

SUMMARY
Micropeptide regulator of β-oxidation (MOXI) is a conserved muscle-enriched protein encoded by an RNA transcript misannotated as non-coding. MOXI localizes to the inner mitochondrial membrane where it associates with the mitochondrial trifunctional protein, an enzyme complex that plays a critical role in fatty acid β-oxidation. Isolated heart and skeletal muscle mitochondria from MOXI knockout mice exhibit a diminished ability to metabolize fatty acids, while transgenic MOXI overexpression leads to enhanced β-oxidation. Additionally, hearts from MOXI knockout mice preferentially oxidize carbohydrates over fatty acids in an isolated perfused heart system compared to wild-type (WT) animals. MOXI knockout mice also exhibit a profound reduction in exercise capacity, highlighting the role of MOXI in metabolic control. The functional characterization of MOXI provides insight into the regulation of mitochondrial metabolism and energy homeostasis and underscores the regulatory potential of additional micropeptides that have yet to be identified.
In Brief
Micropeptide regulator of b-oxidation (MOXI) is encoded by a muscle-enriched RNA transcript misannotated as noncoding. MOXI localizes to the inner mitochondrial membrane where it interacts with the trifunctional protein to modulate fatty acid b-oxidation and exercise capacity.
Graphical Abstract
INTRODUCTION
Advances in computational biology, transcriptome analyses, and proteomic approaches have revealed that a much larger fraction of the genome is transcribed and translated than initially recognized (Guttman et al., 2009; Ingolia et al., 2014). Notably, many small open reading frames (ORFs) that code for functional protein products have been found hidden within transcripts misannotated as non-coding (Andrews and Rothnagel, 2014; Frith et al., 2006; Mackowiak et al., 2015; Makarewich and Olson, 2017). These small proteins, or micropeptides, play essential roles in diverse biological processes, including development (Chng et al., 2013; Galindo et al., 2007; Kondo et al., 2010), DNA repair (Slavoff et al., 2014), calcium homeostasis (Anderson et al., 2015; Anderson et al., 2016; Magny et al., 2013; Nelsonet al., 2016), myoblast fusion (Bi et al., 2017), and metabolism (Leeetal., 2015).
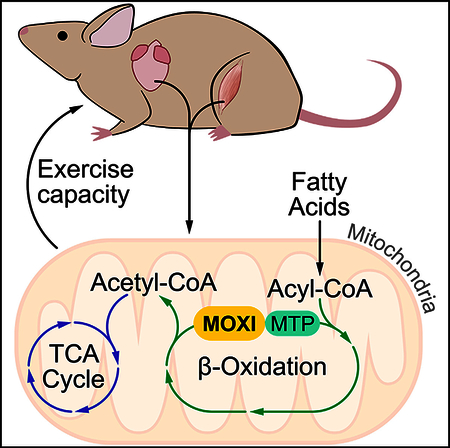
Here, we detail our discovery and analysis of a muscle-enriched protein we termed micropeptide regulator of p-oxidation (MOXI). MOXI is a nuclear encoded peptide that localizes to the inner mitochondrial membrane (IMM), where it specifically interacts with the mitochondrial trifunctional protein (MTP), an IMM localized enzymatic complex that catalyzes the last three steps of long-chain fatty acid (LCFA) oxidation. Mitochondrial fatty acid oxidation is critical for energy homeostasis in skeletal muscle and the heart, and while the major enzymes involved in fatty acid oxidation have been identified, it is still largely unknown how these enzymes are dynamically regulated on a molecular level (Bartlett and Eaton, 2004; Lopaschuk et al., 2010; Nakamura et al., 2014; Rasmussen and Wolfe, 1999; Taegtmeyer et al., 2016). In this regard, our findings indicate that MOXI may fulfill one such role as a regulator of the MTP that helps match the metabolic demand for enhanced fatty acid oxidation in striated muscle during stress conditions.
RESULTS
MOXI Is a Conserved Muscle-Enriched Micropeptide
We discovered MOXI through a screen for putative micropeptides encoded by muscle-enriched transcripts annotated as non-coding using PhyloCSF, a comparative genomics method that identifies the evolutionarily conserved protein coding potential of genomic sequences (Lin et al., 2011). This method revealed a previously unrecognized ORF of 56 amino acids within the muscle-enriched Moxi transcript, annotated as 1500011K16Rik and LINC00116 in the mouse and human genomes, respectively. The murine Moxi transcript is encoded in two exons on chromosome 2 and the entire 168 base pair ORF is contained within the first exon (Figures S1A and S1B). The ORF is highly conserved across species (Figure 1A) and scores positively with PhyloCSF (Figure S1A). The N terminus of the MOXI protein is hydrophobic and is predicted to encode a transmembrane domain, whereas the C-terminal region is highly enriched in basic residues (Figure 1A).
Figure 1. MOXI Is an Evolutionarily Conserved Micropeptide That Is Enriched in Muscle and Localizes to the Mitochondria.
