

Abstract
Purpose
Inherited retinal dystrophies are a clinically and genetically heterogeneous group of disorders. Molecular diagnosis has proven utility for affected individuals. In this study, we report an individual enrolled in the Australian Inherited Retinal Disease Registry and DNA Bank diagnosed with clinical features overlapping between Leber congenital amaurosis and retinitis pigmentosa.
Methods
DNA from the proband was sequenced using a gene panel for inherited retinal disorders, and a single nucleotide polymorphism (SNP) array was conducted to detect the presence of deletions and uniparental disomy.
Results
We identified a novel homozygous variant (c.524dupC, p.(Pro176ThrfsTer7)) in TULP1 resulting from maternal uniparental isodisomy of chromosome 6. The patient had clinical features consistent with biallelic pathogenic variants in TULP1, including congenital nystagmus, night blindness, non-recordable electroretinogram, mild myopia, and mild peripheral pigmentary changes in the fundus.
Conclusions
This is the first report of uniparental disomy 6 and a homozygous variant in TULP1 associated with a rod-cone dystrophy. Molecular diagnosis of inherited retinal dystrophies is essential to inform the mode of transmission and clinical management, and to identify potential candidates for future gene-specific therapies.
Introduction
Inherited retinal dystrophies (IRDs) are a group of disorders characterized by retinal degeneration with changes in the rod or cone photoreceptors. There are currently 301 genes and loci reported for IRD (RetNet), making molecular diagnosis challenging. Retinitis pigmentosa (RP, OMIM 268000) is the most common cause of IRD with a prevalence of 1/3,000 to 1/5,000 people among different populations [1,2]. RP is characterized by the degeneration of the rod and cone photoreceptor cells leading to night blindness, progressive loss of visual fields, and eventually, complete blindness. Leber congenital amaurosis (LCA, OMIM 204000) was first described in 1869 as a congenital severe form of RP [3]. LCA is the most common cause of congenital blindness and is characterized by onset in the first 12 months of life with severe visual loss, retinal dysfunction, congenital nystagmus, and non-recordable electroretinogram. LCA and RP display heterogeneous and overlapping clinical phenotypes, representing a continuum of phenotype with LCA at the severe end and RP at the mild end. Both diseases are genetically heterogeneous: 25 genes are known to be causative for LCA and more than 80 genes for non-syndromic RP (RetNet). Variants in several genes, including TULP1, cause overlapping phenotypes involving LCA and RP [4].
Variants in the TULP1 gene (6q21.3, OMIM 602280) were first identified in patients with RP [5,6] and were subsequently reported in 1–2% of patients with LCA [7]. TULP1 is a member of the tubby-like protein family and contains a highly conserved C-terminal tubby domain. TULP1 is expressed exclusively in the photoreceptor cells of the retina [8]. TULP1 knockout mice display progressive retinal degeneration with rapid loss of photoreceptor cells from 2 weeks of age [9]. The protein is involved in the trafficking of rhodopsin from its synthesis in the inner segments to the outer segments [9] and provides retinoprotection by stimulating phagocytosis of retinal pigment epithelium cells [10]. In this study, we report an individual with a clinical phenotype between LCA and severe early onset RP on the phenotypic continuum, harboring a novel homozygous TULP1 variant arising from maternal uniparental isodisomy of chromosome 6.
Methods
This study was approved by the Sir Charles Gairdner Hospital Human Research Ethics Committee (2001–053) and was conducted in accordance with the National Health and Medical Research Council statement of ethical conduct in research involving humans. The study complied with the tenets of the Declaration of Helsinki and the Association for Research in Vision and Ophthalmology (ARVO) statement on human subjects. Informed written consent was obtained. An individual with isolated non-syndromic retinal degeneration was ascertained with a complete ophthalmologic examination, including best-corrected visual acuity, refraction, fundoscopy, electroretinogram, visual field testing, and macular optical coherence tomography.
Genomic DNA was isolated from peripheral venous blood and stored as detailed previously [11]. Briefly, blood was obtained by venipuncture and collected in EDTA tubes. Genomic DNA was isolated from blood leukocytes using the QIAsymphony DSP DNA Midi extraction kit (Qiagen, Doncaster, Australia) on the QIAsymphony automated nucleic acid extraction platform as per manufacturer’s instructions, and stored at -40 degrees. DNA sourced from the Australian Inherited Retinal Disease Registry and DNA Bank was subjected to targeted next-generation sequencing using a disease-specific IRD next-generation sequencing SmartPanel (250 genes) v9 (Casey Eye Institute, Molecular Diagnostics Laboratory, Portland, OR) [12]. Variants of interest were confirmed with Sanger sequencing. A single nucleotide polymorphism (SNP) array (Illumina Infinium CytoSNP-850K BeadChip; Scoresby, Australia) was conducted to further investigate deletions of the TULP1 gene and uniparental disomy (UPD). Identified variants are available in the ClinVar database.
