

Abstract
Thermochemiluminescence (TCL) is a potentially simple and sensitive detection principle, as the light emission is simply elicited by a thermally-triggered decomposition of a molecule to produce a singlet excited-state product. Here we report about TCL semiconductive Polymer Dots (TCL-Pdots) obtained by doping fluorescent cyano-polyphenylene vinylene (CN-PPV) Pdots with an acridin 1,2-dioxetane derivative. The TCL-Pdots showed remarkable stability over time and minimum leaching of the thermo-responsive species. Furthermore, detectability of TCL-Pdots was improved taking advantage of both the high number of 1,2-dioxetanes entrapped in each nanoparticle (about 20 molecules/Pdot) and the 5-fold enhancement of TCL emission due to energy transfer from the 1,2-dioxetane to the polymer matrix, which itself acted as an energy acceptor. Indeed, upon heating the TCL-Pdots to 110°C, the 1,2-dioxetane decomposes generating an acridanone product in its electronically excited state. The latter transfers its energy to the surrounding CN-PPV chains via Förster mechanism (ϕFRET about 80%), resulting in intense yellow light emission (550 nm wavelength). We next conjugated streptavidin onto the surface of these TCL-Pdots and demonstrated their suitability for use in biological studies. In particular, we used TCL-Pdots as labels in a model non-competitive immunoassay for IgG detection, which has showed a LOD of 13 nM IgG and a dynamic range extending up to 230 nM. By combining the biocompatibility, brightness and tunability of Pdots fluorescence emission with the thermally-triggered reagentless light generation from TCL 1,2-dioxetanes, a broad panel of ultrabright TCL nanosystems could be designed for a variety of bioscience applications, even in multiplexed format.
1. Introduction
Biosensors able to rapidly detect clinical biomarkers in complex biological samples are fundamental for early disease diagnosis and personalized therapy monitoring in point-of-care (POC) settings. Indeed, in the last 20 years, the world market for biosensors and rapid tests has witnessed an apparently endless exponential growth. Predictions for the future estimate a turnover higher than US$ 25.9 billion/year,1 mostly due to the use of biosensors as companion diagnostics in therapy to achieve a precision medicine approach.2,3 Furthermore, such devices can find widespread application also in food safety and quality control, environmental monitoring and, in general, in any condition where a decentralized analysis is required, i.e. at the point of need.
To provide suitable analytical performance, biosensors must be simple to use and, in the meantime, highly sensitive and specific. The transduction principle employed in the biosensor represents a critical factor for its analytical performance and thus for the ability to fulfill the requirements above. Optical transduction has been covering a pivotal role in development of (bio)analytical methods for application in biology and medicine, offering high sensitivity, specificity and wide dynamic range. Furthermore, the high resolution achievable and the nondestructive nature of optical sensors have contributed to their use in in vivo imaging and sensing in living human tissues.4,5 Not least, the combination of optical sensors and optical fibers technology have promoted their diagnostic and surgical application.6,7
Fluorescence (FL)-based biosensors have been successfully used to quantify small organic molecules, proteins and cells in a wide range of applications, from clinical diagnostics and environmental monitoring to cell sorting and bioimaging.8-12 Despite its widespread use, FL detection suffers from several limitations, such as the high background signal (due to the interference from endogenous fluorophores present in the sample matrix and the scattering of excitation light)13 and the low water solubility for many organic FL probes.14 To overcome these limitations, chemical luminescence principles, such as chemiluminescence (CL), bioluminescence (BL) and electrochemiluminescence (ECL), have been successfully employed in biosensors.15-17 They represent powerful detection principles because of their high signal-to-noise ratio, wide linear range and amenability to implementation in miniaturized analytical formats. Indeed, in these detection techniques the light is simply generated by a chemical or electrochemical reaction: the absence of an excitation light source eliminates interferences due to endogenous fluorescent species and light scattering, and the blank signal is mainly due to the thermal noise of the light detector. The instrumentation required for chemical luminescence measurements is very simple, as no lamps, filters or specific cell measurement geometries are required. Nevertheless, these techniques also present some drawbacks, especially for POC applications. Light emission from CL and BL reactions is obtained upon addition of suitable reagents, such as enzyme substrates (e.g. a luciferin substrate for the BL label luciferase) and/or oxidants (e.g. H2O2 for the CL enzyme horseradish peroxidase), which complicates the analytical procedure.
Thermochemiluminescence (TCL) consists of light production due to the decomposition of a thermally unstable molecule (typically a 1,2-dioxetane derivative) upon heating above a threshold temperature. The energy released during the decomposition of the endoperoxide unit is enough to bring one of the two fragments to an electronically excited state, which then decays by emitting a photon.18 Although the TCL principle has been known since the late 80’s, when spiro-adamantane 1,2-dioxetane19 was proposed as a TCL label for the development of immunoassays,20 it was quickly abandoned because of its practical limitations, such as the high temperature (up to 200°C) required to decompose the molecule (which limited the choice of materials for assay format) and the low quantum yield of the TCL reaction, due to the preferential production of the 2-adamantanone fragment in the triplet rather than singlet excited state.
This chemical luminescence phenomenon has been reinvestigated by our group and its applicability as detection principle for bioassays has been revitalized by combining new TCL molecules with silica-based nanoparticle (NP) technology in order to overcome the above reported limitations.21-23 TCL-based methods might benefit from several advantages, namely the high signal-to-noise ratios typical of chemical luminescence-based detection and the reagentless nature of the TCL process itself, which would simplify analytical devices in comparison to CL/BL. Indeed, the detection step of an immunoassay employing TCL biospecific probes only requires heating of the sample to generate a light signal proportional to the amount of the TCL probe. Then, light emission can be measured using simple portable light detectors, such as CMOS or CCD cameras possibly operating in contact mode, e.g. via a fiber optic faceplate.24,25 In addition, by modifying the molecular structure of the 1,2-dioxetane derivative it will be possible to finely tune the TCL properties in terms of TCL quantum yield, emission wavelength and decomposition temperature.26,27 A quite low triggering temperature (e.g. around 100°C) will avoid the use of expensive heat-resistant tubing and materials, also considering that the biosensing part of the device will be disposable, thus it will not sustain several heating cycles.