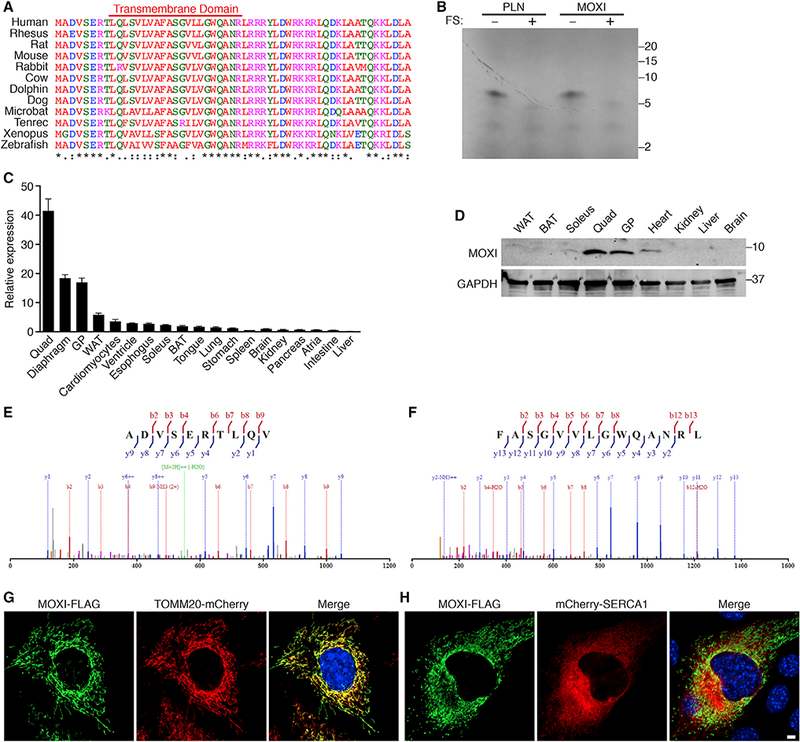
(A) Amino acid sequence alignment of vertebrate MOXI proteins. The colors of the amino acid residues indicate their properties (pink, positive charge; blue, negative charge; red, hydrophobic; green, hydrophilic). The symbols below the alignment represent the biochemical similarity of aligned amino acids with asterisks indicating identical conservation, colons representing high similarity, and periods showing somewhat similar conservation.
(B) In vitro transcription and translation ofthe full-length Moxi or Pln RNAs in the presence of radiolabeled 35S-methionine. Frameshift (FS) mutations introduced within the Moxi and Pln open reading frames abolished micropeptide expression from these RNAs.
(C) Detection of Moxi RNA by qRT-PCR in adult mouse tissues. Quad, quadriceps; GP, gastrocnemius/plantaris; WAT, white adipose tissue; BAT, brown adipose tissue. 18S rRNA was used for normalization and expression is shown relative to liver for n = 3 mice, mean ± SD.
(D) Western blot of adult mouse tissues with a MOXI-specific antibody reveals a band at the predicted size of ~6 kDa.
(E and F) MS/MS spectrum of two unique peptides corresponding to different regions of the MOXI protein identified by mass spectrometry on adult mouse quadriceps muscle.
(G and H) Immunofluorescence of MOXI-FLAG (green) and markers of the mitochondria (TOMM20-mCherry, red) (G) or the sarco/endoplasmic reticulum (mCherry-SERCA1, red) (H) in transfected C2C12 myoblasts. Scale bar, 5 μm.
To determine if the Moxi ORF is translated to a stable micropeptide, we performed in vitro transcription and translation assays using the full-length Moxi RNA transcript in the presence of 35S-methionine (Figure 1B). A single ~6 kilodalton (kDa) micropeptide was produced from the Moxi transcript, whereas a frameshift mutation that disrupted the ORF abolished any detectable expression (Figure 1B). This pattern was identical to that of phospholamban (PLN), a similarly sized 52 amino acid protein. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis revealed that Moxi RNA is highly abundant and enriched in striated muscle (Figure 1C). To further confirm that the Moxi transcript encodes a protein and validate its tissue distribution profile, we raised a polyclonal rabbit antibody against MOXI (Figure S1C). Western blot analysis confirmed that MOXI is strongly expressed in both skeletal muscle and the heart (Figure 1D). MOXI was also identified in adult mouse quadriceps muscle by mass spectrometry using in-gel elastase digestion, which produced two distinct peptide fragments of 1.1 and 1.4 kDa that correspond to unique regions of the protein (Figures 1E and 1F).
MOXI Localizes to the Inner Mitochondrial Membrane
Expression of MOXI as a C-terminal FLAG-fusion protein in C2C12 myoblasts revealed a mitochondrial distribution pattern by immunohistochemistry (Figures 1G and 1H). Co-expression of MOXI-FLAG with an mCherry-tagged mitochondrial membrane marker, translocase of outer mitochondrial membrane 20 (TOMM20), confirmed its mitochondrial localization (Figure 1G). In contrast, co-expression with the endoplasmic reticulum membrane protein sarco/endoplasmic reticulum calcium ATPase (SERCA) revealed distinct and non-overlapping patterns (Figure 1H).
MOXI contains a conserved transmembrane domain comprised of a hydrophobic core that is flanked on both sides by charged amino acid residues (Figure 1A), consistent with sequence determinants of an IMM protein (Stuart and Neupert, 1996). To assess mitochondrial membrane localization, mitochondria were isolated from adult mouse quadriceps muscle and subjected to progressive proteolysis with serial dilutions of proteinase K (PK) (Figure 2A). The outer mitochondrial membrane (OMM) protein TOMM20 was sensitive to proteolysis at much lower PK concentrations than the IMM localized a-subunit of the MTP (HADHA). Similar to HADHA, MOXI was resistant to PK proteolysis at high concentrations of PK and exhibited a degradation pattern consistent with that of an IMM protein (Figure 2A). To further validate its localization to the IMM, mitochondria from quadriceps muscle were subjected to osmotic shock to disrupt the OMM and create mitoplast fractions, which were exposed to PK proteolysis in the presence or absence of detergent (Figure 2B). MOXI exhibited a PK proteolysis resistance pattern similar to that of the IMM marker HADHA, and distinct from the OMM protein TOMM20 (Figure 2B), thus substantiating the sub-mitochondrial localization of MOXI to the IMM.
Figure 2. MOXI Localizes to the Inner Mitochondrial Membrane Where It Interacts with the Mitochondrial Trifunctional Protein.
