

Abstract
Multiple sclerosis (MS) arises from an immune attack on the central nervous system producing demyelination and axonal loss. Clinically the relapsing–remitting course is characterized by subacute onset of neurological symptoms usually with partial or complete recovery, while the progressive course, predominant in the later stages, is characterized by progressive disability in the absence of relapses. A number of disease-modifying treatments have been developed and are increasingly effective at targeting relapses. Early injectable therapies such as interferon and glatiramer acetate are only partially effective, but have a good safety record. Recently, natalizumab, an intravenous therapy, demonstrated increased effectiveness, but side effects complicate its use. The first oral therapy offering good efficacy and convenience, fingolimod, was approved in USA in 2010 and Europe in 2011. BG-12 is a potential novel oral therapy for MS, which has previously been used as a different formulation for psoriasis. It has anti-inflammatory and neuroprotective actions in vitro, which makes it a promising candidate for future therapies. Phase II studies showed that BG-12 reduced MRI inflammatory activity over placebo, which was confirmed in two Phase III studies indicating immune modulation may be its principal action rather than neuroprotection. In these studies, BG-12 reduced relapse rates consistently with variable effects on progression and few serious adverse events. With its favorable efficacy–tolerability profile, BG-12 could offer a substantial step forward for the care for subjects affected by relapsing MS.
Keywords: BG-12, multiple sclerosis, relapses, oral treatments
Treating multiple sclerosis
Multiple sclerosis (MS) is one of the commonest causes of disability in young adults.1 A therapy that could prevent these effects would have a long-term positive impact on people lives.
The pathological characteristics of MS, inflammatory demyelination and axonal loss,2,3 are believed to result from an autoimmune attack on the central nervous system (CNS), this concept represents the driver of much new drug development. Both the innate and adaptive immune systems are implicated. The former represents the first line of defense, but is also implicated in the progressive phase of the disease.4 Adaptive immunity, a driver of the principal model of MS, experimental autoimmune encephalomyelitis (EAE),5,6 in turn is believed to drive predominantly relapse activity.7,8
In the natural history of MS ~85% of subjects initially present with a clinically isolated syndrome (CIS),9 a single clinical episode suggestive of MS. Radiology at diagnosis or a subsequent clinical or radiological event indicates a relapsing–remitting course (RR). The remaining 5%–15% of patients have a progressive course from the beginning termed primary progressive (PP), characterized by slow accumulation of nonrecoverable disability, though this can be difficult to confirm using current diagnostic criteria. This progression can develop in the later stages in about 32%–65% of those with relapsing onset (secondary progressive, SP).10–12 After the onset of progression, the disease follows a uniform course, whether primary or secondary.13,14
It is currently accepted that inflammation, predominantly arising from adaptive immune activation, represents the pathological correlate of the clinical phenomenon of the relapse, whereas neurodegeneration, whether inflammatory (innate) or truly neurodegenerative, is the most relevant pathological process underlying progression.15–20
Relapses
Though the influence of relapses on the final outcome in MS is debated,21 the relapsing course is highly prevalent in early MS and, as the most easily measureable clinical effect of MS, reduction in attack frequencies is used as a way of expressing an impact on disease activity.22 In pharmaceutical trials, it is most commonly expressed as annualized relapse rate (ARR, number of relapses in a period of time extrapolated to 12 months). Their impact on patients and caregiver quality of life (QoL) and their social costs, justifies their use in relapsing MS populations.23 However a remarkable change has occurred in the ARR in untreated trial patients over the last three decades principally due to the availability of licensed therapies.24,25 This progressive reduction in ARR makes it difficult to compare the relevant impacts of therapies on RRMS subjects.
Disability progression
The progressive worsening of disability without recovery represents the phase of MS that has the highest costs for patients, their relatives/caregivers, and society. Epidemiological studies show that after the onset of clinical progression, patient’s disability reaches the Expanded Disability Status Scale (EDSS) score milestone of 6.0 (walking with a cane) in ~8.1 years and the EDSS milestone of 8.0 (bedbound) is reached in ~18.6 years.10,13 During the progressive course of MS, patients are often forced to stop working and the impact on mobility implies secondary complications and an increasing dependence for activities of daily living, with subsequent psychological and financial costs affecting their relatives/caregivers as well as society.26,27
MRI: a biologically plausible outcome measure for MS trials
Over the last three decades, magnetic resonance imaging (MRI) has become the most important paraclinical tool supporting MS diagnosis28,29 and prognosis. MS lesions appear hyperintense on T2 sequences and gadolinium (Gd) contrast administration allows the visualization of blood–brain barrier (BBB) leakage, a sign of active CNS inflammation whereas T1 black holes correlate to an extent with persistent structural damage. The MRI has an emerging role in the earliest phases of MS, identifying clinically silent lesions; allowing MS to be diagnosed even after a single episode.30 Subsequent MRI activity (increased number, volume of T2 lesions or Gd enhancement) and its correlation with relapses,31 allows the diagnostic tool to identify MS as a biologically plausible outcome.32
Reducing ARR during the early phases of MS and any delay in the onset of the progressive course represents two events that need to be impacted to improve the QoL of patients/caregivers, and minimize the social costs of MS. While reducing absolute ARR has an impact on costs, social costs predominate in the latter part of the disease and contribute to the majority of the costs in economic models.33,34 Therefore disability delay is desirable for new therapies aiming at major cost-saving efficacy in cost-based models.
Disease-modifying treatments for MS
Evidence implicating the immune system in the MS pathogenesis led to the concept of “immunomodulatory treatment” for MS. Subsequent to the first trial demonstrating the efficacy of interferon (IFN)β–1b as a disease-modifying treatment (DMT),35 other compounds including other IFNs, glatiramer acetate and latterly natalizumab have been shown to be effective in RRMS patients.36–40 The effectiveness resides in the reduction in ARR, and a reduction in T2, T1 black hole, and Gd-enhancing MRI lesion accumulation; however, their impact on the disease progression aside from an effect on relapses has not been definitively proven (Table 1). In CIS, the IFNs can reduce the probability of progression to clinically definite MS41 and lower relapse rates.42
Table 1.
Consistency of clinical and radiological outcomes between oral therapies compared to current licensed therapies for RRMS; not from comparative studies
| Trial (length years) | Relapses
|
Progression
|
||
|---|---|---|---|---|
| Clinical: ARR | MRI: T2/Gd-enhancing lesions | Clinical: 12 week progression | MRI: T1 black holes | |
| BG-12 DEFINE (2) | Y, ✓ | Y, ✓ | Y | Y |
| BG-12 CONFIRM (2) | Y, ✓ | Y, ✓ | N, # | Y, # |
| Fingolimod FREEDOMS (2) | Y, ✓ | Y, ✓ | Y, ✓ | Y, ✓ |
| Fingolimod TRANSFORMS (1) | Y, ✓ | Y, ✓ | N, ✕ | N, ✕ |
| IFNβ (3) | Y, ✓ | Y, ✓ | N, ✕ | N, ✕ |
| GA (2) | Y, # | N, # | N, ✕ | N, ✕ |
| Natalizumab (2) | Y, ✓ | Y, ✓ | Y, # | N, # |
Notes: Y, positive effect; N, no effect; ✓, positive effect and consistent; #, inconsistent results between outcomes; ✕, no effect in both outcomes.
Abbreviations: ARR, annualized relapse rate; MRI, magnetic resonance imaging; GA, glatiramer acetate; Gd, gadolinium; IFN, interferon; RRMS, relapsing remitting multiple sclerosis.