Nanoparticles made of various materials have attracted great interest in the last decades due to their promising potential for application in biosciences and biomedical research. In recent years, a class of polymeric NPs, namely semiconducting polymer dots (Pdots), has affirmed as highly fluorescent nanomaterials for bioimaging. Pdot-based probes have been successfully used for in vivo dynamic monitoring of small molecules such as HNO and HClO,28-30 in cell sorting31 and for high-resolution fluorescence imaging.32-34 In comparison with organic fluorophores, Pdots are characterized by remarkable photophysical properties such as high photostability and fluorescence quantum yield, fast radiative decay rates and narrow emission bands, which make them particularly suitable for biological and medical applications.35,36 Moreover, by changing the polymer matrix it is possible to tune the wavelength of emitted light, thus creating panels of Pdots covering the entire spectrum from blue to NIR.37-42
Herein, we report for the first time the use of TCL semiconducting polymer dots (TCL-Pdots) as innovative luminescent nanolabels for immunobiosensors. In this label, the fluorescence emission of the Pdot is induced via a Förster Resonance Energy Transfer (FRET) mechanism by the decomposition of TCL molecules entrapped in the nanoparticle, i.e. the TCL-Pdot can be considered a self-fluorescent NP in which heating triggers light emission. With this approach, the NP is actively involved in the enhancement of the TCL emission, rather than merely representing a water-compatible nanocarrier as the previously used silica NPs. Furthermore, many TCL molecules can be entrapped in each NP, thus enabling the amplification of the light signal as a function of the amount of TCL molecules/NP. Using a nanoscale co-precipitation technique43 we obtained cyano-polyphenylene vinylene (CN-PPV)-based Pdots doped with the TCL acridin 1,2-dioxetane derivative 1 (Figure 1). Upon heating above 100°C, compound 1 undergoes a fast thermal decomposition generating two fragments, one of which (ketone 2) is in a singlet excited state. The nanosystem ensures the spatial proximity44 of the excited ketone 2 to the CN-PPV chains, thus leading to a highly efficient FRET process to the surrounding polymer matrix (Figure 1). As a result, the intensity of the TCL emission originating from the decomposition of the 1,2-dioxetane derivative 1 is enhanced thanks to the higher fluorescence quantum yield of CN-PPV (ϕF=60%)45 in comparison to ketone 2 (ϕF=11%).21 After conjugation with Streptavidin (SA), the TCL-Pdots were tested as TCL labelling agent in a model non-competitive immunoassay for detection of Biotinylated immunoglobulin G (IgG).
Fig. 1.
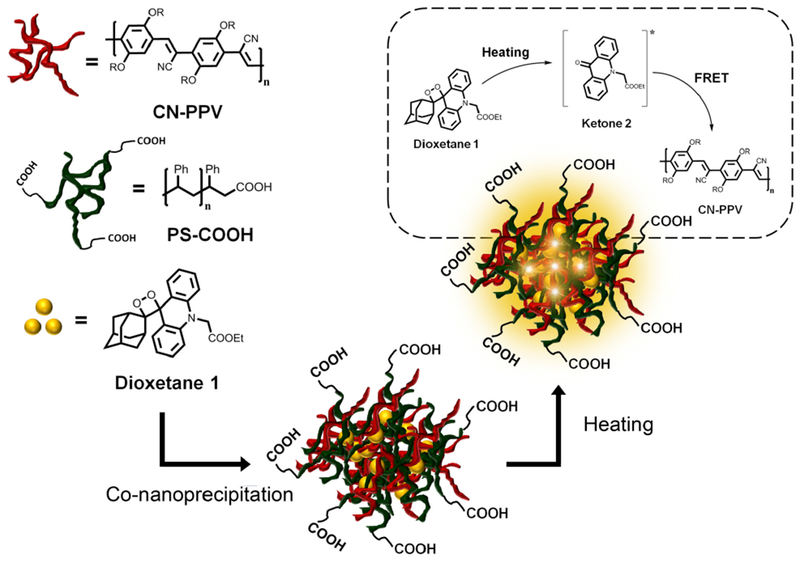
Synthesis of 1,2-dioxetane 1-doped TCL-Pdots through the co-nanoprecipitation method.
2. Results and discussion
2.1. Synthesis and characterization of TCL-Pdot nanoprobes
The 1,2-dioxetane 1-doped Pdots were obtained by a co-nanoprecipitation method (Figure 1),43 in which CN-PPV, carboxylic functionalized polystyrene (PS-COOH) and substrate 1 were first dissolved in anhydrous THF and then quickly injected into Milli-Q water under sonication. Then, the organic solvent was removed by a stream of nitrogen to promote formation of an aqueous solution of Pdots. Different from a previously reported procedure,46 we used PS-COOH rather than PS-PEG-COOH (carboxylic functionalized polystyrene polyethylene glycol) to introduce carboxylic acid units onto the particle surface. The absence of the hydrophilic polyethylene glycol block in the polymers should limit the swelling of Pdots during heating in the presence of water, thus reducing the leakage of 1 from the Pdot at high temperatures.47-49
First attempts to synthesize Pdots have shown a marked dependence of both particle size and amount of entrapped 1,2-dioxetane from the experimental conditions used in the nanoprecipitation step. Thus, aiming at the optimization of Pdots dimension and loading capacity of 1, we conducted a stepwise screening of three relevant variables, namely (a) volume of THF injected, (b) amount of PS-COOH and (c) amount of 1,2-dioxetane 1, and evaluated the effect of these variables on the hydrodynamic diameter measured by dynamic light scattering (DLS) analysis and the TCL emission intensity of Pdots. As reported in Table 1, the Pdot size increases with solvent volume, while the amount of PS-COOH seemed less important in affecting Pdots dimension.
Table 1.
