

Abstract
Almost 100 mutations in the human transthyretin (TTR) gene cause the autosomal dominant disorders of familial amyloidotic polyneuropathy (FAP) and familial amyloidotic cardiomyopathy. While these have been clinically classified as separate disorders, the peripheral and autonomic nervous systems and the heart are frequently involved in the same patient. Deposition of amyloid derived from a kinetically or thermodynamically unstable mutant TTR precursor produces an ascending sensorimotor polyneuropathy with marked autonomic involvement. Since 1990, treatment has been liver transplantation from a donor carrying two wild-type TTR genes, providing a crude form of gene therapy. Multiple studies have shown that small molecules fitting in the T4-binding pocket of TTR can stabilize the molecule, reducing its capacity to release the fibril precursor. Tafamidis is the first molecule to be tested in a placebo-controlled trial in patients with TTR-associated FAP. While the trial did not achieve its primary endpoints, it did stabilize TTR in vivo and had a favorable effect on some aspects of disease progression, particularly when administered early in the course. It may represent an alternative to liver transplantation, particularly in patients with early disease related to the V30M mutation. Longer-term studies are required to determine whether it represents a stabilizing or remittive form of treatment.
Keywords: tafamidis, transthyretin, polyneuropathy, treatment
Introduction
The transthyretin (TTR) amyloidoses are diseases in which misfolded TTR monomers aggregate deposit in a wide range of human tissues producing functional compromise and ultimately failure.1 The deposits may form in virtually any organ but the favored sites are peripheral and autonomic nerves, blood vessels, the walls of the gastrointestinal tract, the heart, the kidneys, vitreous of the eye, and the connective tissue of the carpal ligament. Sites that appear to be spared are the liver (despite the fact that it is the major source of systemic TTR production) and the brain parenchyma. However, cerebral vessels, the leptomeningeal membranes, and the choroid plexus can be sites of TTR fibril deposition in the central nervous system (CNS). The mechanism of tissue injury has traditionally been assumed to be displacement of functional cells by insoluble masses of fibrils, but recent data suggest that an element, perhaps the majority, of tissue damage results from direct toxic effects of oligomers formed from the misfolded TTR monomers or dimers.2
Background
Transthyretin: normal structure, function, biosynthesis
TTR is a normal human serum protein, formerly known as thyroxine-binding pre-albumin, which carries a small portion of circulating thyroxine and all of the serum retinol-binding protein (RBP) charged with retinol.1 In the circulation, the bulk of the protein is an apparently unglycosylated homotetramer of 55 KDa, which is in equilibrium with a small amount of dissociated monomer. Binding of either ligand stabilizes the tetrameric structure, a phenomenon critical in developing the small molecule inhibitor of fibril formation (vide infra: the role of tetramer dissociation in pathogenesis).3 The protein also serves as the major transporter of thyroid hormone into the CNS during development.4 The source of the CNS protein for that function is the choroid plexus, a highly vascular structure in the floor of the cerebral ventricles that synthesizes and filters the cerebrospinal fluid.5,6
The hepatic transcription of the TTR gene is under the control of a group of transcription factors (HNF1, HNF3, and HNF4), which bind to a region just upstream of its promoter.7 HNF6 has a regulatory effect through its binding to HNF3.8 The general transcription factors AP-1 and C/EBP bind to an upstream enhancer site.9 Inflammatory cytokines downregulate the transcription factors with a subsequent relatively rapid reduction in the serum TTR concentration in the course of inflammation.10 Choroid plexus TTR synthesis is not known to be cytokine-responsive.11 Recent data have suggested that TTR may have other functions in neurons (as a neuroprotectant) and in pancreatic β-cells where it may play a role in stimulus response coupling involved in insulin secretion.12,13
The transthyretin amyloidoses
TTR has been associated with three general classes of human disease. Familial amyloidotic polyneuropathy (FAP) and familial amyloidotic cardiomyopathy (FAC) are both caused by amino acid substitutions in the TTR protein. In senile systemic amyloidosis, the amyloid is derived from wild-type TTR and is deposited in tissues of elderly individuals with the most clinically important site being the heart.14 Carpal tunnel syndrome related to wild-type TTR deposition is also common in the elderly and it may or may not be associated with cardiac amyloidosis.15 A systematic examination during autopsy of the gastrointestinal tracts of northern European individuals aged 85 years and over revealed wild-type TTR deposition in 18%.16 The deposition was not associated with any definable clinical symptoms.
Mechanisms of TTR amyloidogenesis
Over the years, work from many laboratories has established that in vitro, under mildly denaturing conditions such as low pH, aggregation and amyloid fibril formation result when monomers released from the native TTR tetramer misfold and form the pathogenic homotypic aggregates. The sequence appears to be tetramer to dimer to native monomers to misfolded monomers to oligomers to protofibrils to fibrils via an energetically downhill polymerization mechanism.17 The enhanced dissociation results in a larger amount of monomer available for aggregation. In the test tube reducing pH to provide mildly denaturing conditions lowers the energetic barrier to dissociation and increased fibril formation even of the wild-type tetramer.18 In the presence of any one of 99 reported substitutions at 67 of the 127 amino acids in the protein, tetramer dissociation resulting in monomer release, misfolding, and the subsequent aggregation required for disease pathogenesis, is accelerated either on the basis of the thermodynamic or kinetic instability of the mutant tetramer.19 It has been inferred that the same process occurs in vivo to provide the aggregation substrate responsible for deposition. The sites of deposition, however, have not yet been associated with definable denaturing conditions.
