

Abstract
The generation of ATP through oxidative phosphorylation is an essential metabolic function for Mycobaterium tuberculosis (Mtb), regardless of the growth environment. The type II NADH dehydrogenase (Ndh-2) is the conduit for electrons into the pathway, and is absent in the mammalian genome, thus making it a potential drug target. Herein, we report the identification of two types of small molecules as selective inhibitors for Ndh-2 through a multicomponent high-throughput screen. Both compounds block ATP synthesis, lead to effects consistent with loss of NADH turnover, and importantly, exert bactericidal activity against Mtb. Extensive medicinal chemistry optimization afforded the best analogue with an MIC of 90 nM against Mtb. Moreover, the two scaffolds have differential inhibitory activities against the two homologous Ndh-2 enzymes in Mtb, which will allow precise control over Ndh-2 function in Mtb to facilitate the assessment of this anti-TB drug target.
Keywords: antimicrobial compounds, drug discovery, Mycobaterium tuberculosis, Ndh-2, oxidative phosphorylation
Mycobacterium tuberculosis resides in a variety of micro-environments, depending on the stage of infection, and to survive it is aided by a remarkable metabolic plasticity, which allows remodeling of energy-utilization and biomass-producing pathways, depending on the environmental conditions and nutrient availability.[1] Recent work suggests that the survival of Mtb under a variety of conditions is completely dependent on the existence of an energized plasma membrane, which couples nutrient oxidation to ATP synthesis through the production of an electrochemical gradient.[2] The discovery that TMC207 (bedaquiline) exerts its antimycobacterial activity through shutdown of ATP production by inhibition of the F-type ATP synthase validates oxidative phosphorylation as a pharmacologic target in Mtb.[3]
Electrons enter the respiratory chain from NADH oxidation, coupled to the reduction of menaquinone. The Mtb genome encodes both type I (nuoA-N) and two type II NADH:menaquinone oxidoreductases (Ndh-2: ndh and ndhA), although nuo appears to be dispensable for growth.[4] The role(s) of each individual copy of Ndh-2 are uncertain, but genetic knockout studies have suggested only ndh is essential for Mtb growth and persistence.[5] Importantly, while common among bacteria and protozoan parasites, these enzymes are absent from the mammalian genome, which further highlights their potential as drug targets.[6] A well-validated, specific Ndh-2 inhibitor would both provide a novel tool to study Ndh-2 function and enable preclinical pharmacological validation of Ndh-2 as a target for TB therapy.
We performed a high-throughput screen for compounds that reduce the production of ATP (through oxidative phosphorylation) by inhibiting Ndh-2. We utilized membrane vesicles derived from mycobacteria and measured ATP production upon the addition of NADH. The NADH is then oxidized through a cascade of redox reactions, resulting in the formation of a proton gradient across the vesicle membrane. This proton gradient drives the synthesis of ATP from ADP and inorganic phosphate by the F1F0 ATP synthase. In this assay, bedaquiline inhibits ATP synthesis with an IC50 of 6.9 nM (Figure S1 in the Supporting Information).[7] In a miniaturized 1536-well-plate format of this assay, we screened over 800000 compounds and identified over 7000 primary hits (Figure S2). Hit compounds were progressed through a variety of triage assays to eliminate false positives, exclude cytotoxic compounds, and to determine the mode of inhibition in the pathway.
Compounds CBR-1825 and CBR-4032 each inhibited electron-transport-dependent ATP production initiated by NADH, as well as NADH oxidation assayed in mycobacterium membranes with IC50 values of 0.5 μM or less (Figure 1 and Figure S3). Furthermore, they were inactive when assayed for succinate-initiated oxidative phosphorylation in membranes (Figure S4). This activity profile suggests that both small molecules block the electron transport chain at Ndh-2. Multiple active structural analogues were identified in the primary screen for both series. The major structural motif for CBR-1825 and related compounds was the thioquinazoline (TQZ) core; CBR-4032 and similar molecules possess a tetrahydroindazole (THI) core. Importantly, both scaffolds showed growth inhibition of Mtb in liquid medium (MIC50 = 0.43 μM and 6.6 μM for CBR-1825 and CBR-4032, respectively) while displaying no gross toxicity towards mammalian cells (Figure 1A,B).
