

Abstract
HIV-1 subtype C (HIV-1C) may duplicate longer amino acid stretches in the p6 Gag protein, leading to the creation of an additional Pro–Thr/Ser–Ala–Pro (PTAP) motif necessary for viral packaging. However, the biological significance of a duplication of the PTAP motif for HIV-1 replication and pathogenesis has not been experimentally validated. In a longitudinal study of two different clinical cohorts of select HIV-1 seropositive, drug-naive individuals from India, we found that 8 of 50 of these individuals harbored a mixed infection of viral strains discordant for the PTAP duplication. Conventional and next-generation sequencing of six primary viral quasispecies at multiple time points disclosed that in a mixed infection, the viral strains containing the PTAP duplication dominated the infection. The dominance of the double-PTAP viral strains over a genetically similar single-PTAP viral clone was confirmed in viral proliferation and pairwise competition assays. Of note, in the proximity ligation assay, double-PTAP Gag proteins exhibited a significantly enhanced interaction with the host protein tumor susceptibility gene 101 (Tsg101). Moreover, Tsg101 overexpression resulted in a biphasic effect on HIV-1C proliferation, an enhanced effect at low concentration and an inhibitory effect only at higher concentrations, unlike a uniformly inhibitory effect on subtype B strains. In summary, our results indicate that the duplication of the PTAP motif in the p6 Gag protein enhances the replication fitness of HIV-1C by engaging the Tsg101 host protein with a higher affinity. Our results have implications for HIV-1 pathogenesis, especially of HIV-1C.
Keywords: human immunodeficiency virus (HIV), infectious disease, viral DNA, viral protein, viral replication, Gag, HIV-1C, NGS, PTAP duplication, Replication advantage
Introduction
Of the various genetic subtypes of HIV-1, subtype C (HIV-1C) is responsible for approximately half of all global infections (1). In HIV-1, subtype-specific sequence variations have been identified in the viral regulatory elements, including the LTR6 (2, 3), TATA box, TAR (4), PBS (5), splice junctions (6), and in the various other cryptic elements (7, 8). Additionally, subtype-specific sequence motifs have been mapped to viral regulatory (9–11), structural (12), and accessory proteins (13, 14). Sequence variations in the viral genome can influence viral biological properties that in turn may influence differences in the prevalence of the viral subtypes. One such genetic variation has been observed in HIV-1 p6 Gag consisting of the amino acid sequence duplication of the Pro–Thr/Ser–Ala–Pro motif, referred to as the PTAP motif (15).
HIV-1 Gag is synthesized as a polyprotein of 55 kDa and is processed by the viral protease into several different proteins, including the matrix (p17), capsid (p24), nucleocapsid (NC, p7), and p6 Gag as well as two spacer peptides SP1 and SP2 (16). The Gag proteins collectively play an important role in viral replication by regulating viral assembly, budding, and maturation of the virions (17). The matrix protein p17 regulates the trafficking of Gag to the plasma membrane and viral assembly (18, 19). The capsid protein p24 forms the structural core of the virion during viral maturation. The NC catalyzes Gag multimerization, regulates Gag trafficking by interacting with various host factors (18, 20, 21), and recruits the viral genomic RNA to the viral particle. The NC by associating with SP2 and p6 recruits Alix, an important member of the host endosomal sorting complex required for transport (ESCRT), thereby regulating viral budding (22). p6, the smallest domain of Gag, plays a major role in viral budding by recruiting the ESCRT machinery by interacting with the host factor tumor susceptibility gene 101 (Tsg101) via the PTAP motif. The C-terminal region of p6, the most polymorphic region of Gag, is translated in two different reading frames: the in-frame Gag p6 and the −1 frame-shifted Gag-Pol p6*. Two major functions are ascribed to p6 Gag, the recruitment of the ESCRT machinery for viral particle formation (23) and the incorporation of Vpr into the viral particles (24, 25).
HIV-1 p6 contains two distinct late assembly (L) domains that regulate budding of the assembled virions at the cell surface. The L-domains function as docking sites for the components of ESCRT. The L-domain activity of HIV-1 p6 is mainly mediated through the PTAP motif that recruits Tsg101 to the site of viral assembly (26–29). The depletion of Tsg101 using siRNA (27) or overexpression of the N-terminal domain of Tsg101 leads to the inhibition of virion release (30). The presence of Tsg101 in the released viral particles has been demonstrated using scanning EM (31) and super-resolution imaging (32). The second L-domain function is mediated through the 36YPLASL41 sequence of p6, consisting of a cryptic YPXnL-type L-domain, representing a binding site for AIP1/Alix (ALG-2 interacting protein 1/X, Alix) (33). Alix is believed to play a supplementary role in viral budding, and its function may not be as critical as that of Tsg101 (34). Alix, however, can facilitate viral budding in the absence of Tsg101 (35).
Amino acid sequence insertion in different regions of Gag has been extensively examined in subtype B (HIV-1B) especially following the administration of anti-retroviral therapy (ART) (36, 37). Of the various Gag amino acid polymorphisms, sequence insertion in p6 Gag, especially in the PTAP motif, is of interest because such modifications have been proposed to modulate viral replication (37–40) and resistance to RT inhibitors (41, 42). The sequence duplication in the PTAP domain may be divided into two categories based on whether or not the core PTAP motif (consisting of the four amino acids proline, threonine, alanine, and proline) is fully duplicated. Based on this classification, a large number of sequence insertions in HIV-1B are characterized by only a partial sequence duplication that does not lead to the creation of an additional PTAP motif. In HIV-1B, although a large number of the sequence insertions lead to the duplication of a 3-amino acid stretch “APP” in p6 Gag and “SPT” in p6 Gag-Pol, only a small fraction of the sequence insertions constitute a complete duplication of the PTAP core motif and the flanking residues (43).
Several studies attempted to understand the biological significance of the PTAP sequence insertion primarily in the context of HIV-1B. The inferences drawn from the studies are conflicting and nonconclusive. Although some studies reported enhanced viral transmission or increased drug resistance due to PTAP sequence modification, other reports failed to find such advantages. The viral strains containing PTAP duplication transmitted more efficiently from ART-treated mothers to their infants suggesting preferential vertical transmission (44). A positive correlation was found between nucleoside-based ART and PTAP duplication in HIV-1B infection (45–48). Infectious clones of subtype B containing a 3-amino acid duplication (APP) in the PTAP motif demonstrated reduced processing of p6 (49). The proposed association between the nature of the amino acid sequence insertion in the PTAP motif and the administration of ART was questioned (43). Furthermore, a few studies failed to find a significant difference in the frequency of the PTAP insertion between ART-naive and drug-exposed arms in subtype B (36). Collectively, the biological significance of the PTAP motif sequence modification in subtype B, where this phenomenon has been examined extensively, remains nonconclusive.
In the backdrop of continued ambiguity surrounding the PTAP sequence modification and the biological significance in HIV-1B, the examination of the phenomenon in HIV-1C could offer a few technical advantages. Importantly, the nature and frequency of PTAP sequence duplication of HIV-1C differ profoundly from that of HIV-1B. Unlike in HIV-1B where the sequence insertions mostly cause only a partial duplication of PTAP, sequence insertions in HIV-1C p6 Gag more commonly lead to complete duplication of the core PTAP motif and the flanking amino acid sequences (50). A publication from Brazil examined PTAP duplication in a clinical cohort of drug-naive and drug-experienced arms representing HIV-1 infection of three different subtypes B, C, and F (15). This study identified a remarkably higher frequency of PTAP duplication in subtype C as compared with the other two subtypes in both the arms of the study. An analysis of the Gag sequences from the extant databases showed that unlike in other subtypes of HIV-1, subtype C has a natural propensity to generate the PTAP motif duplication at a significantly higher frequency and of greater length (Fig. 3). Additionally, the global prevalence of PTAP duplication in HIV-1C increased progressively over the past 30 years (50). Furthermore, sequence insertions of a greater length, especially in drug-naive subjects of HIV-1C, need a more logical and broader explanation. Martins et al. (15) examined the effect of PTAP duplication on the engineered clones of HIV-1B and concluded that PTAP duplication reverses Gag processing defects imposed by protease inhibitor-resistance mutations without affecting viral budding or proliferation. Of note, the proposed gain-of-advantage due to PTAP duplication in drug resistance could be different in HIV-1C, as indeed a large proportion of primary viral isolates acquire a complete PTAP core motif duplication in the absence of ART administration (15). The primary objective of this work was to address the importance of longer sequence insertion in p6 Gag that creates an additional PTAP motif in HIV-1C. The paucity of studies examining the biological significance of a higher frequency of insertions in the p6 Gag of HIV-1C further added to our interest in this study.
Figure 3.

Comparative analysis of the length and frequency association in the PTAP motif of subtypes B and C. Full-length Gag protein sequences belonging to the HIV-1 subtypes B and C were downloaded from the HIV LANL sequence database (accessed in June, 2017). One sequence per patient was selected for the analysis. Of the 3,895 and 1,879 subtype B and subtype C sequences analyzed, 548 and 505 sequences, respectively, contained a sequence insertion in the PTAP motif. The number of sequences containing an insertion is plotted against the length of duplication. The numbers and percentages of sequences with a duplication of 3, 6, 7, 12, and 14 amino acids are depicted (inset). The percentage values represent the proportion of sequences containing a PTAP duplication. The 21 amino acid windows consisting of the PTAP motif and representing the consensus sequences of subtypes B and C are presented below. The original and duplicated amino acid sequences are indicated. The arrows indicate the length of the PTAP motif and the flanking amino acids and the direction of reverse transcription. The duplicated amino acid residues are highlighted in bold. The core PTAP motifs are underlined.
In a longitudinal follow-up study of 3 years (2010–2013) of 65 HIV-1 seropositive subjects, we observed eight of 50 amplified viral sequences (16%) to contain a complete PTAP motif duplication. Using a range of experimental strategies, we demonstrate that PTAP duplication confers a profound replication advantage on the viral strains. We show further that HIV-1C Gag, as compared with that of HIV-1B, differs in the manner in which the cell endosome sorting machinery is exploited.
Results
HIV-1 strains containing the PTAP duplication dominate the viral quasispecies in vivo
In a longitudinal study of two different chronic-phase clinical cohorts of Southern India, and the acute-phase CAPRISA clinical cohort of South Africa (51, 52), we previously reported sequence insertions of variable length in HIV-1C p6 Gag (50). The clinical profile of the eight study subjects, including their HLA profile, has been summarized (Table 1). The sequence insertion generated an additional PTAP motif in p6 Gag. The PTAP motif duplication was found in eight of the 50 subjects by conventional PCR. In two of the eight subjects (2018 and T014), the coexistence of the wildtype (WT, viral strains containing a single-PTAP motif) with the variant type (VT, double-PTAP strains) viral strains was detected. The conventional Sanger sequencing of the PCR products, however, is limited by the inability to detect minority viral variants at a prevalence of 20% or low (53). To this end, we applied the plasmid clone sequencing strategy to three additional samples (T004, 2012, and 2032). The viral strains in all the three subjects contained a 14-amino acid residue PTAP duplication and showed the presence of the WT strains only sporadically by the conventional PCR sequencing strategy.
Table 1.
The clinical profile of the eight study subjects containing a PTAP motif duplication
M indicates the month of sample collection at a 6-month interval from baseline M0. - indicates information is not available. PVL indicates plasma viral load (number of RNA copies/ml).
| Subject ID | Age/gender | Enrollment date | PVL |
CD4 (cells/μl) |
HLA profile |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M0 | M6 | M12 | M0 | M6 | M12 | M18 | M24 | HLA-A | HLA-B | HLA-C | ||||||
| T004 | 29/Female | 12/16/2010 | 33,784 | 36,627 | 112,577 | 553 | 442 | 508 | 359 | 321 | 0201 | 2402 | 5101 | 4006 | 0602 | 1503 |
| 2012 | 28/Female | 1/20/2011 | 6,070 | - | - | 1,087 | 1,214 | 946 | - | 1,187 | 2402 | 2402 | 4402 | 4402 | 0701 | 0701 |
| 2018 | 35/Female | 1/24/2011 | 6,493 | - | - | 825 | 884 | - | - | - | 2402 | 2402 | 1341 | 4006 | 0602 | 1502 |
| 2020 | 38/Male | 2/25/2011 | 70,025 | - | - | 570 | 475 | 484 | - | 422 | 0101 | 0211 | 3501 | 5701 | 0602 | 1510 |
| 2032 | 45/Male | 8/9/2011 | 9,468 | - | - | 671 | 664 | 699 | 539 | 542 | 0101 | 02 | 0705 | 5701 | 0602 | 0701 |
| 2037 | 29/Male | 8/9/2011 | 1,505 | - | - | 732 | 612 | 554 | 470 | 549 | 1101 | 3301 | 5201 | 5201 | 1202 | 1202 |
| T014a | 35/Female | 2/25/2011 | 2,888 | 4,578 | - | 553 | 484 | 376 | 436 | 425 | 1101 | 3301 | 2701 | 5801 | 0202 | 0302 |
| 2006b | 46/Female | 9/26/2011 | 662 | - | - | 659 | - | - | 580 | 563 | 0101 | 2402 | 3901 | 5701 | - | - |
a In subject T014, PTAP duplication was found only at M24, not used in the subsequent analyses.
b Sample from subject 2006 was available only at baseline.
The full-length gag sequences (coordinates 790–2292 as in HXB2) of viral strains derived from the three subjects, all of whom contained a 14-residue PTAP motif duplication, were amplified from the plasma viral RNA at several follow-up time points (M0, M6, M12, and M18 of T004; M6 of 2012; and M0 and M6 of 2032). The PCR product was cloned into Topo TA vector, and the p6 gag gene segment of 20–30 individual plasmid clones for each time point was sequenced using the conventional Sanger sequencing strategy. The plasmid clone sequencing strategy detected the presence of the double-PTAP variants at most of the time points in all three subjects (Figs. S1 and S2). Importantly, the single-PTAP viral strains were either absent or found to be only a minor component in the quasispecies alluding to the intrinsic domination of the double-PTAP viral variants in vivo. In other words, when the viral variants discordant for the PTAP motif duplication coexist in a subject, the double-PTAP viral variants appear to have a replication advantage under the natural conditions. Additionally, the 14 residues of the original and the duplicated PTAP motifs of the viral variants derived from T004 varied considerably from one another and from the subtype C consensus sequence at all the time points. Collectively, the data alluded to a large magnitude of sequence variations within the PTAP motif duplication, the rapidly changing profile of the major viral variants at different time points, and the domination of the double-PTAP containing viral strains in vivo. Although the conventional sequencing strategy appears to have identified the most dominant viral variants in the plasma viral RNA and genomic DNA, this experimental method is clearly insufficient to represent the gigantic viral diversity present in vivo. We therefore used the strategy of the next-generation sequencing to characterize the nature of the viral quasispecies in six of eight study subjects who provided samples at multiple follow-up time points.