Clinical aspects of the transthyretin amyloidoses
Clinically, almost half the reported mutations are associated with deposition in both peripheral nerves and the heart although the dominant clinical presentation is peripheral and autonomic neuropathy, presumably related to a lower threshold for neurologic symptoms given the same degree of cardiac and neuronal tissue deposition. Hence the distinction between FAP and FAC is not absolute and an argument can be made for referring to both conditions as genetically determined TTR amyloidosis rather than the historic designations of FAP and FAC. Approximately 20% of the variants appear to deposit exclusively in one or the other of the major sites. The TTR or host features determining the primary and ultimate sites of deposition are for the most part unknown except in the case of a few highly unstable variants responsible for the oculoleptomeningeal form of the disease.19
The diseases generally behave as autosomal dominants with transmission through either parent, but with greater severity when transmitted from mother to son.20 Aggressiveness of the disease, as assessed by age of onset, appears to be largely a function of the relative thermodynamic stability of the mutant tetramer.19 However, this is not absolute. Portuguese subjects who carry both the highly amyloidogenic V30M TTR mutation and the kinetically stable T119M TTR allele have little in the way of clinical disease.21 Genetic anticipation, ie, earlier onset in succeeding generations carrying the same mutations, best documented for V30M TTR, has been reported for several mutations and unlike Huntington’s disease for example, is not associated with expanded trinucleotide repeats.22,23
Familial amyloidotic polyneuropathy
The V30M TTR mutation is the most common cause of FAP. It was originally described in Portugal, then in Japan and Sweden.24–27 The Japanese carriers have clearly arisen from multiple founders, only one of which shows haplotype identity with the Portuguese subjects.28 The clinical phenotype is similar in Japan and Portugal, but strikingly different from that seen in northern Sweden, where the onset is much later in life and the allele does not appear to be as clinically penetrant.29 These differences have been hypothesized to be related to either genetic background, environmental differences, or some combination of the two.30
Clinically, the disorder is a sensorimotor autonomic polyneuropathy primarily affecting small unmyelinated nerve fibers.31 The disease begins peripherally and progresses proximally with the early phases characterized by primarily sensory symptoms including both dysesthesias and loss of sensation. The occurrence of peripheral ulcers and Charcot joints as severe consequences of loss of sensation is not uncommon. Autonomic manifestations may appear simultaneously with sensory loss or precede it. Impotence, gastrointestinal disturbances, and loss of bladder control are all common. Involvement of the autonomic control of the heart is frequent with orthostatic hypotension reflecting both a failure of the heart to respond to peripheral needs by increasing the cardiac rate and the loss of peripheral vascular tone. The involvement of the autonomic innervation of the gut as well as the infiltration of the intestine with amyloid deposits results in malabsorption with weight loss and general failure to thrive. The recognition that the severity of gastrointestinal involvement had prognostic importance with respect to the outcome of liver transplantation resulted in the development of the modified body mass index (mBMI) as a measure of overall nutrition. This parameter which integrates albumin loss and body mass has become a major criterion for selection for liver transplantation with patients being recommended for transplant prior to the time that there is a significant decrease in the mBMI.32
The transthyretin amyloidoses: therapy
Liver transplantation
For many years treatment was largely symptomatic and attempted to alleviate the symptoms of autonomic and peripheral neuropathy with drugs and the cardiac disease with diuretics and antiarrhythmic measures, either pharmacologic or electrophysiologic (pacemaker placement). In 1990, reasoning that since the liver was the primary source of the mutant TTR in the deposits, it was possible that replacement of that organ, as a form of gene therapy, would stop the production of the mutant protein that ultimately deposited in the target organs.33 Since then, approximately 2000 liver transplants have been performed worldwide.34 With improvements in surgical technique and better patient selection (based primarily on assessment of the mBMI), long-term survival has improved considerably to up to 85%–100% with 10 years of follow-up.35 Disease progression is arrested in approximately 60% of the patients. About 20% improve and another 20% deteriorate. The best results have been obtained in V30M TTR patients. For reasons that are unclear, subjects with other mutations do not do as well, particularly in the presence of heart disease. In some patients, cardiac amyloid may increase post-liver transplant because of the deposition of fibrils derived from wild-type TTR on pre-existing deposits of mutant fibrils, which serve as a template or matrix.36 Therefore, an increasing number of combined heart and liver transplants are being performed.37
In 1995, because of the general shortage of donor livers and the realization that the livers of FAP patients were generally well-functioning, the first “domino” liver transplant, in which a liver from a symptomatic FAP gene carrier was given to a patient with liver failure, was performed.38,39 It was assumed that even though the transplanted liver was producing a mutant TTR, the time necessary to produce TTR amyloid deposition would be longer than the actuarial survival of appropriately selected recipients. This has not turned out to be entirely correct since an increasing number of domino recipients have been found to develop symptomatic or asymptomatic mutant TTR deposition within 5 to 8 years post-transplant.40 While liver transplantation is the current standard of therapy, it is clear that other approaches are both necessary and desirable.
