

Abstract
2-Cys peroxiredoxins (Prxs) modulate hydrogen peroxide (H2O2)-mediated cell signaling. At high H2O2 levels, eukaryotic Prxs can be inactivated by hyperoxidation and are classified as sensitive Prxs. In contrast, prokaryotic Prxs are categorized as being resistant to hyperoxidation and lack the GGLG and C-terminal YF motifs present in the sensitive Prxs. Additional molecular determinants that account for the subtle differences in the susceptibility to hyperoxidation remain to be identified. A comparison of a new, 2.15-Å–resolution crystal structure of Prx2 in the oxidized, disulfide-bonded state with the hyperoxidized structure of Prx2 and Prx1 in complex with sulfiredoxin revealed three structural regions that rearrange during catalysis. With these regions in hand, focused sequence analyses were performed comparing sensitive and resistant Prx groups. From this combinatorial approach, we discovered two novel hyperoxidation resistance motifs, motifs A and B, which were validated using mutagenesis of sensitive human Prxs and resistant Salmonella enterica serovar Typhimurium AhpC. Introduction and removal of these motifs, respectively, resulted in drastic changes in the sensitivity to hyperoxidation with Prx1 becoming 100-fold more resistant to hyperoxidation and AhpC becoming 800-fold more sensitive to hyperoxidation. The increased sensitivity of the latter AhpC variant was also confirmed in vivo. These results support the function of motifs A and B as primary drivers for tuning the sensitivity of Prxs to different levels of H2O2, thus enabling the initiation of variable signaling or antioxidant responses in cells.
Keywords: peroxiredoxin, oxidation-reduction (redox), X-ray crystallography, structural biology, enzyme kinetics, cell signaling, Cys sulfinic acid, protein oxidation, redox biology
Introduction
In contrast to the buildup of free radical oxygen species that cause damage to biomolecules and disease, hydrogen peroxide (H2O2) can be produced in a controlled manner to function as an intracellular signaling molecule (1–3). Peroxiredoxins (Prxs)2 are a family of highly expressed enzymes that directly regulate the levels of H2O2 and enable the cell to respond to different stimuli and oxidative stress (4–7). Oxidized Prxs can also participate in signaling and regulate the activity of transcription factors through thiol-disulfide exchange reactions (8–10). Consistent with the signaling and antioxidant roles of Prxs, dysregulation of these enzymes has been directly linked to a variety of cancers, cardiovascular disease, neurodegenerative diseases, and diabetes (11–13).
In the catalytic cycle of the Prx1-type or typical 2-Cys subclass of Prxs (Prx1–4 in humans) (Fig. 1), the peroxidatic cysteine (Cys-SPH) residue from one subunit of the obligate dimer attacks an H2O2 molecule (4). The dimers of most members of this Prx subclass assemble into a decameric toroid, which is the most active form (14, 15). The reaction with H2O2 results in the formation of the reactive Cys sulfenic acid (Cys-SPOH) species and the release of water. At this point, two competing events can occur depending on how much H2O2 is present. At low H2O2 levels, the resolving cysteine (Cys-SRH), near the C terminus of the adjacent subunit, can react with the Cys-SPOH moiety to form an intersubunit disulfide bond (SP-SR). This process requires the structural rearrangement of the Cys-SPH–containing active-site loop (CP-loop) from a fully folded (FF) to a locally unfolded (LU) conformation and shifts the equilibrium of the oligomeric state toward the dimer (6, 16). The reaction cycle is completed by the reduction of the SP-SR disulfide bond by the thioredoxin-thioredoxin reductase-NADPH system (Trx-TrxR-NADPH).
Figure 1.

Catalytic cycle and hyperoxidation of typical, 2-Cys Prxs. These Prxs exist as obligate homodimers with each subunit (blue or purple) containing Cys-SP (α2 helix) and Cys-SR (near the C terminus) residues (highlighted as yellow circles). For most members of this family, including human Prx1, Prx2, and bacterial AhpC, the dimers can assemble into a decameric toroid with 52-point group symmetry. Reaction of the Cys-SP residue with H2O2 creates the Cys-SPOH and releases a water molecule (not shown for clarity). During normal catalysis and low H2O2 levels, the Cys-SR residue from the adjacent subunit condenses with Cys-SPOH to form an intermolecular disulfide (Cys-SP–SR-Cys), releasing a second water molecule (not shown). For this disulfide bond to occur, the α2 helix, near the GGLG and YF motifs, and the C terminus of the adjacent subunit, must transition from the FF to the LU state. The disulfide can then be reduced by the NADPH-dependent Trx-TrxR system. If the H2O2 levels are high, the Cys-SPOH moiety can alternatively react with a second H2O2 molecule to form Cys-SPO2H. This modification or hyperoxidation can occur on one or both Cys-SP residues of the dimer. The inactivated Prx can be repaired in an ATP-dependent manner by the enzyme sulfiredoxin (Srx).
At high H2O2 levels, the Cys-SPOH intermediate (Fig. 1) can react with another molecule of H2O2 and become sulfinylated, producing Cys sulfinic acid (Cys-SPO2H), a process also called hyperoxidation. This inactivation can switch the activity from a peroxidase to a high-molecular-weight chaperone holdase for some Prxs (17, 18). The loss in peroxidase activity can allow H2O2-mediated signaling to be potentiated through the direct modification of other proteins with reactive Cys thiols (e.g. PTP1b and Nrf2) (6, 19, 20). The hyperoxidized form of the Prx can also function as a cell stress signal that can be resolved by the ATP-dependent reduction of the Cys-SPO2H moiety by sulfiredoxin (17, 21–24).
The susceptibility to hyperoxidation and inactivation is remarkably variable among typical 2-Cys Prxs despite having similar rates for Cys-SPOH formation (kSOH = 105–108 m−1 s−1) (5–7, 25, 26). For example, bacterial Prxs, such as the prototypical Salmonella enterica serovar Typhimurium AhpC, are significantly more resistant to inactivation and represent a category of “robust” Prxs (6). Mammalian Prxs, by comparison, have been classified as “sensitive” to inactivation by the presence of Tyr-Phe (YF) and Gly-Gly-Leu-Gly (GGLG) motifs (Fig. 1) near the active site (6). However, even among the sensitive Prxs, there is a stratification of hyperoxidation sensitivity and differential consequences of gene knockout, supporting their involvement in unique sets of signaling processes and nonredundancy of function (25–27). For example, human mitochondrial Prx3 (hPrx3) is significantly more resistant to hyperoxidation than the cytoplasmic Prx1 and Prx2 even though all three proteins exhibit high sequence identity and contain the YF and GGLG motifs (28).
Detailed kinetic and MS-based studies using chimeras have demonstrated that part of the difference in the resistance to hyperoxidation between hPrx3 and hPrx2 can be attributed to differences in the intermolecular disulfide bond formation rate and the corresponding lifetime of the Cys-SPOH intermediate (25, 26). Importantly, these studies focused on the regions near the GGLG and YF motifs and did not demonstrate full transfer of resistance or sensitivity from one protein to another. These observations suggest that there are likely additional motifs and structural elements that contribute to the molecular basis for fine-tuning the resistance to hyperoxidation of Prxs (29).
In this study, we identified two novel resistance-conferring motifs, motifs A and B, that explain the hierarchical sensitivity of Prxs to hyperoxidation beyond the previously identified GGLG and YF motifs. We demonstrate that hyperoxidation resistance or sensitivity can be manipulated in human Prxs by inserting or disrupting these motifs, respectively. With this approach, we were able to interchange the hyperoxidation sensitivity of human Prx1 and bacterial AhpC. Particularly notable is the increased sensitivity to hyperoxidation in AhpC to the level of human Prxs, which was never achieved before. Thus, our results demonstrate that motifs A and B play a key role in the fine-tuning the sensitivity and resistance of Prxs to hyperoxidation. This control of Prx activity is consistent with the ability of cells to activate specific signaling pathways to appropriately respond to nonstress and stressful levels of H2O2.