NGS analysis confirms the in vivo domination of the double-PTAP viral strains
The next-generation sequencing was applied to plasma viral RNA and blood genomic DNA collected from each subject at three to six follow-up time points spaced at 6-month intervals over a period of 2–3 years representing the chronic phase of the viral infection. Two independent rounds of NGS were performed with minor technical variations. NGS-1 and NGS-2 used gag PCR fragments of an ∼1.5-kb (coordinates 790–2292 as in HXB2) and 2.2-kb (625–2836 as in HXB2) size, respectively, spanning the entire gag region. Additionally, in NGS-2, a 549-bp fragment (1859–2408 as per HXB2) focusing primarily on p6 gag was also subjected to sequencing (Fig. S3, A and B). The mean coverage, the number of sequences that represent each nucleotide after mapping to a reference sequence, varied between the samples and across the length of gag. For instance in NGS-1, the average number of sequence reads obtained per sample was 814,970 with a minimum 30× depth of coverage. The quality of more than 90% of assembled sequences was high (Phred quality score >Q30 or p > 0.001). The NGS data analysis was performed using a custom pipeline (see computational workflow, Fig. S3C).
The results of the NGS analysis performed using the three different PCR fragments were consistent with one another and broadly comparable with the observations of the conventional sequencing strategy described above. Many important inferences could be drawn from the analysis. First, in all six subjects, at all the time points, and in both the DNA and RNA compartments, the single- and double-PTAP viral strains coexisted. Second, in nearly all the samples examined, with the exception of one or two time points, the double-PTAP viral strains demonstrated a near-absolute domination over the single-PTAP strains. For example, in the NGS-1 of plasma viral RNA, at M0 for the subject T004, the double-PTAP strains constituted as high as 99.1% of the total reads, whereas the single-PTAP strains constituted only 0.9% (Fig. 1). In this subject, only at M12 and M18, a significant proportion of single-PTAP strains were observed, whereas the rest of the reads represented the double-PTAP strains. This observation was consistent with the NGS analysis of the genomic DNA compartment (Fig. 1, bottom panel). Finally, the domination of the double-PTAP viruses over single-PTAP strains was evident in all the five other subjects in both the RNA and DNA compartments (Fig. S4). Thus, in natural infection, the double-PTAP viruses appear to outcompete the single-PTAP strains in a profound and consistent fashion.
Figure 1.
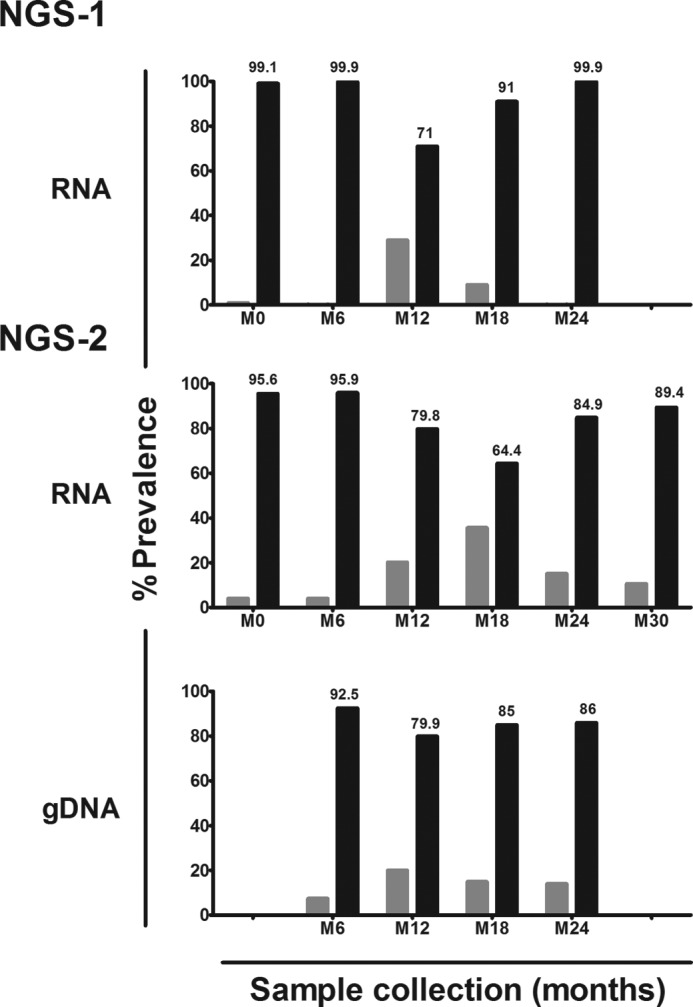
Domination of double-PTAP viral strains in subject T004 in NGS. Two independent rounds of NGS were performed using only plasma viral RNA (NGS-1) and both plasma viral RNA and genomic DNA extracted from peripheral blood (NGS-2). Whereas M0 represents the baseline at which the first sample was collected from the subject, other samples were collected at follow-up times spaced 6 months apart. The dark bars represent the percent prevalence of the double-PTAP forms of the viral quasispecies, and the gray bars represent the WT forms containing a single PTAP motif.
Only a small number of PTAP variant forms dominate the viral quasispecies
Furthermore, each independent bar in the above presentation represented a pool of multiple viral strains collectively making up the single- or double-PTAP viral variant population. We performed an extended analysis to understand the molecular nature of the dominant variant viral strains, constituting the quasispecies at each time point, and to examine how this profile changed with time. Of note, the viral variants were defined only in the context of the PTAP core motif and the flanking sequences of both the original and the duplicated motifs. The relative proportion of all the reads representing a specific genetic variant at a time point was determined. The reads in a sample present at 1% or above were considered as major genetic variants and are presented in a π-format. All the other reads found at less than 1% were pooled into a single group and labeled as the “minority variants” (MV). First, the analysis identified multiple variant forms in both the single- and double-PTAP motifs in all the subjects. In the double-PTAP compartment, only a small number of genetic variants, often less than 10, represented the dominant variants overall; and usually, one to six of these dominant variants constituted the bulk of the viral quasispecies at any given time point (Fig. 2 and Fig. S5). For instance, in subject T004, at M0, 70% of the total cDNA reads of NGS-1 were represented by only two viral variants V1 and V2, both of them containing a PTAP duplication. These two major variant forms differ from each other in a single residue in the original PTAP motif consisting of 14 amino acid residues and in three residues in the duplicated motif (Table 2). The remainder of the 30% of the reads was collectively represented by numerous minority viral variant strains, with each variant individually contributing to the viral quasispecies by less than 1% of the total reads. Second, the profile of the dominant double-PTAP variants was highly dynamic and changed rapidly between the subsequent points spaced 6 months apart. Typically, a double-PTAP form dominant at one time point may disappear at the subsequent point or may be found on the verge of disappearing. Third, when the major variant forms dominant at one time point disappear at the subsequent time point, other variant forms emerge from the minority compartment and occupy this space to become the dominant forms. Variants V3 and V4 become the dominant forms at M6 of NGS-1 in the plasma viral RNA as V1 and V2 have disappeared at this time point. Importantly, V1 and V2 could be still seen in the proviral DNA at M6 (26 and 10%, respectively) and as components of the minority variants (0.0099 and 0.07%, respectively) in the plasma viral RNA. Finally, some of the variant forms that disappeared at one time point may make a reappearance at a later time point to become a dominant form again. For instance, in the NGS-1 of plasma viral RNA for the subject T004, the most dominant viral variant V1 (sequence as mentioned in Fig. S1) with 52% representation at M0 was absent at M6 with <1% prevalence. Interestingly, the same viral variant reappeared at M12 with 35% prevalence but was further reduced to 5% prevalence at M18 and finally to 1% at M24 (Table 2).
Figure 2.

Rapidly changing profile of the double-PTAP forms in subject T004. Each pie chart represents the profile of the PTAP variant forms in the sample at a specific time point. Each slice in a pie chart represents a specific variant at a prevalence of 1% or above the total reads. All the viral variant forms present below 1% prevalence were pooled as the minority variants (MV). NGS-1 and NGS-2 analyses were performed using the plasma viral RNA alone or plasma viral RNA as well as the genomic DNA, respectively. The variant V5 represents the WT-like viral strains that contain a single-PTAP motif. See Table 2 for the sequence information of each of the major viral variants V1–V8.
Table 2.
The percent prevalence of the different viral variant forms of subject T004 at the follow-up time points in the NGS analysis

The phenomenon of dynamic appearance, disappearance, and reappearance of the PTAP variant forms was broadly consistent between NGS-1 and NGS-2 and was represented among all five subjects at most of the time points and in both plasma viral RNA and genomic DNA compartments (Figs. S4 and S5). The phenomenon was not unique to the double-PTAP viral strains but also was manifested by the single-PTAP strains as was evident with the viral strains of subject 2018 (Fig. S5C). Thus, from the present analysis, it appears that HIV-1 establishes a reservoir of a large number of variant viral strains soon after infection, most likely during the acute phase. Subsequently, some of the variant viral strains become dominant forms at a specific time point. These dominant viral forms may enjoy a superior replication advantage within the viral quasispecies and may represent the immune escape mutants dodging the immune response (see under “Discussion”).
PTAP motif duplication of only seven amino acids is the most abundant form in HIV-1C
Of note, it is evident from the NGS analysis (Figs. 1 and 2) that regardless of the differences in the length of the sequence duplication, the double-PTAP viral strains can establish an absolute domination over the single-PTAP strains in a mixed infection. Three of the six study subjects (T004, 2012, and 2032) contained a PTAP duplication of 14 residues, whereas subjects 2018, 2020, and 2037 contained a duplication of 12, 11, and 9 residues, respectively (Fig. S4). This observation raised a question as to the smallest length of PTAP duplication necessary to confer replication fitness advantage on HIV-1C. Because we did not have a sufficient number of gag sequences containing PTAP duplication in our cohort, we analyzed the gag sequences belonging to HIV-1 subtypes B and C downloaded from the LANL HIV sequence database (accessed June, 2017). A total of 3,895 and 1,879 full-length gag sequences of subtypes B and C, respectively, were available for the analysis of which 548 and 505 sequences, respectively, contained PTAP sequence duplications. The sequences of other viral subtypes were not included in the analysis as sufficient numbers representing these subtypes were not available. The number of gag sequences containing a sequence insertion in the PTAP motif was plotted as a function of the duplication length ranging from 3 to 18 residues (Fig. 3). In the absence of other confounding factors and provided the database sequences represent a normal distribution, the abundance of the PTAP sequence duplications is likely to represent the in vivo replication fitness of the viral strains. Important differences are evident between subtypes B and C (Fig. 3). In subtype B, the insertion of a three-amino acid motif “APP” is the most predominant variation representing 57.7% (316 of 548) of all the sequences. Importantly, this is the only type of sequence insertion, among all, that fails to create a new PTAP motif in HIV-1B. In contrast, the sequence insertion of this type that fails to duplicate the core PTAP motif is rare in HIV-1C representing only 3.8% (19 of 505) of the sequences. The second most abundant sequence duplication in HIV-1B (21.4%, 117 of 548) leading to the creation of a new PTAP motif consists of six amino acid residues with an additional amino acid on each side flanking the core motif (EPTAPP). In HIV-1C, in contrast, the duplication of a seven-amino acid sequence is the most abundant insertion (EPTAPPA, 32.3%, 163 of 505) followed by that of six amino acids (EPTAPP, 27.5%, 139 of 505). A seven-amino acid sequence duplication is quite uncommon in HIV-1B (only 3.8%). Although sequences duplications consisting of a larger number of amino acid residues are found in HIV-1C, they represent only a small proportion. The sequence analysis thus identified an important difference in the nature of PTAP duplication between subtypes B and C. In HIV-1C, unlike in HIV-1B, a large majority of the sequence insertions in the PTAP motif leads to the creation of a complete and additional PTAP motif of variable length of which sequence duplications of seven and six amino acids are the most abundant.
Variant shuffling is not associated with CD8 T-cell response targeting the predicted epitopes in the p6 pol region
The restriction on the preferred length of PTAP duplication being seven or six amino acids is suggestive of a possible immune evasion in HIV-1C. Although 59.8% of subtype C gag sequences contained the PTAP duplication of seven or six residues, only 3.4% of the sequences contained a PTAP duplication of eight residues and a total of 30.3% of HIV-1C sequences contained a PTAP duplication of eight residues or longer. The duplication of an immunodominant CTL epitope could attract a stronger immune response. Because a typical CTL epitope is made up of 8–10 residues (54), the duplication of seven or six amino acids is less likely to create an additional epitope, thus minimizing the risk of an augmented immune response. Importantly, the duplication of a seven- or six-amino acid motif can create an additional core PTAP motif, thus taking full advantage of enhanced interaction with the cellular factors mediated through the PTAP motif. This work provides compelling evidence that the ideal length of the PTAP duplication in HIV-1C consists of the four amino acids constituting the core motif flanked by one residue upstream and two downstream (EPTAPPA). Of note, in all the subjects of the present analysis, the viral strains contained a duplication of PTAP sequence larger than seven amino acids (Fig. S4).
Additionally, the NGS analysis and the observed dynamic shuffling of the PTAP variant viral species (Fig. 2) alluded to the involvement of a host restriction mechanism characterized by memory recall. Using on-line prediction tools, we identified a nine-amino acid epitope (AL9, ANSPTSREL, composed of the NL8 epitope with a single residue change) restricted by HLA-C*0602 in p6 pol. Of note, no potential epitope in p6 Gag was identified. Cao et al. (55) previously demonstrated the presence of an NL8 epitope (NL8, NSPTRREL) in p6 pol of HIV-1B restricted by HLA-C*0102 and the destruction of the epitope by the insertion of the APP sequence in the PTAP domain. The ANN and SMM methods of the IEDB tool predicted the AL9 epitope with a percentile rank of 2.6 and with a high-affinity binding of 286 nm. Importantly, the HLA-C*0602 allele was found (Table 1) in four of the study subjects (T004, 2018, 2020, and 2032), and in all these subjects, a higher magnitude of variant shuffling was evident (Figs. 2 and 4 and Figs. S5 and S6). In contrast, subject 2037 lacked the HLA-C*0602 allele, and only one PTAP variant prevailed across all the time points examined (Figs. S5 and S6).
Figure 4.

p6 pol peptide-specific CD8 T-cell immune responses in subject T004. A, each pie chart represents the profile of the p6 pol variant viral forms at a specific time point. Each slice in a pie chart represents a specific variant at a prevalence of 1% or above of the total reads. All the viral variant forms present below 1% prevalence were pooled as the minority variants (MV). NGS-1 and NGS-2 analyses performed using the plasma viral RNA are presented. See Table S2 for the sequences of the major variant viral forms. B, frequency of antigen-specific CD8 T-cell subsets double-positive for IFN-γ secretion and CD107a degranulation as measured using the ICS assay. Stored PBMCs derived from time points M0 and M12 were used in all the three immune assays. C, number of IFN-γ spot-forming units measured using the ELISPOT assay. D, percentage of CD8 T-cells showing a diluted CFSE staining. The dotted lines indicate the cutoff value defined as three times the mean value of the DMSO control. The error bars represent the standard deviation from the mean of responses from three assay replicates.