Pharmacologic therapy
The first pharmacologic approach to therapy was based on the observation that thyroxine (T4), a natural ligand of TTR, stabilized the tetramer and reduced dissociation and aggregation under fibril-forming conditions.41 A subsequent systematic search of molecules that were structurally similar to thyroxine for their capacity to inhibit TTR fibril formation in vitro yielded a set of small molecules including several that were known nonsteroidal anti-inflammatory drugs (NSAIDs) including flufenamic acid and diflunisal.42,43 Their capacity to inhibit fibril formation in the test tube was independent of their capacity to inhibit the cyclooxygenases involved in the production of inflammatory arachidonic acid metabolites. These compounds had the distinct advantage of substantial human exposure and recognized side effects, which included decreased renal blood flow and gastrointestinal bleeding among others. Diflunisal appeared to have the best side effect profile of this group of compounds and is currently in the late stages of a placebo-controlled double-blind trial in FAP.44
Knowledge of the mechanism of stabilization of the tetramer was expanded by studies of mixed tetramers composed of V30M and T119M mutant monomers stimulated by the observation of minimal clinical disease in a V30M carrier who was heterozygous for TTR T119M.21 Examination of aggregation and fibril formation by tetramers comprised of one or more T119M monomers revealed that the latter kinetically stabilized the heterotetramer. This observation suggested that kinetic stabilization of mutant or wild-type human tetramer would reduce the amount of TTR monomer available for misfolding and aggregation in vivo. The heterotetramer effect was confirmed later when attempting to produce transgenic mouse models of the human TTR amyloidoses in the presence of an intact murine ttr gene. As in the case of the human V30M/T119M heteromers, the mutant human–wild-type mouse heterotetramers were kinetically stable, which reduced the concentration of monomers available to misfold unless the human gene was sufficiently overexpressed to exceed the stoichiometric amounts of mouse TTR available for heteromerization.45
Tafamidis (Vyndaqel)
Preclinical
Structure-based design of a molecule that would be soluble under physiologic conditions, have sufficiently high affinity for the T4-binding site to stabilize the tetramer and not have the undesirable properties of inhibiting cyclooxygenase enzymes resulted in the identification of 2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid, common name, tafamidis (Vyndaqel; Pfizer, New York, NY), which has recently been approved by the European Medicines Agency (EMA) for therapy of early stage FAP.46 The structure of tafamidis and how it interacts with the TTR tetramer are shown in Figure 1. The affinities of the drug for the two binding sites of TTR have been determined by two methods yielding relatively similar results: KD1 = 2nM, by subunit dissociation; 3nM by isothermal titration calorimetry (ITC); and KD2 = 154 nM by dissociation, and 278 nM by ITC.47 When incubated with plasma the bulk of the molecule is bound to TTR with a stoichiometry of 0.81, indicating that 0.81 mole of the drug was bound to one molecule of TTR. Complete binding would have a stoichiometry of one since only one of the two T4-binding sites is accessible to the compound. There are few available data examining the binding to other individual plasma proteins. Pharmacokinetic studies suggested that a 20 mg dose orally once a day gives a plasma drug concentration between 5.2 µM (minimum) and 8.4 µM (maximum) in normal volunteers which is adequate to bind serum TTR that is normally present at about 4 µM.
Figure 1.
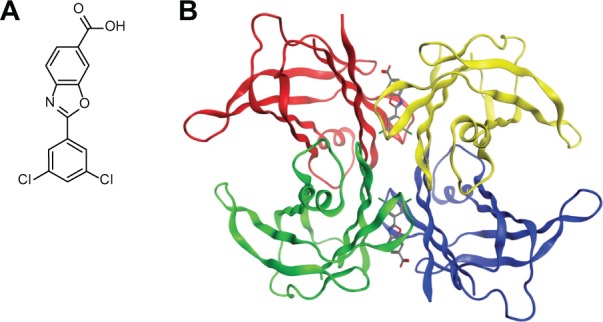
(A) Line drawing of tafamidis. (B) Structural model of how tafamidis is envisioned to bind to TTR.
Note: While two binding sites are shown binding in the first site makes the second site allosterically unavailable. Reprinted from Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. Copyright © 2012,with permission from Elsevier.46
Abbreviation: TTR, transthyretin.