Results
Human Prxs exhibit 10–25-fold differences in resistance to hyperoxidation
As a first step to identifying molecular determinants for the relative sensitivity of human Prxs to hyperoxidation, human Prx1, Prx2, and Prx3 were purified. These proteins were produced in Escherichia coli without any extra residues (e.g. affinity tags) (25, 30). The use of the mature protein is crucial as extra residues and affinity tags can influence the oligomeric state and activity (25, 31). Hyperoxidation of Prxs has been monitored historically through 1D and 2D SDS-PAGE and Western blotting using antibodies against Cys-SO2H/SO3H (28, 32). In our hands, the antibodies preferentially react with the Cys-SO3H moiety and can complicate the interpretation of the species present following the addition of H2O2. Therefore, we used kinetic methods and high-resolution MS to verify the oxidation state.
To measure the hyperoxidation of the Prx variants in this study, the proteins were prereduced with dithiothreitol (DTT), desalted, and then treated with increasing concentrations of H2O2,. The rate of the reaction was determined using the E. coli Trx-TrxR-NAPDH system, and the Chyp1% parameter was calculated (24, 33). Importantly, the Chyp1% parameter quantifies the concentration of H2O2 at which 1% of the Prx molecules are inactivated per catalytic turnover, independently of the concentration and source of the reducing system used. Thus, a higher Chyp1% value indicates a greater resistance to hyperoxidation. Prx1 was 10-fold more resistant than Prx2 (Chyp1% of 50 versus 5 μm) (Fig. S1). In contrast, Prx3 was 25-fold more resistant than Prx2 (Chyp1% of 127 versus 5 μm).
Structure of oxidized hPrx2 highlights regions of interest involved in hyperoxidation
Because we know that the rate of disulfide bond formation greatly influences the sensitivity to hyperoxidation (25, 26), we hypothesized that delineating the structural differences between the reduced and disulfide-bonded forms of Prx2 could be used to identify key amino acids responsible for the differences in hyperoxidation sensitivity. We solved the crystal structure of wildtype (WT) Prx2 in the oxidized, disulfide-bonded form (SP-SR). The electron density for the 2.15-Å–resolution structure (Fig. 2A andTable S1) clearly showed an intermolecular disulfide bond between Cys51-SP and Cys172-SR from the adjacent subunit for six of 10 possible disulfide bonds in the decamer. Importantly, for five of these subunits, we were also able to observe Trp176 packing into a pocket adjacent to the GGLG motif (Fig. 2B). This latter observation contrasts with the majority of oxidized LU Prx structures, prokaryotic and eukaryotic, where only the SP-SR linkage is visible and the remaining residues of the C terminus are disordered (16, 36, 37). The only high-resolution crystal structure available for human Prx2, prior to this study, contained the Cys-SP residue in the hyperoxidized state and the CP-loop in the FF conformation (Fig. 2C), consistent with structures of the reduced form of bacterial Prxs (34, 35).
Figure 2.

Crystal structures of Prx2 help to identify regions implicated in hyperoxidation. A, stereoview of the 2Fo − Fc map contoured at the 1 σ level (chains E and F), including the Cys-SP–SR-Cys disulfide bond between Prx2 subunits. B, active-site structure of oxidized Prx2, Prx2-SPSR. The new structure reveals the necessary reorganization of the active site for disulfide bond formation. In this locally unfolded conformation, the C terminus is present as a coil that positions Trp176 (purple subunit) so that it packs into a crevice generated by the disulfide bond and the GGLG motif (blue subunit). Importantly, Trp176 is >20 Å away from this position in the Prx2-SPO2H structure as shown in the next panel. C, active-site structure of hyperoxidized Prx2 (chains A and B). In this fully folded conformation, the Cys51-SPO2H moiety (cyan) has an electrostatic interaction with Arg127 (Protein Data Bank code 1QMV) (35). The C terminus of the adjacent subunit (pink) places the Cys172-SR residue ∼14 Å away from the Cys51-SP residue. The YF motif (residues 193 and 194) is present in a helix that packs against the GGLG motif. D, summary of structural changes between Prx oxidation states. Left, one Prx dimer is highlighted in its decameric context (other Prx dimers are colored white). Right, location of regions that have the potential to influence the resistance to hyperoxidation. One subunit of the Prx dimer is shown in reference to the adjacent subunit (white) across the dimer-dimer interface of the toroid. Regions 1–3 are shown in green, magenta, and yellow, respectively. Region 1 is located within the β-strand and turn that precede the helix on which the Cys51-SP residue is located. Regions 2 and 3 interact with the same regions on the adjacent subunit. The residues denoted by spheres are those that differ in sequence between Prx1 and Prx2 (see Fig. 3).
Closer examination of the Prx2 structures and a comparison with the disulfide-containing Prx1–sulfiredoxin complex (Fig. S2) illustrates the proximity of the active site to the dimer-dimer interface of the decamer assembly, loop changes at this interface, differences in the position of the CP-loop and the loop containing the Cys-SR residue, and changes in the conformation of the GGLG motif (22). The structural differences identified can be subdivided into three regions (Fig. 2D). Region 1 is present in the β-strand and turn that immediately precedes the α2 helix containing the Cys-SP residue. Region 2 is composed of a surface loop and an α helix, which is adjacent to the active site and associates with its counterpart at the dimer-dimer interface. Region 3 is a linker region that also associates with its counterpart at the dimer-dimer interface. The juxtaposition of regions 2 and 3 and their potential role in hyperoxidation are consistent with the ability of mutations at the dimer-dimer interface to alter the oligomeric state and to affect activity (14, 15, 37). Because the active site must also be in the FF conformation for the Cys-SPOH moiety to be hyperoxidized, it is logical that the same region or regions may play a key role in the inactivation process (6, 34, 38).
Identification of resistance motifs A and B
Using the structural information and regions 1–3 as a guide, we next focused on the key differences in the primary sequence between resistant and sensitive Prx proteins (Fig. 3A) (see Fig. S3 and Table S2 for detailed stepwise rationale description). First, comparisons were made for 10 sensitive and nine resistant Prxs using WebLogo plots using the criterion of the presence or absence of the GGLG and YF motifs (39). The amino acids conserved across both groups were removed as it was assumed that these would not be contributing to differences in hyperoxidation. Second, to narrow the list of potential residues further, residues that were conserved across hPrx1–4 and S. enterica Typhimurium AhpC, the model robust Prx enzyme, were removed (Fig. 3A, remaining residues are indicated by triangles). Third, residues in this region that differed between Prx1 and Prx2 were identified (Fig. 3A, circles).
Figure 3.

Structure and sequence comparisons identify resistance motifs. A, summary of the structural and sequence information that led to the identification of resistance motifs A and B. The bars for regions 1–3 indicate the largest structural changes between the reduced (FF) and oxidized (LU) conformations (see Fig. 2 and Fig. S2). The triangles below the alignment highlight the residues, identified by WebLogo analysis, that differ between sensitive and resistant Prxs (Fig. S3 and Table S2). The circles indicate the residue differences in regions 1–3 for Prx1 and Prx2. The putative resistance motifs A and B are highlighted in red and cyan, respectively. The same coloring scheme for the residues within these motifs is used in the subsequent panels. B, location of motifs A and B in the Prx toroid context (see Fig. 2D for entire toroid). Both the Cys-SP and Cys-SR residues involved in disulfide bonds are colored yellow for the central Prx dimer. One subunit for each of the two adjacent dimers is shown in white. Motif A is predominantly located on the exterior of the toroid. Motif B lines the interior of the toroid. C, proximity of resistance motifs A and B to the Cys-SP residue, the GGLG motif, and the dimer-dimer interface.