Diameter and loading capacity of Pdots obtained in each step of the optimization screening.
| Step I: screening of THF volume injected (mL), keeping constant the amounts of compound 1 (0.5 mg), CN-PPV (0.5 mg) and PS-COOH (0.1 mg) for each batch synthesis. | |||
|
| |||
| Entry | Volume of THF (mL) | Diameter (nm) | TCL signal ± SD (%)c,d |
|
| |||
| 1 | 1 | 16.2 | 2.3 ± 0.8 |
| 2 | 3 | 13.1 | 0.5 ± 0.1 |
| 3 | 5 | 12.7 | 0.8 ± 0.1 |
| 4 | 7 | 21.1 | 5.7 ± 0.4 |
| 5 | 8 | 40.5 | 19.4 ± 1.6 |
| 6 | 9 | 52.3 | 100 ± 2 |
|
| |||
| Step II: screening of the amount of PS-COOH (mg), keeping constant the volume of THF (7 mL) and the amounts of compound 1 (0.5 mg) and CN-PPV (0.5 mg) for each batch synthesis. | |||
|
| |||
| Entry | PS-COOH (mg) | Diameter (nm) | TCL signal ± SD (%)c,d |
|
| |||
| 1 | 0.05 | 21.3 | 18.3 ± 1.3 |
| 2 | 0.1 | 21.1 | 12.6 ± 1.7 |
| 3 | 0.3 | 25.0 | 13.7 ± 0.6 |
| 4 | 0.5 | 32.4 | 100 ± 6 |
| 5 | 0.7 | 20.2 | 21.9 ± 1.4 |
|
| |||
| Step III: screening of the amount of compound 1 (mg), keeping constant the volume of THF (7 mL) and the amounts of PS-COOH (0.5 mg) and CN-PPV (0.5 mg) for each batch synthesis. | |||
|
| |||
| Entry | Compound 1 (mg) | Diameter (nm) | TCL signal ± SD (%)c,d |
|
| |||
| 1 | 0.2 | 43.5 | 75 ± 5 |
| 2 | 0.5 | 32.4 | 100 ± 6 |
| 3 | 0.8 | 28.5 | 12.6 ± 0.9 |
| 4 | 1.2 | 36.5 | 66 ± 4 |
TCL signal was measured through a CCD camera, spotting 3 μL of Pdots solution and acquiring images for 30 min at 110°C.
TCL emission is corrected by Pdots concentration and normalized with respect to the highest value in each series (values reported as mean ± SD of three independent measurements).
The effect of THF volume on the size of Pdots could be explained considering that during the co-precipitation the formation of Pdots is accomplished once almost all the organic solvent is removed from the solution. Thus, the longer evaporation times required for the removal of high THF volumes could favor the aggregation of hydrophobic polymer chains into bigger nanoparticles.50 An opposite tendency in the particle size was observed during the screening of the amount of compound 1.
Indeed, except for the lowest amounts of 1,2-dioxetane (see Entry 1 and 2), the diameter of Pdots was inversely proportional to the amount of doping substrate. This evidence could be ascribed to the establishment of π-staking interactions between dioxetane 1 and CN-PPV during the co-nanoprecipitation step, which might disadvantage the aggregation of polymer chains. Concerning the TCL signal, an overall dependence of the emission intensity from the size of Pdots was observed, since more 1,2-dioxetane molecules were entrapped in larger Pdots.
With the goal to maximize the loading of 1,2-dioxetane 1 (thus the TCL signal) while keeping particles dimensions to relatively low values, to ensure good diffusion rates and avoid aggregation in solution, we chose the best compromise among the variables investigated, namely 7 mL of THF injected and 0.5 mg of both PS-COOH and 1,2-dioxetane 1. Using these experimental conditions, we obtained quite monodispersed NPs with an average hydrodynamic diameter of 32 ± 3 nm, as demonstrated by DLS measures and TEM images (Figure 2a and 2b, respectively).
Fig. 2.

a) Dynamic light scattering analysis. b) TEM image of TCL-Pdots; c) Absorption (yellow line) and emission (orange line) of CN-PPV-based Pdots in water solution; emission (blue line) of 1,2-dioxetane 1 in THF after thermal decomposition.
2.2. TCL emission and FRET experiments
Pdots have been largely used either as direct fluorescent sensors or as donor species in organic dye-coupled systems.51 Here, we took advantage of the broad absorption spectrum and fluorescence properties of CN-PPV-based Pdots to collect the energy from the excited-state species produced by the thermal decomposition of 1,2-dioxetane within the Pdot through a FRET mechanism. Thus, the Pdot represented not only a carrier for 1, but it actively participated in the light generation process.
Considering the large spectral overlap between the TCL emission of ketone 2 (i.e. the excited-state fragment generated after thermal decomposition of 1) and the absorption of CN-PPV-based Pdots (see Figure 2c), we expect an efficient energy transfer to the polymer chains through a Förster mechanism. Indeed, heating of a solution of 1,2-dioxetane 1 in DMSO (Figure 3a, left) and of a water solution of TCL-Pdots (Figure 3a, right) led to clearly different light emissions. The yellow emission (typical of the CN-PPV polymer) observed from TCL-Pdots supported the occurrence of the FRET process from the excited ketone 2 to the CN-PPV polymer within the NPs. A TCL spectrum, in which compound 1 was thermally decomposed in the presence of CN-PPV polymer, was also recorded (for more details see Supporting Information). Both 1,2-dioxetane 1 and CN-PPV polymer were dissolved in THF, then heated above 100°C to record the TCL spectrum. The quick evaporation of the organic solvent THF allowed us to observe the TCL emission in the dry state, as in the TCL-Pdots. As shown in Figure 3b, the TCL spectrum is dominated by the emission of the CN-PPV polymer at about 550 nm due to the FRET process, while a much weaker signal at 400-420 nm derives from the excited ketone 2.
Fig. 3.