Clinical trial
A multicenter double-blind randomized 18-month controlled trial of tafamidis at 20 mg per day in patients judged to have early stage V30M TTR FAP was reported recently.47,48 The trial included 128 patients with 65 receiving the drug and 63 receiving the placebo. The study anticipated a 30% difference in response rates using the Neuropathy Impairment Score–Lower limbs (NIS-LL) to assess response in lower limb neurologic function and the Norfolk Quality of Life Diabetic Neuropathy Questionnaire (TQOL) to evaluate quality of life as primary end point metrics.49,50 These two instruments have been validated for assessing diabetic neuropathy and appeared to be suitably sensitive to evaluate changes in the clinical neuropathic status of the FAP subjects.51 The sample size was chosen to provide 90% power to achieve a P value of 0.05 given the assumed 30% effect size of the therapy on the primary endpoints (NIS-LL and TQOL) and a presumptive dropout rate of 10% related to the ongoing availability of liver transplants for the participants, ie, 88 of the subjects were on the liver transplant wait list at the time of enrolment. Secondary endpoints included the Σ7 NTs nds (summated seven nerve-tested normal deviates, which assesses large nerve function), the Σ3 NTSF nds (summated three nerve-tested small fiber function), and the mBMI (modified body mass index = BMI × serum albumin concentration) a parameter of overall nutrition and autonomic function that has been shown to be a useful prognostic indicator in evaluating FAP patients for liver transplant.35
Fifteen (20%) of the subjects in each treatment arm did not complete the trial because livers became available for transplantation. Five percent of each treatment group withdrew because of adverse effects (see below). Hence, the statistical power of the study was severely compromised in the intention-to-treat (ITT) analysis (only 87/128 starting subjects having complete data sets. The results become more significant in the efficacy-evaluable (EE) analysis, but it is known that such post hoc analyses are subject to overinterpretation. However some clinical epidemiologists note, “… that the intention-to-treat policy may sometimes reduce bias at the cost of removing clinical sensibility from the results”.52 Given these limitations, it is still noteworthy that almost all of the comparisons favored the treated over the placebo group although most of them did not achieve statistical significance particularly when considered in the light of the multiple comparisons performed (Table 1).
Table 1.
Comparison of clinical changes over 18 months in FAP patients receiving tafamidis or placebo
| Intention-to-treat
|
||||||||
|---|---|---|---|---|---|---|---|---|
| # | # completing | NIS-LL* | NIS-LL % responders** | TQOL‡ | Σ3 NTSF‡‡ | Σ7 NTs† | mBMI†† | |
| Tafamidis | 65 | 48 | +2.8 | 45 | +2.5 | +0.3 | +1.5 | +39 |
| Placebo | 63 | 47 | +5.8 | 29 | +8 | +1.6 | +3.1 | −33 |
| P-value | 0.027 | 0.068 | 0.21 | 0.005 | 0.066 | 0.0001 | ||
Notes:
NIS-LL Neuropathy Impairment Score-Lower Limbs: motor, sensory, reflex; the greater the score the greater the impairment;
NIS-LL responder: increase < 2 points in course of study; a primary endpoint in the study;
TQOL Norfolk quality of life diabetic neuropathy questionnaire: higher the score the worse the quality of life; a primary endpoint in the study;
Σ3 NTSF Summated values of three small fiber functions: the higher the number the worse the function; secondary endpoint;
Σ7 NTs: summated values of seven tests of large fiber function: lower is better than higher; secondary endpoint;
mBMI, modified Body Mass Index is calculated by multiplying MBI by serum albumin in g/L developed for the evaluation of FAP patients as candidates for liver transplantation. It is a measure of wasting and malnutrition that appears to be based on gastrointestinal autonomic dysfunction. Higher scores are associated with better metabolic status and predict better outcomes after liver transplantation for FAP. Secondary endpoint.
Abbreviation: #, number of subjects.
From the point of view of the proposed mechanism of action of tafamidis the study was successful with the TTR tetramer stabilized to urea denaturation in the plasma in vitro of 98%–100% of subjects receiving the compound, hence less likely to provide monomer to misfold, aggregate, and deposit. Similarly, from the point of view of overall patient well-being as measured by improvement in the mBMI (a secondary endpoint in the analysis), the difference between the subjects receiving the drug and those on placebo was significant, even after correcting for multiple comparisons. That said, in the comparisons between drug- and placebo-treated individuals, neither primary endpoint NIS-LL nor TQOL differed significantly in the ITT analysis although both were significant in the EE analysis.
How can we account for the failure to achieve a statistically robust result from what should have been a definitive clinical trial? The first and perhaps least likely answer is that the drug does not work. That is probably not true since serum TTR tetramers in patients receiving the compound were stabilized in in vitro testing. The correlation between less monomer and less disease is consistent with in vitro studies of fibril formation. Unfortunately, none of the presently available animal models of the disease is suitable for the analysis of efficacy in terms of reducing deposition or altering clinical manifestations because of either stoichiometric considerations or lack of a defined clinical phenotype. Based on the in vitro studies, reducing the availability of circulating TTR monomer by tetramer stabilization should have some effect on disease development or progression. It is possible that drug-induced reduction of circulating monomer could allow deposits to be mobilized according to some finite “off rate” with the generation of toxic oligomers or intact monomers that could redeposit directly. However, there are no data to suggest that such is the case. The most likely operational explanation is simply that the study was underpowered in the context of the higher than expected frequency of dropouts for liver transplantation and that if the dropout rate were halved all the differences between those receiving tafamidis and the controls would become statistically significant. This assumes however that the proportion of responders and nonresponders in each arm would be the same as the numbers were increased, an assumption that cannot be statistically justified.