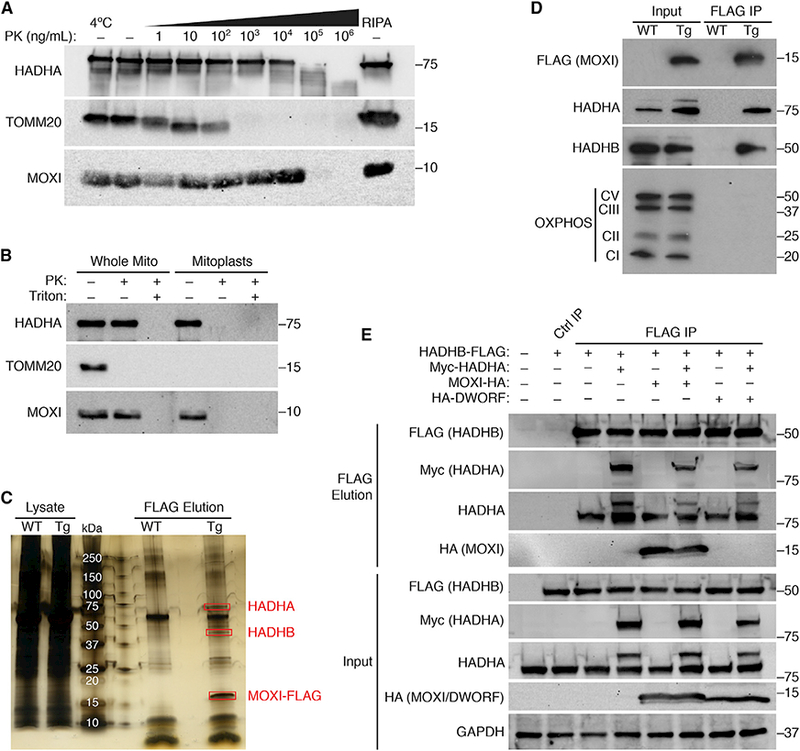
(A) Mitochondria were isolated from adult mouse quadriceps muscle and subjected to proteinase K(PK) digestion at the indicated concentrations at 37°C unless otherwise indicated. Far right, mitochondria lysed in RIPA buffer serves as a control for the total mitochondrial fraction. Western blots were performed for markers of the OMM (TOMM20), IMM (HADHA), or endogenous MOXI protein.
(B) Mouse quadriceps mitochondria were subjected to PK proteolysis to digest exposed proteins in the presence or absence of detergent (Triton). Osmotic shock was used to isolate mitoplasts, which were also subjected to PK digestion. Western blot analysis was performed using antibodies for MOXI or for the sub-mitochondrial markers HADHA and TOMM20.
(C) Silver stain analysis of FLAG immunoprecipitations (IPs) performed in mouse quadriceps muscle homogenates from wild-type (WT) or MCK-MOXI-FLAG transgenic (TG) mice. The 3 highly abundant and unique bands that are only present in the TG sample and indicated in red rectangles were analyzed by LC-MS/MS and identified as HADHA, HADHB, and MOXI-FLAG (see Tables S1–S3).
(D) Western blot analysisof FLAG IPs from quadriceps muscle homogenates of WT and MOXI-FLAGTG mice using antibodies for HADHA, HADHB, FLAG,and an OXPHOS cocktail that detects Complex I, II, III, and V of the OXPHOS complex.
(E) Co-immunoprecipitation (colP) experiments in transfected HEK293 cells using HADHB-FLAG, Myc-HADHA and MOXI-HA or HA-DWORF. Cell lysates were immunoprecipitated using FLAG magnetic beads or a control IgG resin, and bound proteins were eluted using FLAG peptide. Proteins that were pulled down in association with HADHB-FLAG were analyzed by western blot analysis using antibodies for FLAG, Myc, HADHA, or HA. Ctrl IP, control immunoprecipitation. The HADHA antibody recognizes both the endogenous protein (lower molecularweight band) and the transfected Myc-HADHA protein (higher molecularweight band).
MOXI Interacts with the Mitochondrial Trifunctional Protein
To identify protein interaction partners of MOXI, we generated a transgenic (TG) mouse line with striated muscle-specific overexpression of a MOXI-FLAG fusion protein under control of the muscle creatine kinase (MCK) promoter. Proper IMM localization of MOXI-FLAG was validated before immunoprecipitation (IP) experiments were performed (Figures S2A–S2D). Quadriceps muscles of wild-type (WT) and TG mice were homogenized, and lysates were subjected to FLAG IP. Analysis of FLAG-eluted proteins by silver stain revealed three highly abundant proteins that were only present in TG samples (Figure 2C). These unique protein bands were excised and subjected to LC-MS/MS, and peptide mass fingerprinting revealed their identity as MOXI-FLAG and the two subunits of the MTP, the α- (HADHA) and β-subunits (HADHB) (Figure 2C and Tables S1–S3). These results were validated in quadriceps tissue from WT and MOXI-FLAG TG mice by FLAG IP and western blot analysis for HADHA and HADHB (Figure 2D). The interaction of MOXI with the MTP appeared to be specific, as components of the oxidative phosphorylation (OXPHOS) complex, which are highly abundant IMM resident proteins, were excluded from the FLAG elution (Figure 2D). These results were further confirmed in gastrocnemius/plantaris muscles and in the heart of MOXI-FLAG TG mice (Figures S2E and S2F). Additionally, the interaction of MOXI and MTP was confirmed in HEK293 cells transfected with epitope-tagged versions of MOXI (HA), HADHA (Myc), and HADHB (FLAG) (Figure 2E). Cell lysates were subjected to FLAG IP, and bound proteins were eluted using FLAG peptide to purify HADHB-FLAG and its interacting partners. Western blot analysis revealed a strong association of HADHB with both endogenous untagged HADHA and transfected Myc-HADHA (Figure 2E), indicating efficient pull-down of the MTP complex. MOXI-HA was also pulled down in strong association with the MTP, while HA-DWORF, a negative control micropeptide, was not observed in the bound fraction, indicating a specific interaction between MOXI and the MTP (Figure 2E). As the MTP is a critical IMM associated protein that is essential for mitochondrial oxidation of LCFAs, we hypothesized that MOXI plays a role in regulating fatty acid oxidation.