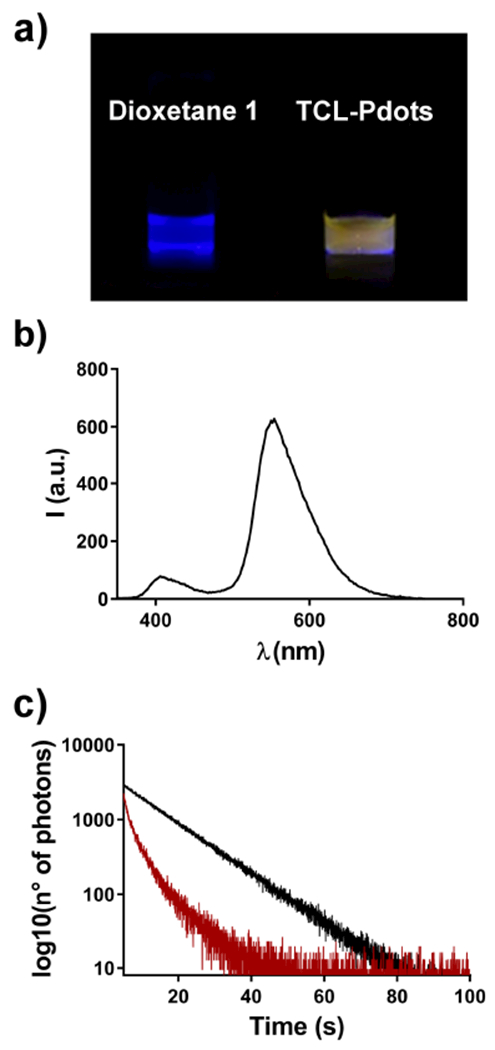
a) Images taken from a solution of 1,2-dioxetane 1 in DMSO (left side) and TCL-Pdots (right side) after heating above 100°C; b) TCL emission spectrum for a solution of dioxetane 1 and CN-PPV polymer in THF; c) Lifetime measurements showing the fluorescence decay of ketone 2 (black line) and 2 in the presence of energy acceptor CN-PPV (red line).
To estimate the efficiency of the FRET process, we measured the fluorescence lifetimes of ketone 2 in NPs with or without CN-PPV polymer. In detail, we conjugated compound 2 with amino-functionalized polystyrene (PS-NH2) and synthesized two different PS-NH2-2-based NPs that either contained or did not contain the energy acceptor polymer CN-PPV (see Supporting Information). The analysis of emission decay profiles (Figure 3c) has shown a remarkable decrease for the lifetime of excited ketone 2 in the presence of CN-PPV. In fact, for NPs containing only ketone 2 we measured a lifetime of 12.9 ns (τ0), which dropped down to 1.3 ns (τ1) when the semiconducting polymer was blended together. Using the equation 1,52 we calculated an efficiency for the energy transfer process (ϕFRET) of about 90%.
| (1) |
As an alternative approach, we also estimated the efficiency of the FRET process by TCL measurements. Upon heating TCL-Pdots at 110°C, we separately measured the light emitted by the excited ketone 2 and the CN-PPV polymer using a CCD camera and bandpass optical filters with transmission at 425 ± 20 nm (ketone 2 emission) and 600 ± 40 nm (CN-PPV emission). By taking also into account the different fluorescence quantum yields of the two species, the comparison of the two emission intensities allowed to estimate a FRET efficiency of about 81 ± 3%. This result is in good agreement with the value of ϕFRET obtained by lifetime measurements.
Based on the estimated ϕFRET value, we calculated an overall ϕTCL value by multiplying ϕFRET to ϕF of CN-PPV and to ϕS (excited singlet state formation quantum yield) reported for adamantylidene adamantane 1,2-dioxetane.53 We obtained ϕTCL of about 0.01, although we expect the actual value to be significantly higher. Indeed, the entrapping of the dioxetane molecule in the nanoparticle should favor the formation of the excited states upon thermolysis and that substitution of the adamantyl moiety with N-substituted acridine-containing 1,2-dioxetanes increases the efficiency of formation of the singlet excited state.54
Once assessed the efficiency of the FRET process, we estimated the number of TCL molecules entrapped in each Pdot. By comparing the TCL signals of TCL-Pdots and 1,2-dioxetane 1 upon normalization for concentration we found a single Pdot to entrap around 20 1,2-dioxetane molecules (see Supporting Information). Considering both the high efficiency of the FRET process (ϕFRET = 81%) and the fluorescence quantum yields of CN-PPV NPs and ketone 2 (60% and 11%, respectively), and taking into account the mass-effect due to the number of TCL molecules entrapped in each Pdot, it can be concluded that entrapment of the 1,2-dioxetane 1 in CN-PPV NPs should significantly enhance the intensity of the TCL emission. Indeed, it can be estimated that the TCL emission of a single TCL-Pdot is 90-fold higher than that of 1,2-dioxetane 1 molecule, thus providing improved label detectability.
2.3. TCL activation parameters for the thermal decomposition of 1,2-dioxetane 1 in Pdots
The thermal decomposition of 1,2-dioxetanes follows a first-order kinetic and can be analyzed according to a standard Arrhenius equation (equation 2) to evaluate the activation parameters of the decomposition reaction. In such equation, k represents the kinetic constant of the thermal decomposition reaction and A and Ea are its preexponential factor and activation energy, respectively.
| (2) |
In previous works regarding acridin-based 1,2-dioxetanes entrapped in silica NPs,21,23,26 we noticed a strong dependence of both the activation energy and preexponential factor of the thermal decomposition reaction on the surrounding environment. Thus, we investigated the effect of the polymeric environment of Pdots on the thermal decomposition of the 1,2-dioxetane 1. By acquiring the emission decay profiles at different temperatures, we obtained the kinetic constants of the TCL process and then we calculated both Ea and lnA from the Arrhenius plot (Figure 4a). We found that the lnA and Ea activation parameters of 1,2-dioxetane 1 in CN-PPV Pdots were 21.1 ± 2.4 kcal mol−1K−1 and 21.4 ± 3.1 respectively. Such values were significantly lower than the values measured for the 1,2-dioxetane 1 in the solid state,27 as already found for compound 1 entrapped in silica NPs. However, the thermal decomposition reaction in silica NPs was much faster than that in Pdots. The emission from TCL-Pdots was still detectable even after 30 min at 110°C (Figure 4b), while the TCL emission in silica NPs totally disappeared in less than 3 min at 90°C.21 On the other hand, the much slower emission decay of TCL-Pdots could be ascribed to the lower value of the preexponential factor, which prevails over the decrease of the Ea term in the Arrhenius equation.
Fig. 4.