Figure 1.
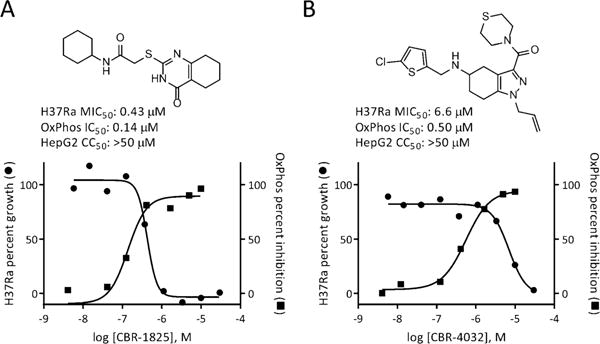
Representative hit compounds from the primary screen and their activities against Mtb H37Ra and oxidative phosphorylation assessed in mycobacterial membranes. A) CBR-1825, a representative of the thioquinazoline scaffold. B) CBR-4032, a representative of the tetrahydroindazole series.
We prioritized medicinal chemistry optimization for the TQZ series due to its high cellular potency. A few key structure–activity relationships were observed during this process (Figure 2 and the Supporting Information). Replacement of the thioether with oxygen, nitrogen, or methylene significantly reduced activity, as did oxidation of the sulfide. Compared to saturated fused rings, a fused phenyl ring was preferred. CBR-5992, a quinazoline compound, was identified with a MIC of 0.67 μM, which makes it two-fold more active than the original hit CBR-1825. Further modifications of the phenyl ring (various substitutions and heterocyclic rings) afford 5-fluoro-substituted analogue CBR-3465 with further improved activity (MIC = 0.16 μM). The next SAR study was conducted around cyclohexane amide side chain in the context of the optimized 5-fluoroquinazoline. While a lot of analogues are not active, fluoride, chloride, and methyl substituents are relatively well tolerated. Among them, introduction of difluorines into the cyclohexane ring afforded the most potent analogue CBR-1922 against virulent Mtb with an MIC of 0.09 μM in growth-inhibition assays. This compound compares favorably with the most advanced inhibitors from two recently described medicinal chemistry efforts targeting Ndh-2: quinolinyl pyrimidines and a quinolone series.[8] The MIC50 values of the most potent compounds from either of these two series ranged from 0.14–1.0 μM. Likewise, no compounds from a suite of phenothiazine-derived compounds gave MIC50 values below 10 μM against aerobic Mtb.[9]
Figure 2.
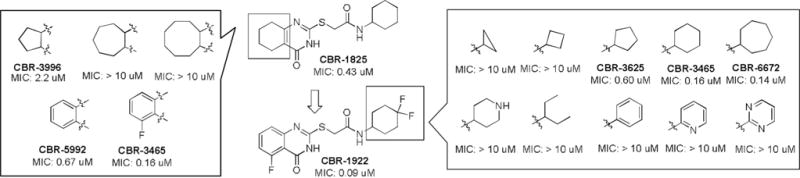
Selected SAR results for the TQZ scaffold.