MOXI Knockout and Transgenic Mice Exhibit Mitochondrial Abnormalities
To assess the function of MOXI in vivo, we generated a loss-of-function mouse model by using the clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) system to disrupt the coding frame of Moxi. A founder with a 1-base pair deletion and 3-base pair insertion that disrupts the ORF, resulting in premature stop codon and an unstable protein, was chosen for further analysis (Figures S3A and S3B). Heterozygous Moxi knockout (KO) mice yielded homozygous mutant offspring at expected Mendelian ratios. Western blot analysis of quadriceps and heart muscle performed with our MOXI-specific antibody showed that the MOXI protein was eliminated in muscle tissues of homozygous mutant KO mice (Figures 3A and 3B). Levels of MOXI overexpression in MOXI TG mice were also assessed by both western blot analysis and qRT-PCR and indicated a high level of expression of the MOXI-FLAG fusion protein, as well as an unanticipated upregulation of the endogenous MOXI protein (Figures 3A-3B and S3C–S3D). It has been demonstrated that mutations in either HADHA or HADHB can result in instability of the MTP complex and reductions in subunit expression levels (Spiekerkoetter et al., 2004a; Ushikubo et al., 1996). Therefore, we assessed whether loss of MOXI protein or its overexpression affected MTP abundance and found no changes in protein or mRNA expression levels of the α- or β-subunits (Figures 3A-3B and S3C–S3D), indicating that MOXI is not required for MTP complex formation or stability.
Figure 3. Consequence of MOXI Loss- and Gain-of-Function.
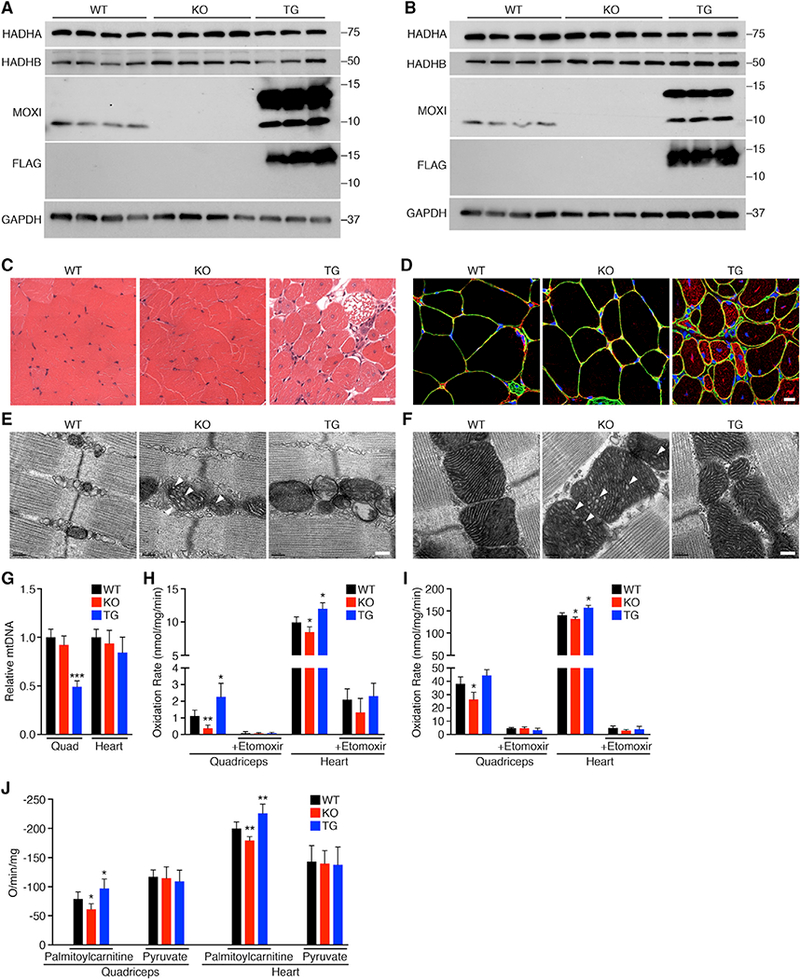
(A and B) Western blot analysis showing the expression level ofMOXI in quadriceps muscle (A) and heart (B) ofWT, KO, and TG mice. The expression and stability of the MTP α-subunit (HADHA) and β-subunit (HADHB) were also analyzed.
(C) Hematoxylin and eosin (H&E) staining on quadriceps muscle sections. Scale bar, 50 μm.
(D) Immunohistochemistryon quadriceps muscle sections. Representative merged images are shown of laminin (green), desmin (red), and DAPI (blue). Scale bar, 10 μm.
(E and F) Transmission electron micrographs of quadriceps muscle (E) and heart (F) from WT, KO, and TG mice. White arrows indicate the disrupted cristae structure observed in KO mice. Representative images from n = 3 mice are shown. Scale bar, 0.2 μm.
(G) Relative mitochondrial DNA (mtDNA) content expressed as a function of total genomic DNA in quadriceps (Quad) and hearts of WT, KO, and TG mice. Data were acquired by qPCR and are represented as mean ± SD, n = 5 mice per genotype, **p < 0.005.
(H and I) Fatty acid oxidation was measured by incubation of quadriceps or heart mitochondria from WT, KO, and TG mice with [14C]-palmitate. Oxidation rates were determined by analyzing captured CO2 (H) and acid soluble metabolites (ASM) (I). Where indicated, the CPT-1 inhibitor etomoxir (40 μM) was used. Data are represented as mean ± SD, n = 8 for WT and KO and n = 6 for TG. *p < 0.05, **p < 0.01.
(J) Oxygen (O) consumption rates were measured in isolated mitochondria from WT, KO, and TG mice using 1 mM malate and either palmitoylcarnitine (25 μM for heart, 50 μM for quad) or pyruvate (100 μM) as metabolic substrates. State 3 respiration was initiated with the addition of ADP at a final concentration of 0.25 mM. Data are presented as mean ± SD, n = 6 per genotype. *p < 0.05, **p < 0.01.