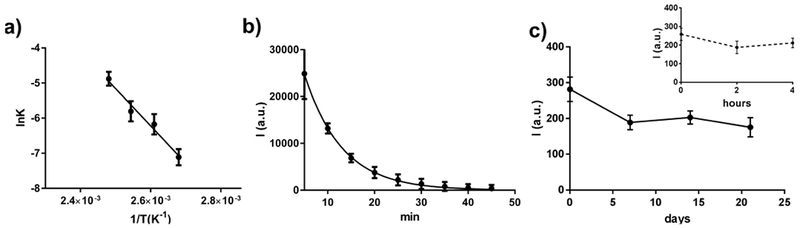
a) Arrhenius plot obtained from thermal decomposition studies of dioxetane 1, displaying the logarithm of kinetic constants at different temperatures against the inverse of temperature; b) TCL emission decay of substrate 1 inside Pdots, occurring at 110°C; c) TCL signal acquired to assess the stability of TCL-Pdots-SA over time, showing the emission of nanoparticles solution kept at 4 (solid line) or 37°C (dashed line) for different times.
2.4. Biofunctionalization of TCL-Pdots with Streptavidin
TCL-Pdots were then conjugated to SA following a previously reported procedure,40 in which 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) promotes a condensation reaction between the carboxylic units of PS-COOH and the amino groups of SA (for more details see Supporting Information). Choosing SA as bioactive moiety, we can exploit the high affinity of SA for Biotin to label biotinylated bioprobes with TCL-Pdots, thus making them detectable by TCL.
2.5. Stability of TCL-Pdots-SA
To be suitable as labels for bioanalytical applications, TCL-Pdots-SA should demonstrate a good stability in storage conditions and throughout all the operational steps. In Figure 4c, we show the changes of TCL signal observed in TCL-Pdots-SA solutions kept at 4°C (storage conditions) or 37°C for different times. It can be clearly seen that TCL-Pdots-SA were quite stable over time. The loss in TCL signal intensity was lower than 30% after 20 days at 4°C and most of the decrease took place in the first days, which could be ascribed to a leak of dioxetane 1 from the hydrophobic NPs. Indeed, we assume that while 1,2-dioxetanes which reside deep in the polymer matrix are held back tightly, the few 1,2-dioxetane molecules entrapped in the outer layer of the Pdot might slowly diffuse into aqueous solution. This aspect will need to be better investigated in future work. Furthermore, the TCL signal did not significantly diminish after 4-h incubation at 37°C.
Comparing these results with the previously described silica-based TCL NPs,23 we concluded that TCL-Pdots showed a remarkably higher stability, thus suggesting their suitability as labels for biosensing. This finding is in line with the lifetimes estimated from the thermal decomposition activation parameters (for example, at 4°C the estimated lifetimes are 180 and 2 days for Pdots and silica NPs, respectively). A further factor that might contribute to the higher stability of TCL Pdots could be related to the high light-harvesting capacity of CN-PPV polymer and the consequent stabilization of entrapped photolabile species.55 Indeed, in previous experiments we observed that dioxetane 1 slowly decomposes upon light irradiation. Thus, the polymer matrix might act as filter, partially shielding the entrapped dioxetanes from external ambient light and contributing to improve its stability.
2.6. Evaluation of TCL-Pdot-SA as universal label for binding assays exploiting biotinylated bioprobes
The applicability of TCL-Pdots-SA in bioassays was tested in a proof-of-concept immunoassay for the determination of biotinylated targets, using a biotin-labelled immunoglobulin G (Biotin-IgG) as a model analyte. We combined TCL-based detection with the biofunctionalized magnetic bead (MB) technology to build up a homogeneous, non-competitive sandwich-type immunoassay (Figure 5a). Briefly, upon blocking with BSA to avoid any non-specific binding, Anti-IgG magnetic beads (Anti-IgG-MBs) were incubated with solutions of the target analyte (Biotinylated IgG) at different concentrations. After a washing step to remove unbound Biotin-IgG, an excess of TCL-Pdots-SA nanoprobes was added to the solutions to bind Biotin-IgG. Upon additional washing, the TCL-Pdots-SA/Biotin-IgG/Anti-IgG-MBs complex was detected by TCL in an aliquot of the solution using a CCD camera. The entire immunoassay analysis was performed in about 4 h, which is a time comparable to common CL- or BL-based immunoassays.
Fig. 5.
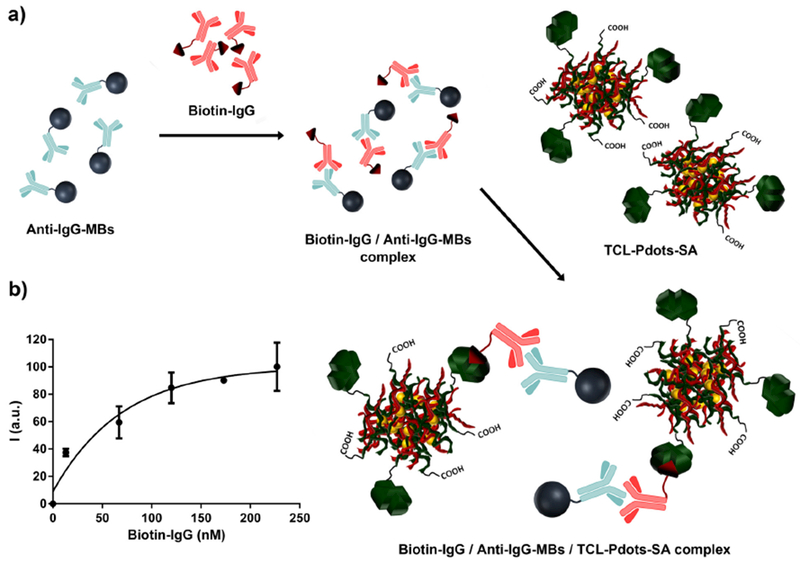
a) Representation of the proof-of-concept non-competitive sandwich-type immunoassay for detection of IgG, b) Calibration curve obtained from the TCL-Pdots-SA based immunoassay. TCL signal was calculated as the mean of three independent measurements.
The sandwich immunoassay developed for detection of Biotinylated IgG has shown a TCL signal proportional to the analyte concentration (Figure 5b) in the nanomolar range. Furthermore, although it was just a proof-of-concept to demonstrate the applicability of TCL-Pdots-SA nanoprobes as labelling reagents in bioanalyses, a quite low limit of detection (i.e. 13 nM of Biotinylated IgG) was reached.