The first oral medication licensed for RRMS patients was fingolimod, as a first-line therapy in RRMS in the USA in 2010 and as a second-line therapy or in active disease in Europe in 2011. Compared to injectable treatments, recent oral DMTs have had a beneficial impact on patients’ QoL, both in terms of side effects and lack of need for injection administration.
The mechanism of action of BG-12
BG-12 is the oral formulation of the dimethylfumarate (DMF) used for the treatment of severe chronic plaque psoriasis as fumaric acid esters (Fumaderm®; Fumapharm, Muri, Switzerland). Fumaderm is a mixture of dimethylester fumarate and salts of ethylhydrogene fumarate. Clinical trials have shown the efficacy and safety of this medication in psoriasis.43–47 BG-12 is a second-generation fumaric acid compound and contains, in contrast to Fumaderm, only DMF in enteric-coated microtablets and is associated with fewer gastrointestinal adverse effects.48 The fumaric acid esters (FAE) have been reported to be effective immunomodulators and have neuroprotective properties; furthermore, the fact that it is an oral treatment with good safety and tolerability profile, makes it a promising treatment option in RRMS.
Pharmacokinetics
BG-12 is an oral formulation of enteric-coated DMF capsules. After the intake DMF is hydrolyzed to monomethylfumarate (MMF) in the small intestine, where it is absorbed, and eventually further metabolized by esterase activity in the hepatic system. This presystemic metabolism explains the negligible blood levels of DMF after oral intake and the transient rise in serum MMF.49 The small amount of DMF not hydrolyzed is converted to MMF by esterases in peripheral blood circulation and inside cells, partially forming glutathione (GSH) conjugates supported by the finding of mercapturic acids of MMF in urine samples of DMF-treated patients.50 It is also partially metabolized to carbon dioxide and water and eliminated mainly through breathing. The serum pharmacokinetic parameters of MMF are influenced by food intake. Fasting normal subjects have a mean time to peak concentration (tmax) of 178 minutes against 600 minutes in fed subjects, the mean peak concentration (Cmax) is respectively 0.8 and 0.46 mg/L−1. The terminal half-life is 56 minutes while the bioavailability, estimated as area under the plot of plasma concentration (AUC) is 1.54 Cl/F (L/min−1).51 There is no evidence for a cytochrome P450-dependent metabolism of FAE.52
Pharmacodynamics
Although MMF and DMF have been shown to affect two pathways, the mechanisms by which FAEs exert their biological effects are not clear.
The first pathway involves the activation of nuclear-factor-E2-related factor-2 (Nrf2) transcriptional pathway (Figure 1) which, entering the nucleus, can induce phase II genes, in particular NADPH:quinone oxidoreductase (NQO1) and GSH.53–55 Activation of this pathway reduces oxidative stress, especially in microglia and astrocytes.56–59
Figure 1.

Neuroprotective mode of action: MMF on Nrf2 Phase II genes activation, such as NQO1 (NAD(P)H:quinone oxidoreductase-1) and glutathione S-transferase.
Abbreviations: ARE, antioxidant-responsive element; MMF, monomethylfumarate; Nrf2, nuclear factor E2-related factor-2; Keap1, Kelch-like ECH-associating protein-1; UL, ubiquitin; Glutat, glutathione S-transferase.
The second pathway involves the activation of nuclear trans-location of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) (Figure 2) which interferes with the production of cytokines and cell growth regulation factors.60–62 This second pathway induces a switch from T-helper (Th) to Th2 phenotype in T cells,51,63 a reduction of B-cell lymphoma 2 expression in B cells,64 and impaired cell differentiation and apoptosis regulation in dendritic cells (DC).65,66 There is a subsequent impaired activation of Th1 cells, reducing the expression of Intercellular Adhesion Molecule 1 (ICAM-1), E-selectin and Vascular cell Adhesion Molecule-1 (VCAM-1) on endothelial cells.63,67
Figure 2.

Anti-inflammatory mode of action: DMF on NF-kB activation.
Abbreviations: DMF, dimethylfumarate; NF-kB, nuclear factor kappa B.
Neuroprotective attributes of BG-12
DMF and its primary metabolite MMF are able to modulate the Nrf2 transcriptional pathway, which is involved in oxidative stress response and immune homeostasis.53,54 Nrf2 is responsible for both constitutive and inducible expression of the antioxidant response element (ARE)-regulated genes, also known as the electrophile response element.55 The activation of genes coding for antioxidant proteins and phase II detoxifying enzymes that include NQO-1, glutathione peroxidase (GPx), ferritin, and heme oxygenase-1 (HO-1), is controlled by this cis-acting transcriptional regulatory element. Data suggest that Nrf2/ARE pathway can protect endothelial cells from oxidant-mediated injury and suppress key redox-sensitive inflammatory responses.54 Previous studies have shown that increasing Nrf2 activity is neuroprotective in mixed neuronal/glial cultures set up to simulate components of stroke damage.56,57,68–70 Nrf2 activation protects the brain from cerebral ischemia in vivo increasing intracellular GSH levels;71 it also regulates expression of inflammatory genes such as monocyte chemoattractant protein-1, VCAM-1, and tumor necrosis factor (TNF)-β through inhibition of p38 mitogen-activated protein kinase.54 Furthermore, enhancing expression of Nrf2-driven genes after brain injury attenuates the loss of endothelial cells, tight junction proteins and reduces BBB permeability and cerebral edema.72 It has been shown that Nrf2 modulates inducible and basal expression of numerous cytoprotective genes.73 Therefore Nfr2 could protect the BBB against injury by either increasing the basal levels of cytoprotective proteins or by increasing the endogenous endothelial cells’ response to injury. Nrf2-mediated neuroprotection involves several neuronal populations and glial cells through NQO1, which is present in these cell populations. Brain astrocytes derived from Nrf2 knockout mice do not induce phase II detoxifying enzyme gene expression and fail to protect neurons from in vitro toxicity paradigms when treated with small molecule inducers of Nrf2 activity.57,58,69 Nrf2 knockout mice show astrogliosis and myelinopathy in the cerebellum.74 In an EAE acute MS model with wild-type and knockout mice for Nrf2, the absence of Nrf2 exacerbated the development of EAE, suggesting Nrf2 activation may attenuate neuroinflammatory-degenerative diseases.75 Recently, one of the pathways influencing the expression of this neuroprotective factor has been reported. Briefly, the activation of Nrf2 depends on modification of free cysteine residues in the Nrf2-binding adaptor protein Keach-like ECH-associated protein 1 (Keap1) at cysteine residue 151. Keap1 targets Nrf2 for ubiquitin-mediated degradation leading to suppression of Nrf2.76 When Keap1 undergoes cysteine 151 modification, it becomes unable to interact with Nrf2, which accumulates in the nucleus inducing the expression of Nrf2-dependent antioxidant and cytoprotective genes.77
Anti-inflammatory attributes of BG-12
It has been assumed that the immunomodulatory effect is mediated by enhancing the Th2 polarization of CD4+ T cells.78 Indeed, MMF induces interleukin (IL)-10, IL-4, and IL-5 expression in peripheral blood mononuclear cells in vitro without modifying IFN-γ, IL-12 and IL-2 levels67 and MMF has been shown to increase in vitro the production of IL-4 and IL-5 in T cells.61 In addition, DMF can inhibit the transcription of pro-inflammatory cytokines such as IL-6, IL-1β, TNF-α51 and promote a downregulation of adhesion molecules ICAM-1 and/or E-selectin.63 DMF can also inhibit TNF-induced CD62E expression in vascular endothelial cells. CD62E is an inducible endothelial adhesion molecule and takes part in mechanisms governing tethering, rolling, and tight adhesion of leukocytes on endothelial cell surfaces. Inhibition of this cascade of events could result in reduced tissue inflammation. The mechanism of action is not fully understood, though DMF might inhibit NF-kB on which CD62E expression depends as DMF appears to have a dose-dependent specificity for NF-kB.79
Clinical efficacy of BG-12 in RRMS
The clinical efficacy of DMF has been assessed in two Phase III trials (Table 2), based on the results of a pilot study and a Phase IIb trial.