In this backdrop, we asked whether immune recognition of an epitope in the original and/or the duplicated sequences of p6 pol could explain the dynamic shuffling of the PTAP variants in our subjects. As a first step, using the NGS sequence data, we determined the sequence variants within p6 pol of the four study subjects (T004, 2020, 2032, and 2037). Although subjects T004 and 2032 contained a PTAP duplication of 14 amino acids each, subjects 2020 and 2037 contained a duplication of 11 and 9 residues, respectively, with the core PTAP motif being intact in all the sequences. The viral variants were designated in the context of the nine-amino acid residues constituting the predicted AL9 epitope in the original as well as the duplicated regions. Similar to that of p6 Gag, the variant profile in p6 pol consisted of several genetic variant strains with only a handful of the variants dominating at a specific time point, and a large majority of the variants were hidden as minority strains (Fig. 4A and Fig. S6). For instance, in subject T004, variant V1 was the most abundant one at M0 (50% of total reads); the variant receded into the minority population (0.01%) at M6 and reappeared with a prevalence of 26% at M12. Other variant forms of this subject and those of the other subjects also demonstrated frequency fluctuations at different time points in a similar fashion (Fig. 4A, Fig. S6, and Table 3).
Table 3.
The percent prevalence of the different viral variant forms in the pol reading frame of subject T004 at the follow-up time points in the NGS analysis

Using frozen PBMC of the study subjects collected at different time points, and synthetic peptides representing dominant epitopes of pol p6 at the corresponding time points (Fig. S7A, inset table), we evaluated cytokine secretion of the CD8 T-cells, CD107a degranulation (a surrogate marker for cell-killing activity), and CD8 T-cell proliferation. A direct association between a variant peptide and immune response is expected to lead to the disappearance of the corresponding viral variant. We used four peptides dominant at months 0 and 12 in subject T004 (ADSPTNGEL, original epitope of V1; ANSPTSREL, duplicated epitope of V1; ANSSASGEL, original epitope of V2 and V3; and ADSPTSREP, duplicated epitope of V3, Fig. S7A, inset table) in the assay and examined CD8 T-cell immune response using PBMC collected at these two time points. In all three assays, the DMSO negative control demonstrated a minimal immune response, and a pool of peptides spanning the p24 region of consensus subtype C Gag protein generated highly induced immune responses confirming the quality of the cells and the assay parameters (Fig. 4B). Importantly, we failed to detect any immune response above the background levels against any of the four variant peptides at either of the time points (Fig. 4, B–D). For instance in the IFN-γ secretion and CD107a degranulation assay, at M0, the percentage values of the CD8 cells double-positive with the DMSO negative control and p24 peptide pool were 0.011 ± 0.004 and 3.24 ± 0.15%, respectively (Fig. 4B). These values following the peptide stimulation were all comparable with that of the DMSO control: 0.008 ± 0.002% (ADSPTNGEL), 0.006 ± 0.002% (ANSPTSREL), 0.008 ± 0.001% (ANSSASGEL), and 0.007 ± 0.001% (ADSPTSREP). The absence of a measurable immune response was evident in this subject also at M12 (Fig. 4B). Likewise, in the ELISPOT assay, the four peptides typically induced only less than 50 IFN-γ–secreting spot-forming units, comparable with the DMSO control, whereas the p24 peptide pool induced over 3,000 spot-forming units per million PBMC (Fig. 4C). In the T-cell proliferation assay, 0.39 ± 0.007, 0.36 ± 0.001, and 3.30 ± 0.77% CD8 T-cells demonstrated CFSE dilution following induction with DMSO, the ANSPTSREL peptide, and the p24 peptide pool, respectively (Fig. 4D).
The peptides used in the above assays were all nine amino acids long. The use of peptides of eight amino acids (DSPTNGEL and NSPTSREL), similar to the previous study (55), did not enhance the immune response (Fig. S7B) suggesting that the length of the peptides was not a factor determining the outcome of the assay. Importantly, we also used two additional peptide pools, one spanning the p6 Gag region (HIV-1C consensus) and the other the p6 pol region (HIV-1B consensus), in the assay but failed to see any immune response in the intracellular cytokine assay (Fig. S7B). The absence of an immune response to either of the peptide pools spanning Gag p6 and pol p6 is suggestive of the epitopes in either of these Gag-Pol regions, including the PTAP peptides used in the above assays being immunologically reactive. As in subject T004, we failed to observe any level of immune response in the other three subjects, 2020, 2032, and 2037. We used peptides specific to the PTAP motifs of each of these subjects (Fig. S7). The PBMC of each of these subjects demonstrated robust responses when induced with the p24 peptide pool (Fig. S7A). Collectively, the ICS, ELISPOT, and the T-cell proliferation assays using the stored PBMC of four different subjects at two different time points unequivocally demonstrated the absence of a CD8 T-cell immune response targeting any epitope in the p6 Gag or p6 pol region, including the PTAP domain. In summary, our data appear to rule out the possibility of the pressure of the immune response underlying the shuffling of the PTAP motif variants in our study subjects.
Double-PTAP viral clone outcompetes the single-PTAP viral strain in a pairwise competition assay
The double-PTAP variant viral forms established an explicit domination in vivo over the single-PTAP forms at all the time points in nearly all the subjects examined in this study (Figs. 1 and 2 and Fig. S4). The NGS analysis limited only to the Gag region of the viral strains ignored the genetic variations at the other locations of the viral genome influencing the overall fitness of the viral strains. Therefore, we constructed a panel of three viral strains using the subtype C molecular clone Indie-C to examine the influence of PTAP-duplication on viral replication fitness in a molecularly defined background (Fig. 5A, schematic diagram). In Fig. 5A, although WT contained only a single PTAP motif, the other two strains VT1 and VT2 contained two different PTAP motifs of 14 amino acids each representing the dominant NGS forms V1 and V3, respectively, of subject T004 RNA at M0 (Table 2). For simplicity, we refer to the single- and double-PTAP viral strains as WT (WT-like) and VT, respectively, throughout this work. The two VT strains were compared independently with the WT strain for the proliferation profile and in a pairwise competition assay. Because the viral strains are genetically identical in the rest of the viral backbone, any biological differences observed between the viral strains can be ascribed to the presence of the PTAP motif duplication. Of note, in all of our study subjects, the core PTAP motif consisting of seven amino acid residues (EPTAPPA) has been highly conserved with amino acid variations seen only in the flanking sequences (Figs. S1, S2, and S5). As we have several follow-up samples from subject T004, we used the PTAP motif sequences of this subject in the molecular validation assays. Given the magnitude of the sequence conservation of the core PTAP motif and the variations seen in the flanking sequences are not likely to influence viral replication, the outcome of the validation assays are expected to be representative of all the viral strains. In CEM-CCR5 cells or CD8-depleted and activated PBMC of healthy subjects, all three viral strains proliferated normally with the peak proliferation observed on day 12 (Fig. 5B). The double-PTAP viral strains VT1 and VT2 secreted significantly higher amounts of p24 as compared with the single-PTAP WT strain at all the time points and in both the cell types, suggesting a superior viral replication that could be ascribed to the presence of the PTAP motif duplication.
Figure 5.

Replication profile of the single- and double-PTAP viral molecular strains of the Indie-C1 molecular clone. A, panel of three PTAP-variant viral strains. WT lacks PTAP duplication. The PTAP sequences of VT1 and VT2 represent the dominant NGS forms of T004 RNA at M0 (see Fig. S1). The viral strains of the panel are genetically identical except for the differences shown in the PTAP motifs. Both the PTAP motif sequences are compared with the HIV-1C consensus sequence in the sequence alignment. The dashes represent sequence deletion, and dots represent sequence identity. B, replication profile of VT1 (top panels, CEM-CCR5 T-cells or CD8-depleted and activated PBMC of a healthy subject) and VT2 (bottom panel, CEM-CCR5 T-cells) strains was compared with that of WT. The cells were infected with 100 TCID50 units of single- or double-PTAP viral strain in duplicate wells, and the secretion of p24 into the medium was monitored up to 21–28 days. The data are presented as the mean of quadruplicate wells ± S.D. and representative of two independent experiments. A two-way ANOVA test was performed to determine the significance of the difference in proliferation between the viral strains. Comparable results were obtained with the PBMC of several other healthy donors. C, pairwise growth competition between single- and double-PTAP viral strains in HTA. The left panel shows the schematic representation of the ratios of the paired viruses used in the assay. Genomic DNA was extracted from the infected cells every week following infection and used in the assay as described under “Materials and methods.” The relative fitness values (depicted above the bars) of the competing viral strains WT versus VT1 (middle panel, days 7, 14, and 21) and WT versus VT2 (right panel, days 7 and 14) in CEM-CCR5 cells are presented. A two-way ANOVA test was performed to determine the statistical significance. Data are representative of two independent experiments.
The replication fitness of the double-PTAP viral strains was compared independently with the single-PTAP WT strain in a pairwise competition assay in CEM-CCR5 cells. The cells were co-infected at a low multiplicity of infection (m.o.i., 0.01) using each viral strain at a 1:1 ratio. Mono-infections were also performed simultaneously for comparison (Fig. 5C, left panel). To determine the absolute proliferation of each viral strain, genomic DNA was extracted at different time points and subjected to the heteroduplex tracking assay (HTA) as described previously for env (56). The HTA was based on the amplification of a p6 gag region of differential size due to the 14-amino acid insertion, 258 and 300 amino acids in single- and double-PTAP strains, respectively. The two p6 gag fragments amplified in PCR are shifted differently by a radiolabeled DNA probe derived from a homologous location of p6 gag of the NL4-3 viral strain of subtype B origin. The heteroduplexes formed between the labeled probe and the amplified DNA fragments of the two competing viral strains migrate differently in the gel (Fig. S8A). The relative fitness of each virus in the competition assay was calculated as the proportion of the band intensities in the mono- versus dual-infections (see the mathematical formula in Fig. S8B). In the assay, at an equivalent m.o.i., the double-PTAP viral strain VT1 out-competed the single-PTAP virus WT as early as at day 7 (p = 0.001) and at the subsequent days 14 and 21 (Fig. 5B, middle panel, p < 0.001). The differences in viral fitness were statistically significant and ascertained that the PTAP motif duplication could confer replication advantage on HIV-1. The viral domination due to the presence of PTAP duplication was reproducible in a competition between WT and VT2 also, regardless of the amino acid sequence variation in the duplicated PTAP motifs between VT1 and VT2, at two different points (Fig. 5B, right panel, p < 0.001). The results of the viral replication kinetics and the pairwise competition assays collectively confirmed that the PTAP motif duplication could confer replication advantage on the double-PTAP viral strains regardless of the genetic variations in the duplicated sequence.
PTAP motif duplication significantly enhances Gag interaction with Tsg101
Given that p6 Gag interacts with Tsg101 via the PTAP motif (27, 57), we asked whether PTAP duplication can enhance the interaction between Gag and Tsg101. We cotransfected HEK293T cells with pCMV-Gag and pCAG-Tsg101-FLAG vectors. Gag represented the consensus sequence of HIV-1C and was codon-optimized for mammalian expression. In the cells, Gag and Tsg101 efficiently co-localized, whereas empty-vector control showed no such pattern (Fig. 6A). Furthermore, we used the proximity ligation assay (PLA) to determine the magnitude of protein–protein interactions between p6 Gag and Tsg101 in quantitative terms (Fig. 6, B and C). A distinct fluorescent signal was evident in cells expressing TSG101 and the single- or double-PTAP Gag proteins but not under control conditions where the anti-Tsg101 primary antibodies were omitted. The mean fluorescence intensity of 200 cells selected randomly for each Gag variant demonstrated a significantly enhanced Gag Tsg101 interaction in the presence of the PTAP motif duplication (Fig. 6B). The mean fluorescence intensity value of the double-PTAP Gag at 35.9 ± 14.4 was significantly higher than that of the single-PTAP Gag protein at 21.5 ± 4.9 (p < 0.0001). The increased fluorescence intensity in the presence of the PTAP motif duplication confirms enhanced interaction between the viral and host proteins. In the assay, in addition to the PTAP variant Gag proteins, additional control Gag proteins with a deletion in the p6 domain (Δp6) or p6 plus p7 domains (Δp15) and Gag mutants that lacked the four-residue core PTAP motif (ΔPTAP) were also used. The Gag proteins fail to interact with Tsg101 when the four amino acids of the PTAP domain in WT Gag are deleted (ΔPTAP). Likewise, Gag lacking the entire p6 (Δp6) or p6 plus p7 (Δp15) domains also failed to interact with Tsg101, ascertaining that the PTAP core motif is critical for Gag interaction with Tsg101 (Fig. 6C). Additionally, the co-immunoprecipitation assay provided convincing evidence that the duplication of the 14-amino acid PTAP motif (VT1) enhances the Gag and Tsg101 interaction significantly (Fig. 6D).
Figure 6.

Immunofluorescence analysis of interaction between Gag and TSG101. A, colocalization of Tsg101 and Gag. Representative confocal images of 293T cells expressing Tsg101-FLAG (red) and pcGag (green). The nuclei are stained with DAPI (blue). Scale bar, 10 μm. Tsg101 shows a significant level of (43%) colocalization with Gag as depicted. Inset on the right, plot shows percent colocalization within the cell marked with the white arrow. B, Gag and Tsg101 interaction in HEK293T cells quantitated using the proximity ligation assay. The cells were cotransfected with plasmids encoding Tsg101 and either the single-PTAP or double-PTAP Gag vector at a ratio of 1:2. The cells were examined by confocal microscopy 36 h after the transfection. No Primary: a negative control without the anti-Tsg101 primary antibody. The fluorescence intensities of 200 cells selected randomly were determined and presented as the mean fluorescence intensity per cell. p < 0.0001 using the Student's t test. C, representative confocal images of 293T cells cotransfected with Tsg101 and pcGag variant expression vectors at a ratio of 1:2. Five different Gag vectors were used in the assay as follows: single-PTAP Gag (WT); double-PTAP Gag (VT1); single-PTAP Gag containing a deletion of PTAP motif (ΔPTAP); p6 domain deleted (Δp6), or both p6 and p7 domains deleted (Δp15). The cells were examined by confocal microscopy 36 h after transfection. The red fluorescent signal is indicative of protein–protein interaction. Blue represents the DAPI staining of the nuclei. The PTAP sequence of the 14-amino acid residues is aligned with subtype C consensus sequence (lower panel). The dots represent sequence identity, and the dashes represent deletion. The PTAP sequences are adapted from the virus of T004 at M0. Scale bar: 20 μm. Right panel, fluorescence intensities of 30 randomly selected cells were determined and presented as the mean fluorescence intensity per cell (n = 30). p < 0.01 using the Student's t test. D, coimmunoprecipitation assay to confirm the interaction between Tsg101 and variant Gag proteins. HEK293T cells were transfected with one of the three variant Gag vectors, WT, VT1, and ΔPTAP Gag. Total cytoplasmic extracts were prepared from the transfected cells, and the endogenously expressed Tsg101 was immunoprecipitated. The immunoprecipitated samples were electroblotted and probed for Gag and Tsg101.