Next, we assessed whether either Ndh-2 inhibitor series is bactericidal. Indeed, compounds from each scaffold produced over 1-log killing within the first three-day treatment, and greater than 4-log killing against cultures over a period of 32 days when tested at ten times the MIC (Figure S5). These data suggest that any assumed activity by the type I Ndh is unable to complement the loss of function of Ndh-2. Moreover, treatment of Mtb with complex I inhibitors, such as rotenone, has little effect on mycobacterial growth or NADH oxidation in purified membranes, thus suggesting that Ndh-2 is the dominant NADH dehydrogenase.[10]
Although our preliminary triage data suggested that both scaffolds block the electron-transport chain (ETC) at Ndh-2, we initiated further target identification studies for confirmation. We attempted to identify the target(s) of the TQZ scaffold by isolating spontaneously arising mutants resistant to it. Single-step selection produced resistant mutants at a frequency of 3 × 10−8. Sequencing of 3 resistant mutants revealed a C-to-G mutation in the promoter region of ndhA (Rv0392c), 44 nucleotides upstream of the start codon. We analyzed gene expression levels of ndh, ndhA, and nuoG in wild-type and one of the resistant strains and found that ndhA was expressed at a level approximately 50-fold higher in the resistant strain than in the wildtype strain (Figure 3A). Determination of MIC50 values of the resistant strains showed a greater than 10-fold decrease in susceptibility to either scaffold, thus suggesting that resistance is conferred by compensatory upregulated expression of ndhA. (Figure 3B and Figure S6).
Figure 3.
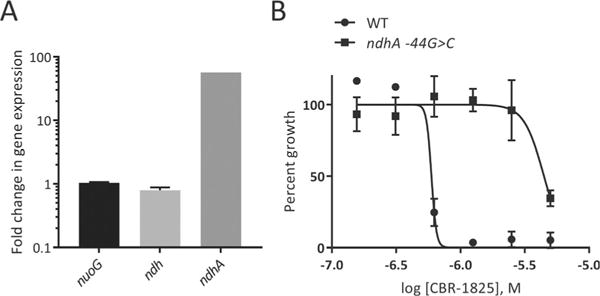
Upregulation of ndhA expression confers resistance to TQZ. A) Gene expression in the H37Ra ndhA-44 G >C background. B) Activity of TQZ against wild-type and ndhA-44 G> C H37Ra strains.
The biochemical and genetic data suggested that the activity of both scaffolds is mediated through one or both type II NADH:quinone oxidoreductases, yet those data do not specifically distinguish the protein target(s) of these inhibitors. The Ndh-2 enzymes Ndh and NdhA in Mtb share high homology (67% identity), are both expressed under aerobic culture conditions, and retain oxidoreductase activity.[11] Little is known though about their respective roles; in organisms with multiple copies of Ndh-2, their roles are often non-redundant.[12] To further examine target engagement, we utilized a temperature-sensitive (TS) Mycobacterium smegmatis mutant to probe the activity of each compound against Ndh and NdhA individually. When grown above the permissive temperature, this TS strain downregulates expression of ndh due to a base substitution of G250 to T of the ndh gene that alters amino acid Gly84 (GGC) to Cys (TGC) of Ndh (Yano and Rubin, unpublished data). When grown at 30°C, this strain shows low susceptibility to either inhibitor series (similar to the WT M.smegmatis), but as the incubation temperature increases to 40°C, ndh expression correspondingly decreases, which results in an more than 8-fold shift in the MIC of the two compounds relative to their potency at the permissive temperature (Table S1 and Figure S7). In addition, the TS mutant can be complemented with Mtb malate dehydrogenase (mdh), which restores growth at 42°C by combining the M. smegmatis malate:menaquinone oxidoreductase (Mqo) activity with Mtb Mdh NADH recycling.[13] When grown at 42°C, a temperature where Ndh-2 activity is minimal and complemented by Mdh/Mqo activity, neither compound inhibited cell growth at 100 μM, thus strongly suggesting that their target is Ndh-2.
This strain also provides a biochemical means to assess inhibitor activity against either Ndh or NdhA by utilizing isolated membrane vesicles. We used the mutant strain complimented with either Mtb ndh or ndhA and prepared membrane vesicles from cultures grown at the non-permissive temperature. In these membrane preparations, the complemented genes are the majority type II NADH dehydrogenases expressed. The TQZ analogue CBR-1825 showed strong potency against both Msmeg and Mtb Ndh (IC50 = 10.7 nM and 64.7 nM, respectively) when expressed inthe temperature-sensitive background and grown under non-permissive conditions (Table 1). Its activity against Mtb NdhA was similarly potent. Unlike the TQZ scaffold, the THI inhibitor CBR-4032 appeared to be specific for Ndh, since its IC50 against NdhA was over 300-fold higher than that for Ndh.
