damaged blood vessels, leads to micro-angiopathies in the brain. Consequently, hemoglobin-derived free heme accumulates in the brain tissue and forms a complex with Aβ. These complexes exhibit a peroxidase activity in the presence of H2O2 and, thus, may considerably contribute to the AD pathology. Accordingly, the in vivo detection of Aβ-heme complexes as well as the evaluation of their contribution to AD pathology should be central aspects of future AD research (Figure 3).
Figure 3.

Pathway for cerebral amyloid β (Aβ)-heme complex formation at sporadic Alzheimer’s disease (AD).
An age-dependent excess Aβ formation and/or its disturbed cleavage result in the formation of fibrils and senile plaques. Yet Aβ also accumulates around small cerebral blood vessels and leads to their destruction. The peptide also readily crosses the blood-brain barrier and accumulates in erythrocytes. A subsequent cerebral hemoglobin release may represent the main precondition for the formation of Aβ-heme complexes which, in the presence of H2O2, exhibit peroxidase activity. This enzymatic activity is known to contribute to several aspects of AD pathology, including neurotransmitter oxidation. Yet, e.g., its effect on plaque formation is still not known. Neuron-derived free heme and leukocyte-derived heme peroxidases may contribute to this pathway.
In fact, e.g., the visualization of cerebral Aβ aggregates by antibodies was already shown to have no influence on the peroxidase activity of Aβ-heme complexes (Pramanik et al., 2013). Thus instead of detecting Aβ (Pramanik et al., 2013), hemoglobin and heme (Slemmon et al., 1994; Atamna and Frey, 2004) or free iron (Smith et al., 1997; Cacciottolo et al., 2016), we suggest the development of tools to directly target Aβ-heme complexes. Thereby both the formation of those complexes as well as their subsequent enzymatic activity should be considered as promising clinical parameters in order to detect early AD pathology. Thereby simplified in vitro approaches may provide a good starting point for the development of suitable antibodies and activity assays (Chiziane et al., 2018) for subsequent clinical application.
Footnotes
Conflicts of interest: The authors have no conflict of interest to report.
Financial support: The work was supported by the Alzheimer Forschung Initiative e.V. (AFI 13810).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review report:
Reviewer: Stavros I. Dimitriadis, Cardiff University, UK.
Comments to authors: The theme of this article is very strong and i am positive to see it publish but it needs to be extended in figures, sections and also in references in order to cover this area in a review mode. Author presents a very nice review of amyloid beta and free heme explaining blood contribution to the pathogenesis of AD. The manuscript is very well written using simple terms and very nice graphics for visualizing the Pathway for cerebral Aβ-heme complex formation at sporadic AD. Moreover, evolutionary conservation of APP-derived Aβ gives a nice picture of alignment or deviations of amyloid beta peptide across species.
Funding: The work was supported by the Alzheimer Forschung Initiative e.V. (AFI 13810).
References
- 1.Akhter F, Chen D, Yan SF, Yan SS. Mitochondrial perturbation in Alzheimer’s disease and diabetes. Prog Mol Biol Transl Sci. 2017;146:341–361. doi: 10.1016/bs.pmbts.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Hilaly YK, Williams TL, Stewart-Parker M, Ford L, Skaria E, Cole M, Bucher WG, Morris KL, Sada AA, Thorpe JR, Serpell LC. A central role for dityrosine crosslinking of amyloid-beta in Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:83. doi: 10.1186/2051-5960-1-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong RA. What causes Alzheimer’s disease? Folia Neuropathol. 2013;51:169–188. doi: 10.5114/fn.2013.37702. [DOI] [PubMed] [Google Scholar]
- 4.Atamna H, Frey WH., 2nd A role for heme in Alzheimer’s disease: heme binds amyloid beta and has altered metabolism. Proc Natl Acad Sci U S A. 2004;101:11153–11158. doi: 10.1073/pnas.0404349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atamna H, Frey WH, 2nd, Ko N. Human and rodent amyloid-beta peptides differentially bind heme: relevance to the human susceptibility to Alzheimer’s disease. Arch Biochem Biophys. 2009;487:59–65. doi: 10.1016/j.abb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Bastianetto S, Ménard C, Quirion R. Neuroprotective action of resveratrol. Biochim Biophys Acta. 2015;1852:1195–1201. doi: 10.1016/j.bbadis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Buss L, Fisher E, Hardy J, Nizetic D, Groet J, Pulford L, Strydom A. Intracerebral haemorrhage in Down syndrome: protected or predisposed? F1000Res. 2016;5:1876. doi: 10.12688/f1000research.7819.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cacciottolo M, Morgan TE, Finch CE. Rust on the brain from microbleeds and its relevance to Alzheimer studies: Invited Commentary on Cacciottolo Neurobiology of Aging, 2016. J Alzheimers Dis Parkinsonism. 2016;6:287. doi: 10.4172/2161-0460.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. 2014;30:271–281. doi: 10.1007/s12264-013-1423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiziane E, Telemann H, Krueger M, Adler J, Arnhold J, Alia A, Flemmig J. Free heme and amyloid-beta: a fatal liaison in Alzheimer’s disease. J Alzheimers Dis. 2018;61:963–984. doi: 10.3233/JAD-170711. [DOI] [PubMed] [Google Scholar]