3. Experimental section
3.1. Materials
All chemicals were purchased from Sigma Aldrich and used without additional purifications. Cyano-polyphenylene vinylene (CN-PPV, MW= 20 - 100 kDa) was purchased from American Dye Source, Inc. while polystyrene derivatives (PS-COOH, 50 kDa and PS-NH2, 6.5 kDa) was purchased from Polymer Source, Inc. Biotinylated Mouse IgG was purchased from Abcam, while Anti-IgG magnetic beads were purchased from New England BioLabs, Inc.
3.2. Synthesis of TCL-Pdots-SA
Substrate 1 was synthesized following a previously reported procedure (for more details see Supporting Information).27 The 1,2-dioxetane 1-doped Pdots were prepared following a co-nanoprecipitation method.43 In a typical procedure, stock solutions of CN-PPV (1 mg/mL), PS-COOH (1 mg/mL) and substrate 1 (2 mg/mL) in THF were prepared. Then, different aliquots from each stock solution were mixed together and THF was added to adjust the final volume. The final THF solution was quickly injected into 10 mL of water under vigorous sonication for about 1 min. For the optimization studies, we kept constant the CN-PPV amount while changing the amount of PS-COOH, dioxetane 1 or THF injected. After sonication, the extra THF was evaporated under nitrogen flow at room temperature and THF-free Pdot solutions were filtrated through a 0.40 μm cellulose membrane filter. TCL-Pdots were bioconjugated to Streptavidin (SA) following a EDC catalyzed condensation reaction between the carboxylic units of polystyrene chains and amino groups of SA protein. Specifically, polyethylene glycol (5% w/v PEG, MW 3350), Streptavidin (2 mg/mL) and a solution of EDC (10 mg/mL in MilliQ water) were added to a solution of TCL-Pdots (80 ppm in MilliQ water) and the pH was adjusted to 7.3 using HEPES buffer (1 M). The mixture was stirred at room temperature for 3 h and then a bovine serum albumin (BSA) solution (10% (w/v)) was added to the Pdots solution and the mixture was stirred for an additional 30 minutes. Finally, Triton X-100 (2.5% (w/v)) was added to the mixture of TCL-Pdots-SA to stabilize the nanoparticles solution. The resulting streptavidinated TCL-Pdots were concentrated by centrifugation through a 100 K molecular cut-off membrane, then purified by size exclusion chromatography using Sephacryl HR-300 gel media. Further details on TCL-Pdots functionalization with Streptavidin are provided in the Supporting Information.
3.3. TCL emission measurements and FRET experiments
The estimation of FRET efficiency by TCL emission measurement was conducted by separately acquiring the light emitted from ketone 2 or CN-PPV, using two different bandpass optical filters centered at 425 nm and 600 nm, respectively. Here, 3 μL of TCL-Pdots-SA solution was spotted onto the mini-heating element (ceramic resistance connected to a power supply) and heated at 110°C, while acquiring the TCL signal through a portable CCD camera. Both the heating pad and the CCD were placed inside a dark box, to avoid the background signal from the external ambient light. Then, the intensities of light at 425 and 600 nm were compared considering the different ϕF of ketone 2 and CN-PPV as well as the spectral response of CCD’s sensor at different wavelengths (for more details see Supporting Information).
For the TCL emission spectrum acquisition, a 200 μL solution of 1,2-dioxetane 1 (6 mM) and CN-PPV (0.07 mM) in THF was prepared. The glass vial containing the mixture was positioned onto a mini-heater element and centered to the spectrofluorimeter’s detector. Then, the solution was heated up to 110°C while acquiring the TCL emission spectrum. Because of the low intensity of TCL signal, the emission spectrum was obtained as sum of multiple scans. The lifetime of ketone 2 was measured from a solution of PS-NH-2 nanoparticles (90 ppm), while the efficiency of the FRET mechanism was determined by analyzing a solution of PS-NH-2/CN-PPV NPs (70 ppm). FRET experiments were performed using a FluoTime 100 spectrometer, setting the time resolution of correlated single photon counting system to 32 ps and using a 375 nm laser for excitation of ketone 2. To prevent photons emitted by excited CN-PPV from being collected, a band pass filter (centered at 425 ± 25 nm) was positioned between the sample and detector. The experimental data were analyzed using the open source software DecayFit. A complete description for the synthesis of ketone 2-doped Pdots is provided in the Supporting Information.
3.4. Determination of the activation parameters (Ea and lnA) of 1,2-dioxetane 1 inside Pdots
Activation parameters of 1,2-dioxetane 1 inside Pdots were calculated by measuring the TCL emission decay kinetics at different temperatures. Specifically, 3 μL of TCL-Pdots solution was spotted onto an aluminum foil covering a homemade mini-heater (4×5 cm2 electrical resistance encapsulated between two kapton layers and connected to a power supply) and the spot was allowed to dry. Then, the foil was heated up to the desired temperature (in the range between 100°C and 130°C), and a sequence of images of TCL emission was acquired, using a CCD camera. For each temperature, the kinetic constant k was calculated by fitting the TCL emission decay profile with the first-order decay equation shown in equation 3, in which ITCL is the TCL signal at time t and (ITCL)0 is the TCL signal at time zero (t0).
| (3) |
Values of the kinetic constants of TCL emission decay are listed in the Supporting Information.
3.5. Non-competitive immunoassay for detection of IgG
For the immunoassay experiments, a solution of Goat Anti-Mouse IgG Magnetic Beads (Anti-IgG MBs, 20 mg/mL) and Biotinylated Immunoglobulin G (Biotin-IgG, 0.5 mg/mL) were used. Specifically, different sample batches were prepared, adding an increasing volume of Biotin-IgG to a constant amount of Anti-IgG MBs, and PBS-1X buffer was added to adjust the final volume of each sample to 500 μL. After 2 h of incubation at 37°C, MBs were washed three times to remove all the free Biotin-IgG, and the same volume of a TCL-Pdots-SA solution was added to each sample. Biotin-IgG MBs were incubated with TCL-Pdots-SA at 37°C for an additional 2 hours, after which MBs were washed three times and dispersed in the elution buffer (solution of glycine, pH = 10) for 15 minutes at room temperature. A BSA solution was used to avoid any non-specific interaction during the incubation step. The calibration curve was obtained by TCL emission measurements, acquiring the light generated from a 10 μL of eluted solution, heated at 110°C for 15 min. Further details on TCL-PDots-SA based immunoassay are provided in the Supporting Information.