Table 2.
| BG-12 phase III trials
| ||||
|---|---|---|---|---|
| At 2 years
|
BG-12 240 mg twice daily vs placebo
|
BG-12 240 mg three times daily vs placebo
|
||
| Clinical outcome measure | Change | Significance | Change | Significance |
| DEFINE study | ||||
| Relapse risk | −49% | P < 0.0001 | −50% | P < 0.0001 |
| ARR | −53% | P < 0.001 | −48% | P < 0.001 |
| Risk of confirmed disability progression | −38% | P < 0.05 | −34% | P < 0.05 |
| MRI outcome measure | ||||
| Risk of new or newly enlarging T2 lesion | −85% | P < 0.0001 | −74% | P < 0.0001 |
| Patients free of new or newly enlarging T2 lesions (placebo 25%) | 45% | ns | 41% | ns |
| Odds of increased Gd-enhancing lesion activity | −90% | P < 0.0001 | −73% | P < 0.0001 |
| Patients free of Gd-enhancing lesion (placebo 62%) | 93% | 86% | ||
| New T1-hypointense lesions | −73% | P < 0.0001 | −63% | P < 0.0001 |
| QoL outcome measure | ||||
| SF-36 | P < 0.001 | P < 0.0001 | ||
| VAS | P = 0.003 | P < 0.0001 | ||
| CONFIRM study | ||||
| ARR | −44% | P < 0.0001 | −51% | P < 0.0001 |
| Proportion of patients experiencing MS relapses | −34% | P < 0.003 | −45% | P < 0.0001 |
| Confirmed disability progression* | −21% | ns | −24% | ns |
| MRI outcome measure | ||||
| Risk of new or newly enlarging T2 lesion | −71% | P < 0.0001 | −73% | P < 0.0001 |
| New T1-hypointense lesions | −57% | P < 0.0001 | −65% | P < 0.0001 |
Note:
At 24 weeks.
Early clinical studies
The initial pilot study80 was performed on 10 subjects with RRMS81 aged 18–55 years with at least one relapse within the 12 months before entering the study, at least one active lesion on brain MRI and an EDSS score between 2.0–6.0. This pilot was a prospective, open-label, baseline-controlled study over 70 weeks. The baseline phase was 6 weeks, followed by an 18-week treatment phase in which subjects received tablets of Fumaderm®, uptitrated over 9 weeks up to 720 mg/day. After a 4-week washout subjects were started on Fumaderm® up to 360 mg/day for 48 weeks. Assessment included: EDSS, ambulation index (AI) and nine-hole peg test (9-HPT) and Gd-MRI scan. Six patients completed the study, one was excluded due to pregnancy at week 46 and three subjects withdrew due to side effects and poor treatment compliance. The primary efficacy outcome was the number and volume of Gd-enhancing lesions. The mean number was reduced from 11.28 at baseline to 0.28 at week 70 (P < 0.02) and the median volume from 244.5 mm3 to 26.1 mm3 at the same time-points (P < 0.018). There was no significant change in the secondary clinical outcomes (EDSS score, AI, and 9-HPT).
The Phase IIb trial82 performed on 257 RRMS patients diagnosed according to the McDonald criteria83 had an initial 24 week multicenter, double-blind placebo-controlled phase when subjects were 1:1:1:1 randomized to receive placebo, BG-12 120 mg once daily, 120 mg three times daily, or 240 mg three times daily. In a subsequent 24-week second phase, subjects were treated with active medication at the same dosage and the placebo group was started with BG-12 at the highest dose. The primary efficacy outcome was the total number of new Gd-enhancing MRI lesions. Secondary endpoints were cumulative number of new Gd-enhancing lesions from week 4 to week 24, the number of new or enlarging T2 lesions and new T1-hypointense lesions. Clinically, the ARR and the proportion of relapse-free patients, as well as safety and tolerability, were assessed. There was a significant relative risk reduction in the high-dose BG-12 group against placebo in new Gd-enhancing lesions: 69% between week 12 and 24 (P < 0.0001); the number of new or enlarging T2 lesions: 48% during the first 24 weeks (P = 0.0006); and new T1-hypointense lesions: 53% during the first 24 weeks (P = 0.014). The ARR and the proportion of relapse-free patients were respectively 0.41 and 68% in the placebo group and 0.28 and 77% in the high-dose BG-12 group (no P value given). The two groups treated with low-dose BG-12 did not show any significant difference versus placebo for all endpoints. The most common adverse events (AEs) reported were flushing and headache (further to MS relapses). AEs were significantly higher in patients receiving the highest BG-12 dose.