Compared to WT mice, both KO and TG animals had normal heart function and cardiac dimensions as assessed by echocardiography (Figures S3E-S3H) and all genotypes had comparable body composition measured by EchoMRI (Figures S3I-S3K). Histologically, quadriceps sections from KO mice appeared normal, while TG animals exhibited signs of myopathy with many fibers containing centralized nuclei (Figure 3C). Additionally, degenerating muscle fibers were present throughout skeletal muscle sections of TG mice, indicating severe pathology (Figure 3C). Immunohistochemistry performed on quadriceps sections showed an accumulation of desmin in skeletal muscle fibers of TG animals, demonstrating a loss of membrane integrity (Figure 3D). Succinate dehydrogenase (SDH) staining of quadriceps muscles from WT, KO, and TG mice revealed no overt differences in relative oxidative capacity among genotypes (Figure S4A); however, TG muscle fibers presented with a ragged appearance, suggesting mitochondrial abnormalities.
We assessed the ultrastructure of quadriceps muscle and heart from WT, KO, and TG mice by electron microscopy (EM) and observed unique mitochondrial abnormalities in KO and TG mice (Figures 3E-3F and S4B-S4C). KO mitochondria presented with an enlarged appearance with swollen and disrupted cristae structure in both quadriceps muscle (Figures 3E and S4B) and heart (Figures 3F and S4C). These features are consistent with previous reports from heterozygous HADHA+/− mice that present with similar mitochondrial abnormalities (Ibdah et al., 2005; Rector et al., 2008). Additionally, EM images from TG mice reveal a clear disorganization of the mitochondrial network with regions of mitochondrial dropout that are most clearly seen in quadriceps muscle (Figures 3E and S4B) and are also present in heart sections (Figures 3F and S4C). Mitochondrial content of quadriceps and heart from WT, KO, and TG mice were assayed by mitochondrial DNA (mtDNA) content and revealed no significant difference between WT and KO animals in either tissue; however, there was a significant reduction in mtDNA content in TG quadriceps muscle (Figure 3G) that correlated with the mitochondrial dropout seen by EM in these animals (Figures 3E and S4B).
MOXI Loss-or Gain-of-Function in Mice Alters Fatty Acid β-Oxidation
To directly measure the effect of MOXI gene deletion or overexpression on LCFA oxidation, ex vivo palmitate oxidation was measured in isolated skeletal muscle and heart mitochondria from WT, KO, and TG mice by assessing the rate of conversion of radiolabeled palmitate into either CO2 (Figure 3H) or acid-soluble metabolites (Figure 3I). Isolated quadriceps and heart mitochondria from KO mice exhibited a diminished ability to metabolize LCFAs compared to WT mice (Figures 3H and 3I), indicating that muscles from these animals have a lower fatty acid oxidation capacity, while TG mitochondria showed enhanced oxidation. There were no observable differences in oxidation rates in the presence of etomoxir, a potent carnitine palmitoyltransferase I (CPT-1) inhibitor, indicating similar levels of contributing non-mitochondrial oxidation between genotypes (Figures 3H and 3I).
To further assess mitochondrial function in our animals, we analyzed ADP-stimulated respiration in isolated mitochondria from quadriceps and heart tissue using either LCFAs (palmitoylcarnitine) or pyruvate as the oxidative substrate. A significant reduction in the rate of state 3 respiration was observed in mitochondria isolated from both quadriceps and heart tissues of KO mice when palmitoylcarnitine was provided as the substrate, while no change was seen when pyruvate oxidation was assessed (Figure 3J), indicating that respiratory function is selectively impaired for fatty acid oxidation. Conversely, palmitoylcarnitine oxidation was selectively enhanced in both skeletal muscle and heart mitochondria from TG mice, while pyruvate oxidation was unchanged (Figure 3J), once again highlighting the substrate-specific effect of MOXI overexpression on mitochondrial respiratory function.
MOXI Loss-of-Function Leads to Reduced LCFA b-Oxidation and an Intolerance to Exercise
To determine the effect of MOXI loss-of-function on substrate utilization in the heart, substrate oxidation was evaluated using 13C-NMR isotopomer analysis in isolated perfused WT and KO heart preparations. TG mice were omitted from these studies due to the severe mitochondrial phenotype exhibited by these animals. Figure S4D shows the metabolism of 13C-enriched substrates provided in the cardiac perfusate. Multiplet ratios in glutamate were analyzed in isolated hearts for the metabolism of [1,6-13C2]glucose, [3-13C]lactate, [3-13C]pyruvate, and [U-13C]LCFA supplied during perfusion (Figure S4D). In this study, the labeling pattern does not allow distinction between glucose, pyruvate, and lactate oxidation, and for this reason, they are collectively termed carbohydrates. The 13C multiplets from glutamate C-2 (Figure 4A), C-4 (Figure 4B), and C-3 (Figure 4C) were analyzed to evaluate substrate oxidation. KO hearts showed a decrease in C-3 and C-4 doublets with a corresponding increase in the C-2 doublet of glutamate (Figures 4A-C). 13C-NMR isotopomer analysis of heart extracts from KO mice showed a decrease in LCFA oxidation and a corresponding increase in carbohydrate oxidation compared to WT hearts (Figures 4D and 4E), demonstrating that KO hearts oxidized significantly more carbohydrates than fatty acids for energy production (Figure 4E). TCA cycle flux (Figure 4F), coronary flow rate (Figure S4E), and oxygen consumption rate (Figure S4F) measured under the metabolic steady state were not significantly different between groups. Consistent with our previous results showing that mitochondria from MOXI KO mice have an impaired fatty acid oxidation capacity, these data show that functioning hearts from KO mice preferentially oxidize carbohydrates over fatty acids as substrates.
Figure 4. 13C-NMR Isotopomer Analysis in WT and KO Hearts and Response of KO Mice to Exercise.