Table 1.
NADH oxidation in M. smegmatis membranes with complemented Mtb ndh or ndhA is inhibited by the TQZ and THI scaffolds.
| Compound |
Mtb Ndh IC50 (nm) |
Msmeg Ndh IC50 (nm) |
Mtb NdhA IC50 (nm) |
|---|---|---|---|
| TQZ | 64.7 | 10.7 | 42.5 |
| THI | 25.0 | 44.3 | 15300 |
Taking these results together, we felt confident that both scaffold series were potent and specific Ndh-2 inhibitors, and we next endeavored to characterize the phenotypic response to loss of function of Ndh-2 in mycobacteria.
The NADH:quinone oxidoreductases are the major enzymes that oxidize NADH to NAD+, and we therefore hypothesized that the immediate consequence of blocking Ndh-2 function would be the prevention of NADH oxidation, resulting in an increase in the ratio of NADH:NAD+. It has previously been shown that increased levels of NADH cofactor can protect InhA from inhibition by isoniazid (INH), likely due to increased competition with the active form of INH, the INH–NAD adduct.[14] Indeed, CBR-1825 abrogated the antimycobacterial activity of INH when the two were tested in combination. Treating Mtb with a sub-lethal concentration of the Ndh-2 inhibitor completely reversed growth inhibition of isoniazid at a concentration that normally prevents proliferation (Figure 4A). These results demonstrate that an increase in the NADH:NAD+ ratio causes resistance to INH, which is consistent with the previous observations in type II NADH dehydrogenase mutants isolated during selection for INH resistance.[13b] Some strains with Ndh mutations are also auxotrophic for serine, possibly due to inhibition of the initial step in serine biosynthesis by elevated levels of NADH.[15] To corroborate this, we found that addition of serine to Ndh-2 inhibitor-treated Mtb cultures could rescue bacterial growth (Figure 4B).
Figure 4.
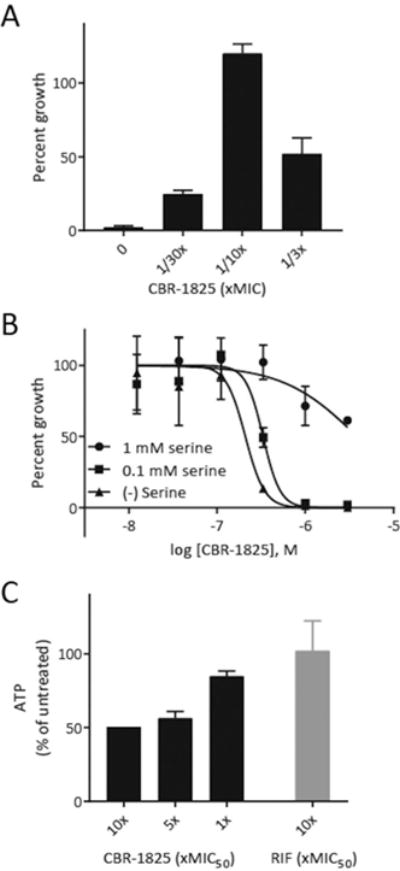
Metabolic and physiologic effects in Mtb produced by Ndh-2 inhibition. A) Treatment with sub-inhibitory concentrations of TQZ blocks growth inhibition by INH at a concentration (250 nm) that normally completely prevents replication in the absence of TQZ. Percent growth was normalized to untreated controls. B) Addition of exogenous serine rescues Mtb treated with an Ndh-2 inhibitor. C) Quantitation of ATP levels in Mtb treated with 10 ×, 5 ×, and 1× the MIC50 of the given inhibitor after 6 h of treatment time.