Phase III studies
The first Phase III study, DEFINE (Determination of the Efficacy and safety of oral Fumarate IN rElapsing-remitting MS), enrolled 1,237 subjects with McDonald-defined RRMS, from 28 countries in a multicenter, randomized, double-blind, placebo-controlled, and dose-comparison study over 96 weeks. Entry criteria were: age 18–55 years, EDSS 0–5.0 inclusive, at least one MS relapse in the 12 months prior the randomization or a Gd-enhancing brain MRI lesion within 6 weeks of randomization. Subjects were randomized 1:1:1 to receive placebo, BG-12 240 mg twice daily or three times daily. After 48 weeks, patients who experienced at least one confirmed MS relapse after the 24th week of treatment or had a disability progression of at least 1.0 EDSS point from baseline (1.5 if baseline EDSS was 0.0) sustained for 12 weeks, were allowed to switch to an open-label rescue MS therapy. Clinical efficacy was assessed using EDSS, MS functional composite (MSFC) and visual function test (VFT) every 12 weeks. Gd-enhancing brain MRI scans were performed at baseline and weeks 24, 48 and 96. Safety assessments were performed at screening, baseline and every 4 weeks. The primary outcome was the reduction of the proportion of subjects relapsing at 2 years; secondary clinical outcomes included ARR and disability progression. Secondary radiological outcomes included the number of new or newly enlarging T2 and the number of Gd-enhancing lesions. The number of new T1-hypointense lesions at 1 and 2 years was a tertiary MRI outcome. Patients treated with the active medication showed a reduction of the relapse risk by 49% and 50% respectively for the low- and high-dose BG-12 compared to placebo at 2 years (P < 0.0001). The ARR was reduced respectively by 53% and 48% (P < 0.001) and the risk of confirmed disability progression was reduced respectively by 38% (P < 0.01) and 34% (P < 0.05).84 Secondary MRI outcomes found that subjects receiving the low- and high-dose BG-12 had respectively 85% and 74% relative risk reduction of new or newly enlarging T2 lesions at 2 years compared with placebo (P < 0.0001) and the 45% and 41% were free of new or newly enlarging T2 lesions compared with 27% of placebo patients. Furthermore, patients receiving the low- and high-dose BG-12 had respectively 90% and 73% reduced odds of increased Gd-enhancing lesion activity at 2 years compared with placebo (P < 0.0001) and 93% and 86% of treated patients were free of Gd-enhancing lesions compared with 62% of placebo patients. The number of new T1-hypointense lesions at 2 years was reduced by 73% in the low dose and 63% in the high dose of BG-12 versus the placebo group (P < 0.0001). A QoL measure, the Short-Form 36 health survey (SF-36), and a visual analogue score (VAS) on well-being were also improved over placebo from week 24 and was maintained throughout the study (SF-36 P < 0.001 and VAS P = 0.003 for BG-12 twice daily vs placebo and SF-36 P < 0.0001 and VAS P < 0.0001 for BG-12 three times daily vs placebo).85
The results of the CONFIRM study (Comparator and an Oral Fumarate In Relapsing–Remitting Multiple Sclerosis), the second of the two Phase III trials, were recently reported.86 In this multicenter, randomized, parallel-group, placebo-controlled and dose-comparison study, 1,430 subjects with RRMS were treated for 100 weeks with either oral BG-12 240 mg three times daily (720 mg total daily dose), two times daily (480 mg total daily dose), placebo or open treatment with GA subcutaneous injection 20 mg/day (used as a study’s reference comparator) with a randomization ratio of 1:1:1:1. Notably the study was not powered to detect a difference between BG-12 and GA therefore unlike with fingolimod vs IFNβ−1a87 there was no direct comparison between BG-12 and GA. Clinical assessment (EDSS, MSFC, and VFT) were performed every 12 weeks and MRI scans at baseline, week 24, 48 and 96. The primary outcome was reduction of the ARR at 2 years. Secondary clinical endpoints included reduction in the proportion of patients experiencing relapses and the 12-week confirmed progression of disability as measured by EDSS. Secondary MRI outcomes included the relative risk reduction of new or newly enlarging T2-hyperintense and new T1-hypointense lesions. There was a significant reduction in ARR relative risk in all the active treatment groups, BG-12 three times daily, twice daily, and GA, versus placebo respectively by 51% (P < 0.0001), 44% (P < 0.0001) and 29% (P < 0.02) at 2 years (Figure 3). Secondary outcome measures showed a significant (P < 0.0001) relative risk reduction in the number of new or newly enlarging T2-hyperintense lesions by 73%, 71% and 54%, respectively; the new T1-hypointense lesions were relatively reduced by 65%, 57% (both P < 0.0001), and 41% (P < 0.003), respectively; the proportion of patients experiencing MS relapses was relatively reduced by 45% (P < 0.0001), 34% (P < 0.003), and 29% (P < 0.01), respectively. Confirmed disability progression at 12 weeks was not statistically significant in all the active treatment groups compared to placebo, with a change of −24%, −21%, and 7%, respectively at 2 years.
Figure 3.
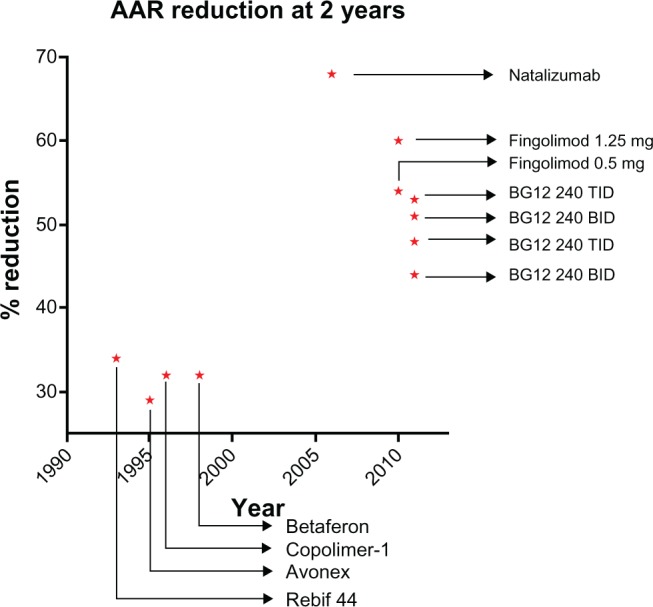
Reduction of ARR at 2 years by BG-12 and DMTs approved for MS treatment, as reported in Phase III clinical trials.
Note: Data obtained from different noncomparative trials.35–39,86,88,105
Abbreviations: ARR, annualized relapse rate; DMT, disease-modifying treatment; MS, multiple sclerosis; BID, twice a day; TID, three times a day.
Tolerability and safety
In the pilot study,80 Fumaderm administration resulted in mild to moderate AEs, the most frequent being gastrointestinal (GI) affecting 7 of 10 treated subjects, though this AE decreased after 6 weeks of treatment one subject discontinued because of this side effect. Flushing was reported in five subjects, increased levels of liver enzymes up to twofold in four subjects, and lymphopenia in three subjects. One of the 10 treated subjects experienced vertigo, headache, increased perspiration, and eosinophilia (up to 15% of white blood cells [WBC]); this progressively resolved during the study.
The phase IIb trial82 reported AEs in most of the BG-12-treated and placebo subjects, 88% and 75%, respectively. The AEs significantly more frequent in the active treatment groups versus placebo were abdominal pain, including upper abdominal pain (respectively 15% versus 3%) and flushing, including hot flushing (53% versus 9%). Serious AEs (SAEs) reported in the BG-12-treated patients included abdominal pain, pelvic inflammatory disease, phlebitis, urinary retention, uterine leiomyoma and vertigo, each of them with a prevalence of 1 in 191 treated subjects. MS relapses were seen in 7% of those on active treatment and 8% in the placebo group. This study confirmed the high rate of GI AEs. These mild to moderate events were mostly represented by nausea, diarrhea and upper abdominal pain, occurring in subjects receiving placebo, BG-12 120 mg once daily, 120 mg three times daily, or 240 mg three times daily in 25%, 30%, 39%, and 41% of patients, respectively. The dose-dependent AEs included headache, fatigue and feeling hot, while during the first 24 weeks of treatment increased levels of transaminase (mostly less than twice the upper limit of normal) were reported. The incidence of infections was 34% in both placebo and active treated patients. Four subjects receiving high dose BG-12 discontinued treatment due to AEs; reasons included flushing, nausea and vomiting. Two subjects receiving the mid-dose BG-12 discontinued treatment due to flushing and increased alanine aminotransferase, three subjects stopped treatment in the low dose BG-12 group; one due to increased alanine aminotransferase concentration and two due to diarrhea. Flushing, headache, nausea, upper abdominal pain and pruritus were more frequent in BG-12- treated subjects during the first treatment period than the second; furthermore flushing and GI symptoms were reduced in frequency from month 1 to month 6.
The two Phase III studies DEFINE and CONFIRM, reported similar safety and tolerability profiles.86,88 In the DEFINE trial the prevalence of AEs (95%–96%) in placebo, BG-12 240 mg twice daily, and BG-12 240 mg three times daily did not show any statistically significant difference between the different treatments. SAEs occurred in 21%, 18%, and 16% of patients, respectively. The SAEs occurring in at least two patients receiving BG-12 were gastroenteritis, gastritis, ovarian cyst, headache, and pneumonia. In general, the common (≥10%) AEs with higher incidence in the BG-12- treated patients, with a threshold of ≥3% versus placebo, were represented in order of decreasing incidence by flushing (35%), diarrhea (17%), nausea (13%), upper abdominal pain (11%), proteinuria (11%), abdominal pain (10%), and vomiting (8%). Causes for drug discontinuation in the two BG-12 groups, 133 patients (16% of total) were represented by all these AEs, with the exception of proteinuria. The two AEs which showed a typical habituation during the treatment period were the flushing and gastrointestinal symptoms, with a marked reduction after the first month of treatment.