Results
The proband was the second child of a non-consanguineous couple of Italian background. The pregnancy was marked by oligohydramnios and intrauterine growth retardation, and the proband was born at 37 weeks gestation by caesarean section with Apgar scores of 9 and 10. Birthweight (1640 g), length (44 cm), and head circumference (30.5 cm) were all below the third percentile. At 3 months, nystagmus, poor pupillary constriction to strong light, and retinal pallor were noted. Magnetic resonance imaging performed at that time was normal. By 17 months of age, the fundi were normal, but an electroretinogram was abnormal with reduced responses under light-adapted conditions and no response under dark-adapted conditions. These findings, in addition to poor night vision, suggested a diagnosis of rod-cone dystrophy. At 2 1/2 years old, the patient had mild myopia (−3.0 D in both eyes). At her last examination, she was 22 years old and had poor night vision and peripheral vision, bilateral visual acuity of 6/76, mild to moderate attenuation of arterioles, mild peripheral bone spicule-like pigmentation in the fundus, and no optic disc atrophy (Figure 1). Optical coherence tomography showed severe thinning at the macula (Figure 2). Her diagnosis was maintained as a rod-cone dystrophy with overlap between LCA and RP. There was no presence of cataracts, keratoconus, or macular edema. Both parents had normal eye examinations with no features of retinal dystrophy. The remaining medical and family histories were unremarkable.
Figure 1.
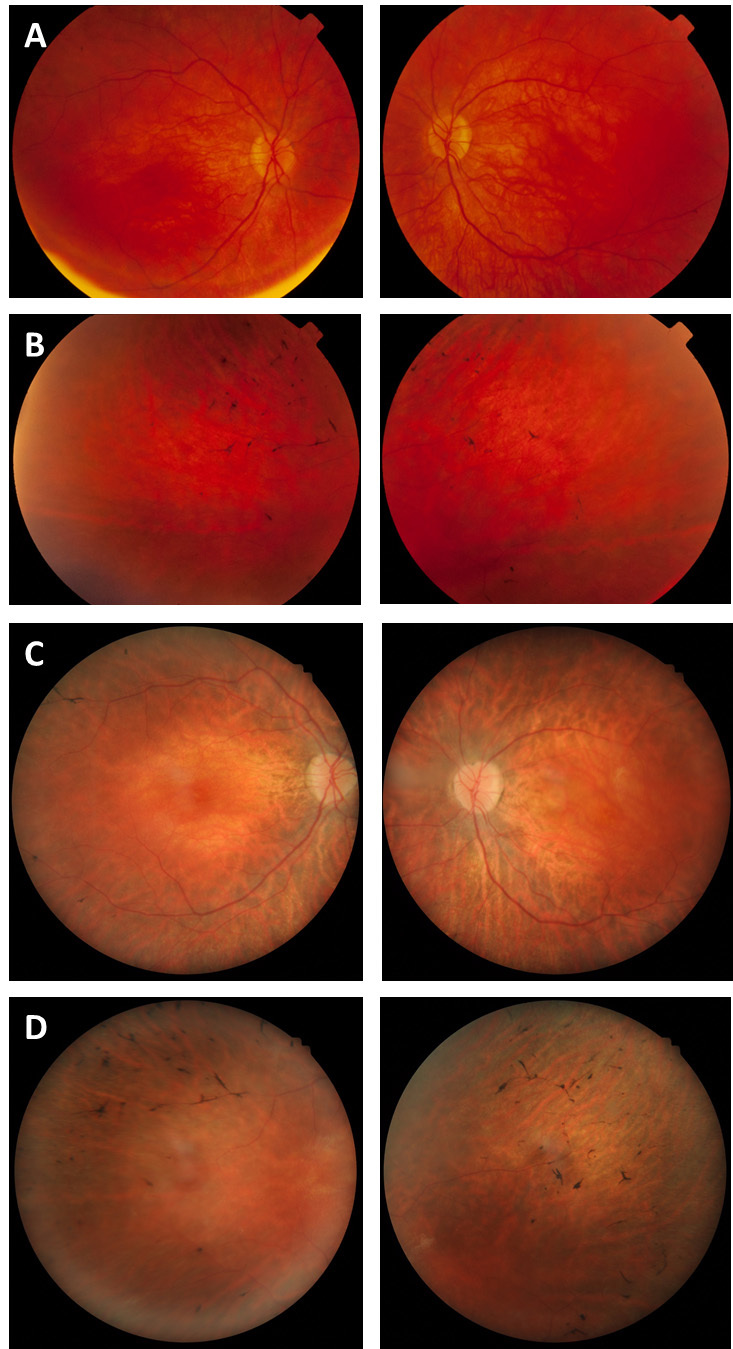
Fundus photography of the proband. Mild macular atrophy (A) with mild peripheral bone spicule-like pigmentation in the periphery (B) at age 14 years in comparison with severe atrophic macular changes, waxy pallor of the optic disc (C), and more pronounced peripheral bone spicule-like pigmentation in the periphery (D) at age 22 years.