With this combinatorial approach, we discovered two putative resistance-conferring motifs. Motif A is DX8(N/G)X10HX27(S/G), and motif B is TX3(S/T). These motifs are located (Fig. 3B) within or in close proximity to the active site and the dimer-dimer interface. The first half of motif A (DX8(N/G)) is associated with the α helix that leads to the Cys-SP residue (Fig. 3C). The GGLG motif is proximal to these features and the Cys-SR residue. The second half of motif A (HX27(S/G)) is also present near the active site and the dimer-dimer interface and adjacent to motif B. Importantly, it made sense to designate motif B separately because these residues were the furthest from the active site and line the interior of the decameric toroid (Fig. 3B), a region known to influence activity based on the presence or absence of affinity tags (31).
To facilitate comparisons and discussion, a shorthand notation will be used to describe the motifs present in different AhpC and Prx variants. The residues in the motif A position will be listed first followed by the motif B residues (motif A-motif B). Table 1 highlights the proteins studied experimentally herein; please see Table S2 for all of the proteins used in the motif identification process. For example, AhpC has both motifs (DGHG-TT); there is a Gly in the second and last position of motif A (Fig. 3A and Table 1). Human Prx1 has a slightly different version of motif A (DNHS-KA) and lacks motif B. Human Prx2, although it is the most sensitive in relative terms, only possesses motif B (NGQA-TS). Interestingly, hPrx3, the isoform most resistant to hyperoxidation among the human Prxs, has a combination of resistance and sensitivity motifs: DNHS-TS like the resistant AhpC and the YF and GGLG sensitivity-conferring motifs that AhpC lacks.
Table 1.
Residues present in the motif A and motif B regions of the peroxiredoxins analyzed in this study
| Protein | Motif A regiona D(N/G)H(S/G) | Motif B region T(S/T) |
|---|---|---|
| AhpC | DGHG | TT |
| hPrx1 | DNHS | KA |
| hPrx2 | NGQA | TS |
| hPrx3 | DNHS | TS |
| hPrx4 | DNQS | TS |
| P1.R1 | DGHS | KA |
| P1.R2 | DNQS | KA |
| P2.R3 | NGQS | KA |
| P2.AB+ | DNHS | TS |
| AhpC.AB− | NNQA | KA |
a The full description of the motifs is as follows. Motif A is DX8(N/G)X10HX27(S/G), and motif B is TX3(S/T). See the main text and Fig. 3A for details and rationales for the variants tested.
Alterations in motif A and motif B change susceptibility to hyperoxidation
To test the putative resistance motifs, we developed a library of constructs for Prx1, Prx2, and AhpC where the residues within the motifs were modified to study their contribution to the sensitivity to hyperoxidation. All proteins were successfully expressed, purified to homogeneity, and confirmed to have comparable CD spectra and thermal stability (Fig. S4 and Table S3), attributes that attest to the maintenance of proper folding. To assess whether the peroxidase activity and the rate of Cys-SPOH formation were affected, the standard horseradish peroxidase (HRP) competition assay was used. All mutants possessed Cys-SPOH formation rates (kSOH) directly comparable with their respective WT protein (∼106–107 m−1 s−1; Table S3). The Chyp1% values were also determined, and a variety of MS approaches were used to cross-validate the oxidation state of each variant.
In an effort to make hPrx1 more resistant, we made a chimera of Prx1, P1.R1. Two amino acids in region 1 were changed. By making the N70G substitution, motif A (DGHS-KA; bold indicates change to the motif) would be similar to motif A in AhpC (Table S2). The Q72E substitution was also made to install an acidic residue (Asp/Glu) found in AhpC and other Prxs (Fig. 3A and Table 1). The resistance of the P1.R1 variant increased dramatically to a level comparable with AhpC (Fig. 4A). Importantly, for enzymes that are highly resistant to hyperoxidation, the Chyp1% value can only be estimated to be greater than the highest concentration of peroxide tested (>5 mm H2O2) as concentrations of peroxide above this result in a spurious consumption of NADPH via TrxR-dependent oxidase activity (24). To confirm resistance to hyperoxidation for P1.R1, the WT and variant proteins were treated with H2O2 and analyzed by electrospray ionization (ESI)-TOF MS (Fig. 4B and Table S4). WT Prx1 was readily hyperoxidized to Cys-SPO2H and Cys-SPO3H. In contrast, the same treatment yielded little of the sulfinic acid moiety for the P1.R1 variant.
Figure 4.

Changes in motifs A and B alter resistance to hyperoxidation in vitro and in vivo. A, assessment of sensitivity to hyperoxidation for Prx1 variants. The Chyp1% value was obtained by generating the plot of the fraction of inactivated Prx molecules per catalytic cycle (finact) versus the H2O2 concentration. Changes in sequence for motif A and B residues (see Table 1) can either sensitize (P1.R2 variant, DNQS-KA; bold indicates change from WT) or prevent hyperoxidation (P1.R1 variant, DGHS-KA) relative to the WT protein (DNHS-KA). The presented data are the mean and S.D. (error bars) for the finact values determined on 3 or more separate days with fresh aliquots of reduced enzyme and each peroxide concentration tested in triplicate (see Fig. S1 and associated method description for details). B, cross-validation of Prx1 variants using ESI-TOF MS. Spectra for samples without and with peroxide treatment (1 mm H2O2 and 10 mm DTT for 3 h) are shown in black and red, respectively. The theoretical and observed mass values for the different oxidation states are presented in Table S4. C, Chyp1% value determination for Prx2 variants, WT (NGQA-TS), P2.R3 (NGQS-KA), and P2.AB+ (DNHS-TS). D, ESI-TOF MS analysis of the Prx2 variants with H2O2 treatment as in B. E, Chyp1% value determination for S. enterica Typhimurium AhpC variants, WT (DGHG-TT) and AhpC.AB− (NNQA-KA). F, relative abundance of the Cys-SP-SO2H/SO3H species determined by LC-MS/MS before and after treatment of AhpC.WT or AhpC.AB− with 3 mm H2O2. G, assessment of AhpC variant inactivation in vivo. The E. coli AhpC knockout strain was transformed with plasmids containing either AhpC.WT or AhpC.AB−. Cells were challenged with two concentrations of cumene hydroperoxide (CHP), rinsed with fresh medium, allowed to recover for 1 h, and plated to determine percent survival. The data are presented as mean ± S.D. (error bars). p values were derived from an unpaired Student's two-tailed t test (each measurement was performed in triplicate with four independent cultures). a.m.u., atomic mass units.
In an effort to make Prx2 like Prx1, we made construct P2.AB+ (DNHS-TS) where the Prx1 version of motif A was inserted into Prx2, which normally only has motif B. The Chyp1% value of the P2.AB+ variant increased to 27 μm, which is close to the value for WT Prx1 (Fig. 4C). ESI-TOF MS analysis of WT Prx2 and the P2.AB+ variant treated with H2O2 (Fig. 4D) showed that the proportion of Cys-SPO2H species decreased for the P2.AB+ variant. Although the conversion of Prx2 to Prx1 was incomplete, these results support that the substitution of motif A into Prx2 makes the protein more resistant to hyperoxidation.
Although Prx1 and Prx2 are already quite sensitive to hyperoxidation, we examined additional variants in an effort to decrease their resistance. We tested construct P1.R2 (DNQS-KA), which swapped the residues in region 2 of Prx1 to those present in Prx2. This meant that there was a Gln instead of a His in the third position of Motif A, causing the motif to be disrupted. These sequence changes resulted in a 25-fold decrease in the Chyp1% value to 2 μm for this Prx1 variant (Fig. 4A); this value is similar to WT Prx2. For Prx2, we made changes in region 3, creating the P2.R3 chimera (NGQS-KA). The loss of motif B resulted in this variant becoming 25-fold more sensitive than WT Prx2 and exhibiting a Chyp1% value of 0.2 μm (Fig. 4C). Interestingly, Prx2 has a Gly in the second position of the motif A region, but it lacks the other residues to complete the motif. Therefore, it appears that these other sequence differences are the reason Prx2 does not have the associated resistance even though the presence of a Gly in this position results in increased resistance for the Prx1 variant P1.R1.