Infections occurred in seven subjects on placebo, 10 on low-dose BG-12 and 8 on high-dose BG-12. Infections in the two BG-12 groups occurred with an incidence of <1% and included gastroenteritis, pneumonia, sinusitis, viral infection, appendicitis, cellulitis, H1N1 influenza, influenza, peritonsillar abscess, postviral fatigue, urinary tract infection, and vulval abscess. The incidence of malignancies was distributed equally between placebo and BG-12 groups. The four reported in the BG-12-treated group were basal cell carcinoma, breast cancer, cervix carcinoma, and transitional cell carcinoma. Laboratory assessments found an 8% incidence of proteinuria in placebo and 11% in BG-12 groups; patients receiving the active treatment also showed a decrease in WBC and lymphocyte counts, which reached a plateau after the first year of treatment, however the mean and median counts were maintained within normal limits. An increase in the liver enzymes was also noted, especially between month 1 and 6, the majority being below the threefold of upper limit value (ULV) and, when higher, never with concurrently elevated bilirubin (≥twofold ULV). MS relapses occurred in 15% of placebo group patients versus 9% of patients receiving either BG-12 dosage. Two patients in each of the BG-12 groups died in road accidents and were not considered related to the treatment. Table 3 reports a summary of AEs in Phase IIb and DEFINE trials.
Table 3.
| Phase IIb trial
|
DEFINE trial
|
||||||
|---|---|---|---|---|---|---|---|
| Placebo (65) | BG-12 120 mg once daily (64) | BG-12 120 mg three times daily (64) | BG-12 240 mg three times daily (63) | Placebo (408) | BG-12 240 mg twice daily (410) | BG-12 240 mg three times daily (416) | |
| AEs | |||||||
| Flushing | 6 (9%) | 34 (53%) | 31 (48%) | 25 (40%) | 20 (5%) | 154 (38%) | 396 (95%) |
| MS relapse | 16 (25%) | 11 (17%) | 20 (31%) | 12 (19%) | 189 (46%) | 111 (27%) | 114 (27%) |
| Headache | 7 (11%) | 5 (8%) | 11 (17%) | 13 (21%) | 80 (20%) | 81 (20%) | 80 (19%) |
| Nasopharyngitis | 10 (15%) | 7 (11%) | 6 (9%) | 7 (11%) | 101 (25%) | 108 (26%) | 109 (26%) |
| Nausea | 5 (8%) | 1 (2%) | 9 (14%) | 10 (16%) | 38 (9%) | 53 (13%) | 54 (13%) |
| Diarrhea | 3 (5%) | 6 (9%) | 5 (8%) | 7 (11%) | 55 (13%) | 62 (15%) | 78 (19%) |
| Pruritus | 5 (8%) | 6 (9%) | 5 (8%) | 6 (10%) | 19 (5%) | 42 (10%) | 34 (8%) |
| Upper abdominal pain | 2 (3%) | 5 (8%) | 4 (6%) | 9 (14%) | 28 (7%) | 40 (10%) | 52 (13%) |
| Hot flushing | 0 | 5 (8%) | 1 (2%) | 6 (10%) | |||
| Abdominal pain | 0 | 2 (3%) | 2 (3%) | 6 (10%) | 22 (5%) | 46 (11%) | 37 (9%) |
| Fatigue | 54 (13%) | 57 (14%) | 63 (15%) | ||||
| Upper respiratory tract infection | 53 (13%) | 63 (15%) | 51 (12%) | ||||
| Urinary infection | 53 (13%) | 55 (13%) | 54 (13%) | ||||
| Back pain | 57 (14%) | 59 (14%) | 46 (11%) | ||||
| Proteinuria | 34 (8%) | 38 (9%) | 50 (12%) | ||||
| Arthralgia | 39 (10%) | 46 (11%) | 37 (9%) | ||||
| Influenza | 39 (10%) | 34 (8%) | 48 (12%) | ||||
| Vomiting | 24 (6%) | 40 (10%) | 30 (7%) | ||||
| ALT < 3 × ULN | 14 (3%) | 26 (6%) | 24 (6%) | ||||
| ALT < 5 × ULN | 7 (2%) | 6 (1%) | 10 (2%) | ||||
| AST < 3 × ULN | 7 (2%) | 8 (2%) | 8 (2%) | ||||
| AST < 5 × ULN | 4 (<1%) | 1 (<1%) | 2 (<1%) | ||||
| WBC counts (<3.0 × 109/L) | 1% | 4% | 4% | ||||
| Lymphocyte counts (<0.5 × 109/L) | <1% | 4% | 4% | ||||
| SAEs | |||||||
| MS relapse | 5 (8%) | 3 (5%) | 6 (9%) | 5 (8%) | 60 (15%) | 39 (10%) | 32 (8%) |
| Abdominal pain | 0 | 0 | 0 | 1 (2%) | |||
| Lower limb fracture | 1 (2%) | 0 | 0 | 0 | |||
| Multiple sclerosis | 0 | 1 (2%) | 0 | 0 | |||
| Pelvic inflammatory disease | 0 | 0 | 1 (2%) | 0 | |||
| Phlebitis | 0 | 0 | 1 (2%) | 0 | |||
| Urinary retention | 0 | 0 | 0 | 1 (2%) | |||
| Uterine leiomyoma | 1 (2%) | 0 | 0 | 0 | |||
| Vertigo | 1 (2%) | 0 | 0 | 0 | |||
| Gastroenteritis | 0 | 4 (<1%) | 1 (<1%) | ||||
| Gastritis | 0 | 0 | 3 (<1%) | ||||
| Ovarian cyst | 1 (<1%) | 1 (<1%) | 2 (<1%) | ||||
| Pneumonia | 1 (<1%) | 2 (<1%) | 0 | ||||
| Basal cell carcinoma | 1 (<1%) | 1 (<1%) | 0 | ||||
| Breast cancer | 1 (<1%) | 0 | 1 (<1%) | ||||
| Cervix carcinoma | 0 | 0 | 1 (<1%) | ||||
| Transitional cell carcinoma | 0 | 1 (<1%) | 0 | ||||
Abbreviations: AEs, adverse events; SAEs, serious adverse events; ALT, alanine transaminase; AST, aspartate transaminase; MS, multiple sclerosis; ULN, upper limit of normal; WBC, white blood cells.