It is also possible that some flaw in the design of the trial or unanticipated bias in the random assignments of patients to different treatment arms could have falsely tilted the outcome in favor of the treatment. In an analysis of the study data, the US Food and Drug Administration (FDA) staff suggested that patients in the tafamidis arm may have had less severe disease than those in the placebo cohort, thus were likely to progress at a slower rate (US FDA document).53 The alternative interpretation of this observation is that subjects with less severe disease are more responsive to therapy.
A more subtle, but possibly related, potential pitfall noted by the FDA was that 58% of the patients were from a single center, an undesirable circumstance in a multicenter trial, and that the patients from that center had better baseline parameters and the best response rates. A possible reason for this circumstance could be investigator bias, which is unlikely since randomization was by computer. More likely is that it reflects the extensive experience of that particular center with earlier identification of gene carriers as relatives of clinically affected individuals at the center contributed a larger number of bona fide early-onset subjects. There could also be differences in responses between the V30M TTR subjects in Portugal from those being treated elsewhere on the basis of genetic or cultural factors. In my view, it is most consistent with the prior suggestion that patients with earlier disease are more responsive to tafamidis therapy. A detailed examination of the subjects at that center shows that at baseline the NIS-LL in the tafamidis cohort had a mean value of 6.7 vs 9.6 in the controls; the TQOL was 20.8 vs 25.5 in the placebo (both consistent with somewhat milder disease), but the mean duration of disease in the drug-treated group was 34.5 months vs 24 months in those receiving placebo. In the other centers, the NIS-LL and TQOL were higher than in the Portuguese subjects, but were more closely matched in the drug- and placebo-treated groups. However more critical was the fact that the mean duration of disease in the tafamidis group was 65 months and in the placebo subjects 51 months in the non-Portuguese subjects, 31.5 months longer than the Portuguese cohort receiving tafamidis and 27 months longer than in the Portuguese controls. It is possible that disease duration determines potentially responsive disease effects not specifically measured in the context of the trial. Thus, it may not be surprising that the response rate in the Portuguese subjects was 61% vs 25% from the other centers and the response to placebo did not differ, ie, 28% and 32%, respectively. Should this be confirmed it would suggest two possibilities regarding responses to tafamidis. First, response rates are higher in earlier disease. Second, it may be that subjects with a longer duration of symptomatic peripheral nerve disease may take longer to respond and/or that very-long-standing deposits may not resolve at all. If that is the case, as suggested by the results of liver transplantation, patients will be left with some degree of irreversible damage, again supporting the wisdom of early treatment. The validity of these suspicions should become evident with further clinical studies.
Tafamidis: toxicity
Data from unpublished preclinical studies presented to the FDA indicate that tafamidis has little or no effect on the hepatic drug detoxifying enzymes CYP1A2, 2C9 or 2D6 in tissue culture and none on 3A4 in vivo. Individuals with moderate liver disease seem to clear the drug from the plasma more quickly than healthy controls. The increased clearance does not appear to require any modification in the recommended 20 mg/day oral dose. In the placebo-controlled trial, the frequency of upper gastrointestinal symptoms was higher in the tafamidis-treated patients than in those receiving placebo and there was an increased frequency of urinary tract infections in those taking the active compound. Some of the infections were considered to be serious. Mechanistically it is not clear why patients taking tafamidis should be subject to an increased occurrence of genitourinary infection. It remains to be seen whether the frequency of these apparent side effects will be the same when the number of recipients of tafamidis increases above the 324 individuals who have received the compound since clinical studies began.
Current therapeutic alternatives
What are the other therapeutic options for FAP, FAC, and SSA patients and their physicians? As we previously noted the current standard of practice for the treatment of FAP worldwide is liver transplantation.54 It clearly has the effect of stabilizing the disease in the majority of patients. It has the additional societal benefit of providing a reasonably well-functioning liver for selected patients requiring a transplant for liver failure related to other forms of disease.
The disadvantages of the procedure are the procedure itself, which has significant short-term morbidity and some degree of mortality, although in recent years perioperative mortality of orthotopic liver transplantation has diminished considerably. The patients require chronic immunosuppressive therapy. The monetary costs of the surgery and the associated short-term supportive care are also a consideration. In addition, the general societal shortage of available livers does not seem to be a solvable problem, although the notion that transplanting an FAP patient also makes a liver available for carefully selected patients with liver failure seems to balance this out.
With respect to outcome the continued risk of cardiac deposition, particularly in non-V30M TTR subjects is both troublesome and perplexing. This problem could be solved with post-transplant tafamidis therapy, but an appropriate placebo-controlled trial must be carried out to determine if this will be the case. Similarly the development of symptomatic (neuropathic) and asymptomatic TTR deposits in patients receiving FAP livers in a domino procedure would seem to provide an excellent clinical setting to test tafamidis in what are essentially stage zero FAP patients. Again, a controlled trial with domino recipients randomized in a tafamidis–placebo protocol with biopsies performed at the time of transplant and at yearly intervals would provide one more tests of the hypothesis that tetramer stabilization in vivo prevents tissue deposition.
