

Erythrokeratodermia (EK) is a rare skin disorder, likely genetic and usually present from infancy.1 There is patchy symmetrical involvement over the body surface, manifested in progressive figurate plaques of hyperkeratosis and more transient areas of erythema. There is significant overlap in the clinical and genetic features of the “variabilis” and “progressiva” forms of EK. Restricted cutaneous syndromes of EK have been described associated with mutations in the connexin (GJB3, GJB4, and GJA1) and loricrin (LOR) genes. The majority of patients with EK, however, have no pathogenic mutations in the GJB genes or LOR.
Three different mutations in the ELOVL4 gene have so far been described in patients presenting with a combination of EK and spinocerebellar ataxia (SCA34).2–4 We describe here a novel variant in ELOVL4 associated with this syndrome. This case draws attention to the importance of assessing subtle chronic neurologic dysfunction in patients presenting with EK and, conversely, being aware of the occurrence of characteristic cutaneous lesions in this newly described syndrome of spinocerebellar ataxia.
Case description and genetic results
A 60-year-old woman was referred for assessment of visual blurring and diplopia. Her cutaneous disorder manifested around age 4 years, with widespread variable itchy areas of erythema and progressive localized skin thickening. She had no success with topical Tazorac and compounded creams. Oral acitretin (25 mg) was helpful but discontinued because of alopecia. Although her visual symptoms had only gradually appeared at age 50 years, she reported noticing ataxia of gait as far back as teenage years. Her parents who were deceased at age 73 years and age 84 years had reported no symptoms of rash or ataxia. Her 5 siblings, similarly, were asymptomatic from the dermatologic and neurologic standpoint. Two children were biologically unrelated (adopted).
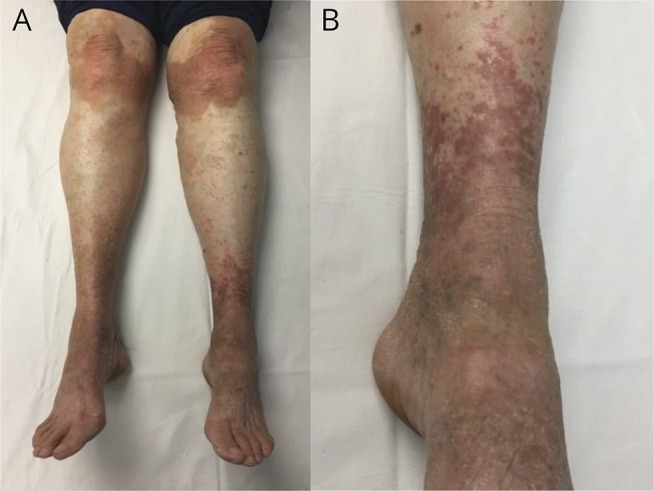
On examination, there were widespread demarcated brown erythematous keratotic plaques typical of EK over her wrists, hands, inner thighs, knees, and ankles (figure, A and B). Neuro-ophthalmic deficits included square wave jerks, saccadic pursuit, and alternating skew deviation with superimposed periodic alternating skew deviation in primary position (period 2.5 minutes). Myotatic reflexes were diffusely reduced (1+) and absent at the ankles. There was moderate rombergism and marked tandem gait ataxia, but no appendicular dysmetria. A skin biopsy showed irregular acanthosis, papillomatosis, and hyperkeratosis, in keeping with EK. MRI of the brain showed only subtle flattening of the ventral pons and mild global cerebellar atrophy. Nerve conduction studies and needle EMG studies were normal.
Figure. Erythrokeratodermia (lower limbs).
Photographs of symmetrical brownish-red hyperkeratosis, most prominent over the knees, lower anterior legs and dorsal foot region. There were similar symmetrical lesions over the inner thigh and more subtle involvement of the dorsal forearm and wrist region.