Figure 2.
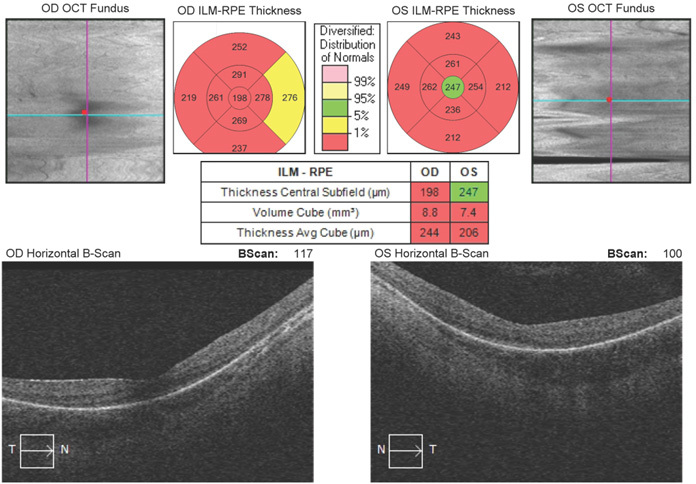
Optical coherence tomography showing severe thinning of the macula in both eyes at age 12 years, as demonstrated by the thickness being less than 1% of normal distribution (red) for both eyes.
A homozygous novel frameshift variant (c.524dupC, p.(Pro176ThrfsTer7)) was identified in the TULP1 gene (NM_003322.3) in the proband (Figure 3A). This novel variant was considered pathogenic due to its frameshifting nature, which is predicted to invoke nonsense-mediated decay with subsequent abolition of the protein product. Targeted sequencing revealed that the mother was heterozygous and the father did not carry the variant. SNP array analyses did not identify a deletion on the other allele. The genotype of the proband and the father were compared: Informative SNPs confirmed paternity and demonstrated no paternal inheritance of the whole of chromosome 6, confirming maternal isodisomy of this entire chromosome (Figure 3B). No evidence for mosaicism of trisomy 6 was detected on buccal swab.
Figure 3.