Therapeutic alternatives in development
There are other potential therapies for the TTR amyloidoses on the horizon. As mentioned previously, there is a current placebo-controlled trial of another TTR tetramer stabilizer, diflunisal, in patients with a variety of TTR mutations, using similar parameters of clinical measurement. This study should be completed at the end of 2012 and the results should be known sometime in 2013.44 A positive result would confirm the principle that tetramer stabilization can suppress clinically important TTR deposition. Whether the drug has a therapeutic and safety profile comparable to that seen in the tafamidis trial remains to be seen. Because of the similar eligibility criteria and clinical metrics it should be relatively easy to compare the results obtained with those published in the tafamidis study.
Several other pharmacologic approaches have been explored in preclinical studies and in Phase I human safety studies. A growing number of compounds have been found to decrease TTR fibril formation in vitro by stabilizing the TTR homotetramer. Small, uncontrolled trials of catechin, epigallocatechin-3-gallate (EGCG), a polyphenol component of green tea and a combination of oral doxycycline (100 mg bid) and tauroursodeoxycholic acid (TUDCA) (250 mg tid), thought to disaggregate TTR deposits have been published.55–57 The notion of treating amyloidosis of all types by a combination of chemical and antibody binding of serum amyloid P-component (SAP) as a means of destabilizing preexisting amyloid deposits has been proposed for many years based on studies in murine models of inflammation associated amyloidosis.58,59 In addition to these traditional small molecule agents, two similar nonpharmacologic approaches designed to inhibit TTR synthesis in the liver, one using antisense, the other using a short-interfering RNA formulation have undergone Phase I testing in humans.60,61
While none of these has progressed far enough in clinical testing to allow a comparison with tafamidis, if they do prove to be effective, there is the strong possibility that the best approach to these diseases might be multimodal, using a combination of small molecule pharmacologic agents and molecular biologically designed macromolecules to achieve an optimal therapeutic result.
The question remains what will be the maximum benefit achievable? Will any of the results of any these modalities resemble those achieved by liver transplantation, ie, arrest of progression but little absolute clinical improvement, only at lower cost? Or will the availability of lower risk therapeutics encourage earlier therapy at a time when disease is reversible as well as preventable? The liver transplant results suggest that at the time of most transplants there is little or no significant off rate of fibrils from the deposits. Perhaps fibrillar deposits in clinically significant sites become increasingly stable with time and the potential for true reversion of pathology is inversely related to the age of the deposits, ie, duration of disease. If that is the case and the safety profiles of the new therapies, like tafamidis, are favorable, it may be worthwhile to treat the mutations rather than the disease. Currently, tafamidis appears to be a start toward that end. Since the absolute number of FAP patients worldwide is relatively small, it appears obvious that all patients receiving tafamidis or any of the emerging therapies should be monitored carefully and the outcomes well documented.
Footnotes
Disclosure
Dr Buxbaum has been a paid consultant for FoldRx Pharmaceuticals, Pfizer Pharmaceuticals (which developed tafamidis as Vyndaquel), Alnylam Pharmaceuticals, and Isis Pharmaceuticals, all of whom have drugs in development or in clinical trials for the transthyretin amyloidoses. He has also held an equity position in FoldRx.
References
- 1.Buxbaum JN. Transthyretin and the Transthyretin Amyloidoses. In: Uversky VN, Fink A, editors. Protein Misfolding, Aggregation, and Conformational Diseases. Santa Cruz, CA: Springer; 2007. pp. 259–283. [Google Scholar]
- 2.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and non-native oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A. 2004;101(9):2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White JT, Kelly JW. Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc Natl Acad Sci U S A. 2001;98(23):13019–13024. doi: 10.1073/pnas.241406698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schreiber G, Southwell BR, Richardson SJ. Hormone delivery systems to the brain – transthyretin. Exp Clin Endocrinol. 1995;103:75–80. doi: 10.1055/s-0029-1211332. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber G. The evolution of transthyretin synthesis in the choroid plexus. Clin Chem Lab Med. 2002;40(12):1200–1210. doi: 10.1515/CCLM.2002.210. [DOI] [PubMed] [Google Scholar]