Because AhpC is the archetypical, hyperoxidation-resistant Prx, we used this enzyme to determine whether the disruption of motifs A and B would result in an increased sensitivity to hyperoxidation. The six residue substitutions in the AhpC.AB− variant (NNQA-KA) resulted in increased sensitivity to hyperoxidation, decreasing the Chyp1% value from >5 mm to 6 μm H2O2 (Fig. 4E), a level directly comparable with the values for Prx1 and Prx2. This increase in sensitivity to hyperoxidation was confirmed by determining the relative abundance of the hyperoxidized species before and after treatment by LC-MS/MS (Fig. 4F and Fig. S5). To test whether the deletion of motifs A and B in AhpC would alter the ability of the enzyme to protect cells from an oxidative stress challenge in vivo, the AhpC deletion strain of E. coli was transformed with a vector encoding either the AB− or the WT variants of AhpC. The cells expressing the AhpC.AB− variant were significantly more sensitive to killing by cumene hydroperoxide (Fig. 4G). Altogether, the in vitro and in vivo data support the important contribution of resistance motifs A and B to the susceptibility of Prxs to inactivation by hyperoxidation.
Discussion
Although several peroxide scavengers exist in cells (e.g. catalase and GSH peroxidase), the unique catalytic cycle of 2-Cys Prxs enables their function as regulators of localized H2O2 levels or molecular chaperones or in the relay of H2O2-induced signal transduction through thiol-disulfide exchange reactions with partner proteins (6, 8–10, 19, 20). The intrinsic sensitivity to oxidation and the repair of hyperoxidized Prxs by sulfiredoxin provide the mechanisms to fine-tune signaling and stress responses (17, 21–24). For example, homeostatic receptor–mediated signaling requires low levels of H2O2 (2). Microorganisms, in contrast, must be able to resist much higher levels of environmental and host-derived reactive oxidants. Thus, the susceptibility to hyperoxidation needs to be stratified. An understanding of the molecular basis for this phenomenon is greatly needed to develop modulators of Prx activity in a variety of diseases.
In this study, we report the identification of two structural and sequence motifs driving the differential hyperoxidation sensitivity in human Prxs. These motifs were identified by comparing the amino acid sequences in the regions that changed the most in the crystal structures of human Prx1 and Prx2 in different oxidation states (Figs. 2 and 3). The alteration or installation of these motifs within Prx1, Prx2, and S. enterica Typhimurium AhpC resulted in dramatic changes in the resistance to hyperoxidation as validated by kinetic and MS-based methods. For example, Prx1 was made as resistant to hyperoxidation as AhpC (∼100-fold change) by modifying the amino acid sequence to mimic motif A in AhpC (Fig. 4). Removal of motifs A and B from AhpC made this variant ∼800-fold more sensitive to hyperoxidation. Both Prx1 and Prx2 could also be made ∼25-fold more sensitive to hyperoxidation by removing motif A or B. This lower level of response is consistent with these Prxs already being quite sensitive to hyperoxidation.
It is also important to acknowledge here that our observation that WT Prx2 is more sensitive to hyperoxidation than Prx1 is the opposite of what was reported by Lee et al. (32). This prior study used antibodies that recognize the Cys-SO2H and Cys-SO3H moieties of 2-Cys Prxs. Our MS analysis of Prx1 after the addition of H2O2 (Fig. 4B) shows increased formation of the sulfonic acid species in Prx1 when compared with Prx2. Thus, our data are consistent with published antibody-based data (which emphasize Cys-SO3H over Cys-SO2H) and highlight the reason why we applied the kinetic Chyp1% method for determining the sensitivity to hyperoxidation by H2O2.
Previous comparisons of eukaryotic and prokaryotic Prxs identified the GGLG and YF motifs as being conserved in sensitive Prxs (6). Hyperoxidation-resistant enzymes like AhpC lack these motifs. The packing of these motifs near the Cys-SP residue (Fig. 2) has been postulated to decrease the rate of intersubunit disulfide bond formation (Cys-SP–SR-Cys). This kinetic pause would lengthen the lifetime of the Cys-SPOH intermediate and increase the probability that a reaction with a second molecule of H2O2 could occur. Recent studies comparing Prx2 and Prx3 demonstrated that Prx3 has a 10-fold faster disulfide bond formation rate and decreased Cys-SPOH lifetime, consistent with the 25-fold higher Chyp1% value we determined (25, 26). Chimeras of Prx2 and Prx3 in regions near the GGLG and YF motifs in these studies were only able to switch the sensitivity to hyperoxidation by ∼50% (2-fold) in either direction. Other mutagenesis and deletion studies of the residues near the YF motif also alter hyperoxidation (40–42). For example, the addition or deletion of the C-terminal helix that contains the YF motif to Schistosoma mansoni Prx1 and Prx2, respectively, alters the sensitivity to hyperoxidation by ∼20-fold. However, because no comparison was made with mammalian Prxs, it is unclear how the magnitude of these changes in sensitivity relates to those in the present study. To our knowledge, the complete reversal in hyperoxidation sensitivity for the motif A and motif B variants of Prx1 and AhpC (Fig. 4) are the first for any Prx.
Motifs A and B are located (Fig. 3C) within or in close proximity to all of the key structural features of human 2-Cys Prxs, including the α helix that contains the Cys-SP residue, the GGLG and YF motifs, and the dimer-dimer interface. Because Prx1–4 all contain the YF and GGLG motifs (Fig. 5), it appears that motifs A and B play a role in modulating the H2O2 reactivity within this group of eukaryotic Prxs. Interestingly, the presence of a modified motif A in the Prx1 P1.R1 variant (Fig. 4, A and B) resulted in an increase in resistance to hyperoxidation similar to AhpC. Moreover, the loss in resistance for AhpC when motifs A and B were disrupted in vitro and in vivo (AhpC.AB−; Fig. 4, E–G) supports that these motifs play a predominant role in determining the sensitivity or resistance of this family of Prxs.
Figure 5.
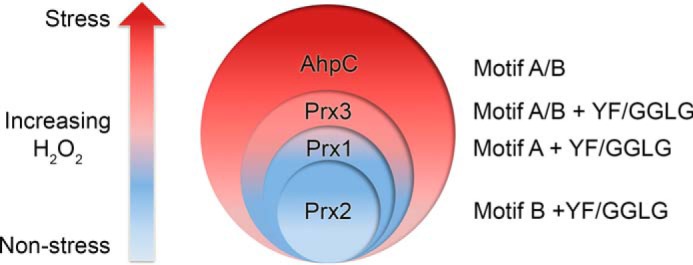
Model for the hierarchy of Prx resistance. Stratification of Prx susceptibility to hyperoxidation is shown. Low levels of H2O2 are associated with nonstress cell signaling, and high levels can lead to oxidative stress–induced signaling. The Prxs most sensitive to hyperoxidation in this study each contain only one resistance motif (Prx1, motif A; Prx2, motif B). Prx3 contains both motifs A and B and is more resistant than Prx1 and Prx2 but is not as resistant as AhpC. AhpC also contains both motifs A and B but lacks the YF and GGLG motifs. Based on the proposed grading of resistance, each eukaryotic Prx will be inactivated at a different threshold of H2O2 concentration, enabling a diversity of cellular responses.
We propose that the fine-tuning of the sensitivity to hyperoxidation for Prxs (Fig. 5) is one mechanism by which the cell differentiates between nonstress and stressful levels of H2O2, thereby leading to the activation of specific signaling pathways. For example, because Prx2 is the most sensitive to inactivation, it most likely influences or controls pathways at the lower end of the H2O2 concentration spectrum. Prx1 would function at the next level and so on for Prx3. Many observations support this notion and the nonredundancy of Prxs within cells: (i) phenotypic differences in Prx1–4 knockout mice, (ii) specificity of interactions with protein binding partners (e.g. Prx1 and Ste20-like kinase (MST1), Prx2 and STAT3, and Prx2 and protein-disulfide isomerase ERp46), and (iii) differential up-regulation in a variety of cancer cell types (8, 43–45).