The CONFIRM study showed similar profiles of AEs and SAEs to the DEFINE study. The prevalence of AEs (placebo 92%; BG-12 twice daily 94%; BG-12 three times daily 92%; GA 87%) and SAEs (placebo 22%; BG-12 twice daily 17%; BG-12 three times daily 16%; GA 17%) were similar. There were no deaths and no malignancies in the BG-12 groups; one malignancy was reported in the placebo group and four in the GA group.86
Current therapeutic landscape for MS
The success of current MS therapies is based on the long-term use of DMT-injectable immunomodulators.89 The first-line injectable drugs IFN-β and GA have been in use for RRMS for approximately 20 years. Despite a suboptimal level of efficacy at ~30% reduction in ARRs the first-line injectable DMTs have shown excellent safety profiles.33–36 More effective drugs with greater safety issues or toxicity, such as natalizumab and mitoxantrone, are largely used as second-line treatments for patients unresponsive to first-line medication, or first-line in those with very active disease or who cannot tolerate injections.37,38,90
A major concern about the use of GA and IFN-β in the longer term is that only 75% of users adhere to treatment over an average of 31 months.91 Reasons for low compliance are the drug route of administration and skin site reactions.92,93 Moreover the injectable IFN-β can cause a transient flu-like syndrome, muscle aches, spasticity and possibly depression that impacts QoL.94,95 Before the advent of oral DMTs, these side effects had to be balanced with the safety issues of natalizumab, including the potentially fatal progressive multifocal leukoencephalopathy (PML), that arises as a result of JC virus reactivation,96 this risk can now be managed with the development of pre-natalizumab treatment JC antibody screening.97,98 Natalizumab has also been associated with the development of neutralizing antibodies, which may persist in up to 6% of patients, potentially causing hypersensitivity reactions and loss of drug efficacy.99 Other options such as mitoxantrone are associated with significant side effects including cardio-toxicity and acute leukemia.100 Thus, though DMTs are effective for people with MS, partial efficacy together with potential side effects mean that their long-term use, which is required for the potential benefits to be realized, can be problematic.
Although the available first-line and second-line parenteral compounds have radically changed the management of MS over the past two decades, the need for new therapies remains. Recently, several oral drugs have provided promising results in Phase II and III trials. Principal among these is fingolimod, which is now licensed and has similar impact on relative ARR reduction to BG-12, but has encountered some safety concerns. Other recent options arising from Phase III trials include cladribine,101 that demonstrated similar efficacy to fingolimod and BG-12 in relative ARR reduction, but has encountered safety issues and requires further study.102 Teriflunomide has produced a relative reduction in ARR similar to IFN-β,103 and has been submitted for review by European Medicines Agency. Laquinimod104 did not show efficacy in a second study and is currently still in trials.
Though oral treatments offer a step forward in therapy for MS, other treatments such as alemtuzumab offer an intermittent dosing approach on a yearly basis together with excellent potential efficacy but side effects are an issue as with other intense therapies, such as autologous stem cell transplantation.
BG-12: a future global oral first line therapy?
In two major Phase III trials, BG-12 has now produced a consistent effect both reducing the frequency of relapses in RRMS subjects, and reducing new T2 and Gd-enhancing MRI lesions. These effects are consistent with its pharmacodynamic effects on the immune system and this view is supported by evidence of a persistent lymphocyte count reduction in both Phase III studies. The effects on progression are less consistent and BG-12 needs to be evaluated in longer term trials, in order to confirm if its in vitro neuroprotective effects are significant in MS. Its safety and tolerability profile is promising. There were low numbers of SAEs and the most frequent AEs decreased during the initial 6 months of treatment in the Phase III studies. Long-term experience with the related fumaric acid compound used for psoriasis is also very promising.
These features give BG-12 a good efficacy–tolerability profile among available and emerging DMTs for RRMS patients (Table 4). The consistent efficacy in two Phase III trials in RRMS; the lack of major safety issues and the oral route of administration optimizing patients’ compliance make BG-12 an excellent potential candidate as a first- and second-line therapy in RRMS.
Table 4.
Relative efficacy (reduction in ARR), safety (major side effects) and ease of use (administration, day-to-day side effects) of existing and emerging therapies currently applying for a USA/EU license; not from comparative studies
| Therapy | Efficacy | Safety | Ease of use |
|---|---|---|---|
| IFNβ three times dailya | 30% | No long-term concerns | Injectable and systemic symptoms |
| GAa | 30% | No long-term concerns | Daily injectable |
| Natalizumaba | 68% | PML risk | Monthly infusion |
| Cladribineb | 55% | Further safety data required by regulators | Intermittent oral dosing |
| Fingolimoda | 55% | Cardiac, macula edema | Daily oral dosing |
| BG-12 | 55% | GI upset, no concerns from prior use in psoriasis | Twice daily oral dosing |
| Teriflunomide | 30% | Minimal, no long-term data | Daily oral dosing |
Notes:
Licensed USA and EU;
refused license in either USA or EU.
Abbreviations: ARR, annualized relapse rate; EU, European Union; IFN, interferon; GI, gastrointestinal; PML, progressive multifocal leukoencephalopathy; USA, United States of America.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Sadovnick AD, Ebers GC. Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci. 1993;20:17–29. doi: 10.1017/s0317167100047351. [DOI] [PubMed] [Google Scholar]
- 2.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 3.Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM. Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis. Ann Neurol. 2000;47:391–395. [PubMed] [Google Scholar]
- 4.Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65:239–248. doi: 10.1002/ana.21640. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- 6.Bartholomäus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462:94–98. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- 7.Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. 1978;38:409–421. [PubMed] [Google Scholar]
- 8.Ozawa K, Suchanek G, Breitschopf H, et al. Patterns of oligodendroglia pathology in multiple sclerosis. Brain. 1994;117:1311–1322. doi: 10.1093/brain/117.6.1311. [DOI] [PubMed] [Google Scholar]
- 9.Richards RG, Sampson FC, Beard SM, Tappenden P. A review of the natural history and epidemiology of multiple sclerosis: implications for resource allocation and health economic models. Health Technol Assess. 2002;6:1–73. doi: 10.3310/hta6100. [DOI] [PubMed] [Google Scholar]
- 10.Cottrell DA, Kremenchutzky M, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. 5. The clinical features and natural history of primary progressive multiple sclerosis. Brain. 1999;122:625–639. doi: 10.1093/brain/122.4.625. [DOI] [PubMed] [Google Scholar]
- 11.Kremenchutzky M, Cottrell D, Rice G, et al. The natural history of multiple sclerosis: a geographically based study. 7. Progressive-relapsing and relapsing-progressive multiple sclerosis: a re-evaluation. Brain. 1999;122:1941–1950. doi: 10.1093/brain/122.10.1941. [DOI] [PubMed] [Google Scholar]
- 12.Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343:1430–1438. doi: 10.1056/NEJM200011163432001. [DOI] [PubMed] [Google Scholar]