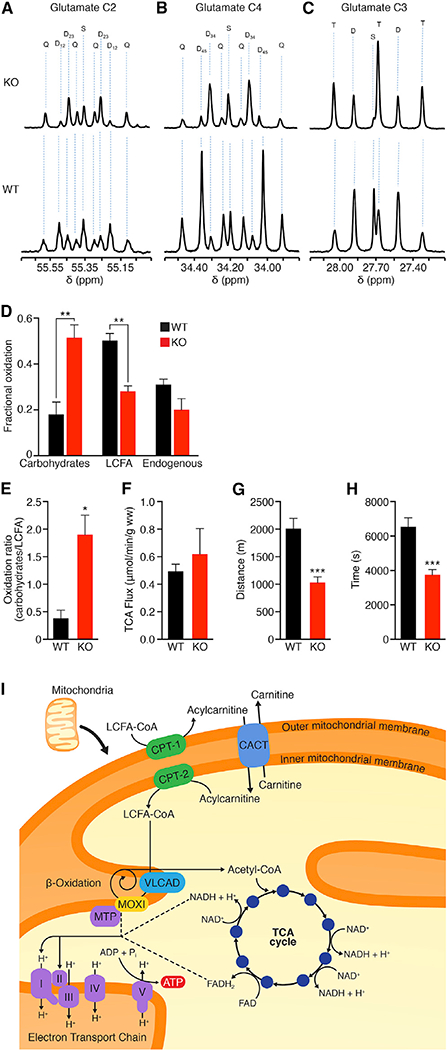
(A-F) Substrate oxidation was evaluated using 13C-NMR isotopomer analysis in isolated hearts from WT and KO mice perfused with 13C-labeled LCFA and carbohydrate substrates. Glutamate C-2 (55.35 ppm) (A), C-4 (34.20 ppm) (B), and C-3 (27.60 ppm) (C) spectra. The letters S, D, T, and Q refer to a singlet, doublet (with the relevant J-coupled spins), triplet (a degenerate doublet of doublets), or Q (quartet or doublet of doublets). Fractional oxidation (D), carbohydrate to long-chain fatty acid oxidation ratio (E), and TCA flux (F) were assessed for n = 4 mice per genotype. Data are presented as mean ± SEM with significance indicated as *p < 0.05, **p < 0.01.
(G and H) Exercise capacity was measured using forced treadmill running to exhaustion. MOXI KO mice ran ~49% less distance (G) and time (H) than WT littermate controls (n = 14). Data are represented as mean ± SD ***p < 0.005. (I) Working model for MOXI function. LCFAs are converted from acyl-CoA molecules into acylcarnitines by carnitine palmitoyltransferase 1 (CPT-1) and shuttled across the mitochondrial membrane by the carnitine-acylcarnitine translocase (CACT) where they are converted back to LCFA-CoA molecules by carnitine palmitoyltransferase 2 (CPT-2). LCFA-CoAs are metabolized by the inner membrane bound enzy mes very long chain acyl-CoA dehydrogenase (VLCAD) and the mitochondrial trifunctional protein (MTP)/MOXI complex to generate acetyl-CoA molecules that then enter the TCA cycle for production of ATP and reducing coenzymes such as NADH and FADH2. These reducing coenzymes can then be utilized by the electron transport chain to produce additional ATP.
β-oxidation is a key metabolic pathway for energy homeostasis during physiological stresses such as exercise when glucose supply becomes limited and several fatty acid oxidation disorders are associated with exercise intolerance (Rinaldo et al., 2002; Spiekerkoetter et al., 2004b; Spiekerkoetter et al., 2005; Spiekerkoetter and Wood, 2010). Therefore, we examined the response of MOXI KO mice to forced treadmill running to assess their ability to respond to physical exercise. KO mice showed a significant reduction in exercise capacity as compared to WT mice and exhibited signs of exhaustion after running only half the distance and time of WT mice (Figures 4G and 4H). WT and KO mice did not lose significant weight during this acute bout of exercise, and there were no differences in blood lactate levels between groups before or after exercise (Figures S4G and S4H). These results further underscore the important role of MOXI in fatty acid oxidation and its requirement for maintaining energy homeostasis during increased metabolic demand (Figure 4I).
DISCUSSION
Fatty acids are a major fuel source required to sustain contractile function in skeletal muscle and the heart, and while the major enzymes involved in β-oxidation have been identified, the precise manner in which they are regulated is still unknown (Bartlett and Eaton, 2004; Lopaschuk et al., 2010; Nakamura et al., 2014; Rasmussen and Wolfe, 1999; Taegtmeyer et al., 2016). Here, we identify MOXI as a muscle-enriched micropeptide that interacts with the MTP in the mitochondrial inner membrane and enhances fatty acid oxidation (Figure 4I), which is required to convert the energy stored in fats into ATP. The importance of MOXI as a regulator of fatty acid oxidation is demonstrated in our loss-of-function studies where hearts from MOXI KO mice switch from using fatty acids to carbohydrates as their preferred substrate (Figures 4A-4E). The discovery of MOXI as a micropeptide capable of enhancing fatty acid oxidation through its interaction with the MTP provides insights into the complex process of metabolic control and suggests that the enzyme complexes involved in ß-oxidation may be regulated by interactions with small regulatory proteins that have yet to be discovered.
In humans, fatty acid oxidation disorders present as a variety of clinical phenotypes and can result in severe disease during the early stages of life due to limited glycogen stores and a high metabolic rate that relies heavily on fatty acid oxidation for energy (Nsiah-Sefaa and McKenzie, 2016). In late-onset ß-oxidation disease, symptoms can include skeletal muscle myopathies and exercise intolerance, cardiovascular disease, and neuropathy (Kompare and Rizzo, 2008). Depending on the gene affected and the exact mutation observed, the severity of fatty acid oxidation disorders varies immensely. Similar to several other gene mutations linked to ß-oxidation disorders (Cox et al., 2001; Spiekerkoetter et al., 2004b; Spiekerkoetter et al., 2005; Spiekerkoetter and Wood, 2010), no overt phenotype is observed in a loss-of-function MOXI mouse model at rest; however, when challenged with physiological stress such as exercise, MOXI KO mice fail to maintain metabolic homeostasis. There is still a great deal to be learned about how MOXI exerts its dynamic control on the MTP complex, and elucidating the precise mechanism by which it induces its effect will be the focus of future work.