HIV-1C Gag can tolerate a higher intracellular concentration of Tsg101 as compared with that of HIV-1B
The results of the above experiments indicated an enhanced level of interaction between Tsg101 and HIV-1C Gag containing an additional PTAP motif. The data, however, do not indicate that the intracellular levels of TSG101 or those of other host factors of the cellular sorting machinery are a limiting factor for viral packaging. It is possible that the duplication of the PTAP motif in HIV-1C is an adaptation that HIV-1C Gag can effectively recruit Tsg101 in the target cell even when the host factor is present at suboptimal concentrations. To this end, we overexpressed Tsg101 in HEK293T, infected the cells with Indie molecular clones containing single- or double-PTAP motifs, and compared the secretion of the viral antigen p24 into the medium (Fig. 7). The HIV-1B reference viral clone NL4-3 was included as a control in the assay as a study previously reported an inhibitory effect of Tsg101 overexpression on HIV-1B viral budding (58). Consistent with the previous report, we found replication of NL4-3 to be the highest under the normal conditions, and the ectopic expression of Tsg101 even at the lowest concentration of 200 ng of the expression vector was inhibitory (Fig. 7, left panel). The secretion of p24 decreased progressively with the increasing concentrations of Tsg101 expression vector suggesting that the concentration of Tsg101 naturally expressed in the cells is adequate for the optimal replication of the HIV-1B viral strain. Goila-Gaur et al. (58) proposed that the overexpression of Tsg101 leads to the perturbation of the cellular endosomal sorting machinery.
Figure 7.
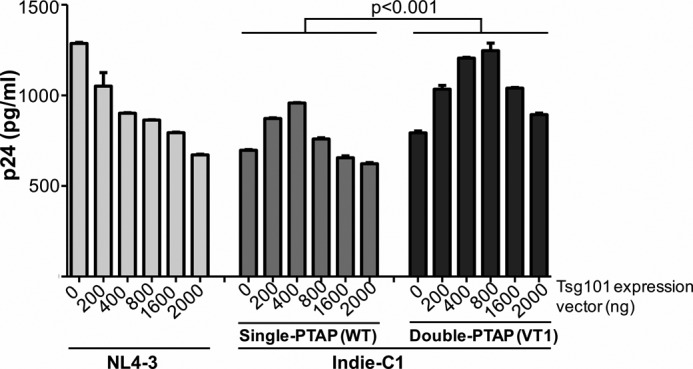
PTAP motif duplication confers higher levels of inducibility on HIV-1C. HEK293T cells were cotransfected with 200 ng of one of the three viral vectors and progressively increasing concentrations of the Tsg101–FLAG expression vector ranging from 200 to 2,000 ng. The secretion of p24 into the medium was quantitated at 48 h and normalized against the β-gal transfection control. Data presented are representative of two independent experiments performed in triplicate wells. Statistical analysis was performed using the two-way ANOVA test.
Importantly, in HIV-1C infection, we found that the impact of Tsg101 overexpression was biphasic proliferation-enhancing at a lower concentration and proliferation-inhibitory only at a higher level (Fig. 7, middle panel). Unlike in HIV-1B infection, at lower concentrations of Tsg101 expression there was a significant increase in the viral proliferation in both single- and double-PTAP viral strains of HIV-1C. The single-PTAP strain produced 697.4 ± 3.5 pg/ml p24 in the absence of Tsg101 overexpression. In the presence of 200 and 400 ng of the Tsg101 expression vector, the p24 levels in the medium increased to 872.1 ± 3.9 and 957.9 ± 1.8 pg/ml, respectively, with both values being statistically significant (p < 0.001). An inhibitory effect of Tsg101 overexpression was manifested on HIV-1C single-PTAP viral strain only at 800 ng and above (Fig. 7, middle panel). Thus, it appears that Gag of HIV-1C, unlike that of HIV-1B, is endowed with a propensity to tolerate a relatively higher concentration of Tsg101. Importantly, the limits of tolerance of the double-PTAP viral strain for the Tsg101 overexpression were significantly higher as compared with the single-PTAP counterpart. The double-PTAP viral strain showed enhanced viral proliferation up to 800 ng of Tsg101 vector, a concentration inhibitory to the single-PTAP strain. Tsg101 became inhibitory to the double-PTAP strain only at 1,600 ng and above (Fig. 7, right panel). Collectively, two important aspects are evident from the data. First, HIV-1C p6 Gag appears to be capable of exploiting Tsg101 at a broader range toward gaining replication advantage as compared with HIV-1B. Second, the ability to utilize Tsg101 and the relative tolerance to the concentration of the host factor are a consequence of the number of PTAP motifs in Gag p6 of HIV-1C. It is possible that the composition of the cellular endosomal sorting machinery assembled by the p6 Gag proteins of the two different viral subtypes may differ significantly underlying the differences noted toward Tsg101 tolerance observed.
Discussion
HIV-1C demonstrates a unique ability to duplicate sequences of biological importance
The viral strains of HIV-1 respond to environmental stresses, such as the immune response or antiretroviral therapy, by introducing amino acid substitutions or causing insertions or deletions of sequences. We demonstrated recently that in two different regions of the viral genome, HIV-1C exploits the phenomenon of sequence duplication to gain replication advantage by increasing the number of the sequence motifs of biological significance.
The first instance of the sequence duplication of biological significance is manifested in the viral promoter (59). Among the HIV-1 subtypes, HIV-1C alone is uniquely capable of duplicating a sequence of 22 bp in the viral enhancer, thus adding an NF-κB–binding site to the already existing three NF-κB motifs in the viral promoter. Importantly, the 4-κB viral strains establish an absolute domination over the 3-κB viral strains in a mixed infection in the proviral DNA and plasma viral RNA compartments and have been spreading in India at a faster rate (2). The presence of the 4-κB viral strains has been confirmed in South Africa and Brazil subsequently (60).
The second instance of the ability of HIV-1C to duplicate the sequence motifs of biological importance is in p6 Gag. In HIV-1C, the duplication of the core PTAP motif and the flanking amino acid residues is seen at a significantly higher frequency as compared with other subtypes such as A and D, whereas subtype B occupies an intermediary position between these extremes (50). The double-PTAP strains represented 16% (8/50) of prevalence in two Southern Indian clinical cohorts. The prevalence of the double-PTAP strains is much larger in three other cohorts of HIV-1C, 29.3% (22/75) in the CAPRISA clinical cohort of South Africa (50) and 30% (212/706) in a South African clinical cohort (61). The prevalence of double-PTAP HIV-1C strains in a Brazilian cohort was found to be 23% (52/228) and 54% (33/61) in drug-naive and drug-exposed subjects, respectively (15). The African epidemic, as compared with that of India, is older at least by a decade and is probably underlying the differences in the prevalence of PTAP duplication observed.
The phenomenon of PTAP duplication in HIV-1C appears to differ from that of other viral subtypes in two main properties: the length of duplication and its frequency. Unlike in HIV-1B, and possibly in the other non-C subtypes, the process of PTAP motif duplication is largely complete in HIV-1C. From the analysis of the gag sequences (Fig. 3), it appears that in HIV-1C, PTAP duplication of seven amino acids (EPTAPPA, 32.3% of all sequences) is the most common sequence insertion followed by that of six amino acids (EPTAPP, 27.5%). In HIV-1C, 94.9% of p6 Gag sequences are represented by the duplications of six amino acids or more, and in HIV-1B, only 37.9% of sequences represent such duplications. In HIV-1B, in contrast, a sequence insertion of only three amino acids (APP, 57.7%) is the most common phenomenon not leading to the creation of an additional PTAP motif. Incomplete PTAP motif duplication in non-C subtypes (15, 36, 41) led to many hypotheses and controversies to explain how this phenomenon would confer replication advantage on these HIV-1 subtypes (15, 39, 48, 62). In this study, we took advantage of the occurrence of complete PTAP duplication in HIV-1C to examine the biological significance of this phenomenon influencing viral replication fitness. Interestingly, in the eight subjects of this study, PTAP duplication ranged from 9 to 14 amino acids, longer than the most common duplication length observed in the sequence analysis (Fig. 3). Additional studies will be required to see whether the Indian HIV-1C strains have a propensity to insert PTAP duplications of a longer size.
The high prevalence of complete duplication of the PTAP motif in HIV-1C, even in drug-naive subjects, raises the question of the biological significance of this phenomenon for HIV-1C, and by extension to the other viral subtypes. The present analysis, employing three different sequencing strategies, demonstrates for the first time that the length of PTAP duplication is highly stable and dominant over long periods of 2–3 years in the chronic phase of the viral infection. However, we could not infer an association between the double-PTAP viral strains and disease progression due to the paucity of information regarding the date of viral infection. For the same reason, we also could not perform a stringent statistical evaluation on the confounding variables such as the time of infection, HLA, CD4 counts, viral load, etc. Nevertheless, a few precautions were taken to minimize the influence of some of the confounding factors. For instance, at the time of recruitment, the study subjects were all drug-naïve; they were believed to have acquired the virus through heterosexual transmission, and they were free from opportunistic infections and AIDS-related symptoms. Given these limitations, the results of this study should be considered as inferential evidence suggesting a positive evolutionary selection of the variant viral strains of HIV-1C in drug-naive subjects.
Dynamic shuffling of the viral variants
The NGS analysis of the primary viral isolates of six study subjects unequivocally demonstrated a profound and significant replication advantage for the HIV-1 strains containing the PTAP duplication in natural infection (Fig. 2 and Fig. S4). The double-PTAP viral strains established an absolute domination over the single-PTAP strains in all six subjects, at all the time points, in both the DNA and RNA compartments. Thus, our study provides compelling experimental evidence that in natural infection PTAP duplication confers a significant replication advantage on HIV-1 strains.
Importantly, the analytical strength of NGS also permitted the examination of the nature of the viral quasispecies containing single- and double-PTAP motifs at a higher resolution. Indeed, both the single- and double-PTAP compartments contained a very large number of variant viral strains. However, only a small number of the viral variants, typically less than 10 variants, were found at an abundance of 1% or higher of the reads at any point, of which usually one to six variants dominated the viral quasispecies (Fig. 2 and Table 2). Furthermore, the profile of the dominant viral variants changed dynamically at a rapid rate. A viral variant that was seen as a dominant species at a specific time point was found to be a minority species or to disappear completely at a subsequent time point of the study. Alternatively, in some cases, the variant viruses were also seen to reappear at later time points in the study, following their initial disappearance. A previous study reported the phenomenon of the major viral variant fluctuation in the context of HIV-1B Tat (63). The dynamic nature of the variant shuffling can only be indicative of a stably established viral reservoir and an active host restriction mechanism. The present analysis is suggestive of HIV-1 establishing a large reservoir of variant viral strains soon after the primary infection, most probably during the acute phase. Subsequently, some of the variant viral strains become the dominant variants at a specific time point either as these forms enjoy a superior replication advantage within the viral quasispecies and/or they represent the immune escape mutants dodging the immune response at a specific time.
The shuffling of the PTAP variant viral strains may be explained using three different models that need not necessarily be mutually exclusive: inter-viral strain competition, an active immune response, and viral latency. The double-PTAP viral strains demonstrate a decisive domination over the single-PTAP counterparts when both the forms coexist in the same subject. In contrast, competition between the double-PTAP strains is not expected to be decisive and therefore cannot explain the precipitate disappearance of a specific variant at one point followed by its reappearance at a subsequent point. Importantly, we provided convincing experimental evidence for the absence of immune response to p6 Gag and p6 pol. Nevertheless, we found a strong immune response against p24 of Gag in all the assays (Fig. 4 and Fig. S7). A physical linkage between the PTAP domain in p6 and an array of dominant epitopes in p24, p17, and other antigens of the virus, can probably explain the removal of the dominant PTAP-variant viral strains in a PTAP-independent manner. In the biological space available within the confines imposed by the viral set point, the vacuum created by the removal of the dominant viral strain is occupied by a different variant viral strain becoming the dominant variant subsequently, and the process is repeated over and over again. The subsequent reappearance of a previously dominant viral strain can be explained when the corresponding latent virus from a large pool of latent reservoir is activated to repopulate the compartment. The current NGS analysis shows that only approximately 10 PTAP variant strains are dominant at any point suggesting that only a handful of several thousand strains constituting the repertoire of the PTAP variants may enjoy replication fitness and therefore can proliferate. Thus, our model takes into account all the experimental observations and offers an explanation for the shuffling of the PTAP variant viral strains.
Biological significance of PTAP duplication for HIV-1C
The biological significance of PTAP duplication and other sequence insertions in Gag has been examined predominantly in the context of HIV-1B. A large majority of these publications (41, 45, 46, 48, 64) identified an association between drug resistance and sequence insertions in Gag. In our cohorts, however, all the six primary viral isolates were derived from drug-naive subjects, and drug-resistance mutations were not identified in the pol sequences (PR, RT, RNase H, and IN) in any of these viral strains (data not shown). The presence of an additional and functional PTAP motif is expected to enhance the association between p6 Gag and the ESCRT machinery. From the literature, no direct evidence has been available for an increased association between TSG101, a key member of the ESCRT machinery, and the number of functional PTAP motifs in p6 Gag, other than a previous demonstration of a progressive reduction in the association between p6 Gag and Tsg101 when one or both the PTAP motifs were removed from the viral protein (29). Using a panel of Gag expression vectors and the proximity ligation assay that can detect protein–protein interactions at high sensitivity, we demonstrated a significantly enhanced interaction between TSG101 and a Gag variant containing a 14-residue PTAP duplication (Fig. 6, B and C). The mutant Gag proteins lacking at least one functional PTAP motif (ΔPTAP, Δp6, and Δp15) failed to interact with TSG101. To gain a deeper insight into the interaction between p6 Gag and TSG101, we overexpressed the host protein and examined the effect on the protein–protein interactions. Goila-Gaur et al. (58) previously demonstrated a strong inhibitory effect on the viral proliferation even at the lowest level of TSG101 overexpression in the context of HIV-1B. Consistent with the previous report, we observed a distinctly negative impact of TSG101 overexpression on the proliferation of the NL4-3 viral strain (Fig. 7). Surprisingly, Tsg101 overexpression demonstrated a biphasic effect on the HIV-1C viral strain, an enhancing effect at low levels, and an inhibitory effect only at higher concentrations (Fig. 7). Importantly, the double-PTAP strain demonstrated tolerance to higher levels of TSG101 overexpression. Thus, it appears that Gag of HIV-1C, unlike that of HIV-1B, is endowed with a propensity to tolerate a relatively higher concentration of Tsg101 to gain replication advantage. It is possible that the composition of the cellular endosomal sorting machinery assembled by the p6 Gag proteins of the two HIV-1 subtypes may vary significantly underlying the differences noted toward Tsg101 tolerance. In the natural circumstances, under suboptimal conditions, especially in a target cell not optimally activated to support viral proliferation effectively, the variant viral strains of HIV-1C containing PTAP duplications may recruit Tsg101 more efficiently, thus gaining a selective advantage. Advantages due to an additional PTAP motif might be cell-type–dependent, as demonstrated previously (30, 65). Additionally, because the PTAP motif can either positively (66) or negatively (28) regulate Gag ubiquitination, the presence of two PTAP motifs may affect this process and as a consequence viral proliferation. Work is presently in progress to examine whether the augmented recruitment of the ESCRT machinery by the double-PTAP motif could lead to an enhanced viral budding from cells. The preliminary data surprisingly did not find enhanced viral budding (data not shown), consistent with the data of Martins et al. (62), who also failed to find an augmented viral budding due to PTAP motif duplication.