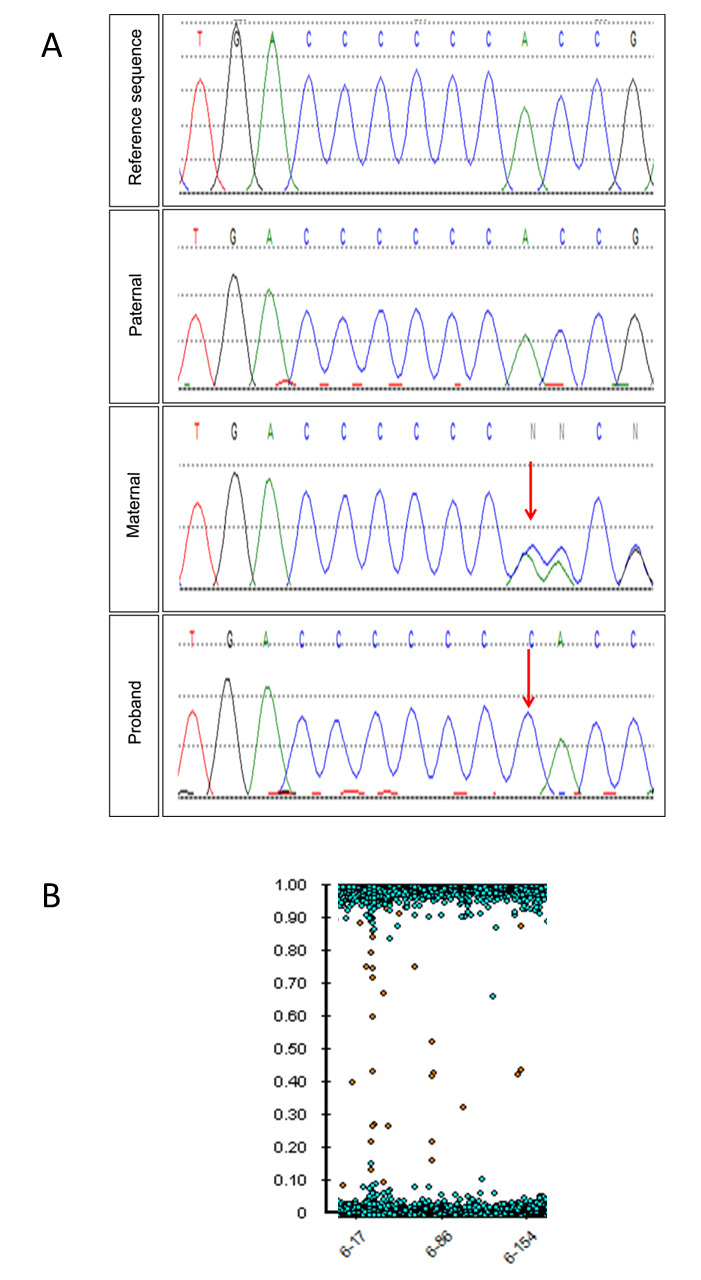
Genetic analysis of the proband. A: Sanger sequencing electropherograms showing (top to bottom) TULP1 reference sequence (NM_003322.3), paternal electropherogram showing wild-type sequence, maternal electropherogram revealing heterozygous duplication, c.524dupC (arrow), and proband electropherogram showing homozygous duplication (arrow) of the preceding C nucleotide. B: Single nucleotide polymorphism (SNP) array showing the absence of heterozygous calls on chromosome 6, corresponding to uniparental isodisomy.
Additionally, a novel heterozygous variant (c.1267_1656delinsA, p.(Ala423AsnfsTer11)) in the RP1 gene (OMIM 603937; NM_006269.1) was identified in the proband and her mother. Truncations occurring between codons 264 and 499 are associated with autosomal recessive RP [13]. This is supported by the fact that the proband’s mother does not have retinal degeneration. Although it is unlikely to be a primary cause of disease, we cannot rule out a contributory effect to disease based on the mutation load [14].
Discussion
TULP1 variants have been associated with milder forms of LCA [7] and severe juvenile-onset forms of RP [15,16], and intrafamilial variable expressivity has been reported [16]. Biallelic variants in TULP1 are usually associated with congenital nystagmus, a non-recordable electroretinogram, relative preservation of visual acuity and visual field in the first decade of life, night blindness, myopic refractive errors, and peripheral involvement of the retina resembling the early aspects of RP [7,16]. Similarly, the individual described in this study had congenital nystagmus, night blindness, visual acuity of 6/76 in both eyes at age 22 years, mild myopia, and mild peripheral pigmentary changes in the fundus.
UPD is a rare event in which an individual with a diploid genome carries either two homologs of a pair of chromosomes from one parent (uniparental heterodisomy) or two copies of a single chromosome from one parent (uniparental isodisomy) [17]. The most common mechanism is trisomic rescue. Disease phenotypes can occur as a result of UPD through different mechanisms: genomic imprinting, mosaicism, or unmasking of autosomal recessive genetic variants with both alleles carrying a deleterious variant inherited from one parent. Paternal UPD6 is associated with transient neonatal diabetes mellitus and is caused by abnormal imprinting of PLAGL1 (OMIM 603044; NM_001080951.2) and HYMAI (OMIM 606546; NR_002768.2) [18]. In contrast, maternal UPD6 is much rarer with only 15 cases reported. There is a possible association with intrauterine growth retardation, which was noted in the individual on whom we report [19]. The underlying mechanism behind intrauterine growth retardation is not known. The extent of UPD involvement in IRD is unknown, as UPD often goes unnoticed unless revealed by genetic investigation of recessive disease [20]. UPD has previously been reported in patients with IRD: UPD1 in patients with LCA and variants in RPE65 (OMIM 180069; NM_000329.2) [21], RP without hearing loss and variants in USH2A (OMIM 608400; NM_007123.5) [22], and Stargardt disease and variants in ABCA4 (OMIM 601691; NM_000350.2) [23,24]; UPD2 in patients with RP and variants in MERTK (OMIM 604705; NM_006343.2) [21] and RP and variants in FAM161A (OMIM 613596; NM_001201543.1) [25]; UPD6 in a patient with cone dysfunction and variants in TULP1 [26]; and UPD14 in a patient with achromatopsia and variants in GNAT2 (OMIM 139340; NM_005272.3) [27]. This is the first report of a patient with UPD6 and variants in TULP1 associated with a rod-cone dystrophy.