4. Conclusions
In summary, we described the synthesis of the first TCL-Pdot nanoprobe for biosensors and binding bioassays. Following a co-nanoprecipitation method, we obtained quite monodisperse CN-PPV-based TCL-Pdots entrapping the 1,2-dioxetane 1 as TCL molecule, which were subsequently functionalized with SA to obtain TCL-Pdot-SA nanolabels suitable for the detection of biotinylated targets. We demonstrated that in the TCL-Pdots a highly efficient FRET process takes place from the embedded TCL molecules to the surrounding CN-PPV polymer matrix, which resulted in a bright TCL emission thanks to the strong fluorescence of CN-PPV. Furthermore, TCL-Pdots-SA are remarkably stable, showing a minimal TCL signal loss under storage conditions as well as during performance of the assay. We tested the nanoprobes in a proof-of-concept non-competitive immunoassay for detection of Biotinylated IgG, which showed a LOD in the order of 10 nM Biotinylated IgG. We therefore propose TCL-Pdots based systems as a valid alternative to the classical luminescent probes used so far in bioassays. In fact, taking advantage of the peculiar characteristics of Pdots (high fluorescence quantum yields, tunability of the emission spectrum, biocompatibility) and TCL-based detection (high signal/noise ratio, no reagents required for triggering the luminescent reaction, simple instrumentation since only a mini-heater pad and a portable CCD are required for light measurement) a broad panel of bright TCL nanoprobes could be designed and used to develop highly sensitive immunoassays.
Supplementary Material
Acknowledgements
A.R gratefully thanks Chun-Ting Kuo for performing TEM analysis of TCL-Pdots nanoparticles D.T.C. gratefully acknowledge support from the National Institutes of Health (R01EB021150).
Footnotes
Electronic Supplementary Information (ESI) available: Synthesis of 1,2-dioxetane 1, TCL-Pdots-SA and 2-doped nanoparticles, spectroscopic characterization of both CN-PPV polymer and 1,2-dioxetane 1 and estimation of the number of TCL substrates per NP, kinetic constants for thermal decomposition of 1 and determination of ϕFRET through TCL emission measurements, non-competitive TCL-Pdots-SA immunoassay. See DOI: 10.1039/b000000x/
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Experts I, Biosensors - A Global Market Overview, 2017, http://industry-experts.com/verticals/biotechnology/biosensors-a-global-market-overview.
- 2.Roda A, Michelini E, Caliceti C, Guardigli M, Mirasoli M and Simoni P, Analytical and Bioanalytical Chemistry, 2018, 410, 669–677. [DOI] [PubMed] [Google Scholar]
- 3.Nayak S, Blumenfeld NR, Laksanasopin T and Sia SK, Analytical Chemistry, 2017, 89, 102–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narsaiah K, Jha SN, Bhardwaj R, Sharma R and Kumar R, Journal of Food Science and Technology, 2012, 49, 383–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson RH, Vishwanath K and Mycek M-A, Advances in Physics: X, 2016, 1, 523–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alemohammad H, Opto-mechanical Fiber Optic Sensors: Research, Technology, and Applications in Mechanical Sensing, Elsevier Science, 2018. [Google Scholar]
- 7.Heijmans JAC, Cheng LK and Wieringa FP, 4th European Conference of the International Federation for Medical and Biological Engineering, Berlin, Heidelberg, 2009, pp. 2330–2334. [Google Scholar]
- 8.Geddes C, Reviews in Fluorescence 2016, Springer International Publishing, 2017. [Google Scholar]
- 9.Strianese M, Staiano M, Ruggiero G, Labella T, Pellecchia C and D’Auria S, in Fluorescence-Based Biosensors, ed. Bujalowski WM, Humana Press, Totowa, NJ, 2012, pp. 193–216. [DOI] [PubMed] [Google Scholar]
- 10.Gong X, Cai J, Zhang B, Zhao Q, Piao J, Peng W, Gao W, Zhou D, Zhao M and Chang J, J. Mater. Chem. B, 2017, 5, 5079–5091. [DOI] [PubMed] [Google Scholar]
- 11.Goldys E, Fluorescence Applications in Biotechnology and Life Sciences, Wiley, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Wolfbeis OS, Chem. Soc. Rev, 2015, 44, 4743–4768. [DOI] [PubMed] [Google Scholar]
- 13.Crosignani V, Dvornikov A, Aguilar JS, Stringari C, Edwards R, Mantulin WW and Gratton E, Journal of biomedical optics, 2012, 17, 116023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fei X and Gu Y, Progress in Natural Science, 2009, 19, 1–7. [Google Scholar]
- 15.Mirasoli M, Guardigli M, Michelini E and Roda A, Journal of Pharmaceutical and Biomedical Analysis, 2014, 87, 36–52. [DOI] [PubMed] [Google Scholar]
- 16.Roda A, Mirasoli M, Michelini E, Fusco MD, Zangheri M, Cevenini L, Roda B and Simoni P, Biosensors and Bioelectronics, 2016, 76, 164 – 179. [DOI] [PubMed] [Google Scholar]
- 17.Kirschbaum SEK and Baeumner AJ, Analytical and bioanalytical chemistry, 2015, 407, 3911–3926. [DOI] [PubMed] [Google Scholar]
- 18.Turro NJ, Lechtken P, Schore NE, Schuster G, Steinmetzer HC and Yekta A, Accounts of Chemical Research, 1974, 7, 97–105. [Google Scholar]
- 19.Wieringa J, Strating J, Wynberg H and Adam W, Tetrahedron Letters, 1972, 13, 169 – 172. [Google Scholar]
- 20.Hummelen JC, Luider TM and Wynberg H, in Bioluminescence and Chemiluminescence Part B, Academic Press, 1986, vol. 133 of Methods in Enzymology, pp. 531 – 557. [DOI] [PubMed] [Google Scholar]
- 21.Roda A, Di Fusco M, Quintavalla A, Guardigli M, Mirasoli M, Lombardo M and Trombini C, Analytical Chemistry, 2012, 84, 9913–9919. [DOI] [PubMed] [Google Scholar]
- 22.Di Fusco M, Guardigli M, Lombardo M, Mirasoli M, Quintavalla A, Roda A and Trombini C, Method for the production of thermochemiluminescent silica nanoparticles and their use as markers in bioanalytic methods, 2014, https://www.google.com/patents/WO2014024106A1?cl=en, WO Patent App. PCT/IB2013/056,340.