Possible impact of PTAP duplication on the HIV-1C epidemic
Given that “the transmission bottleneck” allows only a single or a small number of viral strains to initiate a new infection (67, 68), the double-PTAP viral strains of HIV-1C representing larger than 80–99% of the viral reads are likely to be transmitted at a significantly higher rate to the new host (69, 70). If this prediction holds true, the PTAP variant viral strains are expected to expand rapidly replacing the WT viral strains in the coming years. In an analysis using Gag sequences from the extant database, we found that the percent prevalence of the PTAP VT strains of HIV-1C increased from 17.6 to 31% between 1996 and 2015 unlike that of HIV-1B, which remained stable (50).
Our data raise several important questions that we could not address in this work. In this study, representative viruses only from one of the subjects were used in the functional assays. Whether subject-specific sequences, position, and length of the PTAP duplications would affect the phenotype of the double-PTAP viruses are currently being investigated. Nevertheless, given the absolute level of conservation of the seven core amino acid residues of the core PTAP motif (EPTAPPA), among all the primary viral isolates of this study, the specific PTAP sequences used here are expected to be representative. The analysis of the gag sequences downloaded from the extant databases showed that the most common length of the PTAP motif duplication is of seven or six amino acids in HIV-1C (Fig. 3). It is necessary to examine whether PTAP motif duplication consisting of sequences longer than seven amino acids would confer a stronger advantage on the viral molecular clones. The functional significance of incomplete PTAP duplication in non-C subtypes needs to be elucidated. Furthermore, the molecular mechanisms underlying the sequence duplications need to be identified. In HIV-1C, there are similarities between the sequence duplication of the NF-κB site in the viral promoter (2) and the PTAP duplication in p6 Gag (50). Both of the events confer a great replication advantage on the variant viral strains in natural infection, independently. The PTAP motif duplication differs from that of NF-κB motif duplication by being highly variable especially in the nine residues flanking the core PTAP motif. The reverse transcriptase is the single important viral factor common to both of these phenomena, thus suggesting subtype-specific differences in the RT function possibly influencing these processes. However, none of the viral isolates of the six subjects of this study containing the PTAP duplications possessed the NF-κB duplication in the LTR. The lower rate of prevalence of the NF-κB and PTAP duplication individually in the population could be the reason why these two events were not seen to overlap in our study subjects.
In summary, the results presented here demonstrate that the duplication of PTAP motif enhances the replication fitness of HIV-1C viruses by recruiting a higher level of Tsg101. Future studies are currently in progress to validate the underlying mechanisms that confer replication fitness on HIV-1C due to the PTAP sequence duplication. It would be of great importance to monitor through further longitudinal clinical studies, if the viral strains with variant-PTAP expand in the population in the years to come.
Materials and methods
Ethics statement
Ethical approval for the study was granted by the Institutional Review Board of Y. R. Gaitonde Centre for AIDS Research and Education (YRGCARE), Chennai, India. YRG CARE maintained the clinical cohorts, screened the potential subjects, counseled, and recruited the study participants for this study. A written informed consent was obtained from all the subjects enrolled in the study. The Human Ethics and Biosafety Committee of Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore, India, reviewed the proposal and approved the study.
Study participants
Using the clinical records available at YRGCARE over the past several years, 65 seropositive subjects were enrolled in two different clinical cohorts, called the YRGCARE (n = 30, Chennai, Tamil Nadu) and Nellore (n = 35, Nellore, Andhra Pradesh) cohorts. The clinical profiles of all 65 study subjects have been reported previously (50). The clinical profiles of the eight study subjects used in this analysis are presented, including their HLA profile (Table 1). For the longitudinal study spanning over a period of 2–3 years, the clinical samples were collected at an interval of 6 months. The primary inclusion criteria required that the CD4 counts should be above 500 cells/μl at the time of enrollment and that the subjects should be drug-naive and free of AIDS-related clinical symptoms. The exclusion criteria included the presence of opportunistic infections, signs of acute systemic illness, and prior antiretroviral treatment. The study participants consisted of only adult subjects over 18 years of age, representing both genders. The study participants are believed to have acquired the infection primarily through heterosexual transmission.
Clinical procedures
A single vial of 20 ml of peripheral blood was collected from each participant at 6-month intervals from 2010 to 2013. The blood samples were processed on the same day of collection. The PBMC and plasma samples were stored in 1-ml aliquots in a liquid nitrogen container or a deep freezer, respectively. The genomic DNA was extracted from 200 μl of the whole blood using a commercial DNA extraction kit (QIAmp Blood Mini Kit, catalog no. 69504, Qiagen India, New Delhi, India), was eluted in a 50-μl volume, and was stored in a deep freezer until use. The CD4 T-cell count was determined using the FACSCount reagent kit (catalog no. 340167, BD Biosciences) and the FACSCount control kit (catalog no. 340166, BD Biosciences) following the manufacturer's instructions. The samples were analyzed on a FACSCalibur flow cytometer (BD Biosciences). The plasma viral RNA load was determined using the m2000rt real-time PCR machine (Abbott Molecular Inc., Des Plaines, IL).
RNA isolation and RT-PCR
RNA was extracted from 150 μl of the stored plasma samples using a commercial viral RNA isolation kit (NucleoSpin® RNA Virus, Ref. No. 740956.50, MACHEREY-NAGEL GmbH & Co. KG, Germany). In the case of clinical samples that failed the PCR, an alternative kit was used to extract the viral RNA from 1 ml of plasma (the NucliSENS miniMAG nucleic acid extraction kit, Ref. No. 200293, BioMerieux, France). The cDNA was synthesized using random hexamers and a commercial kit (SuperScript® III Reverse Transcriptase, catalog no. 18080-051, Invitrogen). The reaction vials were incubated at 25 °C for 10 min and 50 °C for 50 min. The reactions were terminated by incubating the samples at 85 °C for 5 min followed by RNase H treatment. The cDNA was used for the amplification of Gag.
Gag amplification, cloning, and sequence analysis
The full-length gag or p6 gag sequence was amplified from the plasma RNA or genomic DNA samples (using 200–300 ng of DNA as a template) using a nested-PCR strategy and a commercial long-range PCR kit (XT-20 PCR system, Merck Genie, India) on the I-Cycler (Bio-Rad). Two different nested-PCR strategies were used to amplify full-length gag for the purpose of sequence determination by the conventional method and a third strategy that involves the amplification of a larger gag region along with the flanking sequences for the purpose of NGS. The PCR primers were positioned in the highly conserved regions of gag and the flanking regions. The details of the amplification and sequencing primers for gag are summarized elsewhere (50). The carryover contamination was prevented by adherence to strict procedural and physical safeguards that included reagent preparation and PCR setup, amplification, and post-PCR processing of samples in separate rooms. The PCR products were purified using a commercial DNA purification kit (catalog no. YDF100, Real Biotech Corp., Taiwan) and subjected to sequencing. After confirmation of the authenticity, the PCR products were cloned using TOPO TA cloning system (pCR2.1-TOPO vector) as per the manufacturer's instructions (catalog no. 4530-20, Invitrogen). Positive bacterial clones were identified by blue-white screening. The recombinant clones were identified by restriction digestion analysis, and 20–30 plasmid clones representing each clinical sample were sequenced. The sequencing was performed on ABI PRISM® 3130xl Genetic Analyzer (Applied Biosystems, Illinois) using the ABI PRISMTM dye terminator cycle sequencing ready reaction kit at the Central Instruments Facility, MBGU, Jawaharlal Nehru Centre for Advanced Scientific Research. The sequences were assembled using Vector NTI Contig Express software (Invitrogen BioServices India Pvt. Ltd.) and aligned to subtype C gag consensus sequence and manually edited as required.
Next-generation sequencing
The Gag PCR products amplified from viral strains containing the PTAP duplication were subjected to the NGS analysis. The Illumina MiSeq platform was used for the NGS analysis of the Gag PCR products amplified from the plasma RNA or the genomic DNA samples. The NGS analysis was performed in two independent rounds. The first round of NGS analysis was performed using the complete Gag amplicon of 1,516 bp (HXB2 coordinates 790–2306) amplified using Set-I primers (50). For the second round of NGS analysis, a larger gag region of 2,211 bp (HXB2 coordinates 625–2836), including longer flanking sequences on either side of the Gag, was amplified using Set-III primers. Additionally, a smaller gag region of only 549 bp consisting of the PTAP domain was also amplified using the primers of Set-IV (HXB2 coordinates 1859–2408) and are included in NGS-2. The three amplified gag fragments of 1,516, 2,211, or 549 bp were used for the preparation of pair-ended indexed libraries for Illumina MiSeq. The quality of the amplicons was determined using agarose gel electrophoresis, and the quantity was determined using the Qubit® 2.0 Fluorometer (Invitrogen). Five nanograms of each sample were fragmented using the Nextera XT DNA sample preparation kit (catalog no. FC-131-1096).The average size of the fragmented amplicons was 250–300 bp. The fragmented DNA was further amplified using a limited-cycle PCR program to add the Illumina sequencing indices and molecular tag sequences required for the cluster formation. Size-exclusion PCR cleanup was performed using AMPure XP beads to purify the DNA library and remove very short library fragments from the population. The samples were normalized using Library Normalization Additives 1 and Library Normalization Beads 1. For the preparation of cluster generation and sequencing, equal volumes of normalized libraries were pooled, and the pools were diluted 25-fold using pre-chilled hybridization buffer HT1. The diluted sample was heat-denatured at 96 °C for 2 min, and 1% denatured PhiX beads were added as a sequencing internal control to the denatured library pool. All the steps were performed as per the manufacturer's instructions (Illumina, San Diego) and Illumina MiSeq protocol. Samples were loaded to a 300-cycle MiSeq cartridge and sequenced.
The data analysis was performed using a custom pipeline, and a computational workflow as depicted (Fig. S3). In the first step, the input paired-end reads were merged using the PEAR with default parameters (71). The merged reads were initially mapped using BWA-MEM (72) to a reference sequence that contains the complete 42-bp insertion of the PTAP region. The gag sequence of subject T004 was used as the reference. A consensus sequence for the sample was obtained using these initial alignments by taking the dominant allele at each position in the reference. The merged reads were then mapped to the consensus sequence using BWA-MEM. The haplotypes in the sample and the number of reads supporting each one of them were counted from these alignments using custom scripts. Specifically, only the haplotypes within a 90-bp window flanking the 42-bp region were counted. These haplotypes in the nucleotide space were converted to haplotypes in amino acid space in both Gag and Pol reading frames, and the relative abundance of each haplotype was computed. The sequence output files of the NGS data have been submitted to NCBI Sequence Read Archive. The sequences are available under the accession numbers SRX2596628 to SRX2596659.
Analysis of the length of the PTAP motif duplication
Full-length Gag amino acid sequences, one sequence per patient and excluding the problematic sequences, belonging to the subtypes B and C were downloaded in October, 2016, from the HIV Los Alamos National Laboratory (HIV-LANL) sequence database. The sequences were aligned using ClustalW multiple sequence alignment program. The sequences containing a duplication of any length were identified and used in the analysis. Of note, the percentage of sequences of a specific length of duplication was determined relative to the total number of sequences containing PTAP duplication and not the total number of the gag sequences.
Prediction of the potential HLA-binding epitopes
We used the on-line IEDB resource (http://www.iedb.org/)7 to predict the peptides that possess a potential to bind the HLA alleles. The amino acid sequences of p6 Gag or p6 pol inferred from the NGS analysis were used as the query sequences. The IEDB recommended consensus tool (73) that combines predictions from ANN (74, 75), SMM (76), and comblib (77) was chosen as the prediction method.
Engineering of a gag chimera Indie-C1 molecular clone
The Indie-C1 molecular clone (the prototype HIV-1C viral strain, accession number AB023804) was subjected to three successive molecular manipulations to graft the gag ORF from a clinical sample into the viral molecular clone using unique restriction sites. First, a single EcoRI site present on the vector backbone in the multiple cloning site was eliminated by digesting the Indie-C1 plasmid with EcoRI, Klenow-filling, and then self-ligating. Second, a silent EcoRI site was engineered at position 2,801 using the overlap PCR strategy to generate an intermediate vector pIndie_EcoRI2801. The grafting of the silent EcoRI site into the vector places the entire gag ORF between two unique RE sites, EcoRI and KasI. The infectivity of the recombinant viral clone pIndie_EcoRI2801 was confirmed by infecting CEM-CCR5 T-cells with the virus packaged in 293T cells. Third, using KasI and EcoRI, the native gag ORF was deleted from pIndie_EcoRI2801 and replaced by a 12-bp MluI linker (5′-GGCGCCACGCGTGAATTC 3′, the RE site is underlined) containing a unique MluI site positioned between the two RE sites. The resulting viral vector p824-pIndieΔgagKME served as a parental vector for the grafting of the gag ORF from clinical samples using the two unique RE sites KasI and EcoRI with the MluI site functioning as a spacer. The infectivity of the vector p824 was confirmed after back-cloning of the autologous gag sequence between KasI and EcoRI sites and infecting TZM-bl and CEM-CCR5 cells.
Construction of the PTAP duplication panel
A panel of three viral clones that differ from one another in the PTAP duplication sequence was generated using the parental vector pIndieΔgag KME (p824). Although one of the viral clones contains a single PTAP motif, the other two variant type clones (VT1 and VT2) contain two different sequence insertions representing the PTAP duplication of 14 amino acid residues. The two sequences have been derived from the two dominant variant forms, V1 and V2, of subject T004 at M0. The 42-bp tandem duplicated sequences, consisting of the PTAP motif, were grafted into the Indie gag sequence using overlap PCR, and the recombinant Gag fragments were cloned into p824 using the KasI and EcoRI sites. The recombinant clones were confirmed by sequencing. The three plasmid clones in the panel were genetically identical except for the differences in the PTAP motif.