Interestingly, a survey of the posttranslational modifications reported in the PhosphoSite database suggests that the phosphorylation of Ser112 of Prx2 in motif B and Ser106 of Prx1 in motif A and acetylation of Lys109 of Prx1 within the motif B region could represent an additional level of control for the sensitivity and inactivation of Prxs by hyperoxidation (46), although this has yet to be tested. It is tempting to speculate that targeting motif A or B pharmacologically could be used to tune Prx activity and to achieve a desired therapeutic effect. Perhaps increasing the sensitivity to hyperoxidation of Prxs in cancer cells will make current chemotherapeutics that generate reactive oxygen species more efficacious. Making parasitic and bacterial Prxs more sensitive to inactivation could also be an avenue for the development of new antibiotics. In conclusion, the results presented herein support that motifs A and B are predominant modulators for controlling the sensitivity to hyperoxidation for the typical 2-Cys Prxs and the stratification between and within resistant and sensitive Prx enzyme groups.
Experimental procedures
Protein preparation and purification
The human Prx1 and Prx2 constructs were expressed without a His tag using the pET17b vector (Novagen) and BL21(DE3)-gold E. coli cells (New England Biolabs) and purified using a combination of hydrophobic interaction, ion-exchange, and size-exclusion chromatography columns as described previously (22, 25). Human Prx3 was also produced in the mature form within E. coli but relied upon the use of the pTYB21 intein system (New England Biolabs). The genes for the chimeras of Prx1/Prx2 and the S. enterica Typhimurium AhpC.AB− variant were synthesized and codon-optimized by GenScript. The Prx1/2 chimeras were purified using the same protocols as the WT proteins. S. enterica Typhimurium AhpC was expressed and purified as reported (30). The AhpC.AB− variant was expressed from the pTrcHisA vector using the AhpC knockout E. coli strain JW5098-2 as host. Importantly, all Prx proteins were purified in the oxidized state (disulfide (SS) bond between Cys-SP and Cys-SR residues) to prevent further oxidation during storage at −80 °C. E. coli thioredoxin and thioredoxin reductase were expressed and purified as reported (47). The purity and oxidation state of all proteins were verified by SDS-PAGE and MS. All proteins were flash frozen with liquid nitrogen and stored at −80 °C until use. The extinction coefficients and molecular weight values for each protein were determined using ProtParam on the ExPASy Bioinformatics Resource Portal.
CD analysis
All proteins were prereduced with 10 mm DTT (Fisher Scientific) for 30 min, and Bio-Spin disposable chromatography columns containing Bio-Gel P-6 Gel resin (Bio-Rad) were used for desalting. Post-desalting, the proteins were diluted to final concentrations of ∼20 μm with the final concentration being determined by measuring absorbance at 280 nm. CD spectra were collected three times over wavelengths from 250 to 180 nm and averaged. Molar ellipticity and helical content were calculated from the averaged data. Melting points of proteins were determined by monitoring the molar ellipticity at 220 nm as the temperature was increased from 20 to 75–85 °C. Each CD experiment was repeated two to three times, with each repeat being on separate days, and averaged.
Determination of kSOH using HRP competition assay
To confirm that the residue substitutions installed into the panel of Prx variants did not affect their peroxidase activity, the second-order rate constant for Prx oxidation (kSOH) for each Prx variant was determined by monitoring the ability of the prereduced enzyme, as described above, to compete with HRP in the reduction of H2O2 (33). Briefly, in the wells of a 96-well UV half-area plate (Corning), six to eight different concentrations (2–16 μm) of purified Prx protein were combined with 15 μm HRP (Sigma), and 10 μl of 45 μm H2O2 was added to start the reaction (100-μl total volume). Within 60 s of peroxide addition, the plate was put in a plate reader, and absorbance was monitored at a fixed wavelength of 403 nm. Experiments were done in triplicate for each Prx concentration and repeated on 3 or more days with fresh aliquots of enzymes. HRP reacts with H2O2 to form compound I, which can be measured as a decrease in absorbance. When the Prx outcompetes HRP for the H2O2 (which must be at a lower concentration than HRP), there is less compound I formed. To transform the raw data into the kSOH, the decrease in HRP oxidation at each Prx concentration was plotted against Prx concentration. The slope of the line gives the second-order rate constant. The decrease in HRP oxidation can be calculated by the following equation and the rate of reaction of H2O2 with HRP (kHRP is 1.7 × 107 m−1 s−1).
| (Eq. 1) |
Crystallographic analysis of human Prx2 (SS)
Vapor diffusion crystallization conditions for Prx2-SS were initially identified from the JCSG+ Suite (Qiagen). Several rounds of optimization included protein concentration, drop size, temperature, buffer concentration and pH, and precipitant concentration. The condition that yielded the best diffracting crystals was: sitting drop size of 14 μl (7 μl of 10 mg/ml protein and 7 μl of well solution; 500-μl well), 6.1% PEG 3350, 0.1 m sodium acetate, pH 4.1, and 0.2 m zinc acetate at 25 °C. The crystals were transferred to synthetic mother liquor containing 10% PEG 3350, 0.2 m zinc acetate, 0.1 m sodium acetate, pH 4.1, and 5% glycerol. The amount of glycerol was slowly increased to 25% as a cryoprotectant by serially changing half of the solution with synthetic mother liquor containing 5% more glycerol over 30 min. Data were collected at the Brookhaven National Synchrotron Light Source (NSLS) on the X25 beamline (wavelength, 1.1 Å) and merged and scaled using HKL3000 (48). Data collection and refinement statistics are given in Table S1. The data were truncated at 2.15-Å resolution due to an abrupt loss in completeness beyond this resolution. Phasing was performed by molecular replacement using PHASER within the PHENIX suite (49). A dimer from the Prx2-SO2H decamer crystal structure was used as the search model (Protein Data Bank code 1QMV) (35). This search model was significantly modified, however, by the removal of the active-site helix region (residues 42–68), the GGLG motif region (residues 88–100), and the C terminus (residues 150–198) to prevent model bias during structure solution and rebuilding. A toroid of five dimers was found within the asymmetric unit. All omitted regions were iteratively rebuilt using Coot and refined with PHENIX (50). The final model contained 1683 of 1970 (85.4%) residues, four zinc ions, and 95 water molecules. The model was refined with individual B-values and validated using MolProbity (51). The following residues were not observed in the electron density: chain A, 2, 3, and 166–198; chain B, 2, 3, 165–170, and 180–198; chain C, 2, 146–150, and 165–198; chain D, 2, 3, 148–150, and 165–198; chain E, 2, 3, and 181–198; chain F, 2, 3, and 181–198; chain G, 2, 3, and 168–198; chain H, 2, 3, 149–150, and 173–198; chain I, 2, 3, 148–150, and 180–198; chain J, 2, 3, and 180–198. Two intermolecular SS bonds are possible for each dimer. The following numbers of disulfide bonds were observed between the following: chains A and B, one SS; chains E and F, two SS; chains G and H, one SS; and chains I and J, two SS. The following chains also showed clear electron density for the coiled-region containing Trp176: chains B, E, F, I, and J. Final Rwork/free values (20.7/25.7%) are consistent with the resolution of the structure and the large proportion of the structure with high mobility. The Ramachandran plot indicated that the residues were present in the following regions: 94.5% most favored, 4.5% additionally allowed, and 1% disallowed. Additional statistics supporting the quality of the model are: 1.4% rotamer outliers and clashscore of 6. Structural comparisons were done using the PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC. The model and diffraction data for the Prx2 disulfide structure were deposited to the Protein Data Bank under code 5IJT.