In summary, the discovery of MOXI highlights the role of a previously overlooked small protein in regulating fatty acid oxidation and metabolic responsiveness. Similar to other recently discovered micropeptides (Anderson et al., 2015; Anderson etal., 2016; Nelson et al., 2016), MOXI appears to exert its biological activity by engaging with and modulating a much larger protein complex. The small size of MOXI and its distinct amino acid sequence likely contribute to its capacity to fine-tune the activity of the MTP and regulate such a complex biological system. Furthermore, our findings underscorethe likelihood that additional transcripts currently classified as non-coding are misannotated and contain small ORFs that encode functional proteins. These micropeptides could serve as critical regulators of essential biological processes, and the prospect of identifying these proteins provides exciting opportunites for future discoveries.
EXPERIMENTAL PROCEDURES
Additional details and resources used in this work can be found in Supplemental Experimental Procedures.
Animals
All animal workdescribed in this manuscript has been approved and conducted under the oversight ofthe UT Southwestern Institutional Animal Care and Use Committee. All mouse lines were generated on a pure C57BL/6N background. All functional experimentswere performed using 12-to16-week-old malemice.
MOXI Antibody Production
Acustom polyclonal antibodywas derived againstthe predicted MOXI protein by New England Peptide. Rabbits were immunized with a synthetic peptide with the following sequence: RYLDWRKRRLQDKL. Sera were collected and affinity purified against the peptide immunogen.
Mitochondrial Isolations for Functional Analysis
Tissues were homogenized in ice cold isolation buffer(10mM MOPS,210mM mannitol, 70 mM sucrose, and 1 mM EDTA, pH 7.4). Mitochondriawere resuspended in isolation buffer for mitochondrial respiratory function analysis or STE buffer (250 mM sucrose, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4) for fatty acid oxidation assays. Additional information can be found in Supplemental Experimental Procedures.
Long-Chain Fatty Acid Oxidation Assays
Isolated mitochondria were incubated in oxidation reaction buffer (100 mM sucrose, 10 mM Tris-HCl, 5 mM KH2PO4, 0.2 mM EDTA, 80 mM KCl, 1 mM MgCl2, 2 mM L-carnitine, 0.1 mM malate, 0.05 mM coenzyme A, 2 mM ATP, 1 mM DTT, and 0.7% BSA/0.1 mM palmitate/0.4 μCi 14C-palmitate) for 1 hr then transferred to tubes containing 1 M perchloric acid. Fully oxidized CO2 was captured on Whatman filter paper discs treated with 1 M NaOH and acid-soluble metabolites (ASMs) were analyzed by scintillation counting. Additional details can be found in Supplemental Experimental Procedures.
Mitochondrial Respiratory Function Analysis
Isolated heart mitochondria were diluted to 0.25 mg/mL (heart) or 0.5 mg/mL (quadriceps) in respiratory buffer composed of 10 mM MOPS, 210 mM mannitol, 70 mM sucrose, and 5 mM K2HPO4 at pH 7.4 and incubated with the indicated substrates. For measurements with quadriceps mitochondria, 1.25 mM MgCl2 was also included in the respiratory buffer. State 3 respiration was initiated after 2 min with ADP at a final concentration of 0.25 mM. Rates of mitochondrial respiration were evaluated using a Neofox oxygen chamber. Detailed information can be found in Supplemental Experimental Procedures.
13C-NMR IsotopomerAnalysis
Hearts were cannulated via aorta and perfused for 30 min at 100 cm H2O pressure at 37°C with a modified Krebs-Henseleit (KH) buffer containing 8 mM [1,6-13C]glucose, 1.2 mM [3-13C]lactate, 0.12 mM [3-13C]pyruvate, 0.4 mM [U-13C]long-chain fatty acids (LCFA), 0.75% bovine serum albumin (BSA), and 2.5 mM CaCl2 equilibrated with 95:5 O2:CO2. After 30 min of perfusion, hearts were snap-frozen in liquid nitrogen. Proton-decoupled 13C-NMR spectra of heart extracts were acquired at 600 MHz spectrometer (Bruker Corporation, USA) equipped with 5 mm cryoprobe. Additional information can be found in Supplemental Experimental Procedures.
Treadmill Exercise
Mice were run on Exer-3/6 treadmill apparatus (Columbus Instruments) with mild electrical stimulus. The treadmill was set to ramp from 0 to 10 m/min over a period of 5 min and then stay at 10 m/min for an additional 5 min. The treadmill speed then incrementally increased (1 m/min every 5 min) to a maximum speed of20 m/min until exhaustion. Exhaustionwasdefined by failure to run for greater than 10 s.
Quantification and Statistical Analysis
Statistical comparisons between groups were evaluated by Student’s t test using Prism 6 (GraphPad). Data are presented as mean ± SD or SEM as indicated in each figure legend. Statistical significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001; n indicatesthe numberof biological replicates within each group.
Supplementary Material
Highlights.