- 6.Redzic ZB, Segal MB. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Deliv Rev. 2004;56(12):1695–1716. doi: 10.1016/j.addr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Costa RH, Grayson DR, Darnell JE. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and α1-antitrypsin genes. Mol Cell Biol. 1989;9:1415–1425. doi: 10.1128/mcb.9.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samadani U, Costa RH. The transcriptional activator hepatocyte nuclear factor 6 regulates liver gene expression. Mol Cell Biol. 1996;16(11):6273–6284. doi: 10.1128/mcb.16.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa RH, Grayson DR. Site-directed mutagenesis of hepatocyte nuclear factor (HNF) binding sites in the mouse transthyretin (TTR) promoter reveal synergistic interactions with its enhancer region. Nucleic Acids Res. 1991;19(15):4139–4145. doi: 10.1093/nar/19.15.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian X, Samadani U, Porcella A, Costa RH. Decreased expression of hepatocyte nuclear factor 3 alpha during the acute-phase response influences transthyretin gene transcription. Mol Cell Biol. 1995;15(3):1364–1376. doi: 10.1128/mcb.15.3.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickson PW, Aldred AR, Marley PD, Bannister D, Schreiber G. Rat choroid plexus specializes in the synthesis and the secretion of transthyretin (prealbumin). Regulation of transthyretin synthesis in choroid plexus is independent from that in liver. J Biol Chem. 1986;261(8):3475–3478. [PubMed] [Google Scholar]
- 12.Li X, Buxbaum JN. Transthyretin and the brain re-visited: is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol Neurodegener. 2011;6:79. doi: 10.1186/1750-1326-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Refai E, Dekki N, Yang SN, et al. Transthyretin constitutes a functional component in pancreatic beta-cell stimulus-secretion coupling. Proc Natl Acad Sci U S A. 2005;102(47):17020–17025. doi: 10.1073/pnas.0503219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christmansson L, Betsholtz C, Gustavsson Å, Johansson B, Sletten K, Westermark P. The transthyretin cDNA sequence is normal in transthyretin-derived senile systemic amyloidosis. FEBS Lett. 1991;281:177–180. doi: 10.1016/0014-5793(91)80387-i. [DOI] [PubMed] [Google Scholar]
- 15.Stein K, Storkel S, Linke RP, Goebel HH. Chemical heterogeneity of amyloid in the carpal tunnel syndrome. Virchows Arch A Pathol Anat Histopathol. 1987;412(1):37–45. doi: 10.1007/BF00750729. [DOI] [PubMed] [Google Scholar]
- 16.Rocken C, Saeger W, Linke RP. Gastrointestinal amyloid deposits in old age. Pathol Res Pract. 1994;190:641–649. doi: 10.1016/S0344-0338(11)80742-9. [DOI] [PubMed] [Google Scholar]
- 17.Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43(23):7365–7381. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 18.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31(36):8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 19.Sekijima Y, Wiseman RL, Matteson J, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Sousa A, Coelho T, Sequeiros J. Amyloid and Amyloidosis. Kluwer; 1991. Parental transmission and age of onset in Familial Amyloidotic Polyneuropathy (Portuguese type) pp. 691–693. [Google Scholar]
- 21.Coelho T, Carvalho M, Saraiva M, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET 119. J Rheumatol. 1993;20:179. [Google Scholar]
- 22.Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89(3):353–356. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- 23.Sousa A, Coelho T, Lobato L, Sequeiros J. Anticipation of Age of Onset in Familial Amyloidotic Polyneuropathy (Portuguese type) Philadelphia, PA: Kluwer Academic Publishers; 1990. pp. 694–697. [Google Scholar]
- 24.Andrade C. A peculiar form of peripheral neuropathy. Familial atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 25.Costa PP, Figueira AS, Bravo FR. Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proc Natl Acad Sci U S A. 1978;75:4499–4503. doi: 10.1073/pnas.75.9.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araki S, Mawatari S, Ohta M, Nakajima A, Kuroiwa Y. Polyneuritic amyloidosis in a Japanese family. Arch Neurol. 1968;18(6):593–602. doi: 10.1001/archneur.1968.00470360015001. [DOI] [PubMed] [Google Scholar]
- 27.Drugge U, Andersson R, Chizari F, et al. Familial amyloidotic polyneuropathy in Sweden: a pedigree analysis. J Med Genet. 1993;30:388–392. doi: 10.1136/jmg.30.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshioka K, Furuya H, Sasaki H, Saraiva MJM, Costa PP, Sakaki Y. Haplotype analysis of familial amyloidotic polyneuropathy. Evidence for multiple origins of the Val-Met mutation most common to the disease. Hum Genet. 1989;82:9–13. doi: 10.1007/BF00288262. [DOI] [PubMed] [Google Scholar]
- 29.Holmgren G, Costa PM, Andersson C, et al. Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet. 1994;31:351–354. doi: 10.1136/jmg.31.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardell L, Holmgren G, Steen L, Fredrikson M, Axelson O. Occupational and other risk factors for clinically overt familial amyloid polyneuropathy. Epidemiology. 1995;6:598–601. doi: 10.1097/00001648-199511000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086–1097. doi: 10.1016/S1474-4422(11)70246-0. [DOI] [PubMed] [Google Scholar]
- 32.Suhr OB, Ericzon BG, Friman S. Long-term follow-up of survival of liver transplant recipients with familial amyloid polyneuropathy (Portuguese type) Liver Transpl. 2002;8(9):787–794. doi: 10.1053/jlts.2002.34386. [DOI] [PubMed] [Google Scholar]