Amino acid sequence alignments and identification of putative motifs
Protein sequences of WT proteins were retrieved from the UniProt database. The alignments of the amino acids sequences were performed with Clustal Omega, visualized using ESPript3 (52, 53). WebLogo plots were generated using WebLogo3 using the alignments produced by Clustal Omega (39).
Hyperoxidation assay: Determination of Chyp1%
All proteins were prereduced with 10 mm DTT for 30 min on ice, and Bio-Spin disposable chromatography columns containing Bio-Gel P-6 Gel resin were used to remove the DTT immediately prior to kinetic experiments. The Chyp1% value, the concentration of peroxide at which 1% of the Prx molecules are inactivated per catalytic cycle, was determined by the following method described in detail by Nelson et al. (24, 33). NADPH oxidation was monitored at 340 nm where a decrease in absorbance occurs upon NADPH oxidation. Initially, the concentrations of the E. coli Trx, E. coli TrxR, Prxs tested, and H2O2 were varied to optimize for minimal background H2O2 oxidation and a reaction time of 40 min. A fresh dilution from a stock of H2O2 was made daily. This dilution was calibrated using an extinction coefficient of 43.6 m−1 cm−1 at 240 nm (33). The final concentrations within the reaction were: Prx, 0.25–2.5 μm; Trx, 5 or 10 μm; TrxR, 0.2 or 0.5 μm; NADPH (Roche Applied Science), 150 μm, H2O2 (Fisher Scientific), 50–5000 μm. H2O2 was added last to start the reaction. This setup was also analyzed without Prx at each peroxide concentration to monitor background NADPH oxidation. This mixture was incubated at 25 °C for the duration of the reaction.
The kinetic relationships governing the reactions and inactivation are represented by the following equations for the fraction inactivated, finact.
| (Eq. 2) |
The rates of the reaction are as follows: kSS, rate of disulfide bond formation; kSO2H, rate of inactivation and Cys-SPO2H formation. It is assumed that the LU and FF populations of the enzyme are in rapid equilibrium; thus, the numerator and denominator can be divided by [EFF], and [ELU]/[EFF] can be replaced with the equilibrium constant KLU.
| (Eq. 3) |
When the fraction inactivated is less than 10%, the first term in the denominator is small compared with the second term, so the equation can be simplified further.
| (Eq. 4) |
One can obtain the numerator and denominator terms experimentally over a range of peroxide concentrations using the following procedure. The initial 2 min of the reaction are used to determine the linear portion of the data, which estimates the kSSKLU. The exponential decay of the rate plot approximates the kSO2H and is determined by using a direct fit to an equation containing both linear and exponential decay terms as follows.
| (Eq. 5) |
Absorbance at time t is y; k is the exponential rate of decay in activity; and a, b, and c are all floating terms to allow for the best direct fit. Per Nelson et al. (24, 33), k can only accurately be obtained if there is “sufficient curvature in line so that a simple linear fit of the data gives R < 0.995.” All values reported in this study meet this requirement.
Ultimately, using the acquired experimental approximation for the rates represented in Equation 4, one can show the relationship as follows.
| (Eq. 6) |
To determine the Chyp1% value, the initial linear rate that approximates the rate of disulfide formation must be pulled out from the raw data for each peroxide concentration (see Fig. S1, A and B). For the most sensitive Prxs, higher H2O2 concentrations caused this rate to decrease due to some inactivation occurring even at the shortest time points. Thus, for calculations, the rate for the most reliable, lowest H2O2 concentration was used to divide the exponential rates at all H2O2 concentrations. Next, the rate of hyperoxidation must be determined by calculating the exponential decay rate of the raw data, also at each peroxide concentration (Fig. S2, A and C). For sensitive Prxs, this rate would increase with increased H2O2 concentration.
The finact value was then plotted against peroxide concentration (Fig. S2D). The reciprocal of the slope of the line produced gives the Chyp100% value so the Chyp1% is obtained by dividing by 100. For the robust Prxs tested, the y-intercept was at or near 0 in all cases. For sensitive Prxs, there were often nonzero intercepts (<10%), indicating that some of the Prx molecules were inactivated during the mixing time and the start of recording. Importantly, these intercepts were consistent with the sensitivities of the proteins, meaning the most sensitive had the highest intercept and so forth, further validating the Chyp1% calculations. The presented data are the mean and S.D. for the finact values determined on 3 or more separate days with fresh aliquots of reduced enzyme and each peroxide concentration tested in triplicate.
Chemicals and reagents for MS experiments
All solvents for MS analysis, 0.1% formic acid in water and 0.1% formic acid in acetonitrile, were of LC-MS grade purchased from J. T. Baker Chemicals (Avantor Performance Materials, Center Valley, PA). Sequencing-grade Asp-N protease was purchased from Promega (Madison, WI), and iodoacetamide was obtained from Sigma-Aldrich. Unless stated otherwise, all other chemicals were extra-pure grade or cell culture–tested.
ESI-TOF MS
Orthogonal validation of the hyperoxidation sensitivity for the Prx1 variants was done using ESI-TOF MS. Prereduced protein (50 μm) was treated with 1 mm H2O2 for 3 h at room temperature in the presence of dithiothreitol (10 mm) to enable catalytic cycling. The sample was then put over two Bio-Spin disposable chromatography columns containing Bio-Gel P-6 Gel resin equilibrated with 7 mm ammonium bicarbonate, pH 6.9. Subsequently, the sample was injected and analyzed. The ESI-TOF MS data were recorded in positive ion mode on an Agilent 6120 system. Deconvolution of the averaged MS spectra was done using Agilent MassHunter Workstation software version B.02.00. The analytical parameters were as follows: capillary voltage, 3500 V; nebulizer gas pressure, 30 p.s.i. gage; drying gas flow, 5 liters/min; fragmentor voltage, 200 V; skimmer voltage, 65 V; and gas temperature, 325 °C.
Nano-LC-MS/MS analysis
For sample preparation, aqueous AhpC protein was incubated with 30 mm iodoacetamide in the dark at room temperature for 30 min. The same volume of 2× digestion buffer consisting of 100 mm Tris-HCl with 1 mm zinc acetate, pH 8.0, was added and then incubated at 37 °C overnight with water-reconstituted Asp-N at a ratio of 1:50. Digested peptides were subjected to desalting using Pierce C18 Spin Columns (Thermo Scientific, Rockford, IL). The eluent was dried under vacuum and suspended in 5% (v/v) acetonitrile containing 1% (v/v) formic acid for LC-MS/MS analysis. The LC-MS/MS system consisted of a Q Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific) and a Dionex Ultimate-3000 nano-UPLC system (Thermo Scientific) using a Nanospray Flex Ion Source (Thermo Scientific). An Acclaim PepMap 100 (C18, 5 μm, 100 Å, 100 μm × 2 cm) trap column and an Acclaim PepMap RSLC (C18, 2 μm, 100 Å, 75 μm × 15 cm) analytical column were used for the stationary phase. Good chromatographic separation was observed with a 90-min linear gradient consisting of mobile phases A (5% acetonitrile with 0.1% formic acid) and B (80% acetonitrile with 0.1% formic acid) where the gradient was from 4% B at 0 min to 45% B at 75 min. MS spectra were acquired by data-dependent mode consisting of MS/MS scans of the 20 most intense ions from the full MS scan with dynamic exclusion option of 10 s. Spectra were searched using the Sequest HT algorithm within Proteome Discoverer version 2.1 (Thermo Scientific) in combination with the AhpC FASTA database of WT and mutant proteins. Search parameters were as follows: parent mass error tolerance of 10 ppm, fragment mass error tolerance of 0.02 Da (monoisotopic), and variable modifications on cysteine with carbamidomethyl of +57.021 Da, dioxidation of +31.990 Da, and trioxidation of +47.985 Da.