The clinical presentation suggested the possibility of SCA34. ELOVL4 gene sequencing (Prevention Genetics, Marshfield, WI) identified a variant (c.698C>T; p.Thr233Met), which has not been reported in the literature or public databases. The amino acid substitution programs CADD (27.9), PolyPhen-2 (1.0), and MutationTaster (1.0) predict the variant to be damaging, and the Sorting Intolerant From Tolerant (SIFT) program predicts it to be tolerated (table). It was not possible to test parents, but 2 clinically unaffected siblings were tested and did not carry this variant. The patient was started on baclofen, starting at 5 mg tid and titrating up to 15 mg tid over 1 month. She reported significant improvement in the perception of oscillopsia and the duration and frequency of bouts of diplopia. She also elected to try application of beeswax cream (reported to contain 30-32 carbon very long chain fatty acids) on her legs for 2 months, which was of no benefit.
Table.
Clinical and MRI features of patients with ELOVL4 mutations associated with clinical features of SCA34


Discussion
This case broadens the genetic mutation profile of EK by reporting a patient with a fourth variant in the ELOVL4 gene associated with SCA34 (table). The ELOVL4 gene catalyzes the rate-limiting reaction in elongation of fatty acids with a chain length greater than C26. It is ubiquitously expressed, but particularly enriched in the retina. It is critical in the development of the outer layer of the epidermis, the stratum corneum.5 Rare diseases associated with dysregulation of ELOVL4 in addition to SCA34 include 2 autosomal recessive conditions: hereditary macular degeneration (Stargardt disease) and a syndrome of ichthyosis, spastic quadriplegia, and mental retardation.6
The clinical presentation of our patient is quite similar to what was reported in a large French Canadian family with 32 affected members.2 In this large SCA34 family, cutaneous involvement began in infancy, with a preponderance of hyperkeratosis over erythema. Gait ataxia was noted to start usually around age 50 years. Our patient may be unique in reporting prominent oculomotor symptoms. A periodic alternating skew deviation was significantly improved when treated with baclofen.
The possibility of SCA34 should be considered in patients with EK, and clinicians should be aware that cerebellar manifestations are relatively late and variable, whereas cutaneous involvement has an onset early in childhood. SCA34 should thus be considered in the differential diagnosis of genetic neurocutaneous disorders. Oculomotor manifestations should be specifically assessed because they may be improved with treatment.
Author contributions
P.R. Bourque: analysis and interpretation and production and revision of the manuscript. J. Warman Chardon: analysis and interpretation and critical revision of the manuscript. D.A. Lelli: acquisition and interpretation of neuro-ophthalmology data and critical revision of the manuscript. L. LaBerge: acquisition and revision of dermatology data and critical revision of the manuscript. C. Kirshen: acquisition and revision of clinical dermatology data and critical revision of the manuscript. S.H. Bradshaw: acquisition and interpretation of dermatopathology data and critical revision of the manuscript. T. Hartley: preparation of the table and assessment of genetic classification. K.M. Boycott: study supervision, critical revision of the manuscript, and overview of genetic data acquisition and interpretation.
Study funding
No targeted funding reported.
Disclosure
All authors have no financial disclosures relevant to this research and publication. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Ishida-Yamamoto A. Erythrokeratodermia variabilis et progressiva. J Dermatol 2016;43:280–285. [DOI] [PubMed] [Google Scholar]
- 2.Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 2014;71:470–475. [DOI] [PubMed] [Google Scholar]
- 3.Bourassa C, Raskin S, Serafini S, Teive HA, Dion PA, Rouleau GA. A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol 2015;72:942–943. [DOI] [PubMed] [Google Scholar]
- 4.Ozaki K, Doi H, Misui J, et al. A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 2015;72:797–805. [DOI] [PubMed] [Google Scholar]
- 5.Cameron DJ, Tong Z, Yang Z, et al. Essential role of ELOVL4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci 2007;3:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldahmesh MA, Mohamed JY, Alkuraya HS, et al. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 2011;89:745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]