Cell culture
CEM-CCR5 (CEM.NKR-CCR5, AIDS Reagent Program at National Institutes of Health) cells were propagated in complete RPMI 1640 medium (catalog no. R4130, Sigma), supplemented with 10% fetal bovine serum (RM10435, HiMedia Laboratories), 2 mm glutamine (G8540, Sigma), 100 units/ml penicillin G (P3032, Sigma), and 100 g/ml streptomycin (S9137, Sigma). The peripheral blood mononuclear cells were isolated from 10 ml of fresh blood, collected from healthy donors, by density-gradient centrifugation using HiSep LSM-1077 (LS001, HiMedia Laboratories, Mumbai, India). The CD8 cells were depleted from the PBMC using the StemSep Human CD8+ depletion kit (catalog no. 14662, Stem Cell Technologies, Vancouver, Canada). PBMC were cultured in complete RPMI 1640 medium supplemented with 20 units/ml IL-2 (H7041, Sigma) and 5 μg/ml PHA-P (L1668, Sigma) for 72 h. PBMC were subsequently used for viral infection in a medium supplemented only with 20 units/ml IL-2 (H7041, Sigma). The TZM-bl cells (AIDS Reagent Program, National Institutes of Health), HEK293T (ATCC, CRL-3216), and HEK293 (ATCC, CRL-1573) cells were grown in Dulbecco's modified Eagle's medium (D1152, Sigma) supplemented with 10% FBS.
Synthetic peptides
Peptides of greater than 90% purity were custom-synthesized by a commercial vendor (Genemed Synthesis, Inc.). The 15-mer peptide pools spanning the p24 and p6 Gag domains represent the consensus HIV-1C Gag peptide set (catalog no. 8118, AIDS Reagent Program, National Institutes of Health) and the p6 pol peptide pool of the HIV-1B consensus (catalog no. 6208). All the antibodies were obtained from BD Biosciences. The live–dead staining dye (L10120) was purchased from Molecular Probes.
Flow cytometry analyses of the T-cell responses in PBMC
The PBMC collected at various time points were stored in liquid nitrogen until used. The PBMC were thawed in the presence of 20 μg/ml DNase (DN25, Sigma) and rested overnight at a density of 2 million cells per ml in complete RPMI 1640 medium without IL-2. The intracellular cytokine staining (ICS) assay was performed as reported previously (78) with a few modifications. We first optimized the number of cells needed for the assays, the duration of stimulation, the time of addition of brefeldin A and monensin, and the concentration of peptides needed for the stimulation. We titered all the fluorescent-labeled antibodies and the live–dead stain for the experiments. Briefly, 0.5 million cells were plated in V-bottom 96-well plates and stimulated in vitro for 6 h with 2.5 μg/ml of each peptide in the presence of anti-CD28 (catalog no. 555725) and anti-CD49d (catalog no. 555501) at a concentration of 1 μg/ml each. The cells were stimulated in complete RPMI 1640 medium supplemented with 20 μg/ml DNase, and the final concentration of DMSO in the all the samples was adjusted to 0.1% v/v. The anti-CD107a-PE (catalog no. 555801), brefeldin A (catalog no. B7651, Sigma), and monensin (catalog no. 554724, BD Biosciences) were added to the medium right at the beginning of the stimulation phase. After 6 h of stimulation, the cells were washed once with PBS and incubated in the presence of APC-conjugated live–dead staining dye for 30 min at room temperature. The cells were then surface-stained with anti-CD3-APC-H7 (catalog no. 641397) and anti-CD8-PE-Cy5 (catalog no. 555636) for 20 min at 4 °C. Following the fixing and permeabilization using Cytofix/Cytoperm (catalog no. 554722, BD Biosciences), the intracellular IFN-γ was stained with anti-IFN–γ-FITC (catalog no. 340449) for 20 min in Perm/Wash buffer (catalog no. 554723, BD Biosciences) at room temperature. The stained cells were resuspended in 200 μl of 0.4% paraformaldehyde and incubated at 4 °C for 1 h before acquisition.
The T-cell proliferation assay was performed using CFSE as reported previously with slight modifications (79). The PBMC were suspended in PBS supplemented with 5% FBS (v/v) at a density of 10 million cells per ml in 15-ml conical-bottom tubes. To each tube 100 μl of 10 μm CFSE (catalog no. 21888, Sigma) solution (diluted from 1 mm stock in PBS) was added, and the samples were vortexed immediately to facilitate uniform labeling of the cells. The cells were allowed to stand for 4 min at room temperature and diluted with 10 volumes of PBS containing 5% FBS, and centrifuged at 2,000 rpm for 4 min. The cell pellets were then washed twice with PBS containing 5% FBS and resuspended at a density of 5 × 106 cells/ml in complete RPMI 1640 medium without DNase. The cell suspension deposited in a 96-cluster flat-bottom plate, 0.5 million cells/well, was stimulated with DMSO (0.05% v/v, negative control) or 1 μg/ml of the peptide in the absence of any co-stimulation or IL-2. The peripheral wells of the plate were not used in the assay. After 6 days of incubation, the cells were washed and stained with live–dead APC stain, anti-CD3-APC-H7, and anti-CD8-PE-Cy5 using the protocol described above for the ICS technique. The cells were resuspended in 200 μl of 0.4% paraformaldehyde solution.
For both the ICS and CFSE dilution assays, the cells were transferred to 5-ml round-bottom tubes containing 100 μl of sheath fluid, and 0.2–0.3 million events were acquired using a FACS Aria III instrument (BD Biosciences). The CD8 T-cell population was identified as CD3+ CD8+ subset of the live cells that excluded the live–dead stain. The live CD8 T-cell subset was further analyzed for IFN-γ and/or CD107a phenotype for the ICS assay and the dilution of CFSE in the T-cell proliferation assay.
IFN-γ ELISPOT assay
The PBMC were thawed and prepared for flow cytometry as described above. The cells were seeded at a concentration of 0.2 × 106 cells/well in the ELISPOT plates pre-coated with anti-IFN-γ antibody (catalog no. 552138, BD Biosciences). The cells were stimulated with 2.5 μg of individual peptides or a p24 peptide pool (positive control) for 24 h. A negative control of three wells lacking the peptides was included in each assay. After 24 h of incubation, the cells were removed, and the wells were washed twice in distilled water for 4 min each time. A biotinylated detection antibody was added to the wells and incubated. The wells were washed extensively, and a streptavidin-HRP conjugate was added. The plate was developed using chromogenic 3-amino-9-ethylcarbazole substrate and dried at room temperature until the spots developed. The spots were counted manually using an upright microscope.
Preparation of the viral stocks
The viral stocks were prepared in HEK293T cells by transfecting the cells with plasmid DNA representing different viral molecular clones using a standard calcium phosphate transfection method (80). HEK293T cells were seeded in 90-mm dishes at a density of 2 × 106 cells. Twenty four hours later, at a cell confluency of 30%, the cells were transfected with 15 μg of the viral plasmid DNA along with 0.1 μg of the pCMV-LIG expression vector, the latter as an internal control for the transfection efficiency. The culture medium was changed after 6 h of incubation, and 7 ml of fresh medium were added to the dishes. The culture supernatant containing the virus was harvested at 72 h, centrifuged at 2,000 × g to pellet the cell debris, passed through a 0.45-μm filter, and stored in multiple 1-ml aliquots at −80 °C until use. Viral supernatants used for the HTA study were treated with DNase I (catalog no. D5025, Sigma) for 1 h at 37 °C to eliminate any contaminating plasmids. The concentration of the viral core protein p24 in the viral stocks was measured using a commercial ELISA kit (catalog no. NEK050B, HIV-1 p24 ELISA kit, PerkinElmer Life Sciences) following the manufacturer's instructions. The levels of the Gaussia luciferase secreted into the culture supernatant was monitored using the BioLux Gaussia luciferase assay kit (catalog no. E3300L, New England Biolabs). The luciferase assay was performed using a SpectraMax L Luminescence 96-well Microplate Reader (MDS, Inc., model no: serial number Lu 03094) by mixing 20 μl of the culture supernatant and an equal volume of the 1× BioLuxGLuc substrate reagent. The assays were performed in triplicate wells, and every experiment was repeated at least two times.
Viral titer determination
The tissue culture infectious dose (TCID50) titer of the viral stocks was determined using β-gal assay in TZM-bl cells. Briefly, 104 TZM-bl cells were seeded in 100 μl of DMEM in a flat-bottom 96-well culture plate. After 24 h, 50 μl of 4-fold dilution series of the viral stocks were added to appropriate wells in complete DMEM supplemented with 25 μg/ml DEAE-dextran (catalog no. D9885, Sigma). Following 6 h of incubation, the medium in the wells was replaced with 100 μl of complete DMEM, and the plates were incubated for 48 h at 37 °C in the presence of 5% CO2. β-Galactosidase expression was examined on day 3. The culture medium in each well was replaced with 100 μl of 1× PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.47 mm KH2PO4, pH 7.4) followed by the fixing of the cells. To each well, 100 μl of a fixing solution (1% formaldehyde and 0.2% glutaraldehyde in 1× PBS) was added, and the plates were incubated for 5 min at room temperature. The cells were washed twice with 1× PBS and stained with 100 μl of the freshly prepared β-gal staining solution (4 mm potassium ferrocyanide, 4 mm potassium ferricyanide, 2 mm MgCl2, and 1 mm X-gal in PBS), and the plates were incubated for 4 h at 37 °C. The wells were washed twice with 1× PBS, and the blue-stained cells were counted manually under a low-resolution microscope. The infectious units of each viral stock were determined by multiplying the cell count with the dilution factor.
Viral proliferation and the replication kinetics
CEM-CCR5 cells or PBMC (0.3 × 106) were infected with WT and variant type (14 amino acid insertions) using 500 IU of Gag chimera virus stocks. Before viral infection, the PBMC were CD8 cell-depleted and activated with 5 μg/ml PHA in complete RPMI 1640 medium supplemented with 20 units/ml IL-2 for 72 h. Following viral infection, the cells were incubated with the viruses in complete RPMI 1640 medium supplemented with 10 μg/ml DEAE-dextran for 6 h at 37 °C in the presence of 5% CO2. Post-infection, cells were washed three times with PBS, suspended in complete RPMI 1640 medium, and incubated. Supernatants from the third wash were saved for p24 ELISA or luciferase assay. Cells were monitored; the medium was replenished twice a week, and freshly activated PBMC from the same donor were added to the cultures as required. The culture supernatant was collected at regular intervals for p24 estimation for 1 month. The p24 ELISA was performed at each time point, and the viral growth curve was constructed for each mono-infection. p24 was monitored using a commercial kit (PerkinElmer Life Sciences).
Pairwise viral competition assay
Dual-infection/competition experiments were performed in CEM-CCR5 cells. The competition assay involved three separate infections as depicted in Fig. 5C. The cells, 2 × 106 cells/assay, were infected with single- or double-PTAP (VT1 or VT2) viral strains (see schematic diagram, Fig. 5). The infectious titers of the viral stocks were determined in TZM-bl cells as described above, and the competing viruses were mixed at an m.o.i. of 0.01 each (1:1). Additionally, mono-infections were also included in the assays for comparison (500 infectious units/assay). Six hours following the infection, the cells were washed three times with PBS and incubated in RPMI 1640 complete medium. Secretion of the p24 or luciferase into the culture medium was monitored periodically as described above. Every week following the infection, a fixed number of cells were harvested, and fresh cells were added to continue the culture. The genomic DNA was extracted from the cells using a commercial kit (QIAmp Blood Mini Kit, catalog no. 69504, Qiagen, Germany), and 250 ng of the DNA were used as template in the PCR for the p6 Gag-HTA.
The relative replicative fitness of the competing viral strains was evaluated essentially as described previously (81) and as depicted (Fig. S8). Briefly, the replicative fitness of a viral strain was calculated as the ratio of the band intensities in a coinfection to that in the mono-infection that in turn was compared with the sum of the two ratios of the two competing viruses. For instance, the production of the single-PTAP virus was calculated as a ratio of the band intensities of the coinfection “c” to that of the mono-infection “a.” The relative fitness of the single-PTAP viral strain “W” was then expressed as the ratio of single-PTAP band comparison (c/a) to the sum of single-PTAP and double-PTAP band ratios (c/a + b/d). A similar strategy was used to determine the relative fitness of the double-PTAP viral strain “V,” which was expressed as the ratio of single-PTAP band comparison (b/d) to the sum of single-PTAP and double-PTAP band ratios (c/a + b/d).
Heteroduplex tracking assay
The p6 Gag-HTA consisted of a PCR that amplified the fragments of 300 or 258 bp spanning the p6 region, from viral strains containing or lacking the 14-amino acid insertions. A primer pair N1812 and N1435 was used for the amplification. A homologous fragment of 270 bp amplified from NL4-3 (subtype B) gag with the primer pair N1812 and N1435 was used as the probe in HTA to form heteroduplex complexes between the single-PTAP and the double-PTAP (VT) PCR fragments. The identity of the two competing viral strains could be unambiguously distinguished when the heteroduplexes are resolved in a polyacrylamide gel (Fig. S8) and quantitated using a phosphorimager. The forward primer N1812 was end-labeled using T4 polynucleotide kinase (New England Biolabs) in a solution containing 30 μCi of [γ-32P]dATP (catalog no. LCP-101, Board of Radiation and Isotope Technology, India). The mixture was incubated for 30 min at 37 °C, and the enzyme was heat-inactivated at 65 °C for 20 min. The oligonucleotide was passed twice through a pre-packed Sephadex G-50 column. The radiolabeled N1812 forward primer, in combination with the unlabeled N1435 reverse primer, was used for the amplification of the p6 Gag fragment. The PCR-amplified fragments resolved on a 1% agarose gel and purified using a commercial kit (QIAquick gel extraction kit, Qiagen) were used as probes in the HTA. For the HTA, the PCR products of the mono-infections and dual-infections were column-purified (QIAquick PCR purification kit, Qiagen, Germany), and the DNA concentration of each sample was determined using UV spectrophotometry as well as confirmed by agarose gel electrophoresis. A typical HTA reaction of a 25-μl volume consisted of 100 ng of the amplified DNA hybridized to ∼8,000 cpm of the probe in the annealing buffer (10 mm Tris-HCl, pH 7.8, 100 mm NaCl, and 2 mm EDTA). The samples were incubated at 95 °C for 3 min and then rapidly annealed by placing the reaction vials on wet ice. After an incubation of 30 min on ice, 5 μl of HTA loading dye (50% glycerol, 0.02 m Tris-Cl, 0.5 m DTT, 0.25% bromphenol blue, 0.25% xylene cyanol) was added to each tube, and the tubes were vortexed and centrifuged at 10,000 × rpm for 1 min. The entire HTA reaction mix was applied to an 8% nondenaturing polyacrylamide gel of 0.75-mm thickness, and the gel was allowed to stand for 10 min and was electrophoresed in the presence of Tris borate/EDTA buffer for ∼6 h at 250 V using the Protean II xi electrophoresis system (Bio-Rad). The gel was placed on Whatman chromatography paper, wrapped in a plastic film, dried on a gel dryer at 80 °C for 1 h, and scanned using a phosphorimager (FLA-5000, Fujifilm).The band intensities were quantified using the ImageJ software.