- 13.Kremenchutzky M, Rice GPA, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129:584–594. doi: 10.1093/brain/awh721. [DOI] [PubMed] [Google Scholar]
- 14.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129:606–616. doi: 10.1093/brain/awl007. [DOI] [PubMed] [Google Scholar]
- 15.Felts PA, Baker TA, Smith KJ. Conduction in segmentally demyelinated mammalian central axons. J Neurosci. 1997;17:7267–7277. doi: 10.1523/JNEUROSCI.17-19-07267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waxman SG. Demyelinating diseases – new pathological insights, new therapeutic targets. N Engl J Med. 1998;338:323–325. doi: 10.1056/NEJM199801293380610. [DOI] [PubMed] [Google Scholar]
- 17.Lovas G, Szilágyi N, Majtényi K, Palkovits M, Komoly S. Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain. 2000;123:308–317. doi: 10.1093/brain/123.2.308. [DOI] [PubMed] [Google Scholar]
- 18.Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 19.Pelletier D, Nelson SJ, Oh J, et al. MRI lesion volume heterogeneity in primary progressive MS in relation with axonal damage and brain atrophy. J Neurol Neurosurg Psychiatr. 2003;74:950–952. doi: 10.1136/jnnp.74.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocca MA, Iannucci G, Rovaris M, Comi G, Filippi M. Occult tissue damage in patients with primary progressive multiple sclerosis is independent of T2-visible lesions – a diffusion tensor MR study. J Neurol. 2003;250:456–460. doi: 10.1007/s00415-003-1024-1. [DOI] [PubMed] [Google Scholar]
- 21.Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133:1914–1929. doi: 10.1093/brain/awq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sormani MP, Li D, Bruzzi P, Stubinski B, et al. Combined MRI lesions and relapses as a perfect surrogate for disability in multiple sclerosis. Neurology. 2011;77:1684–1690. doi: 10.1212/WNL.0b013e31823648b9. [DOI] [PubMed] [Google Scholar]
- 23.Aronson KJ. Quality of life among persons with multiple sclerosis and their caregivers. Neurology. 1997;48:74–80. doi: 10.1212/wnl.48.1.74. [DOI] [PubMed] [Google Scholar]
- 24.Inusah S, Sormani MP, Cofield SS, et al. Assessing changes in relapse rates in multiple sclerosis. Mult Scler. 2010;16:1414–1421. doi: 10.1177/1352458510379246. [DOI] [PubMed] [Google Scholar]
- 25.Nicholas R, Straube S, Schmidli H, Schneider S, Friede T. Trends in annualized relapse rates in relapsing remitting multiple sclerosis and consequences for clinical trial design. Mult Scler. 2011;17:1211–1217. doi: 10.1177/1352458511406309. [DOI] [PubMed] [Google Scholar]
- 26.Naci H, Fleurence R, Birt J, Duhig A. Economic burden of multiple sclerosis: a systematic review of the literature. Pharmacoeconomics. 2010;28:363–379. doi: 10.2165/11532230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 27.Daugherty KK, Butler JS, Mattingly M, et al. Factors leading patients to discontinue multiple sclerosis therapies. J Am Pharm Assoc. 2005;45:371–375. doi: 10.1331/1544345054003804. [DOI] [PubMed] [Google Scholar]
- 28.Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157–1174. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charil A, Yousry TA, Rovaris M, et al. MRI and the diagnosis of multiple sclerosis: expanding the concept of ‘no better explanation’. Lancet Neurol. 2006;5:841–852. doi: 10.1016/S1474-4422(06)70572-5. [DOI] [PubMed] [Google Scholar]
- 30.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sormani MP, Bonzano L, Roccatagliata L, Cutter GR, Mancardi GL, Bruzzi P. Magnetic resonance imaging as a potential surrogate for relapses in multiple sclerosis: a meta-analytic approach. Ann Neurol. 2009;65:268–275. doi: 10.1002/ana.21606. [DOI] [PubMed] [Google Scholar]
- 32.Chataway J, Nicholas R, Todd S, et al. A novel adaptive design strategy increases the efficiency of clinical trials in secondary progressive multiple sclerosis. Mult Scler. 2011;17:81–88. doi: 10.1177/1352458510382129. [DOI] [PubMed] [Google Scholar]
- 33.Green G, Todd J. ‘Restricting choices and limiting independence’: social and economic impact of multiple sclerosis upon households by level of disability. Chronic Illn. 2008;4:160–172. doi: 10.1177/1742395307087457. [DOI] [PubMed] [Google Scholar]
- 34.Tremlett HL, Luscombe DK, Wiles CM. Prescribing for multiple sclerosis patients in general practice: a case–control study. J Clin Pharm Ther. 2001;26:437–444. doi: 10.1046/j.1365-2710.2001.00377.x. [DOI] [PubMed] [Google Scholar]
- 35.The IFNB Multiple Sclerosis Study Group Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 36.Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG) Ann Neurol. 1996;39:285–294. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- 37.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 38.PRISMS Study Group Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing-remitting multiple sclerosis. Lancet. 1998;352:1498–1504. [PubMed] [Google Scholar]
- 39.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 40.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 41.Comi G, De Stefano N, Freedman MS, et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012;11:33–41. doi: 10.1016/S1474-4422(11)70262-9. [DOI] [PubMed] [Google Scholar]
- 42.Kinkel RP, Dontchev M, Kollman C, Skaramagas TT, O’Connor PW, Simon JH. Association between immediate initiation of intramuscular interferon beta-1a at the time of a clinically isolated syndrome and long-term outcomes: a 10-year follow-up of the Controlled High-Risk Avonex Multiple Sclerosis Prevention Study in Ongoing Neurological Surveillance. Arch Neurol. 2012;69:183–190. doi: 10.1001/archneurol.2011.1426. [DOI] [PubMed] [Google Scholar]
- 43.Hohlfeld R, Barkhof F, Polman C. Future clinical challenges in multiple sclerosis: relevance to sphingosine 1-phosphate receptor modulator therapy. Neurology. 2011;76:S28–S37. doi: 10.1212/WNL.0b013e31820db40f. [DOI] [PubMed] [Google Scholar]
- 44.Altmeyer PJ, Matthes U, Pawlak F. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J Am Acad Dermatol. 1994;30:977–981. doi: 10.1016/s0190-9622(94)70121-0. [DOI] [PubMed] [Google Scholar]
- 45.Altmeyer P, Hartwig R, Matthes U. Efficacy and safety profile of fumaric acid esters in oral long-term therapy with severe treatment refractory psoriasis vulgaris. A study of 83 patients. Hautarzt. 1996;47:190–196. doi: 10.1007/s001050050401. [DOI] [PubMed] [Google Scholar]
- 46.Carboni I, De Felice C, De Simoni I. Fumaric acid esters in the treatment of psoriasis: an Italian experience. J Dermatolog Treat. 2004;15:23–26. doi: 10.1080/09546630310019346. [DOI] [PubMed] [Google Scholar]
- 47.Litjens NH, Nibbering PH, Barrois AJ. Beneficial effects of fumarate therapy in psoriasis vulgaris patients coincide with downregulation of type 1 cytokines. Br J Dermatol. 2003;148:444–451. doi: 10.1046/j.1365-2133.2003.05153.x. [DOI] [PubMed] [Google Scholar]
- 48.Fumapharm/Biogen Idec. BG12: BG00012, BG12/oral fumarate, FAG-201, second-generation fumarate derivative. Drugs R D. 2005;6:229–230. doi: 10.2165/00126839-200506040-00005. [DOI] [PubMed] [Google Scholar]
- 49.Werdenberg D, Joshi R, Wolffram S, Merkle HP, Langguth P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm Drug Dispos. 2003;24:259–273. doi: 10.1002/bdd.364. [DOI] [PubMed] [Google Scholar]
- 50.Rostami-Yazdi M, Clement B, Schmidt TJ, Schinor D, Mrowietz U. Detection of metabolites of fumaric acid esters in human urine: implications for their mode of action. J Invest Dermatol. 2009;129:231–234. doi: 10.1038/jid.2008.197. [DOI] [PubMed] [Google Scholar]
- 51.Litjens NHR, Rademaker M, Ravensbergen B, et al. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur J Immunol. 2004;34:565–575. doi: 10.1002/eji.200324174. [DOI] [PubMed] [Google Scholar]
- 52.Mrowietz U, Christophers E, Altmeyer P. Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol. 1999;141:424–429. doi: 10.1046/j.1365-2133.1999.03034.x. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci. 2005;83:313–328. doi: 10.1093/toxsci/kfi027. [DOI] [PubMed] [Google Scholar]