Cell viability assay using AhpC knockout E. coli cells
The JW5098-2 AhpC knockout strain of E. coli (kanamycin-resistant), originally from the Coli Genetic Stock Center at Yale University (CGSC strain number 8713), was transformed with the pTrcHisA vector (ampicillin-resistant) containing either WT or the AB− variant of S. enterica Typhimurium AhpC (98.4% sequence identity to E. coli AhpC). Importantly, the genes were cloned such that no affinity tag or additional residues were added to the proteins. Starter cultures (5 ml) were grown overnight in minimal medium (M9 salts, glycerol, MgSO4, CaCl2, casamino acids, and B vitamin complex mixture) containing kanamycin and chloramphenicol at 37 °C. Cultures (50 ml) were inoculated with 1 ml of overnight culture and grown to an A600 of 0.4 with shaking at 200 rpm. Protein expression was induced by the addition of 1 mm lactose for 2 h as induction by isopropyl 1-thio-β-d-galactopyranoside was found to be toxic to the cells like the study by Dvorak et al. (54). One milliliter of each replicate culture (n = 4) was treated with either 10 μl of DMSO as a control or cumene hydroperoxide in DMSO (final concentration, 2 and 4 mm) for 10 min at room temperature. The cells were centrifuged (5000 rpm for 3 min) and washed with minimal medium. The cells were then resuspended in 1 ml of minimal medium, serially diluted (10−3, 10−4, 10−5, and 10−6), plated in triplicate, and grown overnight at 37 °C. Cell counts were recorded for each dilution; only the dilutions that gave cell counts between 10 and 100 colonies per replicate were used for data processing.
Author contributions
J. A. B., J. A. R., L. B. P., C. M. F., and W. T. L. conceptualization; J. A. B., A. C. H., J. L., J. A. R., A. H. G., J. E. C., L. B. P., C. M. F., and W. T. L. data curation; J. A. B., A. C. H., J. L., J. A. R., A. H. G., J. E. C., D. P., L. B. P., C. M. F., and W. T. L. formal analysis; J. A. B., K. J. N., A. C. H., J. L., J. A. R., A. H. G., J. E. C., D. P., L. B. P., C. M. F., and W. T. L. validation; J. A. B., K. J. N., A. C. H., J. L., J. A. R., A. H. G., J. E. C., D. P., L. B. P., C. M. F., and W. T. L. investigation; J. A. B., A. C. H., J. L., J. A. R., A. H. G., J. E. C., L. B. P., C. M. F., and W. T. L. visualization; J. A. B., K. J. N., A. C. H., J. L., J. A. R., A. H. G., J. E. C., D. P., L. B. P., C. M. F., and W. T. L. methodology; J. A. B., K. J. N., A. C. H., J. L., J. A. R., A. H. G., J. E. C., C. M. F., and W. T. L. writing-original draft; J. A. B., K. J. N., A. C. H., J. L., J. A. R., A. H. G., J. E. C., D. P., L. B. P., C. M. F., and W. T. L. writing-review and editing; K. J. N., D. P., L. B. P., C. M. F., and W. T. L. supervision; L. B. P., C. M. F., and W. T. L. funding acquisition; C. M. F. and W. T. L. project administration.
Supplementary Material
Acknowledgments
The Crystallography and Computational Biosciences Shared Resource and the Proteomics and Metabolomics Shared Resource are supported by the Wake Forest Baptist Comprehensive Cancer Center (National Institutes of Health Grant P30CA012197). We also acknowledge the Kimbrell funds, which supported the acquisition of MS instrumentation in the Furdui laboratory. Diffraction data presented in this publication were collected on beamline X25 at the National Synchrotron Light Source, which was supported by the United States Department of Energy and the National Institutes of Health. We thank Drs. P. Andrew Karplus and James West for helpful comments on the manuscript.
This work was supported by National Institutes of Health Grants GM072866 (to W. T. L.) and GM050389 and GM119227 (to L. B. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1–S4.
The atomic coordinates and structure factors (code 5IJT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Prx
- peroxiredoxin
- AhpC
- alkylhydroperoxide reductase C
- Cys-SPH
- peroxidatic cysteine
- Cys-SRH
- resolving cysteine
- Cys-SPOH
- Cys sulfenic acid
- Cys-SPO2H
- Cys sulfinic acid
- Cys-SPO3H
- Cys sulfonic acid
- FF
- fully folded
- HRP
- horseradish peroxidase
- LU
- locally unfolded
- SS
- disulfide
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- hPrx
- human Prx
- ESI
- electrospray ionization.
References
- 1. Winterbourn C. C. (2013) The biological chemistry of hydrogen peroxide. Methods Enzymol. 528, 3–25 10.1016/B978-0-12-405881-1.00001-X [DOI] [PubMed] [Google Scholar]
- 2. Choi M. H., Lee I. K., Kim G. W., Kim B. U., Han Y. H., Yu D. Y., Park H. S., Kim K. Y., Lee J. S., Choi C., Bae Y. S., Lee B. I., Rhee S. G., and Kang S. W. (2005) Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 435, 347–353 10.1038/nature03587 [DOI] [PubMed] [Google Scholar]
- 3. Rhee S. G., and Kil I. S. (2016) Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: linking mitochondrial function to circadian rhythm. Free Radic. Biol. Med. 99, 120–127 10.1016/j.freeradbiomed.2016.07.029 [DOI] [PubMed] [Google Scholar]
- 4. Perkins A., Nelson K. J., Parsonage D., Poole L. B., and Karplus P. A. (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435–445 10.1016/j.tibs.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rabilloud T., Heller M., Gasnier F., Luche S., Rey C., Aebersold R., Benahmed M., Louisot P., and Lunardi J. (2002) Proteomics analysis of cellular response to oxidative stress: evidence for in vivo overoxidation of peroxiredoxins at their active site. J. Biol. Chem. 277, 19396–19401 10.1074/jbc.M106585200 [DOI] [PubMed] [Google Scholar]
- 6. Wood Z. A., Poole L. B., and Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 10.1126/science.1080405 [DOI] [PubMed] [Google Scholar]
- 7. Woo H. A., Chae H. Z., Hwang S. C., Yang K.-S., Kang S. W., Kim K., and Rhee S. G. (2003) Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653–656 10.1126/science.1080273 [DOI] [PubMed] [Google Scholar]
- 8. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 10.1038/nchembio.1695 [DOI] [PubMed] [Google Scholar]
- 9. Veal E. A., Findlay V. J., Day A. M., Bozonet S. M., Evans J. M., Quinn J., and Morgan B. A. (2004) A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol. Cell 15, 129–139 10.1016/j.molcel.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 10. Vivancos A. P., Castillo E. A., Biteau B., Nicot C., Ayté J., Toledano M. B., and Hidalgo E. (2005) A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 8875–8880 10.1073/pnas.0503251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park J.-G., Yoo J.-Y., Jeong S.-J., Choi J.-H., Lee M.-R., Lee M.-N., Lee J. H., Kim H. C., Jo H., Yu D.-Y., Kang S. W., Rhee S. G., Lee M.-H., and Oh G. T. (2011) Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E–deficient mice. Circulation Res. 109, 739–749 10.1161/CIRCRESAHA.111.245530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park M. H., Jo M., Kim Y. R., Lee C. K., and Hong J. T. (2016) Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 163, 1–23 10.1016/j.pharmthera.2016.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milev N. B., and Reddy A. B. (2015) Circadian redox oscillations and metabolism. Trends Endocrinol. Metab. 26, 430–437 10.1016/j.tem.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barranco-Medina S., Lázaro J.-J., and Dietz K.-J. (2009) The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 583, 1809–1816 10.1016/j.febslet.2009.05.029 [DOI] [PubMed] [Google Scholar]