Immunofluorescent staining
For the immunofluorescent staining, the cells were seeded on glass coverslips coated with 0.2% gelatin, grown to 30–40% confluency, fixed, and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. The cells were washed three times for 5 min each in 500 μl of 1× PBS. For blocking, 5% fetal calf serum was used. The cells were stained for Gag and Tsg-101 by incubating the cells with a pool of the primary antibodies overnight at 4 °C, mouse anti-p24 mAb at 1 μg/ml concentration (raised in-house, Clone-G1.5) and rabbit anti-FLAG monoclonal antibodies, 1:600 dilution (catalog no. F7425, Sigma), the latter to detect TSG101. The antibodies were diluted in 1× PBS with 0.01% Triton X-100. The cells were washed three times for 5 min each in 500 μl of 1× PBS with 0.01% Triton X-100. Secondary antibodies used were goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 568 at 1:500 dilution (Molecular Probes, Invitrogen, catalog no. A-21202 and A-11010, respectively). The cells were washed twice with PBS and mounted in 70% glycerol supplemented with DAPI for the imaging analysis. Imaging was performed using a Carl Zeiss LSM510 Meta confocal laser-scanning microscope using a Plan Apochromat X63/1.4, oil immersion objective and analyzed using the LSM Image Examiner software (Carl Zeiss, Inc.).
In situ PLA
A commercial kit (Duolink In Situ Orange Starter Kit Mouse/Rabbit, catalog no. DUO92102, Sigma) was used for the PLA. HEK293T cells were seeded on glass coverslips, grown to 40–50% confluency, and transfected with a pool of pcGag (500 ng) and Tsg101-FLAG (1,000 ng) vectors. The transfection efficiency was normalized using pCMV_LIG vector-expressing luciferase. The cells, 36 h post-transfection, were fixed with 4% paraformaldehyde for 20 min and permeabilized as described above. The cells were washed three times for 5 min each in 500 μl of 1× PBS. The cells were incubated in the blocking solution provided in the kit for 1 h. This was followed by the incubation of the cells with a pool of the primary antibodies overnight at 4 °C as described above. The cells were washed three times for 5 min each in 500 μl of 1× PBS with 0.01% Triton X-100. The PLA was performed as per the manufacturer's instructions. Briefly, the cells were incubated with a pool of two different PLA probes (the PLA Probe Anti-Mouse MINUS; DUO92004 and PLA Probe Anti-Rabbit PLUS; DUO92002) in a 40-μl reaction volume for 1 h at 37 °C and washed twice for 5 min each in 500 μl of wash buffer A. The ligation (Ligase 1 unit/μl) and amplification (polymerase 10 units/μl) reactions were performed as per instructions using the Duolink In Situ Detection reagents Orange (catalog no. DUO92007). The samples were mounted using the mounting medium supplemented with DAPI supplied in the PLA kit and imaged using an LSM510 confocal microscope. From each coverslip, 15–20 representative fields were imaged. The intensity of the signals from the PLA positive spots was analyzed manually using the ImageJ software. The Tsg101-FLAG expression plasmid was a kind gift from Prof. Wes Sundquist, Department of Biochemistry, University of Utah School of Medicine, Salt Lake City.
Co-immunoprecipitation assay
HEK293T cells were transfected with 3 μg of one of the three Gag variant vectors, WT Gag (single-PTAP), VT1 Gag (double-PTAP), and ΔPTAP (PTAP-deleted) vector. Total cytoplasmic extracts were prepared from the cells following 48 h of transfection, and the endogenously expressed Tsg101 was immunoprecipitated using a mouse mAb (sc-7964, Santa Cruz Biotechnology). The immunoprecipitated products were resolved on a 12% SDS-PAGE and electrotransfered to PVDF membranes. The blots were probed for Gag using rabbit polyclonal antibody (National Institutes of Health 4250, AIDS reagent program) and re-probed with a different anti-Tsg101 mAb (MA1-23296, ThermoFisher Scientific) to confirm the presence of Tsg101 in the immunoprecipitates.
Tsg101 overexpression analysis
HEK 293T cells (0.5 × 106) were seeded in a 12-well plate and transfected the following day. The cells were cotransfected with 200 ng of one of the three infectious HIV-1 molecular clones (single-PTAP or double-PTAP Indie or NL4-3) and progressively increasing concentrations of the Tsg101-FLAG expression vector ranging from 200 to 2,000 ng. The amount of DNA was normalized for each transfection using the pcDNA3.1(+) empty plasmid. Additionally, 100 ng of a β-galactosidase expression vector, pCMV–β-gal, was transfected as an internal control in all the wells. The medium was changed 6 h following the transfection. The secretion of p24 into the medium was quantitated at 48 h post-transfection and normalized against the β-galactosidase transfection control.
HLA typing and CTL immune–escape analysis
High-resolution HLA typing of a subset of the subjects was performed using the genomic DNA extracted from the stored PBMC of a select subset of the study participants at a four-digit resolution. The HLA typing was performed at the Rotary Blood Bank, Bangalore, India, using the Micro SSP Generic HLA class I kit (catalog no. SSP1L One Lambda, ThermoFisher Scientific), and the data were analyzed using the HLA Fusion 2012/03 software (One Lambda, ThermoFisher Scientific).
Author contributions
S. Sharma, P. S. A, and M. M., conceptualization, data curation, investigation, validation, writing original draft, reviewing and editing of the article; S. Saravanan, K. G. M., P. B., S. Solomon, and I. H., providing resources; V. R., R. V. S., and J. J., data curation and validation; S. G. A., S. P., and C. R., investigation and methodology; U. R., conceptualization, funding acquisition, validation, writing, reviewing and editing of the article.
Supplementary Material
Acknowledgments
The following reagents were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program and NIAID: CEM.NKR CCR5+ cells from Dr. Alexandra Trkola and TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.
This work was supported in part by Grant DST/INT/SAFR/MEGA-P(5)/2011 from Department of Science and Technology, Government of India (to U. R.), and intramural funds from Jawaharlal Nehru Centre for Advanced Scientific Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the Food and Drug Administration (FDA).
This article contains Figs. 1–8.
The sequence output files of the NGS data have been submitted to NCBI Sequence Read Archive under accession numbers SRX2596628 to SRX2596659.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- LTR
- long terminal repeat
- ANOVA
- analysis of variance
- DAPI
- 4,6-diamidino-2-phenylindole
- NGS
- next-generation sequencing
- ICS
- intracellular cytokine staining
- PBMC
- peripheral blood mononuclear cell
- IFN
- interferon
- CFSE
- carboxyfluorescein succinimidyl ester
- HTA
- heteroduplex tracking assay
- m.o.i.
- multiplicity of infection
- PTAP
- Pro–Thr/Ser–Ala–Pro
- NC
- nucleocapsid
- ESCRT
- endosomal sorting complex required for transport
- ART
- anti-retroviral therapy
- VT
- variant type
- PLA
- proximity ligation assay
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- MV
- minority variant
- pol
- polymerase.
References
- 1. Hemelaar J., Gouws E., Ghys P. D., Osmanov S., and WHO-UNAIDS Network for HIV Isolation and Characterisation (2011) Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 25, 679–689 10.1097/QAD.0b013e328342ff93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bachu M., Yalla S., Asokan M., Verma A., Neogi U., Sharma S., Murali R. V., Mukthey A. B., Bhatt R., Chatterjee S., Rajan R. E., Cheedarla N., Yadavalli V. S., Mahadevan A., Shankar S. K., et al. (2012) Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J. Biol. Chem. 287, 44714–44735 10.1074/jbc.M112.397158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siddappa N. B., Dash P. K., Mahadevan A., Desai A., Jayasuryan N., Ravi V., Satishchandra P., Shankar S. K., and Ranga U. (2005) Identification of unique B/C recombinant strains of HIV-1 in the southern state of Karnataka, India. AIDS 19, 1426–1429 10.1097/01.aids.0000180795.49016.89 [DOI] [PubMed] [Google Scholar]
- 4. Ramírez d A., Soriano V., Alcamil J., and Holguín A. (2006) New findings on transcription regulation across different HIV-1 subtypes. AIDS Rev. 8, 9–16 [PubMed] [Google Scholar]
- 5. De Baar M. P., De Ronde A., Berkhout B., Cornelissen M., Van Der Horn K. H., Van Der Schoot A. M., De Wolf F., Lukashov V. V., and Goudsmit J. (2000) Subtype-specific sequence variation of the HIV type 1 long terminal repeat and primer-binding site. AIDS Res. Hum. Retroviruses 16, 499–504 10.1089/088922200309160 [DOI] [PubMed] [Google Scholar]
- 6. Bilodeau P. S., Domsic J. K., and Stoltzfus C. M. (1999) Splicing regulatory elements within tat exon 2 of human immunodeficiency virus type 1 (HIV-1) are characteristic of group M but not group O HIV-1 strains. J. Virol. 73, 9764–9772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang S. Y., Apichartpiyakul C., Kuiken C. L., Essex M., and Lee T. H. (1999) Sequence features downstream of the primer-binding site of HIV type 1 subtype E shared by subtype G and a subset of subtype A. AIDS Res Hum Retroviruses 15, 1703–1706 10.1089/088922299309748 [DOI] [PubMed] [Google Scholar]
- 8. Chang S. Y., Sutthent R., Auewarakul P., Apichartpiyakul C., Essex M., and Lee T. H. (1999) Differential stability of the mRNA secondary structures in the frameshift site of various HIV type 1 viruses. AIDS Res. Hum. Retroviruses 15, 1591–1596 10.1089/088922299309892 [DOI] [PubMed] [Google Scholar]
- 9. Mishra M., Vetrivel S., Siddappa N. B., Ranga U., and Seth P. (2008) Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann. Neurol. 63, 366–376 10.1002/ana.21292 [DOI] [PubMed] [Google Scholar]
- 10. Ranga U., Shankarappa R., Siddappa N. B., Ramakrishna L., Nagendran R., Mahalingam M., Mahadevan A., Jayasuryan N., Satishchandra P., Shankar S. K., and Prasad V. R. (2004) Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J. Virol. 78, 2586–2590 10.1128/JVI.78.5.2586-2590.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sood V., Ranjan R., and Banerjea A. C. (2008) Functional analysis of HIV-1 subtypes B and C HIV-1 Tat exons and RGD/QGD motifs with respect to Tat-mediated transactivation and apoptosis. AIDS 22, 1683–1685 10.1097/QAD.0b013e3282f56114 [DOI] [PubMed] [Google Scholar]
- 12. Neogi U., Sood V., Banerjee S., Ghosh N., Verma S., Samrat S., Sharma Y., Saxena A., Husain S., Ramachandran V. G., Das S., Sreedhar K. V., Goel N., Wanchu A., and Banerjea A. C. (2009) Global HIV-1 molecular epidemiology with special reference to genetic analysis of HIV-1 subtypes circulating in North India: functional and pathogenic implications of genetic variation. Indian J. Exp. Biol. 47, 424–431 [PubMed] [Google Scholar]
- 13. Neogi U., Sahoo P. N., Kumar R., De Costa A., and Shet A. (2011) Characterization of HIV type 1 subtype C protease gene: selection of L63P mutation in protease inhibitor-naive Indian patients. AIDS Res. Hum. Retroviruses 27, 1249–1253 10.1089/aid.2011.0078 [DOI] [PubMed] [Google Scholar]
- 14. Bano A. S., Sood V., Neogi U., Goel N., Kuttiat V. S., Wanchu A., and Banerjea A. C. (2009) Genetic and functional characterization of HIV-1 VprC variants from North India: presence of unique recombinants with mosaic genomes from B, C and D subtypes within the ORF of Vpr. J. Gen. Virol. 90, 2768–2776 10.1099/vir.0.011080-0 [DOI] [PubMed] [Google Scholar]
- 15. Martins A. N., Arruda M. B., Pires A. F., Tanuri A., and Brindeiro R. M. (2011) Accumulation of P(T/S)AP late domain duplications in HIV type 1 subtypes B, C, and F derived from individuals failing ARV therapy and ARV drug-naive patients. AIDS Res. Hum. Retroviruses 27, 687–692 10.1089/aid.2010.0282 [DOI] [PubMed] [Google Scholar]
- 16. Swanstrom R., and Wills J. W. (1997) in Retroviruses (Coffin J. M., Hughes S. H., and Varmus H. E., eds) pp. 263–311, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 17. Bell N. M., and Lever A. M. (2013) HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 21, 136–144 10.1016/j.tim.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 18. Saad J. S., Miller J., Tai J., Kim A., Ghanam R. H., and Summers M. F. (2006) Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U.S.A. 103, 11364–11369 10.1073/pnas.0602818103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghanam R. H., Samal A. B., Fernandez T. F., and Saad J. S. (2012) Role of the HIV-1 matrix protein in Gag intracellular trafficking and targeting to the plasma membrane for virus assembly. Front. Microbiol. 3, 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Resh M. D. (2005) Intracellular trafficking of HIV-1 Gag: how Gag interacts with cell membranes and makes viral particles. AIDS Rev. 7, 84–91 [PubMed] [Google Scholar]
- 21. Jouvenet N., Simon S. M., and Bieniasz P. D. (2009) Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U.S.A. 106, 19114–19119 10.1073/pnas.0907364106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Usami Y., Popov S., Popova E., Inoue M., Weissenhorn W., and G. Göttlinger H. (2009) The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 37, 181–184 10.1042/BST0370181 [DOI] [PubMed] [Google Scholar]
- 23. Gottlinger H. G., Dorfman T., Sodroski J. G., and Haseltine W. A. (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U.S.A. 88, 3195–3199 10.1073/pnas.88.8.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paxton W., Connor R. I., and Landau N. R. (1993) Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of Gag and mutational analysis. J. Virol. 67, 7229–7237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Checroune F., Yao X. J., Göttlinger H. G., Bergeron D., and Cohen E. A. (1995) Incorporation of Vpr into human immunodeficiency virus type 1: role of conserved regions within the P6 domain of Pr55gag. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10, 1–7 [PubMed] [Google Scholar]
- 26. Bieniasz P. D. (2009) The cell biology of HIV-1 virion genesis. Cell Host Microbe 5, 550–558 10.1016/j.chom.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garrus J. E., von Schwedler U. K., Pornillos O. W., Morham S. G., Zavitz K. H., Wang H. E., Wettstein D. A., Stray K. M., Côté M., Rich R. L., Myszka D. G., and Sundquist W. I. (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107, 55–65 10.1016/S0092-8674(01)00506-2 [DOI] [PubMed] [Google Scholar]