- 23.Di Fusco M, Quintavalla A, Lombardo M, Guardigli M, Mirasoli M, Trombini C and Roda A, Analytical and Bioanalytical Chemistry, 2015, 407, 1567–1576. [DOI] [PubMed] [Google Scholar]
- 24.Mirasoli M, Bonvicini F, Dolci LS, Zangheri M, Gallinella G and Roda A, Analytical and Bioanalytical Chemistry, 2013, 405, 1139–1143. [DOI] [PubMed] [Google Scholar]
- 25.Roda A, Michelini E, Zangheri M, Di Fusco M, Calabria D and Simoni P, TrAC Trends in Analytical Chemistry, 2016, 79, 317 – 325. [Google Scholar]
- 26.Di Fusco M, Quintavalla A, Trombini C, Lombardo M, Roda A, Guardigli M and Mirasoli M, The Journal of Organic Chemistry, 2013, 78, 11238–11246. [DOI] [PubMed] [Google Scholar]
- 27.Andronico LA, Quintavalla A, Lombardo M, Mirasoli M, Guardigli M, Trombini C and Roda A, Chemistry - A European Journal, 2016, 22, 18156–18168. [DOI] [PubMed] [Google Scholar]
- 28.Sun K, Tang Y, Li Q, Yin S, Qin W, Yu J, Chiu DT, Liu Y, Yuan Z, Zhang X and Wu C, ACS Nano, 2016, 10, 6769–6781. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Wu L, Wu I-C and Chiu DT, RSC Advances, 2016, 6, 103618–103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu L, Wu I-C, DuFort CC, Carlson MA, Wu X, Chen L, Kuo C-T, Qin Y, Yu J, Hingorani SR and Chiu DT, Journal of the American Chemical Society, 2017, 139, 6911–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuo C-T, Gallina ME, Ye F, Johnson ES, Sun W, Zhao M, Yu J, Wu I-C, Fujimoto B, DuFort CC and et al. , Nature Communications, 2016, 7, 11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Li R, Liu Z, Sun K, Sun Z, Chen D, Xu G, Xi P, Wu C and Sun Y, Advanced Materials, 2017, 29, 1604850. [DOI] [PubMed] [Google Scholar]
- 33.Sun W, Yu J, Deng R, Rong Y, Fujimoto B, Wu C, Zhang H and Chiu DT, Angewandte Chemie International Edition, 2013, 52, 11294–11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu C, Jin Y, Schneider T, Burnham DR, Smith PB and Chiu DT, Angewandte Chemie International Edition, 2010, 49, 9436–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu C and Chiu DT, Angewandte Chemie International Edition, 2013, 52, 3086–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu J, Rong Y, Kuo C-T, Zhou X-H and Chiu DT, Analytical Chemistry, 2017, 89, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liou S-Y, Ke C-S, Chen J-H, Luo Y-W, Kuo S-Y, Chen Y-H, Fang C-C, Wu C-Y, Chiang C-M and Chan Y-H, ACS Macro Letters, 2016, 5, 154–157. [DOI] [PubMed] [Google Scholar]
- 38.Rong Y, Wu C, Yu J, Zhang X, Ye F, Zeigler M, Gallina ME, Wu I-C, Zhang Y, Chan Y-H and et al. , ACS Nano, 2013, 7, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu I-C, Yu J, Ye F, Rong Y, Gallina ME, Fujimoto BS, Zhang Y, Chan Y-H, Sun W, Zhou X-H and et al. , Journal of the American Chemical Society, 2015, 137, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rong Y, Yu J, Zhang X, Sun W, Ye F, Wu I-C, Zhang Y, Hayden S, Zhang Y and Wu C. t., ACS Macro Letters, 2014, 3, 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen D, Wu I-C, Liu Z, Tang Y, Chen H, Yu J, Wu C and Chiu DT, Chem. Sci, 2017, 8, 3390–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Yu J, Rong Y, Ye F, Chiu DT and Uvdal K, Chem. Sci, 2013, 4, 2143–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C, Peng H, Jiang Y and McNeill J, The Journal of Physical Chemistry B, 2006, 110, 14148–14154. [DOI] [PubMed] [Google Scholar]
- 44.Genovese D, Rampazzo E, Bonacchi S, Montalti M, Zaccheroni N and Prodi L, Nanoscale, 2014, 6, 3022–3036. [DOI] [PubMed] [Google Scholar]
- 45.Ye F, Wu C, Jin Y, Wang M, Chan Y-H, Yu J, Sun W, Hayden S and Chiu DT, Chemical communications, 2012, 48, 1778–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun W, Ye F, Gallina ME, Yu J, Wu C and Chiu DT,Analytical Chemistry, 2013, 85, 4316–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Owens DE, Jian Y, Fang JE, Slaughter BV, Chen Y-H and Peppas NA, Macromolecules, 2007, 40, 7306–7310. [Google Scholar]
- 48.Gandhi A, Paul A, Sen SO and Sen KK, Asian Journal of Pharmaceutical Sciences, 2015, 10, 99 – 107. [Google Scholar]
- 49.Wu C and McNeill J, Langmuir, The ACS Journal of Surfaces and Colloids, 2008, 24, 5855–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miladi K, Sfar S, Fessi H and Elaissari A, in Nanoprecipitation Process: From Particle Preparation to In Vivo Applications, ed. Vauthier C and Ponchel G, Springer International Publishing, Cham, 2016, pp. 17–53. [Google Scholar]
- 51.Jin Y, Ye F, Zeigler M, Wu C and Chiu DT, ACS Nano, 2011, 5, 1468–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lakowicz J, Principles of Fluorescence Spectroscopy, Springer; US, 2007. [Google Scholar]
- 53.Schuster GB, Turro NJ, Steinmetzer HC, Schaap AP, Faler G, Adam W and Liu JC, Journal of the American Chemical Society, 1975, 97, 7110–7118. [Google Scholar]
- 54.Lee C and Singer LA, Journal of the American Chemical Society, 1980, 102, 3823–3829. [Google Scholar]
- 55.Reisch A and Klymchenko AS, Small, 12, 1968–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.