- 11.Cullen KM, Kócsi Z, Stone J. Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging. 2006;27:1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K, Broich K, Cavedo E, Crutch S, Dartigues JF, Duyckaerts C, Epelbaum S, Frisoni GB, Gauthier S, Genthon R, Gouw AA, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323. doi: 10.1016/j.jalz.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh C, Seal M, Mukherjee S, Ghosh Dey S. Alzheimer’s disease: a heme-Abeta perspective. Acc Chem Res. 2015;48:2556–2564. doi: 10.1021/acs.accounts.5b00102. [DOI] [PubMed] [Google Scholar]
- 14.Habib LK, Lee MT, Yang J. Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J Biol Chem. 2010;285:38933–38943. doi: 10.1074/jbc.M110.132860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayakumar R, Kusiak JW, Chrest FJ, Demehin AA, Murali J, Wersto RP, Nagababu E, Ravi L, Rifkind JM. Red cell perturbations by amyloid beta-protein. Biochim Biophys Acta. 2003;1622:20–28. doi: 10.1016/s0304-4165(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 16.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loughran NB, O’Connor B, O’Fágáin C, O’Connell MJ. The phylogeny of the mammalian heme peroxidases and the evolution of their diverse functions. BMC Evol Biol. 2008;8:101. doi: 10.1186/1471-2148-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu N, Li J, Tian R, Peng YY. Key roles of Arg(5), Tyr(10) and his residues in Abeta-heme peroxidase: relevance to Alzheimer’s disease. Biochem Biophys Res Commun. 2014;452:676–681. doi: 10.1016/j.bbrc.2014.08.130. [DOI] [PubMed] [Google Scholar]
- 19.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 20.Martino Adami PV, Galeano P, Wallinger ML, Quijano C, Rabossi A, Pagano ES, Olivar N, Reyes Toso C, Cardinali D, Brusco LI, Do Carmo S, Radi R, Gevorkian G, Castano EM, Cuello AC, Morelli L. Worsening of memory deficit induced by energy-dense diet in a rat model of early-Alzheimer’s disease is associated to neurotoxic Abeta species and independent of neuroinflammation. Biochim Biophys Acta. 2017;1863:731–743. doi: 10.1016/j.bbadis.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 21.Mendel TA, Wierzba-Bobrowicz T, Lewandowska E, Stepien T, Szpak GM. The development of cerebral amyloid angiopathy in cerebral vessels. A review with illustrations based upon own investigated post mortem cases. Pol J Pathol. 2013;64:260–267. doi: 10.5114/pjp.2013.39334. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee S, Seal M, Dey SG. Kinetics of serotonin oxidation by heme-Abeta relevant to Alzheimer’s disease. J Biol Inorg Chem. 2014;19:1355–1365. doi: 10.1007/s00775-014-1193-7. [DOI] [PubMed] [Google Scholar]
- 23.Pramanik D, Dey SG. Active site environment of heme-bound amyloid beta peptide associated with Alzheimer’s disease. J Am Chem Soc. 2011;133:81–87. doi: 10.1021/ja1084578. [DOI] [PubMed] [Google Scholar]
- 24.Pramanik D, Ghosh C, Mukherjee S, Dey SG. Interaction of amyloid β peptides with redox active heme cofactor: Relevance to Alzheimer’s disease. Coord Chem Rev. 2013;257:81–92. [Google Scholar]
- 25.Ringman JM, Schulman H, Becker C, Jones T, Bai Y, Immermann F, Cole G, Sokolow S, Gylys K, Geschwind DH, Cummings JL, Wan HI. Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch Neurol. 2012;69:96–104. doi: 10.1001/archneurol.2011.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salazar C, Valdivia G, Ardiles ÁO, Ewer J, Palacios AG. Genetic variants associated with neurodegenerative Alzheimer disease in natural models. Biol Res. 2016;49:14. doi: 10.1186/s40659-016-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmaier AH. Alzheimer disease is in part a thrombohemorrhagic disorder. J Thromb Haemost. 2016;14:991–994. doi: 10.1111/jth.13277. [DOI] [PubMed] [Google Scholar]
- 28.Slemmon JR, Hughes CM, Campbell GA, Flood DG. Increased levels of hemoglobin-derived and other peptides in Alzheimer’s disease cerebellum. J Neurosci. 1994;14:2225–2235. doi: 10.1523/JNEUROSCI.14-04-02225.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tycko R. Molecular structure of aggregated amyloid-beta: insights from solid-state nuclear magnetic resonance. Cold Spring Harb Perspect Med. 2016;6:8. doi: 10.1101/cshperspect.a024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Dam D, Van Dijck A, Janssen L, De Deyn PP. Neuropeptides in Alzheimer’s disease: from pathophysiological mechanisms to therapeutic opportunities. Curr Alzheimer Res. 2013;10:449–468. doi: 10.2174/1567205011310050001. [DOI] [PubMed] [Google Scholar]
- 32.Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc Natl Acad Sci U S A. 2016;113:E4976–4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T, Jo H, DeGrado WF, Hong M. Water distribution, dynamics, and interactions with Alzheimer’s beta-amyloid fibrils investigated by solid-state NMR. J Am Chem Soc. 2017;139:6242–6252. doi: 10.1021/jacs.7b02089. [DOI] [PMC free article] [PubMed] [Google Scholar]