- 54.Chen X-L, Dodd G, Thomas S, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 55.Satoh T, Okamato SI, Cui J, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proc Natl Acad Sci U S A. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 57.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide three times daily microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 59.Wierinckx A, Brevé J, Mercier D, Schultzberg M, Drukarch B, Van Dam A-M. Detoxication enzyme inducers modify cytokine production in rat mixed glial cells. J Neuroimmunol. 2005;166:132–143. doi: 10.1016/j.jneuroim.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Treumer F, Zhu K, Gläser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol. 2003;121:1383–1388. doi: 10.1111/j.1523-1747.2003.12605.x. [DOI] [PubMed] [Google Scholar]
- 61.de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw-Kraan T, Ottenhoff TH, Nibbering PH. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26:2067–2074. doi: 10.1002/eji.1830260916. [DOI] [PubMed] [Google Scholar]
- 62.Kolenko V, Bloom T, Rayman P, Bukowski R, Hsi E, Finke J. Inhibition of NF-kappa B activity in human T lymphocytes induces caspase-dependent apoptosis without detectable activation of caspase-1 and -3. J Immunol. 1999;163:590–598. [PubMed] [Google Scholar]
- 63.Vandermeeren M, Janssens S, Borgers M, Geysen J. Dimethylfumarate is an inhibitor of cytokine-induced E-selectin, VCAM-1, and ICAM-1 expression in human endothelial cells. Biochem Biophys Res Commun. 1997;234:19–23. doi: 10.1006/bbrc.1997.6570. [DOI] [PubMed] [Google Scholar]
- 64.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 65.Zhu K, Mrowietz U. Inhibition of dendritic cell differentiation by fumaric acid esters. J Invest Dermatol. 2001;116:203–208. doi: 10.1046/j.1523-1747.2001.01159.x. [DOI] [PubMed] [Google Scholar]
- 66.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med. 1998;188:2175–2180. doi: 10.1084/jem.188.11.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asadullah K, Schmid H, Friedrich M, et al. Influence of monomethyl-fumarate on monocytic cytokine formation – explanation for adverse and therapeutic effects in psoriasis? Arch Dermatol Res. 1997;289:623–630. doi: 10.1007/s004030050251. [DOI] [PubMed] [Google Scholar]
- 68.Murphy TH, De Long MJ, Coyle JT. Enhanced NAD(P)H:quinone reductase activity prevents glutamate toxicity produced by oxidative stress. J Neurochem. 1991;56:990–995. doi: 10.1111/j.1471-4159.1991.tb02019.x. [DOI] [PubMed] [Google Scholar]
- 69.Duffy S, So A, Murphy TH. Activation of endogenous antioxidant defenses in neuronal cells prevents free radical-mediated damage. J Neurochem. 1998;71:69–77. doi: 10.1046/j.1471-4159.1998.71010069.x. [DOI] [PubMed] [Google Scholar]
- 70.Shih AY, Johnson DA, Wong G, et al. Coordinate regulation of glutathi-one biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McMahon M, Itoh K, Yamamoto M, Chanas SA, et al. The Cap‘n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 74.Hubbs AF, Benkovic SA, Miller DB, et al. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 77.Linker RA, Lee D-H, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 78.Ockenfels HM, Schultewolter T, Ockenfels G, Funk R, Goos M. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br J Dermatol. 1998;139:390–395. doi: 10.1046/j.1365-2133.1998.02400.x. [DOI] [PubMed] [Google Scholar]
- 79.Loewe R, Valero T, Kremling S, et al. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006;66:11888–11896. doi: 10.1158/0008-5472.CAN-06-2397. [DOI] [PubMed] [Google Scholar]
- 80.Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13:604–610. doi: 10.1111/j.1468-1331.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 81.Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 82.Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 83.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 84.Gold R, Kappos L, Bar-Or A, et al. Clinical efficacy of BG-12, an oral therapy, in relapsing-remitting multiple sclerosis: data from the phase 3 DEFINE trial. Mult Scler. 2011;17:S34. [Google Scholar]
- 85.Kappos L, Gold R, Arnold DL, et al. BG-12 effects on patient-reported outcomes in relapsing-remitting multiple sclerosis: results from the DEFINE study. Mult Scler. 2012;17:S488–S489. [Google Scholar]
- 86.Biogen idec press release Positive results from Phase 3 CONFIRM clinical trial show efficacy and safety of Oral BG-12 in multiple sclerosis. Detailed CONFIRM Data Presented at 2012 American Academy of Neurology Annual Meeting; April 24, 2012; [Accessed on September 1, 2012]. Available from: http://www.biogenidec.com/press_release_details.aspx?ID=5981&ReqId=1686377. [Google Scholar]
- 87.Cohen JA, Barkhof F, Comi G, TRANSFORMS Study Group Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 88.Selmaj K, Gold R, Kappos L, et al. Safety and Tolerability of BG-12 in the Phase 3 DEFINE Trial in Patients with Relapsing-Remitting Multiple Sclerosis. Poster 994. Amsterdam, The Netherlands: 5th Joint Triennial Congress of the European and Americas Committees for Treatment and Research in Multiple Sclerosis; 2011. [Google Scholar]
- 89.Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78:1315–1322. doi: 10.1212/WNL.0b013e3182535cf6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hartung H-P, Gonsette R, König N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 91.Rice GPA, Incorvaia B, Munari L, et al. Interferon in Relapsing-remitting Multiple Sclerosis. Chichester, UK: John Wiley & Sons, Ltd; 2006. [Google Scholar]
- 92.Tremlett HL, Oger J. Interrupted therapy: stopping and switching of the beta-interferons prescribed for MS. Neurology. 2003;61:551–554. doi: 10.1212/01.wnl.0000078885.05053.7d. [DOI] [PubMed] [Google Scholar]
- 93.O’Connor P, Filippi M, Arnason B, et al. BEYOND Study Group 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8:889–897. doi: 10.1016/S1474-4422(09)70226-1. [DOI] [PubMed] [Google Scholar]
- 94.Goeb J-L, Even C, Nicolas G, Gohier B, Dubas F, Garré JB. Psychiatric side effects of interferon-beta in multiple sclerosis. Eur Psychiatry. 2006;21:186–193. doi: 10.1016/j.eurpsy.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 95.Meca-Lallana JE, de Mingo-Casado P, Amorin-Díaz M, Martínez-Navarro ML, Barreiro AF. Effects of glatiramer acetate on spasticity in previously interferon-beta-treated and treatment-naive patients with relapsing-remitting multiple sclerosis: a prospective, nonrandomized, open-label, uncontrolled, observational pilot study. Clin Ther. 2010;32:1061–1066. doi: 10.1016/j.clinthera.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 96.Clifford DB, De Luca A, DeLuca A, et al. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- 97.Laroni A, Giacomazzi CG, Grimaldi L, et al. Urinary JCV-DNA Testing during natalizumab treatment may increase accuracy of PML risk stratification. J Neuroimmune Pharmacol. 2012;7:665–672. doi: 10.1007/s11481-012-9366-z. [DOI] [PubMed] [Google Scholar]
- 98.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 99.Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–1403. doi: 10.1212/01.wnl.0000277457.17420.b5. [DOI] [PubMed] [Google Scholar]
- 100.Marriott JJ, Miyasaki JM, Gronseth G, O’Connor PW. Evidence report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2010;74:1463–1470. doi: 10.1212/WNL.0b013e3181dc1ae0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 102.EMEA assessment report. February 17, 2011. Procedure No. EMEA/H/C/2202. [Accessed on September 1, 2012]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002202/WC500104529.pdf.
- 103.O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 104.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 105.Kappos L, Radue EW, O’Connor P, et al. FREEDOMS Study Group A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]