- 15. Parsonage D., Youngblood D. S., Sarma G. N., Wood Z. A., Karplus P. A., and Poole L. B. (2005) Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 44, 10583–10592 10.1021/bi050448i [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall A., Nelson K., Poole L. B., and Karplus P. A. (2011) Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 15, 795–815 10.1089/ars.2010.3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lowther W. T., and Haynes A. C. (2011) Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid. Redox Signal. 15, 99–109 10.1089/ars.2010.3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jang H. H., Lee K. O., Chi Y. H., Jung B. G., Park S. K., Park J. H., Lee J. R., Lee S. S., Moon J. C., Yun J. W., Choi Y. O., Kim W. Y., Kang J. S., Cheong G. W., Yun D. J., et al. (2004) Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117, 625–635 10.1016/j.cell.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 19. Östman A, Frijhoff J., Sandin Å., and Böhmer F.-D. (2011) Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 150, 345–356 10.1093/jb/mvr104 [DOI] [PubMed] [Google Scholar]
- 20. Leichert L. I., and Dick T. P. (2015) Incidence and physiological relevance of protein thiol switches. Biol. Chem. 396, 389–399 10.1515/hsz-2014-0314 [DOI] [PubMed] [Google Scholar]
- 21. Biteau B., Labarre J., and Toledano M. B. (2003) ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984 10.1038/nature02075 [DOI] [PubMed] [Google Scholar]
- 22. Jönsson T. J., Johnson L. C., and Lowther W. T. (2008) Structure of the sulphiredoxin–peroxiredoxin complex reveals an essential repair embrace. Nature 451, 98–101 10.1038/nature06415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang T.-S., Jeong W., Woo H. A., Lee S. M., Park S., and Rhee S. G. (2004) Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 279, 50994–51001 10.1074/jbc.M409482200 [DOI] [PubMed] [Google Scholar]
- 24. Nelson K. J., Parsonage D., Karplus P. A., and Poole L. B. (2013) Evaluating peroxiredoxin sensitivity towards inactivation by peroxide substrates. Methods Enzymol. 527, 21–40 10.1016/B978-0-12-405882-8.00002-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haynes A. C., Qian J., Reisz J. A., Furdui C. M., and Lowther W. T. (2013) Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J. Biol. Chem. 288, 29714–29723 10.1074/jbc.M113.473470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peskin A. V., Dickerhof N., Poynton R. A., Paton L. N., Pace P. E., Hampton M. B., and Winterbourn C. C. (2013) Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 288, 14170–14177 10.1074/jbc.M113.460881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 10.1038/nchembio.85 [DOI] [PubMed] [Google Scholar]
- 28. Cox A. G., Pearson A. G., Pullar J. M., Jönsson T. J., Lowther W. T., Winterbourn C. C., and Hampton M. B. (2009) Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 421, 51–58 10.1042/BJ20090242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perkins A., Poole L. B., and Karplus P. A. (2014) Tuning of peroxiredoxin catalysis for various physiological roles. Biochemistry 53, 7693–7705 10.1021/bi5013222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poole L. B., and Ellis H. R. (1996) Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC Proteins. Biochemistry 35, 56–64 10.1021/bi951887s [DOI] [PubMed] [Google Scholar]
- 31. Cao Z., Bhella D., and Lindsay J. G. (2007) Reconstitution of the mitochondrial PrxIII antioxidant defence pathway: general properties and factors affecting PrxIII activity and oligomeric state. J. Mol. Biol. 372, 1022–1033 10.1016/j.jmb.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 32. Lee W., Choi K.-S., Riddell J., Ip C., Ghosh D., Park J.-H., and Park Y.-M. (2007) Human Peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of Cys83 underscored the structural and functional differences between Prx1 and Prx2. J. Biol. Chem. 282, 22011–22022 10.1074/jbc.M610330200 [DOI] [PubMed] [Google Scholar]
- 33. Nelson K. J., and Parsonage D. (2011) Measurement of peroxiredoxin activity. Curr. Protoc. Toxicol. Chapter 7, Unit 7.10 10.1002/0471140856.tx0710s49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perkins A., Parsonage D., Nelson K. J., Ogba O. M., Cheong P. H., Poole L. B., and Karplus P. A. (2016) Peroxiredoxin catalysis at atomic resolution. Structure 24, 1668–1678 10.1016/j.str.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schröder E., Littlechild J. A., Lebedev A. A., Errington N., Vagin A. A., and Isupov M. N. (2000) Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure 8, 605–615 10.1016/S0969-2126(00)00147-7 [DOI] [PubMed] [Google Scholar]
- 36. Cao Z., Tavender T. J., Roszak A. W., Cogdell R. J., and Bulleid N. J. (2011) Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a non-disulfide-bonded intermediate in the catalytic cycle. J. Biol. Chem. 286, 42257–42266 10.1074/jbc.M111.298810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wood Z. A., Poole L. B., Hantgan R. R., and Karplus P. A. (2002) Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 41, 5493–5504 10.1021/bi012173m [DOI] [PubMed] [Google Scholar]
- 38. Perkins A., Nelson K. J., Williams J. R., Parsonage D., Poole L. B., and Karplus P. A. (2013) The sensitive balance between the fully folded and locally unfolded conformations of a model peroxiredoxin. Biochemistry 52, 8708–8721 10.1021/bi4011573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crooks G. E., Hon G., Chandonia J.-M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jara M., Vivancos A. P., and Hidalgo E. (2008) C-terminal truncation of the peroxiredoxin Tpx1 decreases its sensitivity for hydrogen peroxide without compromising its role in signal transduction. Genes Cells 13, 171–179 10.1111/j.1365-2443.2007.01160.x [DOI] [PubMed] [Google Scholar]
- 41. Koo K. H., Lee S., Jeong S. Y., Kim E. T., Kim H. J., Kim K., Song K., and Chae H. Z. (2002) Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch. Biochem. Biophys. 397, 312–318 10.1006/abbi.2001.2700 [DOI] [PubMed] [Google Scholar]
- 42. Sayed A. A., and Williams D. L. (2004) Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J. Biol. Chem. 279, 26159–26166 10.1074/jbc.M401748200 [DOI] [PubMed] [Google Scholar]
- 43. Kim S.-U., Park Y.-H., Min J.-S., Sun H.-N., Han Y.-H., Hua J.-M., Lee T.-H., Lee S.-R., Chang K.-T., Kang S. W., Kim J.-M., Yu D.-Y., Lee S.-H., and Lee D.-S. (2013) Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-κB-mediated iNOS induction and microglial activation. J. Neuroimmunol. 259, 26–36 10.1016/j.jneuroim.2013.03.006 [DOI] [PubMed] [Google Scholar]
- 44. Morinaka A., Funato Y., Uesugi K., and Miki H. (2011) Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene 30, 4208–4218 10.1038/onc.2011.139 [DOI] [PubMed] [Google Scholar]
- 45. Pace P. E., Peskin A. V., Han M. H., Hampton M. B., and Winterbourn C. C. (2013) Hyperoxidized peroxiredoxin 2 interacts with the protein disulfide-isomerase ERp46. Biochem. J. 453, 475–485 10.1042/BJ20130030 [DOI] [PubMed] [Google Scholar]
- 46. Hornbeck P. V., Chabra I., Kornhauser J. M., Skrzypek E., and Zhang B. (2004) PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4, 1551–1561 10.1002/pmic.200300772 [DOI] [PubMed] [Google Scholar]
- 47. Lin Z., Johnson L. C., Weissbach H., Brot N., Lively M. O., and Lowther W. T. (2007) Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc. Natl. Acad. Sci. U.S.A. 104, 9597–9602 10.1073/pnas.0703774104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 49. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 51. Davis I. W., Murray L. W., Richardson J. S., and Richardson D. C. (2004) MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 10.1093/nar/gkh398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dvorak P., Chrast L., Nikel P. I., Fedr R., Soucek K., Sedlackova M., Chaloupkova R., de Lorenzo V., Prokop Z., and Damborsky J. (2015) Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21(DE3) carrying a synthetic metabolic pathway. Microb. Cell Fact. 14, 201 10.1186/s12934-015-0393-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.