Shutdown of electron transport to the proton-pumping terminal electron acceptors will disrupt the proton motive force, which is utilized by the F1F0-ATP synthase to catalyze ATP synthesis.[16] Consistent with this notion, the inhibition of Ndh-2 also decreased intracellular ATP levels in a dose-dependent fashion (Figure 4C).
The mycobacterium ETC and the ATP synthetic machinery driven by it represent a hub of vulnerability for the pathogen.[17] Furthermore, due to the druggability of targets in this pathway, as evidenced here and elsewhere, it remains an opportunity for anti-TB drug discovery.[3,18] By using a pathway-based screen in mycobacterial membranes, we identified inhibitors that block oxidative phosphorylation and prevent Mtb growth through disruption of Ndh-2 activity. Inhibition of Ndh-2 not only prevents ATP synthesis through blockade of the ETC, but also disturbs the intracellular redox balance, which leads to an array of metabolic consequences. The identification of inhibitors from a biochemical screen that have cellular activity is especially encouraging, given past low success rates in these screens in the antimicrobial field.[19] The scaffolds identified here act on Ndh and NdhA differently even though their protein sequences are highly conserved. We believe that it is reasonable to suggest that the TQZ and THI scaffolds occupy the quinone and NADH binding sites, respectively, given their structural resemblance to the quinone and adenine molecules that normally reside in those positions.[20] Structural analysis coupled with kinetic enzymatic analysis of the inhibitors would be the best way to gain more insight into their inhibitory mechanism.[21] Future studies will also include utilizing our Ndh-2 inhibitors to explore the roles of Ndh-2 and Nuo in vivo, and will allow us to inactivate both or one copies of Ndh-2, respectively.
Supplementary Material
Acknowledgments
The authors thank Drs. Cliff Barry and Helena Boshoff for technical support or helpful discussion. This work was supported in part by National Key R&D Program of China 2017YFA0505400, Strategic Priority Research Program of CAS, XDPB0304, CAS Pioneer Hundred Talents Program, the Bill and Melinda Gates Foundation, the Global Alliance for TB Drug Development, and NIH Grants AI026170 (WRJ) and AI051519 (WRJ). We also acknowledge Jennifer Yano for her technical assistance.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201800260.
Conflict of interest The authors declare no conflict of interest.
Contributor Information
Dr. Michael B. Harbut, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Dr. Baiyuan Yang, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Dr. Renhe Liu, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Dr. Takahiro Yano, Department of Medicine, University of Pennsylvania Philadelphia, PA 19104 (USA)
Dr. Catherine Vilchèze, Department of Medicine, University of Pennsylvania Philadelphia, PA 19104 (USA)
Dr. Bo Cheng, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Dr. Jonathan Lockner, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Dr. Hui Guo, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101 (China)
Dr. Chenguang Yu, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Prof. Scott G Franzblau, Institute for Tuberculosis Research, University of Illinois at Chicago, Chicago, IL 60612 (USA)
Dr. H. Mike Petrassi, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Prof. William R. Jacobs, Jr., Howard Hughes Medical Institute, Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, NY 10461 (USA)
Prof. Harvey Rubin, Jr., Department of Medicine, University of Pennsylvania Philadelphia, PA 19104 (USA)
Dr. Arnab K. Chatterjee, California Institute for Biomedical Research, La Jolla, CA 92037 (USA)
Prof. Feng Wang, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101 (China), California Institute for Biomedical Research, La Jolla, CA 92037 (USA).