Molecular diagnosis in patients with IRD has proven clinical utility. The differential diagnosis for LCA/RP can be extensive, and an early molecular diagnosis can inform whether the IRD is part of a syndrome or a neurometabolic disease, and thus, influence clinical management. LCA and RP can be inherited in an autosomal dominant or recessive manner; therefore, molecular diagnosis allows the provision of accurate genetic counseling, including information about risk of recurrence and reproductive options. Finally, advances in gene-specific therapy and stem cell therapy could lead to new therapeutic avenues for patients with IRD, for which identification of causative genetic mechanisms will be a pivotal prerequisite.
Acknowledgments
This work was supported by funding from Retina Australia, and the National Health & Medical Research Council (NHMRC) of Australia (#1116360). The authors acknowledge the assistance of the Western Australian DNA Bank, the support of the Department of Medical Technology and Physics, Sir Charles Gairdner Hospital and the use of the services and facilities of the Casey Eye Institute Molecular Diagnostics Laboratory and Molecular Vision Laboratory (which has no relationships with the journal Molecular Vision).
References
- 1.Boughman JA, Conneally PM, Nance WE. Population genetic studies of retinitis pigmentosa. Am J Hum Genet. 1980;32:223–35. [PMC free article] [PubMed] [Google Scholar]
- 2.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;(233):1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 3.Leber T. Ueber Retinitis pigmentosa und angeborene Amaurose. Graefes Arch Clin Exp Ophthalmol. 1869;15:1–25. [Google Scholar]
- 4.den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, Hehir-Kwa JY, Veltman JA, Arends ML, Meitinger T, Musarella MA, van den Born LI, Fishman GA, Maumenee IH, Rohrschneider K, Cremers FP, Koenekoop RK. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007;48:5690–8. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 5.Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet. 1998;18:174–6. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, Kovats SG, Lee JJ, Penchaszadeh GK, Ott J, Jacobson SG, Gilliam TC. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet. 1998;18:177–9. doi: 10.1038/ng0298-177. [DOI] [PubMed] [Google Scholar]
- 7.Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23:306–17. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda S, He W, Ikeda A, Naggert JK, North MA, Nishina PM. Cell-specific expression of tubby gene family members (tub, Tulp1,2, and 3) in the retina. Invest Ophthalmol Vis Sci. 1999;40:2706–12. [PubMed] [Google Scholar]
- 9.Hagstrom SA, Adamian M, Scimeca M, Pawlyk BS, Yue G, Li T. A role for the Tubby-like protein 1 in rhodopsin transport. Invest Ophthalmol Vis Sci. 2001;42:1955–62. [PubMed] [Google Scholar]
- 10.Caberoy NB, Maiguel D, Kim Y, Li W. Identification of tubby and tubby-like protein 1 as eat-me signals by phage display. Exp Cell Res. 2010;316:245–57. doi: 10.1016/j.yexcr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Roach JN, McLaren TL, Paterson RL, O’Brien EC, Hoffmann L, Mackey DA, Hewitt AW, Lamey TM. Establishment and evolution of the Australian Inherited Retinal Disease Register and DNA Bank. Clin Experiment Ophthalmol. 2013;41:476–83. doi: 10.1111/ceo.12020. [DOI] [PubMed] [Google Scholar]