- 33.Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341:1113–1116. doi: 10.1016/0140-6736(93)93127-m. [DOI] [PubMed] [Google Scholar]
- 34.Wilczek HE, Larsson M, Ericzon BG. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR) Amyloid. 2011;18(Suppl 1):188–190. doi: 10.3109/13506129.2011.574354072. [DOI] [PubMed] [Google Scholar]
- 35.Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235(5):479–485. doi: 10.1111/j.1365-2796.1994.tb01106.x. [DOI] [PubMed] [Google Scholar]
- 36.Yazaki M, Tokuda T, Nakamura A, et al. Cardiac amyloid in patients with familial amyloidotic polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun. 2000;274:702–706. doi: 10.1006/bbrc.2000.3203. [DOI] [PubMed] [Google Scholar]
- 37.Barreiros AP, Post F, Hoppe-Lotichius M, et al. Liver transplantation and combined liver-heart transplantation in patients with familial amyloid polyneuropathy: a single-center experience. Liver Transpl. 2010;16(3):314–323. doi: 10.1002/lt.21996. [DOI] [PubMed] [Google Scholar]
- 38.Ando Y, Ericzon BG, Suhr OB, Tashima K, Ando M. Reuse of a Japanese familial amyloidotic polyneuropathy patient’s liver for a cancer patient: the domino liver transplantation procedure. Intern Med. 1997;36(11):847. doi: 10.2169/internalmedicine.36.847. [DOI] [PubMed] [Google Scholar]
- 39.Monteiro E, Perdigoto R, Furtado AL. Liver transplantation for familial amyloid polyneuropathy. Hepatogastroenterology. 1998;45(23):1375–1380. [PubMed] [Google Scholar]
- 40.Stangou AJ, Heaton ND, Hawkins PN. Transmission of systemic transthyretin amyloidosis by means of domino liver transplantation. N Engl J Med. 2005;352(22):2356. doi: 10.1056/NEJM200506023522219. [DOI] [PubMed] [Google Scholar]
- 41.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baures PW, Oza VB, Peterson SA, Kelly JW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Med Chem. 1999;7(7):1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 43.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 44.Berk JL, Dyck PJ, Obici L, et al. The diflunisal trial: update on study drug tolerance and disease progression. Amyloid. 2011;18(Suppl 1):191–192. doi: 10.3109/13506129.2011.574354073. [DOI] [PubMed] [Google Scholar]
- 45.Reixach N, Foss TR, Santelli E, Pascual J, Kelly JW, Buxbaum JN. Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic. A lesson in the generation of transgenic models of diseases involving oligomeric proteins. J Biol Chem. 2008;283(4):2098–2107. doi: 10.1074/jbc.M708028200. [DOI] [PubMed] [Google Scholar]
- 46.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy. Neurology. 2012;79:1–8. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dyck PJ, Litchy WJ, Daube JR, et al. Individual attributes versus composite scores of nerve conduction abnormality: sensitivity, reproducibility, and concordance with impairment. Muscle Nerve. 2003;27(2):202–210. doi: 10.1002/mus.10320. [DOI] [PubMed] [Google Scholar]
- 50.Vinik EJ, Hayes RP, Oglesby A, et al. The development and validation of the Norfolk QOL-DN, a new measure of patients’ perception of the effects of diabetes and diabetic neuropathy. Diabet Technol Ther. 2005;7(3):497–508. doi: 10.1089/dia.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 51.Kim DH, Zeldenrust SR, Low PA, Dyck PJ. Quantitative sensation and autonomic test abnormalities in transthyretin amyloidosis polyneuropathy. Muscle Nerve. 2009;40(3):363–370. doi: 10.1002/mus.21332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feinstein AR. Clinical Epidemiology: The Architecture of Clinical Research. Philadelphia, PA: W.B. Saunders Company; 1985. [Google Scholar]
- 53.Katz R, Jillapalli D, Luan J. Peripheral and central nervous System Drugs Advisory Committee Meeting, Background package 24 May 2012. Available at www.fda.gov/downloads/AdvisorycommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdviso-ryCommittee/UCM304830.pdf.
- 54.Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78(9):637–643. doi: 10.1212/WNL.0b013e318248df18. [DOI] [PubMed] [Google Scholar]
- 55.Kristen AV, Lehrke S, Buss S, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol. 2012;101(10):805–813. doi: 10.1007/s00392-012-0463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19(Suppl 1):34–36. doi: 10.3109/13506129.2012.678508. [DOI] [PubMed] [Google Scholar]
- 57.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 58.Botto M, Hawkins PN, Bickerstaff MC, et al. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med. 1997;3(8):855–859. doi: 10.1038/nm0897-855. [DOI] [PubMed] [Google Scholar]
- 59.Pepys MB, Herbert J, Hutchinson WL, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417(6886):254–259. doi: 10.1038/417254a. [DOI] [PubMed] [Google Scholar]
- 60.Alvarez R, Borland T, Chen Q, et al. ALN-TTR, an RNAi therapeutic for the treatment of Transthyretin-mediated amyloidosis. Amyloid. 2010;17(s1):51–52. [Google Scholar]
- 61.Benson MD, Pandey S, Witchell D, et al. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid. 2011;18(Suppl 1):55. doi: 10.3109/13506129.2011.574354021. [DOI] [PubMed] [Google Scholar]