References
- 1.Dartois V, Barry CE., 3rd Bioorg Med Chem Lett. 2013;23:4741–4750. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook GM, Hards K, Vilcheze C, Hartman T, Berney M. Microbiol Spectr. 2014 doi: 10.1128/microbiolspec.MGM2-0015-2013. [DOI] [PMC free article] [PubMed]
- 3.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 4.Sassetti CM, Boyd DH, Rubin EJ. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 5.Awasthy D, Ambady A, Narayana A, Morayya S, Sharma U. Gene. 2014;550:110–116. doi: 10.1016/j.gene.2014.08.024. [DOI] [PubMed] [Google Scholar]
- 6.Bald D, Villellas C, Lu P, Koul A. mBio. 2017;8:e00272–17. doi: 10.1128/mBio.00272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. Nat Chem Biol. 2007;3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 8.a) Shirude PS, Paul B, Roy Choudhury N, Kedari C, Bandodkar B, Ugarkar BG. ACS Med Chem Lett. 2012;3:736–740. doi: 10.1021/ml300134b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hong WD, Gibbons PD, Leung SC, Amewu R, Stocks PA, Stachulski A, Horta P, Cristiano MLS, Shone AE, Moss D, Ardrey A, Sharma R, Warman AJ, Bedingfield PTP, Fisher NE, Aljayyoussi G, Mead S, Caws M, Berry NG, Ward SA, Biagini GA, O’Neill PM, Nixon GL. J Med Chem. 2017;60:3703–3726. doi: 10.1021/acs.jmedchem.6b01718. [DOI] [PubMed] [Google Scholar]
- 9.Warman AJ, Rito TS, Fisher NE, Moss DM, Berry NG, O’Neill PM, Ward SA, Biagini GA. J Antimicrob Chemother. 2013;68:869–880. doi: 10.1093/jac/dks483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao SP, Alonso S, Rand L, Dick T, Pethe K. Proc Natl Acad Sci USA. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. Proc Natl Acad Sci USA. 2005;102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin SS, Gross U, Bohne W. Mol Microbiol. 2011;82:209–221. doi: 10.1111/j.1365-2958.2011.07807.x. [DOI] [PubMed] [Google Scholar]
- 13.a) Molenaar D, van der Rest ME, Drysch A, Yucel R. J Bacteriol. 2000;182:6884–6891. doi: 10.1128/jb.182.24.6884-6891.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vilcheze C, Weisbrod TR, Chen B, Kremer L, Hazbon MH, Wang F, Alland D, Sacchettini JC, Jacobs WR., Jr Antimicrob Agents Chemother. 2005;49:708–720. doi: 10.1128/AAC.49.2.708-720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen M, Quemard A, Broussy S, Bernadou J, Meunier B. Antimicrob Agents Chemother. 2002;46:2137–2144. doi: 10.1128/AAC.46.7.2137-2144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugimoto E, Pizer LI. J Biol Chem. 1968;243:2081–2089. [PubMed] [Google Scholar]
- 16.Hards K, Robson JR, Berney M, Shaw L, Bald D, Koul A, Andries K, Cook GM. J Antimicrob Chemother. 2015;70:2028–2037. doi: 10.1093/jac/dkv054. [DOI] [PubMed] [Google Scholar]
- 17.Cook GM, Hards K, Dunn E, Heikal A, Nakatani Y, Greening C, Crick DC, Fontes FL, Pethe K, Hasenoehrl E, Berney M. Microbiol Spectr. 2017 doi: 10.1128/microbiolspec.TBTB2-0014-2016. [DOI] [PMC free article] [PubMed]
- 18.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SP, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, Lee J, Brodin P, Cho SN, Nam K, Kim J. Nat Med. 2013;19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 19.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. Nat Rev Drug Discovery. 2015;14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 20.Heikal A, Nakatani Y, Dunn E, Weimar MR, Day CL, Baker EN, Lott JS, Sazanov LA, Cook GM. Mol Microbiol. 2014;91:950–964. doi: 10.1111/mmi.12507. [DOI] [PubMed] [Google Scholar]
- 21.Yano T, Rahimian M, Aneja KK, Schechter NM, Rubin H, Scott CP. Biochemistry. 2014;53:1179–1190. doi: 10.1021/bi4013897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.