- 12.Chiang JP, Lamey T, McLaren T, Thompson JA, Montgomery H, De Roach J. Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert Rev Mol Diagn. 2015;15:1269–75. doi: 10.1586/14737159.2015.1081057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LJ, Lai TY, Tam PO, Chiang SW, Zhang X, Lam S, Lai RY, Lam DS, Pang CP. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2010;51:2236–42. doi: 10.1167/iovs.09-4437. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, Bieg A, Gliem M, Charbel Issa P, Holz FG, Baig SM, Hellenbroich Y, Galvez A, Platzer K, Wollnik B, Laddach N, Ghaffari SR, Rafati M, Botzenhart E, Tinschert S, Borger D, Bohring A, Schreml J, Kortge-Jung S, Schell-Apacik C, Bakur K, Al-Aama JY, Neuhann T, Herkenrath P, Nurnberg G, Nurnberg P, Davis JS, Gal A, Bergmann C, Lorenz B, Bolz HJ. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8:e78496. doi: 10.1371/journal.pone.0078496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.den Hollander AI, van Lith-Verhoeven JJ, Arends ML, Strom TM, Cremers FP, Hoyng CB. Novel compound heterozygous TULP1 mutations in a family with severe early-onset retinitis pigmentosa. Arch Ophthalmol. 2007;125:932–5. doi: 10.1001/archopht.125.7.932. [DOI] [PubMed] [Google Scholar]
- 16.Mataftsi A, Schorderet DF, Chachoua L, Boussalah M, Nouri MT, Barthelmes D, Borruat FX, Munier FL. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:5160–7. doi: 10.1167/iovs.06-1013. [DOI] [PubMed] [Google Scholar]
- 17.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6:137–43. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 18.Mackay DJ, Coupe AM, Shield JP, Storr JN, Temple IK, Robinson DO. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet. 2002;110:139–44. doi: 10.1007/s00439-001-0671-5. [DOI] [PubMed] [Google Scholar]
- 19.Leung WC, Lau WL, Lo TK, Lau TK, Lam YY, Kan A, Chan K, Lau ET, Tang MH. Two IUGR foetuses with maternal uniparental disomy of chromosome 6 or UPD(6)mat. J Obstet Gynaecol. 2017;37:113–5. doi: 10.1080/01443615.2016.1242558. [DOI] [PubMed] [Google Scholar]
- 20.Engel E. Uniparental disomies in unselected populations. Am J Hum Genet. 1998;63:962–6. doi: 10.1086/302074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson DA, McHenry CL, Li Y, Richards JE, Othman MI, Schwinger E, Vollrath D, Jacobson SG, Gal A. Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK, respectively. Am J Hum Genet. 2002;70:224–9. doi: 10.1086/338455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivolta C, Berson EL, Dryja TP. Paternal uniparental heterodisomy with partial isodisomy of chromosome 1 in a patient with retinitis pigmentosa without hearing loss and a missense mutation in the Usher syndrome type II gene USH2A. Arch Ophthalmol. 2002;120:1566–71. doi: 10.1001/archopht.120.11.1566. [DOI] [PubMed] [Google Scholar]
- 23.Fingert JH, Eliason DA, Phillips NC, Lotery AJ, Sheffield VC, Stone EM. Case of Stargardt disease caused by uniparental isodisomy. Arch Ophthalmol. 2006;124:744–5. doi: 10.1001/archopht.124.5.744. [DOI] [PubMed] [Google Scholar]
- 24.Riveiro-Alvarez R, Valverde D, Lorda-Sanchez I, Trujillo-Tiebas MJ, Cantalapiedra D, Vallespin E, Aguirre-Lamban J, Ramos C, Ayuso C. Partial paternal uniparental disomy (UPD) of chromosome 1 in a patient with Stargardt disease. Mol Vis. 2007;13:96–101. [PMC free article] [PubMed] [Google Scholar]
- 25.Carmichael H, Shen Y, Nguyen TT, Hirschhorn JN, Dauber A. Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clin Genet. 2013;84:213–22. doi: 10.1111/cge.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roosing S, van den Born LI, Hoyng CB, Thiadens AA, de Baere E, Collin RW, Koenekoop RK, Leroy BP, van Moll-Ramirez N, Venselaar H, Riemslag FC, Cremers FP, Klaver CC, den Hollander AI. Maternal uniparental isodisomy of chromosome 6 reveals a TULP1 mutation as a novel cause of cone dysfunction. Ophthalmology. 2013;120:1239–46. doi: 10.1016/j.ophtha.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Wiszniewski W, Lewis RA, Lupski JR. Achromatopsia: the CNGB3 p.T383fsX mutation results from a founder effect and is responsible for the visual phenotype in the original report of uniparental disomy 14. Hum Genet. 2007;121:433–9. doi: 10.1007/s00439-006-0314-y. [DOI] [PubMed] [Google Scholar]