- 28. Martin-Serrano J., Zang T., and Bieniasz P. D. (2001) HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7, 1313–1319 10.1038/nm1201-1313 [DOI] [PubMed] [Google Scholar]
- 29. VerPlank L., Bouamr F., LaGrassa T. J., Agresta B., Kikonyogo A., Leis J., and Carter C. A. (2001) Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. U.S.A. 98, 7724–7729 10.1073/pnas.131059198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demirov D. G., Ono A., Orenstein J. M., and Freed E. O. (2002) Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. U.S.A. 99, 955–960 10.1073/pnas.032511899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Engelenburg S. B., Shtengel G., Sengupta P., Waki K., Jarnik M., Ablan S. D., Freed E. O., Hess H. F., and Lippincott-Schwartz J. (2014) Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 343, 653–656 10.1126/science.1247786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prescher J., Baumgärtel V., Ivanchenko S., Torrano A. A., Bräuchle C., Müller B., and Lamb D. C. (2015) Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog. 11, e1004677 10.1371/journal.ppat.1004677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strack B., Calistri A., Craig S., Popova E., and Göttlinger H. G. (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114, 689–699 10.1016/S0092-8674(03)00653-6 [DOI] [PubMed] [Google Scholar]
- 34. Sundquist W. I., and Kräusslich H. G. (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2, a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Usami Y., Popov S., and Göttlinger H. G. (2007) Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 81, 6614–6622 10.1128/JVI.00314-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holguín A., Alvarez A., and Soriano V. (2005) Differences in the length of gag proteins among different HIV type 1 subtypes. AIDS Res. Hum. Retroviruses 21, 886–893 10.1089/aid.2005.21.886 [DOI] [PubMed] [Google Scholar]
- 37. Marlowe N., Flys T., Hackett J. Jr., Schumaker M., Jackson J. B., Eshleman S. H., HIV Prevention Trials Network and Adult Clinical Trials Groups. (2004) Analysis of insertions and deletions in the gag p6 region of diverse HIV type 1 strains. AIDS Res. Hum. Retroviruses 20, 1119–1125 10.1089/aid.2004.20.1119 [DOI] [PubMed] [Google Scholar]
- 38. Flys T., Marlowe N., Hackett J., Parkin N., Schumaker M., Holzmayer V., Hay P., and Eshleman S. H. (2005) Analysis of PTAP duplications in the gag p6 region of subtype C HIV type 1. AIDS Res. Hum. Retroviruses 21, 739–741 10.1089/aid.2005.21.739 [DOI] [PubMed] [Google Scholar]
- 39. Rossi A. H., Rocco C. A., Mangano A., Sen L., and Aulicino P. C. (2013) Sequence variability in p6 gag protein and gag/pol coevolution in human immunodeficiency type 1 subtype F genomes. AIDS Res. Hum. Retroviruses 29, 1056–1060 10.1089/aid.2012.0311 [DOI] [PubMed] [Google Scholar]
- 40. Song Y. H., Meng Z. F., Xing H., Ruan Y. H., Li X. P., Xin R. L., Ma P. F., Peng H., and Shao Y. (2007) Analysis of HIV-1 CRF07_BC gag p6 sequences indicating novel deletions in the central region of p6. Arch. Virol. 152, 1553–1558 10.1007/s00705-007-0973-6 [DOI] [PubMed] [Google Scholar]
- 41. Peters S., Muñoz M., Yerly S., Sanchez-Merino V., Lopez-Galindez C., Perrin L., Larder B., Cmarko D., Fakan S., Meylan P., and Telenti A. (2001) Resistance to nucleoside analog reverse transcriptase inhibitors mediated by human immunodeficiency virus type 1 p6 protein. J. Virol. 75, 9644–9653 10.1128/JVI.75.20.9644-9653.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simon V., and Ho D. D. (2003) HIV-1 dynamics in vivo: implications for therapy. Nat. Rev. Microbiol. 1, 181–190 10.1038/nrmicro772 [DOI] [PubMed] [Google Scholar]
- 43. Ibe S., Shibata N., Utsumi M., and Kaneda T. (2003) Selection of human immunodeficiency virus type 1 variants with an insertion mutation in the p6(gag) and p6(pol) genes under highly active antiretroviral therapy. Microbiol. Immunol. 47, 71–79 10.1111/j.1348-0421.2003.tb02788.x [DOI] [PubMed] [Google Scholar]
- 44. Colgrove R. C., Millet A., Bauer G. R., Pitt J., and Welles S. L. (2005) Gag-p6 Tsg101 binding site duplications in maternal-infant HIV infection. AIDS Res. Hum. Retroviruses 21, 191–199 10.1089/aid.2005.21.191 [DOI] [PubMed] [Google Scholar]
- 45. Brumme Z. L., Chan K. J., Dong W. W., Wynhoven B., Mo T., Hogg R. S., Montaner J. S., O'Shaughnessy M. V., and Harrigan P. R. (2003) Prevalence and clinical implications of insertions in the HIV-1 p6Gag N-terminal region in drug-naive individuals initiating antiretroviral therapy. Antivir. Ther. 8, 91–96 [PubMed] [Google Scholar]
- 46. Fun A., Wensing A. M., Verheyen J., and Nijhuis M. (2012) Human immunodeficiency virus Gag and protease: partners in resistance. Retrovirology 9, 63 10.1186/1742-4690-9-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallego O., de Mendoza C., Corral A., and Soriano V. (2003) Changes in the human immunodeficiency virus p7-p1-p6 gag gene in drug-naive and pretreated patients. J. Clin. Microbiol. 41, 1245–1247 10.1128/JCM.41.3.1245-1247.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lastere S., Dalban C., Collin G., Descamps D., Girard P. M., Clavel F., Costagliola D., Brun-Vezinet F., and NARVAL Trial Group (ANRS 088) (2004) Impact of insertions in the HIV-1 p6 PTAPP region on the virological response to amprenavir. Antivir. Ther. 9, 221–227 [PubMed] [Google Scholar]
- 49. Tamiya S., Mardy S., Kavlick M. F., Yoshimura K., and Mistuya H. (2004) Amino acid insertions near Gag cleavage sites restore the otherwise compromised replication of human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 78, 12030–12040 10.1128/JVI.78.21.12030-12040.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sharma S., Aralaguppe S. G., Abrahams M. R., Williamson C., Gray C., Balakrishnan P., Saravanan S., Murugavel K. G., Solomon S., and Ranga U. (2017) The PTAP sequence duplication in HIV-1 subtype C Gag p6 in drug-naive subjects of India and South Africa. BMC Infect. Dis. 17, 95 10.1186/s12879-017-2184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chopera D. R., Cotton L. A., Zawaira A., Mann J. K., Ngandu N. K., Ntale R., Carlson J. M., Mlisana K., Woodman Z., de Assis Rosa D., Martin E., Miura T., Pereyra F., Walker B. D., Gray C. M., et al. (2012) Intersubtype differences in the effect of a rare p24 gag mutation on HIV-1 replicative fitness. J. Virol. 86, 13423–13433 10.1128/JVI.02171-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ntale R. S., Chopera D. R., Ngandu N. K., Assis de Rosa D., Zembe L., Gamieldien H., Mlotshwa M., Werner L., Woodman Z., Mlisana K., Abdool Karim S., Gray C. M., Williamson C., and CAPRISA 002 Study Team (2012) Temporal association of HLA-B*81:01- and HLA-B*39:10-mediated HIV-1 p24 sequence evolution with disease progression. J. Virol. 86, 12013–12024 10.1128/JVI.00539-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guimarães A. P., Sa-Filho D. J., Sucupira M. C., Janini L. M., and Diaz R. S. (2008) Profiling resistance-related mutations in the protease region of the pol gene: single genome sequencing of HIV in plasma and peripheral blood mononuclear cells. AIDS Res. Hum. Retroviruses 24, 969–971 10.1089/aid.2008.0020 [DOI] [PubMed] [Google Scholar]
- 54. Josephs T. M., Grant E. J., and Gras S. (2017) Molecular challenges imposed by MHC-I restricted long epitopes on T cell immunity. Biol. Chem. 398, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 55. Cao J., McNevin J., McSweyn M., Liu Y., Mullins J. I., and McElrath M. J. (2008) Novel cytotoxic T-lymphocyte escape mutation by a three-amino-acid insertion in the human immunodeficiency virus type 1 p6 Pol and p6 Gag late domain associated with drug resistance. J. Virol. 82, 495–502 10.1128/JVI.01096-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Abraha A., Troyer R. M., Quiñones-Mateu M. E., and Arts E. J. (2005) Methods to determine HIV-1 ex vivo fitness. Methods Mol. Biol. 304, 355–368 [DOI] [PubMed] [Google Scholar]
- 57. Pornillos O., Higginson D. S., Stray K. M., Fisher R. D., Garrus J. E., Payne M., He G. P., Wang H. E., Morham S. G., and Sundquist W. I. (2003) HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 162, 425–434 10.1083/jcb.200302138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goila-Gaur R., Demirov D. G., Orenstein J. M., Ono A., and Freed E. O. (2003) Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J. Virol. 77, 6507–6519 10.1128/JVI.77.11.6507-6519.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bachu M., Mukthey A. B., Murali R. V., Cheedarla N., Mahadevan A., Shankar S. K., Satish K. S., Kundu T. K., and Ranga U. (2012) Sequence insertions in the HIV type 1 subtype C viral promoter predominantly generate an additional NF-κB binding site. AIDS Res. Hum. Retroviruses 28, 1362–1368 10.1089/aid.2011.0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boullosa J., Bachu M., Bila D., Ranga U., Süffert T., Sasazawa T., and Tanuri A. (2014) Genetic diversity in HIV-1 subtype C LTR from Brazil and Mozambique generates new transcription factor-binding sites. Viruses 6, 2495–2504 10.3390/v6062495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Neogi U., Engelbrecht S., Claassen M., Jacobs G. B., van Zyl G., Preiser W., and Sonnerborg A. (2016) Mutational heterogeneity in p6 Gag late assembly (L) domains in HIV-1 subtype C viruses from South Africa. AIDS Res. Hum. Retroviruses 32, 80–84 10.1089/aid.2015.0266 [DOI] [PubMed] [Google Scholar]
- 62. Martins A. N., Waheed A. A., Ablan S. D., Huang W., Newton A., Petropoulos C. J., Brindeiro R. D., and Freed E. O. (2016) Elucidation of the molecular mechanism driving duplication of the HIV-1 PTAP late domain. J. Virol. 90, 768–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meyerhans A., Cheynier R., Albert J., Seth M., Kwok S., Sninsky J., Morfeldt-Månson L., Asjö B., and Wain-Hobson S. (1989) Temporal fluctuations in HIV quasispecies in vivo are not reflected by sequential HIV isolations. Cell 58, 901–910 10.1016/0092-8674(89)90942-2 [DOI] [PubMed] [Google Scholar]
- 64. Gallego O., d Mendoza C., Labarga P., Altisent C., González J., García-Alcalde I., Valer L., Valencia E., and Soriano V. (2003) Long-term outcome of HIV-infected patients with multinucleoside-resistant genotypes. HIV Clin. Trials 4, 372–381 10.1310/X618-KWKJ-WCTQ-LQ2L [DOI] [PubMed] [Google Scholar]
- 65. Dettenhofer M., and Yu X. F. (1999) Proline residues in human immunodeficiency virus type 1 p6(Gag) exert a cell type-dependent effect on viral replication and virion incorporation of Pol proteins. J. Virol. 73, 4696–4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hahn S., Setz C., Wild J., and Schubert U. (2011) The PTAP sequence within the p6 domain of human immunodeficiency virus type 1 Gag regulates its ubiquitination and MHC class I antigen presentation. J. Immunol. 186, 5706–5718 10.4049/jimmunol.1003764 [DOI] [PubMed] [Google Scholar]
- 67. Keele B. F., Giorgi E. E., Salazar-Gonzalez J. F., Decker J. M., Pham K. T., Salazar M. G., Sun C., Grayson T., Wang S., Li H., Wei X., Jiang C., Kirchherr J. L., Gao F., Anderson J. A., et al. (2008) Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 105, 7552–7557 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Salazar-Gonzalez J. F., Salazar M. G., Keele B. F., Learn G. H., Giorgi E. E., Li H., Decker J. M., Wang S., Baalwa J., Kraus M. H., Parrish N. F., Shaw K. S., Guffey M. B., Bar K. J., Davis K. L., et al. (2009) Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206, 1273–1289 10.1084/jem.20090378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Garcia P. M., Kalish L. A., Pitt J., Minkoff H., Quinn T. C., Burchett S. K., Kornegay J., Jackson B., Moye J., Hanson C., Zorrilla C., and Lew J. F. (1999) Maternal levels of plasma human immunodeficiency virus type 1 RNA and the risk of perinatal transmission. Women and Infants Transmission Study Group. N. Engl. J. Med. 341, 394–402 10.1056/NEJM199908053410602 [DOI] [PubMed] [Google Scholar]
- 70. Quinn T. C., Wawer M. J., Sewankambo N., Serwadda D., Li C., Wabwire-Mangen F., Meehan M. O., Lutalo T., and Gray R. H. (2000) Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N. Engl. J. Med. 342, 921–929 10.1056/NEJM200003303421303 [DOI] [PubMed] [Google Scholar]
- 71. Zhang J., Kobert K., Flouri T., and Stamatakis A. (2014) PEAR: a fast and accurate illumina paired-end reAd mergeR. Bioinformatics 30, 614–620 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li H., and Durbin R. (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim Y., Ponomarenko J., Zhu Z., Tamang D., Wang P., Greenbaum J., Lundegaard C., Sette A., Lund O., Bourne P. E., Nielsen M., and Peters B. (2012) Immune epitope database analysis resource. Nucleic Acids Res. 40, W525–W530 10.1093/nar/gks438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., and Nielsen M. (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–W512 10.1093/nar/gkn202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nielsen M., Lundegaard C., Worning P., Lauemøller S. L., Lamberth K., Buus S., Brunak S., and Lund O. (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 10.1110/ps.0239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Peters B., and Sette A. (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6, 132 10.1186/1471-2105-6-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sidney J., Assarsson E., Moore C., Ngo S., Pinilla C., Sette A., and Peters B. (2008) Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4, 2 10.1186/1745-7580-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lamoreaux L., Roederer M., and Koup R. (2006) Intracellular cytokine optimization and standard operating procedure. Nat. Protoc. 1, 1507–1516 10.1038/nprot.2006.268 [DOI] [PubMed] [Google Scholar]
- 79. Quah B. J., Warren H. S., and Parish C. R. (2007) Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2, 2049–2056 10.1038/nprot.2007.296 [DOI] [PubMed] [Google Scholar]
- 80. Chen C., and Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752 10.1128/MCB.7.8.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ariën K. K., Troyer R. M., Gali Y., Colebunders R. L., Arts E. J., and Vanham G. (2005) Replicative fitness of historical and recent HIV-1 isolates suggests HIV-1 attenuation over time. AIDS 19, 1555–1564 10.1097/01.aids.0000185989.16477.91 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.