MOXI is a muscle-enriched protein encoded by an RNA that is annotated as non-coding
MOXI interacts with the mitochondrial trifunctional protein and enhances β-oxidation
MOXI loss- or gain-of-function in mice alters β-oxidation and substrate utilization
MOXI knockout mice exhibit a reduced capacity for exercise
ACKNOWLEDGMENTS
Wethank J. Cabrera for graphics, H. Sadek fors cientific discussion and insight, W. Tan for echocardiography, J. Karch for providing protocols and advice, and the following Core Facilities at the University of Texas Southwestern Medical Center: The Molecular Pathology Core under the direction of J. Richardson; the Electron Microscopy Core under the direction of K. Luby-Phelps; and the Proteomics Core under the direction of H. Mirzaei. This work was supported by grants from the NIH (HL-130253, HL-138426, HD-087351, DK-099653, AR-067294, P41-EB015908, and R37-HL034557), American Heart Association (16POST31100009 to K.K.B.), Fondation Leducq Networks of Excellence, and the Robert A. Welch Foundation (grant 1–0025 to E.N.O.). C.A.M. was supported bya National Heart, Lung, and Blood Institute, NIH, Ruth L. Kirschstein National Research Service Award (NRSA) (F32HL129674).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, C.A.M., K.K.B., A.Z.M., C.R.M., L.I.S., and E.N.O.; Methodology, C.A.M., A.Z.M., K.K.B., S.B., A.M.S., J.R.M., G.S., and C.K.; Visualization, C.A.M., K.K.B., and A.Z.M.; Writing, C.A.M., R.B.D., and E.N.O., Supervision, C.R.M., L.I.S., R.B.D., and E.N.O.; Project Administration, C.A.M. and R.B.D.; Funding Acquisition, C.A.M., K.K.B., C.R.M., L.I.S., R.B.D., and E.N.O.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, four figures, and three tables, and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.058.
REFERENCES
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, and Olson EN (2015). A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 760, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, and Olson EN (2016). Widespread control ofcalcium signaling by a family ofSERCA-inhibiting micropeptides. Sci. Signal 9, ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SJ, and Rothnagel JA (2014). Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet 15, 193–204. [DOI] [PubMed] [Google Scholar]
- Bartlett K, and Eaton S (2004). Mitochondrial beta-oxidation. Eur. J. Biochem 271, 462–69. [DOI] [PubMed] [Google Scholar]
- Bi P, Ramirez-Martinez A, Li H, Cannavino J, McAnally JR, Shelton JM, Sanchez-Ortiz E, Bassel-Duby R, and Olson EN (2017). Control of muscle formation by the fusogenic micropeptide myomixer. Science 356, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng SC, Ho L, Tian J, and Reversade B (2013). ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev. Cell 27, 672–680. [DOI] [PubMed] [Google Scholar]
- Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR, and Wood PA (2001). Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet 10, 2069–2077. [DOI] [PubMed] [Google Scholar]
- Frith MC, Forrest AR, Nourbakhsh E, Pang KC, Kai C, Kawai J, Carninci P, Hayashizaki Y, Bailey TL, and Grimmond SM (2006). The abundance of short proteins in the mammalian proteome. PLoS Genet. 2, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, and Couso JP (2007). Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 5, e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, Matern D, Rinaldo P, and Cline JM (2005). Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 128, 1381–1390. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJ, Jackson SE, Wills MR, and Weissman JS (2014). Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8, 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompare M, and Rizzo WB (2008). Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol 15, 140–149. [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, and Kageyama Y (2010). Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329, 336–339. [DOI] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, and Cohen P (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, and Kellis M (2011). PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, and Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev 90, 207–258. [DOI] [PubMed] [Google Scholar]
- Mackowiak SD, Zauber H, Bielow C, Thiel D, Kutz K, Calviello L, Mastrobuoni G, Rajewsky N, Kempa S, Selbach M, and Obermayer B (2015). Extensive identification and analysis of conserved small ORFs in animals. Genome Biol. 16, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA, and Couso JP (2013). Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science 341,1116–1120. [DOI] [PubMed] [Google Scholar]
- Makarewich CA, and Olson EN (2017). Mining for Micropeptides. Trends Cell Biol. 27, 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura MT, Yudell BE, and Loor JJ (2014). Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res 53, 124–144. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, et al. (2016). A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsiah-Sefaa A, and McKenzie M (2016). Combined defects in oxidative phosphorylation and fatty acid ß-oxidation in mitochondrial disease. Biosci. Rep 36, e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen BB, and Wolfe RR (1999). Regulation offatty acid oxidation in skeletal muscle. Annu. Rev. Nutr 19, 463–484. [DOI] [PubMed] [Google Scholar]
- Rector RS, Payne RM, and Ibdah JA (2008). Mitochondrial trifunctional protein defects: clinical implications and therapeutic approaches. Adv. Drug Deliv. Rev 60,1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo P, Matern D, and Bennett MJ (2002). Fatty acid oxidation disorders. Annu. Rev. Physiol 64, 477–502. [DOI] [PubMed] [Google Scholar]
- Slavoff SA, Heo J, Budnik BA, Hanakahi LA, and Saghatelian A (2014). A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J. Biol. Chem 289, 10950–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiekerkoetter U, and Wood PA (2010). Mitochondrial fatty acid oxidation disorders: pathophysiological studies in mouse models. J. Inherit. Metab. Dis 33, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, and Strauss AW (2004a). General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alterTFPcomplex expression and subunit turnover. Pediatr. Res 55, 190–196. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Tokunaga C, Wendel U, Mayatepek E, Exil V, Duran M, Wijburg FA, Wanders RJ, and Strauss AW (2004b). Changes in blood carnitine and acylcarnitine profiles of very long-chain acyl-CoA dehydrogenase-deficient mice subjected to stress. Eur. J. Clin. Invest 34, 191–196. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Tokunaga C, Wendel U, Mayatepek E, Ijlst L, Vaz FM, van Vlies N, Overmars H, Duran M, Wijburg FA, et al. (2005). Tissue carnitine homeostasis in very-long-chain acyl-CoA dehydrogenase-deficient mice. Pediatr. Res 57, 760–764. [DOI] [PubMed] [Google Scholar]
- Stuart RA, and Neupert W (1996). Topogenesis ofinner membrane proteins of mitochondria. Trends Biochem. Sci 21, 261–267. [PubMed] [Google Scholar]
- Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, et al. ; American Heart Association Council on Basic Cardiovascular Sciences (2016). Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ. Res 118, 1659–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushikubo S, Aoyama T, Kamijo T, Wanders RJ, Rinaldo P, Vockley J, and Hashimoto T (1996). Molecular characterization of mitochondrial trifunctional proteindeficiency:formation oftheenzyme complexis important for stabilization of both alpha- and beta-subunits. Am. J. Hum. Genet 58, 979–988. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.